
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the role of acidity on the surface exchange reaction in mixed conductors: what is the effect of surface hydration?†
David M.
Schwenkel
 a,
Roger A.
De Souza
a and
George F.
Harrington
*ab
a,
Roger A.
De Souza
a and
George F.
Harrington
*ab
aInstitute of Physical Chemistry, RWTH Aachen University, Aachen, Germany. E-mail: gh778@bath.ac.uk
bDepartment of Chemistry, University of Bath, Bath, UK
First published on 20th February 2025
Abstract
An increasingly robust body of evidence attests that the kinetics of the oxygen exchange reaction at the surface of mixed ionic–electronic conducting oxides can be modified by infiltrating binary oxides. Furthermore, a clear relationship has been found between the reaction rate and the acidity of the surface binary oxide. Nevertheless, the underlying mechanism is still poorly understood. In this study we investigate the effect of acidic and basic infiltrated species (SiO2 and CaO) on SrTi0.65Fe0.35O3−δ (STF), a perovskite-structured, technologically relevant MIEC. From these experiments and an analysis of literature data, we demonstrate that a model based on electron-transfer as the rate-determining step and a modified surface electron concentration is quantitatively inconsistent with the data. Consequently, we propose instead that water species, present at trace levels in the conditions of the experiments, play a decisive role in the surface exchange kinetics and their modification through acidic or basic infiltrated species.
Introduction
Despite its technological importance, the oxygen surface exchange reaction remains poorly understood for the general class of materials termed mixed ionic–electronic conducting oxides (MIEC oxides). At first sight, this is surprising: the reaction involves only a simple, small molecule and a solid. A deeper consideration reveals that this is not at all surprising: the reaction has a complex and multistep nature, involving the adsorption and dissociation of molecular species, charge transfer to the adsorbates from the solid, and the incorporation of adsorbates into the oxide lattice.1 For MIEC oxides, the concentration of reaction intermediates and the rate-determining step (RDS) remain elusive or controversial.2 This lack of understanding hinders the design of mixed conducting oxides with superior surface exchange rates.In response to the complexity and multistep nature of the reaction, researchers have devoted efforts over the last few decades to developing empirical correlations between the surface exchange rate and material properties that are easier to measure or calculate, with the ultimate aim of tailoring surface exchange rates via compositional design. These material properties include the concentration of oxygen vacancies and electrons,2 the oxygen diffusion coefficient,3 the bulk oxygen p-band centre or bulk vacancy formation energy,4 and the gap between the Fermi level and the conduction band.5 The benefit of correlating these properties to the surface exchange rate is that they are based on bulk properties of the material and, therefore, are easy to measure reliably. Each of these correlations, however, suffer from the same limitation: they are based on the bulk properties of the MIEC rather than on its surface properties. This assumes that there is a simple, unique relationship between the bulk chemistry and the kinetics of the surface reactions, and therefore does not provide any mechanistic insight into the exchange reaction nor does it account for the variability in surface exchange rates observed for nominally the same compositions within the literature.4
Recently, the Smith acidity of surface oxides was identified by Nicollet et al. as a sensitive descriptor that shows a strong correlation with the surface exchange rate.6 This descriptor has the clear benefit of being based on surface chemistry rather than bulk properties. Specifically for Pr-substituted ceria (PCO), Nicollet et al.6 demonstrated that binary oxides infiltrated onto the surface modified the surface exchange rate, kchem, by up to 5 orders of magnitude. The change in kchem was correlated with the acidity of the infiltrated oxide: oxides that are more basic than PCO (Gd2O3, CaO, Li2O) led to an increase in kchem, while oxides that are more acidic (Al2O3, SiO2) led to a decrease. Furthermore, despite such a large change in kchem, no change was observed in the activation energy, EA, strongly implying no change in the mechanism of the surface exchange reaction.
Since Nicollet et al.6 proposed the link between surface acidity and the oxygen exchange rate, there have been numerous corroborating studies. Riedl et al., for example, experimentally reproduced the same effect, which was initially shown using porous ceramics, on freshly fabricated surfaces of PCO thin films using in situ pulsed laser deposition (iPLD).7 Several studies have reproduced similar effects in perovskite-structured La0.6Sr0.4CoO3−δ (LSC).8–11 Furthermore, in a survey of the literature, it was shown that the surface exchange rate correlates with the Smith acidity for the native surfaces of the perovskite- and double perovskite-structured cobaltates (LaCoO3, La0.6Sr0.4CoO3−δ, La0.6Sr0.4Co0.2Fe0.8O3−δ, Sm0.5Sr0.5CoO3−δ, PrBaCo2O5+δ and GdBaCo2O5+δ).12 In addition, the concept of acidity has also been used to explain a long-standing controversy regarding the effect of Sr-compounds on the surface exchange of LSC: at higher temperatures SrO, a basic oxide improves the surface exchange, while at low temperatures it forms acidic SrSO4 and SrCO3 phases which reduce the exchange rate.13 Finally, it has been demonstrated that successive infiltration of basic species is effective at recovering surfaces after degradation from poisons on high temperature devices such as silica and chromia.14–17
These reports highlight the significance of having a strong correlation between a readily available descriptor of the surface chemistry and the surface exchange rate. It can be used to explain inconsistencies in experimental datasets caused by the presence of common surface contaminants (SiO2, Cr2O3, CaO, Al2O3, CO2, SO4), and it constitutes a largely unexplored route for designing new MIEC materials for electrochemical devices, with superior surface exchange rates and chemical stability. While current experimental evidence for this correlation is robust, the underlying mechanism that links the acidity to the exchange rate is unclear.
The mechanism proposed by Nicollet et al. assumes electron transfer as the RDS and requires the degree of band bending at the surface of the MIEC to vary, owing to a pinning of the Fermi level to that of the binary oxide on the surface.6 For basic oxides, the bands of the MIEC oxide bend downwards leading to an accumulation of surface electrons, while for acidic oxides, the bands bend upwards leading to a depletion of electrons. This picture was found to be in qualitative agreement with the experimental data.
In this study, we examined the role of acidity on the surface exchange rate of SrTi0.65Fe0.35O3−δ (STF). STF was selected as a model system to study because (1) it is a technically relevant perovskite structured MIEC,18,19 (2) a point-defect model of the bulk is well established,20 and (3) electron transfer has been reported as the RDS from several independent studies.5,21–23 We measured the surface exchange rate, kchem, of porous STF using electrical conductivity relaxation (ECR), focussing on the effects of infiltrated basic (CaO) and acidic (SiO2) oxides. Through these new data, we explore the limitations of the band-bending model, and propose a yet unconsidered effect: that of H2O-based species on the surface.
Experimental
SrTi0.65Fe0.35O3−δ powder was prepared by a solid-state route. Commercially obtained precursors, SrCO3 (99.99% purity, Alfa Aesar), TiO2 (99.995% purity, Alfa Aesar) and Fe2O3 (99.999% purity, Acros Organics) were milled in a planetary ball mill (Fritsch Pulverisette 7) in a PTFE container with 5 mm ZrO2 balls in ethanol for 6 h at 850 rpm. The powder was then dried, uniaxially pressed into pellets, and sintered at 1300 °C for 6 h. The pellets were checked for phase purity by X-ray diffraction (XRD, Bruker D2 Phaser), reground in a ZrO2 pestle and mortar, and ball milled again under the same conditions. After drying, the powder was sieved to restrict the aggregate size to between 20 μm and 100 μm, uniaxially pressed, and sintered at 1100 °C for 6 h to obtain a porous ceramic body.XRD measurments were repeated and the resultant patterns refined using the Le Bail method (using the FullProf software) to confirm the phase purity and the cubic perovskite structure in the Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group (ESI, Fig. S1†). The density of the ceramics was calculated to be 70.6% from geometric measurements and the refined lattice parameter (3.902 Å) from XRD. The average and distribution of grain sizes was determined by scanning electron microscopy (SEM; JSM-7800F, JEOL, Japan) to be (0.53 ± 0.32) μm (ESI, Sections 2 and 3†). X-ray energy dispersive spectroscopy (XEDS; Oxford Instruments) confirmed the desired stoichiometry had been obtained (ESI, Table S1†).
m space group (ESI, Fig. S1†). The density of the ceramics was calculated to be 70.6% from geometric measurements and the refined lattice parameter (3.902 Å) from XRD. The average and distribution of grain sizes was determined by scanning electron microscopy (SEM; JSM-7800F, JEOL, Japan) to be (0.53 ± 0.32) μm (ESI, Sections 2 and 3†). X-ray energy dispersive spectroscopy (XEDS; Oxford Instruments) confirmed the desired stoichiometry had been obtained (ESI, Table S1†).
In preparation for electrical conductivity relaxation (ECR) measurments, the porous ceramic pellet was sectioned into bar shaped samples using a diamond wire saw (4240, Well Diamond Wire Saws, Germany) with dimensions of approximately 2 mm × 3 mm × 20 mm. The surfaces of the bars were lightly sanded with 800 grit SiC paper to remove blemishes and then cleaned for 10 min in an ultrasonic bath of ethanol, followed by acetone, and then ethanol again. Four gold wire contacts were wrapped around each bar sample, painted lightly with gold paste (Heraeus, Germany), and sintered at 800 °C for 2 h to ensure good connectivity. All samples were cut from the same ceramic pellet to minimise any variability in surface contamination and in microstructure.
CaO and SiO2 binary oxides were infiltrated into the porous bars using solutions of Ca(NO)2·4H2O (99%, Emsure) dissolved in ethanol and tetraethoxysilane (99.9% Alfar Aesar), respectively. The STF samples were infiltrated with 0.5 at% of the respective cations. This was done using ≈60 μL of a 0.2 mol L−1 solution for a sample mass of ≈0.4 g. Prior to measurement using ECR, the infiltrated samples were calcined within the tube furnace of the ECR set-up at 625 °C under pO2 of 0.1 atm for 1 h to form the infiltrated oxide.
ECR measurements were performed in a tube furnace within a quartz tube under a flowing gas atmosphere. The sample temperature was monitored by an S-type thermocouple located adjacent to the sample. Once the sample temperature was considered sufficiently stable, the pO2 was rapidly changed from 0.2 atm to 0.1 atm (or vice versa) using a bank of mass flow controllers and a four-way valve. High purity O2 and N2 gases (5.0 purity, Westfalen) were used, and the total flow rate kept to 50 sccm. For measurments under nominally dry conditions, gases were used directly from the cylinders. For measurement under wet conditions, the water vapour pressure in the atmospheres was controlled by bubbling the gases through two temperature-controlled reservoirs after mixing. Each mixture is oversaturated in the first reservoir and then set to the desired water vapour pressure in the second. The gas lines containing humidified gases were heated to prevent condensation. The resistance of the samples was monitored using a source meter (Keithley 2611B) operating in a four-wire galvanostatic mode while ensuring the voltage across the sample did not exceed 0.5 V.
Results and discussion
The surface exchange coefficient kchem of porous STF was measured as a function of temperature from electrical conductivity relaxation (ECR) experiments between oxygen partial pressures of 0.2 and 0.1 atm. Due to the high porosity (29.4%) and small average grain size (0.53 μm) of the samples [see the ESI, Section 3†], the conductivity relaxation can be considered entirely surface limited and fitted to directly extract kchem (ESI, Section 4†), as previously demonstrated for mixed ionic–electronic conducting oxides.6,24Small, systematic differences in kchem were observed between oxidising and reducing steps in pO2 (ESI, Fig. S6†), as expected (because the data refer to the final pO2), and here an average of these values is presented. All measurements were made for both decreasing and increasing temperature to ensure reversibility, and all samples were measured prior to any infiltration to ensure repeatability and establish a consistent baseline for the unmodified STF surface. kchem for the unmodified STF was found to agree well with literature values (ESI, Fig. S7†).
After assessing kchem of the unmodified STF surface, the samples were infiltrated with 0.5 at% of either a basic oxide, CaO, or an acidic oxide, SiO2. Fig. 1a compares kchem values obtained for the unmodified and infiltrated surfaces. CaO infiltration led to a 5.2 times increase in kchem, while SiO2 infiltration led to a decrease to 0.35 of that of the unmodified surface, together altering the surface exchange rate by over an order of magnitude. As shown on Fig. 1b, the activation energy, EA, remains essentially constant within experimental uncertainty, while the pre-exponential displays a clear trend with the Smith acidity of the surface species. These findings are consistent with those previously reported for PCO.6,15 Systematic changes in the conductivity of the samples with the acidity were also observed (ESI, Fig. S9†), also consistent with previous work.6
 | ||
| Fig. 1 Influence of acidic and basic species on the oxygen surface exchange coefficient, kchem, of STF measured by ECR. (a) kchem as a function of temperature. The error on kchem are smaller than the data points. (b) Pre-exponential (squares) and EA (circles) as a function of the Smith acidity of the infiltrated species or uninfiltrated surface. The semi-transparent points indicate the pre-exponential if EA is kept constant. | ||
We now compare our data with literature values25 for STF. To do so, we need to convert our chemical exchange coefficients to tracer exchange coefficients using the thermodynamic factor for oxygen, (ESI, Section 6†) which was calculated from the defect-chemical model.20 In Fig. 2a we find very good agreement between our data and literature data in terms of activation enthalpy, with the absolute magnitude of 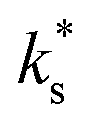 being a factor of 5–8 times higher (which may be due to an overestimated surface area in our case). Importantly, one finds that the data fall on the low-temperature branch of the surface exchange data. For many oxides,25–31 a change in the activation enthalpy of surface exchange, from a higher value at higher temperatures to a lower value at lower temperatures, is observed. In Fig. 2b and c we reproduce surface exchange data for the MIEC systems, LSCF and CGO, that show this behaviour. In this way, we show that our STF data are consistent with literature data for the same system, and generally consistent in terms of the overall surface exchange behaviour of MIEC oxides.
being a factor of 5–8 times higher (which may be due to an overestimated surface area in our case). Importantly, one finds that the data fall on the low-temperature branch of the surface exchange data. For many oxides,25–31 a change in the activation enthalpy of surface exchange, from a higher value at higher temperatures to a lower value at lower temperatures, is observed. In Fig. 2b and c we reproduce surface exchange data for the MIEC systems, LSCF and CGO, that show this behaviour. In this way, we show that our STF data are consistent with literature data for the same system, and generally consistent in terms of the overall surface exchange behaviour of MIEC oxides.
 | ||
| Fig. 2 Tracer surface exchange coefficients for (a) SrTi0.65Fe0.35O3−δ,25 (b) La0.6Sr0.4Fe0.8Co0.2O3−δ,26,27 and (c) Gd0.1Ce0.9O2−δ.28,29 The open symbols are taken from 18O tracer diffusion measurements in the literature, the solid lines are theoretical descriptions using eqn (1), and the dashed lines are linear fits of the low temperature regimes. In (a) the closed green squares are taken from ECR measurements in this study and converted to k* using a thermodynamic factor (see ESI, Section 6†) and the dashed line is a fit to the low temperature data by Argirusis et al.25 | ||
Limitations of the electron transfer model
The findings presented here, and in recently published works,6–8,10,12–15 represent an increasingly robust body of experimental evidence correlating the Smith acidity of surface species to the surface exchange rate. What is less certain, is the underlying mechanism that is responsible for this empirical relationship. To explore this further, we consider, as Nicollet et al. did,6 that the tracer surface exchange coefficient for semiconducting oxides, such as STF and PCO, can be described by:2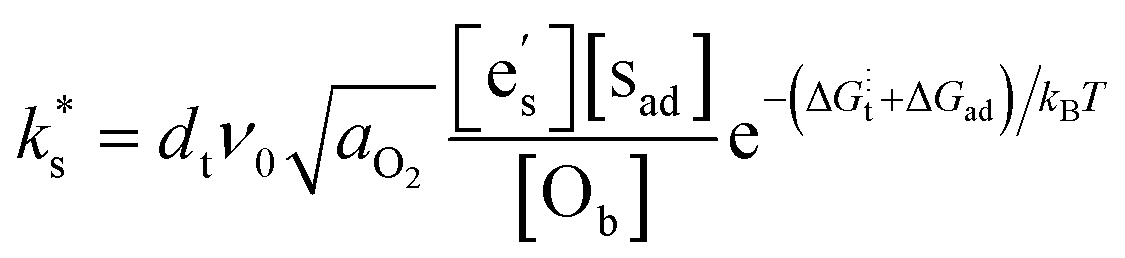 | (1) |
This expression assumes the rate-determining step (RDS) is electron transfer from the conduction band at the surface of the oxide to an adsorbed oxygen species, as expected to be the case for STF.5,21,22dt is the distance over which the electron transfer takes place, ν0 is the attempt frequency, and aO2 is the oxygen activity. 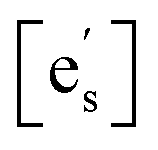 , [sad], and [Ob] are the site fractions of electrons at the surface, of surface adsorption sites, and of oxide ions in the bulk, respectively. ΔG⋮t is the Gibbs activation energy for electron transfer, which could either be the difference in the energy barrier between the conduction band and the adsorbed oxygen Fermi level, or an activation barrier that must be overcome. ΔGad is the Gibbs energy of oxygen adsorption from the gas onto an adsorption site.
, [sad], and [Ob] are the site fractions of electrons at the surface, of surface adsorption sites, and of oxide ions in the bulk, respectively. ΔG⋮t is the Gibbs activation energy for electron transfer, which could either be the difference in the energy barrier between the conduction band and the adsorbed oxygen Fermi level, or an activation barrier that must be overcome. ΔGad is the Gibbs energy of oxygen adsorption from the gas onto an adsorption site.
From experimental data (obtained in this study and in other studies6,7), the change in 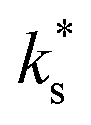 upon infiltration clearly originates from the pre-exponential terms, which has been shown to change in magnitude over a range of ∼101 for STF and ∼105 for PCO with the acidity of the surface species. As it seems unlikely that the distance, attempt frequency, oxygen activity, or bulk oxygen concentration could be varied by such an amount at the surface, attention naturally turns to the surface electron concentration and the concentration of (suitable) adsorption sites.
upon infiltration clearly originates from the pre-exponential terms, which has been shown to change in magnitude over a range of ∼101 for STF and ∼105 for PCO with the acidity of the surface species. As it seems unlikely that the distance, attempt frequency, oxygen activity, or bulk oxygen concentration could be varied by such an amount at the surface, attention naturally turns to the surface electron concentration and the concentration of (suitable) adsorption sites.
Drawing on the equivalence of Smith acidity and position of the Fermi level for oxides,32 Nicollet et al. postulated that pinning of the Fermi level and band bending at the surface cause an accumulation/depletion of electrons in the form of a space-charge zone.6 The site fraction of electrons at the surface, 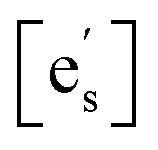 , can be related to the degree of band bending, i.e. the space-charge potential ϕ0, through a Maxwell–Boltzmann expression,
, can be related to the degree of band bending, i.e. the space-charge potential ϕ0, through a Maxwell–Boltzmann expression,
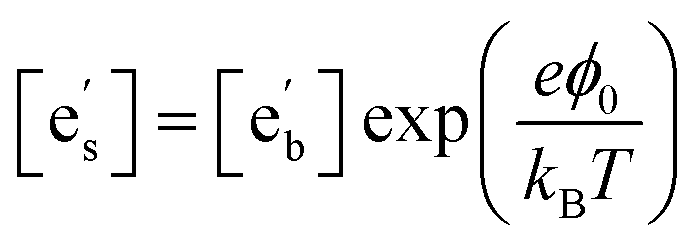 | (2) |
Despite these strengths, we argue that this model is not indicative of the underlying mechanism based on three observations. (1) The model predicts that EA should increase with acidic species and decrease with basic species (ESI, Section 6†), yet in this work and in the literature the activation energy remains constant within experimental error. (2) Riedl et al. reported XPS studies on the surface of PCO films with a basic oxide coating that did not show any increase in the concentration of Pr3+ species, which would be expected if electron accumulation due to band bending was occurring.7 (3) In terms of the activation energy, our data for STF (1.1 eV) agrees very well with literature data for STF (1.0 eV), as published by Argirusis et al.,25 for the low-temperature regime. It is, however, the high-temperature regime (>700 °C) that can be well described with eqn (1) (see Fig. 2a), i.e. with a model assuming electron transfer to be the RDS. 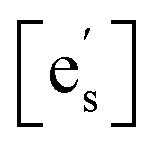 and [Ob] are taken from the defect model,20,33 and the
and [Ob] are taken from the defect model,20,33 and the  term, expanded to
term, expanded to 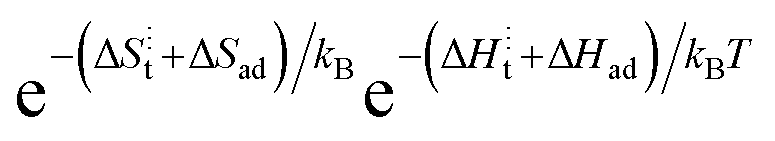 , can be well described with the standard ΔS⋮t + ΔSad = −4kB and a fitted ΔH⋮t + ΔHad of 0.9 eV, which is reasonable considering the value for weakly acceptor doped STO (1.15 eV).2 Hence, our data evidently does not refer to the regime in which electron transfer is supposedly the RDS and to which band bending can affect the availability of electrons. As we argue in the next section, there is evidence to suggest that the dominant mechanism responsible for surface exchange in the ‘low’ temperature section involves water species from the gas atmosphere.
, can be well described with the standard ΔS⋮t + ΔSad = −4kB and a fitted ΔH⋮t + ΔHad of 0.9 eV, which is reasonable considering the value for weakly acceptor doped STO (1.15 eV).2 Hence, our data evidently does not refer to the regime in which electron transfer is supposedly the RDS and to which band bending can affect the availability of electrons. As we argue in the next section, there is evidence to suggest that the dominant mechanism responsible for surface exchange in the ‘low’ temperature section involves water species from the gas atmosphere.
Case for water species
Eqn (1) has been shown to describe quantitatively as a function of T and pO2 for diverse MIEC systems, including acceptor-doped SrTiO3, Gd0.1Ce0.9O1.95, and La0.6Sr0.4Fe0.8Co0.2O3−δ.2 In each case, it was shown that the literature data could only be described by eqn (1) above a certain temperature, Ttrsn, below which the activation energy decreases and
quantitatively as a function of T and pO2 for diverse MIEC systems, including acceptor-doped SrTiO3, Gd0.1Ce0.9O1.95, and La0.6Sr0.4Fe0.8Co0.2O3−δ.2 In each case, it was shown that the literature data could only be described by eqn (1) above a certain temperature, Ttrsn, below which the activation energy decreases and 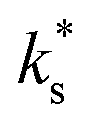 is observed to be larger than that predicted by eqn (1), as shown for La0.6Sr0.4Fe0.8Co0.2O3−δ and Gd0.1Ce0.9O1.95 in Fig. 2b and c, respectively. This trend is also reproduced in the STF system, as shown in Fig. 2a. Such a change in the activation energy is indictive of a change in the RDS within a mechanism or a change to a different dominant mechanism.
is observed to be larger than that predicted by eqn (1), as shown for La0.6Sr0.4Fe0.8Co0.2O3−δ and Gd0.1Ce0.9O1.95 in Fig. 2b and c, respectively. This trend is also reproduced in the STF system, as shown in Fig. 2a. Such a change in the activation energy is indictive of a change in the RDS within a mechanism or a change to a different dominant mechanism.
It has been argued previously that the change in activation energy is caused by the presence of adsorbed water, possibly in the form of surface hydroxyl (OH) species, due to trace amounts of water vapour in gases used in tracer experiments.2,34 Support for this hypothesis comes from multiple studies where oxygen exchange has observed to be faster in the presence of H2O.35–37 This argument has been strengthened by tracer diffusion data for weakly acceptor-doped STO single crystals exchanged under either nominally dry or wet atmospheres. Kler et al. observed higher exchange rates and lower activation energies for wet atmospheres, which converged with the data under nominally dry atmospheres at higher temperatures.38
We propose, based on these arguments and on the limitations of the electron-transfer model, that trace amounts of water vapour in the gas atmosphere during our experiments, and potentially previous studies on PCO,6,7 play a decisive role on the surface exchange kinetics. It may be expected that removing the trace amounts of water from the gas atmosphere would result in a lower 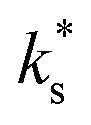 and higher activation energy, which would be consistent with the data at high temperature and indicative of electron transfer being the RDS. It is important to note, however, that a transition between high and low temperature regions has been observed even when the water partial pressure in the gas atmosphere is very low. Kler et al. performed isotope exchange experiments on (110) and (111) oriented STO single crystals, where the H2O partial pressures (pH2O) was kept below 1 × 10−8 bar and observed a transition between high and low temperature regions with Ttrsn = 737 °C.30 It would be, therefore, extremely challenging to dry gases beyond this level for ECR experiments.
and higher activation energy, which would be consistent with the data at high temperature and indicative of electron transfer being the RDS. It is important to note, however, that a transition between high and low temperature regions has been observed even when the water partial pressure in the gas atmosphere is very low. Kler et al. performed isotope exchange experiments on (110) and (111) oriented STO single crystals, where the H2O partial pressures (pH2O) was kept below 1 × 10−8 bar and observed a transition between high and low temperature regions with Ttrsn = 737 °C.30 It would be, therefore, extremely challenging to dry gases beyond this level for ECR experiments.
From Fig. 2, one might expect that any measurments of the surface exchange below 700–500 °C to fall within this low temperature regime, unless the atmosphere is kept exceptionally dry. To investigate this hypothesis further, we performed ECR measurments on STF under varying pH2O. Due to the challenge of experimentally drying gases to below ppb levels, we instead opted to increase the pH2O above our nominally ‘dry’ conditions. The clearest indication that water in the gas atmosphere alters the exchange kinetics would be a shift in Ttrsn to lower temperatures at higher pH2O. Unfortunately, the range of temperatures that we can perform ECR measurments is limited because the relaxation becomes too fast at high temperatures to accurately monitor with our setup.
Fig. 3a shows kchem of non-infiltrated STF measured under three conditions: ‘dry’ (pH2O unknown), wet#1 (pH2O = 10 mbar), and wet#2 (pH2O = 30 mbar). Only minimal changes were observed for kchem comparing between all three conditions. These deviations between nominally ‘dry’ and the two wet conditions are larger with decreasing temperatures, resulting in a small, albeit repeatable, change in the activation energy from (1.25 ± 0.02) eV to (1.45 ± 0.06) eV. This may be due to hydration of the bulk, resulting in a change in the sub-surface oxygen vacancy and electron concentration, rather than just hydroxylation of the surface.
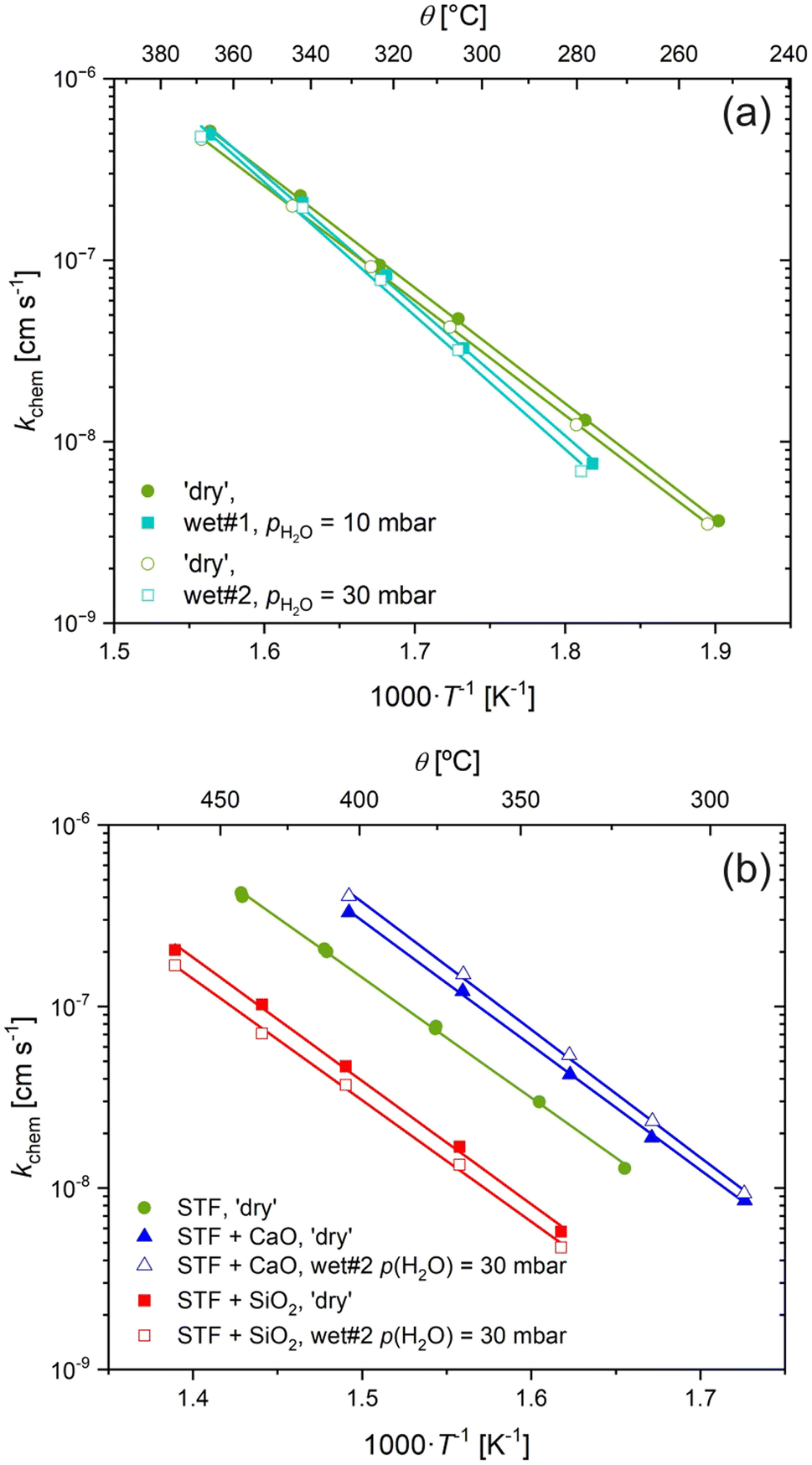 | ||
| Fig. 3 Oxygen surface exchange coefficent, kchem, of STF measured by ECR under ‘dry’ and wet conditions. (a) Uninfiltrated STF for nominally dry gas and under wet atmospheres. (b) Influence of acidic and basic species on kchem under ‘dry’ and wet conditions. | ||
An increase in the magnitude of kchem and a substantial decrease in the activation energy would be expected if there was a change in mechanism from electron transfer under ‘dry’ conditions to one involving water species under wet conditions. It is, however, clear from the data in Fig. 3a, that this is not what is observed. This suggests either that there is no effect or that water is dominating oxygen surface exchange in our ECR measurements in all conditions, including the ‘dry’ conditions, such that an increase in pH2O does not lead to change in the surface water species involved in the surface exchange reaction, as the surface sites are saturated at all levels of pH2O investigated.
To further investigate the role of water species on the surface exchange reaction with infiltrated acidic/basic species, we repeated the ECR measurements on STF before and after infiltration with a basic oxide, CaO, and an acidic oxide, SiO2 under ‘dry’ and wet conditions. The resultant kchem values are shown in Fig. 3b. Under wet conditions, the basic and acidic species still lead to dramatic increases and decreases in the surface exchange reaction, respectively. The magnitude of the change is approximately the same, or even slightly higher under wet atmospheres compared to ‘dry’, although this may be within the experimental uncertainty. It is clear from these results that the mechanism by which acidic/basic infiltrated species lead to dramatic changes in the surface exchange reaction plays a decisive role in wet conditions as well as in ‘dry’ conditions.
Thus, on the one hand, our 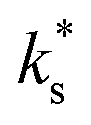 data falls on the low-temperature branch, strongly suggesting a different mechanism to that operative for the high-temperature branch, which we can describe using the electron transfer model. Furthermore, the presence of trace amounts of water has been heavily implicated in the change from high-temperature to low-temperature branch.34,38 On the other hand, no significant effect of varying the pH2O on kchem were observed (Fig. 3), which may seemingly contradict the hypothesis that water is playing a role in the surface exchange. This does not, however, present a logical inconsistency, as it is important to note that water is present in all conditions, including the nominally ‘dry’ conditions, and that there is a finite number of surface sites where absorption may occur, resulting in surface saturation at very low pH2O. Such Langmuir-type behaviour, where the equilibrium fractional surface coverage is unity over a broad pressure range if the adsorption rate is sufficiently high, has been shown for adsorbed water on Y2O3-doped ZrO2 and Al2O3 by Raz et al.39 They concluded that a full surface coverage of water species would be present on most oxides below ∼450 °C.
data falls on the low-temperature branch, strongly suggesting a different mechanism to that operative for the high-temperature branch, which we can describe using the electron transfer model. Furthermore, the presence of trace amounts of water has been heavily implicated in the change from high-temperature to low-temperature branch.34,38 On the other hand, no significant effect of varying the pH2O on kchem were observed (Fig. 3), which may seemingly contradict the hypothesis that water is playing a role in the surface exchange. This does not, however, present a logical inconsistency, as it is important to note that water is present in all conditions, including the nominally ‘dry’ conditions, and that there is a finite number of surface sites where absorption may occur, resulting in surface saturation at very low pH2O. Such Langmuir-type behaviour, where the equilibrium fractional surface coverage is unity over a broad pressure range if the adsorption rate is sufficiently high, has been shown for adsorbed water on Y2O3-doped ZrO2 and Al2O3 by Raz et al.39 They concluded that a full surface coverage of water species would be present on most oxides below ∼450 °C.
Although we cannot at this stage comment on the mechanism, it is not unreasonable to consider that water may be playing a role in the surface exchange. At low temperatures, such as those used in the present study, there are numerous studies demonstrating faster oxygen exchange when H2O is present in the gas atmosphere. In some examples, it has been shown that oxygen exchange happens faster from H2O than molecular oxygen,40–42 while other work has shown that the presence of water may assist in the exchange of molecular oxygen.43,44
It is clear that the surface exchange is still strongly affected by the acidity on an infiltrated species under these wet conditions. As discussed above, we argue that a rate-determining step involving electron transfer, as described by eqn (1) and where the electron concentration is modified by the acidity of the surface species according to eqn (2) is not indicative of the underlying mechanism. Instead, the presence of acidic or basic oxides on the surface may shift the adsorption/desorption equilibrium of oxygen and water species (O2, O, H2O, OH) on the surface. Recent work by Siebenhofer et al. support this line of reasoning. They performed a combination of experimental and computational studies on PCO and LSC showing that the acidity, which they also correlate to the ionic potential, of infiltration or adsorbed species modifies the energy of adsorbed O2−2 and O2−1 species, which is caused by a change in the surface dipole.10 In the work by Siebenhofer et al. the absorption of water species was not considered, but here we argue that considering that in the temperature range being investigated, surfaces are almost certainly hydrated and that H2O offers a competing pathway for oxygen exchange, this may have an effect or even play a decisive role.
Conclusions
We have investigated the role of acidic and basic oxides on the oxygen surface exchange of STF as a model MIEC. We have reproduced the previously reported effect of infiltrated acidic/basic species on STF under both nominally dry and wet conditions, demonstrating a total change in the surface exchange kinetics of one order of magnitude. Through our experiments and an analysis of the literature data, we reason that a model considering a change in the surface electron concentration is insufficient to describe the underlying mechanism. We instead propose that the presence of water species in the gas atmosphere, which has so far been overlooked, may act as a critical factor in the exchange kinetics, and their interaction with acidic/basic surface species may be the cause for the substantial changes seen in reaction rates.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Discussions with Clement Nicollet, Julius Dąbrowa, Andreas Falkenstein, and Bernd Huppertz are all gratefully acknowledged. G. F. H. gratefully acknowledges funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement number [101031819 – OPTICS]. This project has received funding from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) 463184206 (SFB 1548, FLAIR: Fermi Level Engineering Applied to Oxide Electroceramics).References
- R. Merkle and J. Maier, How is oxygen incorporated into oxides? A comprehensive kinetic study of a simple solid-state reaction with SrTiO3 as a model material, Angew. Chem., Int. Ed., 2008, 47, 3874–3894, DOI:10.1002/anie.200700987.
- R. A. De Souza, Limits to the rate of oxygen transport in mixed-conducting oxides, J. Mater. Chem. A, 2017, 5, 20334–20350, 10.1039/c7ta04266c.
- J. A. Kilner, R. A. De Souza and I. C. Fullarton, Surface exchange of oxygen in mixed conducting perovskite oxides, Solid State Ionics, 1996, 86–88, 703–709, DOI:10.1016/0167-2738(96)00153-1.
- Y. L. Lee, J. Kleis, J. Rossmeisl, S. H. Yang and D. Morgan, Prediction of solid oxide fuel cell cathode activity with first-principles descriptors, Energy Environ. Sci., 2011, 4, 3966–3970, 10.1039/c1ee02032c.
- W. Jung and H. L. Tuller, A New Model Describing Solid Oxide Fuel Cell Cathode Kinetics: Model Thin Film SrTi1-xFexO 3-δ Mixed Conducting Oxides–a Case Study, Adv. Energy Mater., 2011, 1, 1184–1191, DOI:10.1002/aenm.201100164.
- C. Nicollet, C. Toparli, G. F. Harrington, T. Defferriere, B. Yildiz and H. L. Tuller, Acidity of surface-infiltrated binary oxides as a sensitive descriptor of oxygen exchange kinetics in mixed conducting oxides, Nat. Catal., 2020, 3, 913–920, DOI:10.1038/s41929-020-00520-x.
- C. Riedl, M. Siebenhofer, A. Nenning, G. E. Wilson, J. Kilner, C. Rameshan, A. Limbeck, A. K. Opitz, M. Kubicek and J. Fleig, Surface Decorations on Mixed Ionic and Electronic Conductors: Effects on Surface Potential, Defects, and the Oxygen Exchange Kinetics, ACS Appl. Mater. Interfaces, 2023, 15, 26787–26798, DOI:10.1021/acsami.3c03952.
- M. Siebenhofer, A. Nenning, G. E. Wilson, J. A. Kilner, C. Rameshan, M. Kubicek, J. Fleig and P. Blaha, Electronic and ionic effects of sulphur and other acidic adsorbates on the surface of an SOFC cathode material, J. Mater. Chem. A, 2023, 11, 7213–7226, 10.1039/d3ta00978e.
- M. Siebenhofer, C. Riedl, A. Nenning, S. Raznjevic, F. Fellner, W. Artner, Z. Zhang, C. Rameshan, J. Fleig and M. Kubicek, Crystal-Orientation-Dependent Oxygen Exchange Kinetics on Mixed Conducting Thin-Film Surfaces Investigated by In Situ Studies, ACS Appl. Energy Mater., 2023, 6, 6712–6720, DOI:10.1021/acsaem.3c00870.
- M. Siebenhofer, A. Nenning, C. Rameshan, P. Blaha, J. Fleig and M. Kubicek, Engineering surface dipoles on mixed conducting oxides with ultra-thin oxide decoration layers, Nat. Commun., 2024, 15, 1–10, DOI:10.1038/s41467-024-45824-9.
- A. Merieau, M. Siebenhofer, C. Böhme, M. Kubicek, O. Joubert, J. Fleig and C. Nicollet, Oxygen surface exchange kinetics of La1−xSrxCoO3-δ thin-films decorated with binary oxides: links between acidity, strontium doping, and reaction kinetics, J. Mater. Chem. A, 2024, 12, 13960–13969, 10.1039/d3ta07422f.
- C. Nicollet and H. L. Tuller, Perspective on the Relationship between the Acidity of Perovskite Oxides and Their Oxygen Surface Exchange Kinetics, Chem. Mater., 2022, 34, 991–997, DOI:10.1021/acs.chemmater.1c03140.
- M. Siebenhofer, C. Riedl, A. Nenning, W. Artner, C. Rameshan, A. K. Opitz, J. Fleig and M. Kubicek, Improving and degrading the oxygen exchange kinetics of La0.6Sr0.4CoO3−δ by Sr decoration, J. Mater. Chem. A, 2023, 11, 12827–12836, 10.1039/d2ta09362f.
- H. G. Seo, A. Staerz, D. S. Kim, D. Klotz, C. Nicollet, M. Xu, J. M. LeBeau and H. L. Tuller, Reactivation of chromia poisoned oxygen exchange kinetics in mixed conducting solid oxide fuel cell electrodes by serial infiltration of lithia, Energy Environ. Sci., 2022, 15, 4038–4047, 10.1039/d1ee03975j.
- H. G. Seo, A. Staerz, D. S. Kim, J. M. LeBeau and H. L. Tuller, Tuning Surface Acidity of Mixed Conducting Electrodes: Recovery of Si-Induced Degradation of Oxygen Exchange Rate and Area Specific Resistance, Adv. Mater., 2023, 35, 2208182, DOI:10.1002/adma.202208182.
- H. G. Seo, A. Staerz, G. Dimitrakopoulos, D. Kim, B. Yildiz and H. L. Tuller, Degradation and recovery of solid oxide fuel cell performance by control of cathode surface acidity: Case study – Impact of Cr followed by Ca infiltration, J. Power Sources, 2023, 558, 1–8, DOI:10.1016/j.jpowsour.2022.232589.
- H. G. Seo, H. Kim, W. C. Jung and H. L. Tuller, Reversal of chronic surface degradation of Sr(Ti,Fe)O3 perovskite-based fuel cell cathodes by surface acid/base engineering, Appl. Catal., B, 2024, 355, 124172, DOI:10.1016/j.apcatb.2024.124172.
- S.-L. Zhang, D. Cox, H. Yang, B.-K. Park, C.-X. Li, C.-J. Li and S. A. Barnett, High stability SrTi1−xFexO3−δ electrodes for oxygen reduction and oxygen evolution reactions, J. Mater. Chem. A, 2019, 7, 21447–21458, 10.1039/C9TA07548H.
- J. H. Zhang, F. Z. Han, C. X. Li and S. L. Zhang, A-site deficient Sr0.9Ti0.3Fe0.7O3−δ perovskite:A high stable cobalt-free oxygen electrode material for solid oxide electrochemical cells with excellent electrocatalytic activity and CO2 tolerance, J. Eur. Ceram. Soc., 2022, 42, 5801–5812, DOI:10.1016/j.jeurceramsoc.2022.06.047.
- A. Rothschild, W. Menesklou, H. L. Tuller and E. Ivers-Tiffée, Electronic Structure, Defect Chemistry, and Transport Properties of SrTi1-xFexO3-y Solid Solutions, Chem. Mater., 2006, 18, 3651–3659, DOI:10.1021/cm052803x.
- R. Merkle and J. Maier, Oxygen incorporation into Fe-doped SrTiO3: Mechanistic interpretation of the surface reaction, Phys. Chem. Chem. Phys., 2002, 4, 4140–4148, 10.1039/b204032h.
- R. Merkle, R. A. De Souza and J. Maier, Optically Tuning the Rate of Stoichiometry Changes: Surface-Controlled Oxygen Incorporation into Oxides under UV Irradiation, Angew. Chem., Int. Ed., 2001, 40, 2126–2129, DOI:10.1002/1521-3773(20010601)40:11<2126::AID-ANIE2126>3.0.CO;2-K.
- V. Metlenko, W. Jung, S. Bishop, H. L. Tuller and R. De Souza, Oxygen diffusion and surface exchange in the mixed conducting oxides SrTi1-yFeyO3-δ, Phys. Chem. Chem. Phys., 2016, 4, 1166–1169, 10.1039/C6CP05756J.
- R. Ganeshananthan and A. V. Virkar, Measurement of Surface Exchange Coefficient on Porous La0.6Sr0.4CoO3−δ Samples by Conductivity Relaxation, J. Electrochem. Soc., 2005, 152, A1620, DOI:10.1149/1.1940828.
- C. Argirusis, F. Jomard, S. F. Wagner, W. Menesklou and E. Ivers-Tiffée, Study of the oxygen incorporation and diffusion in Sr(Ti0.65Fe0.35)O3 ceramics, Solid State Ionics, 2011, 192, 9–11, DOI:10.1016/j.ssi.2010.02.016.
- S. J. Benson, R. J. Chater and J. A. Kilner, Oxygen diffusion and surface exchnage in the mixed conducting perovskite La0.4Sr0.4Fe0.8Co0.2O3-d, in Proceedings of the Third International Symposium on Ionic and Mixed Conducting Ceramics, 1998, pp. 596–609 Search PubMed.
- V. Thoréton, M. Niania, J. Druce, H. Tellez and J. A. Kilner, Oxygen Diffusion in Ceramic Mixed Conducting La0.6Sr0.4Co0.2Fe0.8O3−δ : The Role of Grain and Twin Boundaries, J. Electrochem. Soc., 2022, 169, 044513, DOI:10.1149/1945-7111/ac6396.
- P. S. Manning, J. D. Sirman and J. A. Kilner, Oxygen self-diffusion and surface exchange studies of oxide electrolytes having the fluorite structure, Solid State Ionics, 1996, 93, 125–132, DOI:10.1016/s0167-2738(96)00514-0.
- S. P. Waldow, B. J. Statham, H. F. Wardenga, T. E. Weirich, A. Klein and R. A. De Souza, Oxygen Surface Exchange and Tracer Diffusion in Differently Oriented Thin Films of Gd-Doped CeO2, ACS Appl. Mater. Interfaces, 2020, 12, 36768–36777, DOI:10.1021/acsami.0c09605.
- J. Kler, J. M. Börgers and R. A. De Souza, Surface Space-Charge Potential and Oxygen Surface Exchange Coefficient of SrTiO3: The Effect of Surface Orientation, Adv. Mater. Interfaces, 2023, 10, 2201434, DOI:10.1002/admi.202201434.
- P. S. Manning, J. D. Sirman, R. A. De Souza and J. A. Kilner, The kinetics of oxygen transport in 9.5 mol% single crystal yttria stabilised zirconia, Solid State Ionics, 1997, 100, 1–10, DOI:10.1016/S0167-2738(97)00345-7.
- J. Portier, P. Poizot, J. M. Tarascon, G. Campet and M. A. Subramanian, Acid-base behavior of oxides and their electronic structure, Solid State Sci., 2003, 5, 695–699, DOI:10.1016/S1293-2558(03)00031-1.
- M. Kuhn, J. J. Kim, S. R. Bishop and H. L. Tuller, Oxygen nonstoichiometry and defect chemistry of perovskite-structured BaxSr1-xTi1-yFeyO3-y/2+δ solid solutions, Chem. Mater., 2013, 25, 2970–2975, DOI:10.1021/cm400546z.
- N. Sakai, K. Yamaji, T. Horita, Y. P. Xiong, H. Kishimoto and H. Yokokawa, Effect of Water on Oxygen Transport Properties on Electrolyte Surface in SOFCs, J. Electrochem. Soc., 2003, 150, A689, DOI:10.1149/1.1568938.
- M. J. Pietrowski, R. A. De Souza, M. Fartmann, R. Ter Veen and M. Martin, Oxygen isotope transport properties of yttria-stabilized zirconia (YSZ) in O2- and H2O-containing atmospheres, Fuel Cells, 2013, 13, 673–681, DOI:10.1002/fuce.201300087.
- J. H. Joo, R. Merkle and J. Maier, Effects of water on oxygen surface exchange and degradation of mixed conducting perovskites, J. Power Sources, 2011, 196, 7495–7499, DOI:10.1016/j.jpowsour.2011.04.032.
- S. J. Cooper, M. Niania, F. Hoffmann and J. A. Kilner, Back-exchange: A novel approach to quantifying oxygen diffusion and surface exchange in ambient atmospheres, Phys. Chem. Chem. Phys., 2017, 19, 12199–12205, 10.1039/c7cp01317e.
- J. Kler and R. A. De Souza, Hydration Entropy and Enthalpy of a Perovskite Oxide from Oxygen Tracer Diffusion Experiments, J. Phys. Chem. Lett., 2022, 13, 4133–4138, DOI:10.1021/acs.jpclett.2c00970.
- S. Raz, K. Sasaki, J. Maier and I. Riess, Characterization of adsorbed water layers on Y2O3-doped ZrO2, Solid State Ionics, 2001, 143, 181–204, DOI:10.1016/S0167-2738(01)00826-8.
- M. Kessel, R. A. De Souza, H. I. Yoo and M. Martin, Strongly enhanced incorporation of oxygen into barium titanate based multilayer ceramic capacitors using water vapor, Appl. Phys. Lett., 2010, 97, 10–13, DOI:10.1063/1.3460156.
- A. Nenning, E. Navickas, H. Hutter and J. Fleig, Water-Induced Decoupling of Tracer and Electrochemical Oxygen Exchange Kinetics on Mixed Conducting Electrodes, J. Phys. Chem. Lett., 2016, 7, 2826–2831, DOI:10.1021/acs.jpclett.6b00778.
- V. Thoréton, M. Niania and J. Kilner, Kinetics of competing exchange of oxygen and water at the surface of functional oxides, Phys. Chem. Chem. Phys., 2021, 23, 2805–2811, 10.1039/d0cp04953k.
- A. Staykov, S. Fukumori, K. Yoshizawa, K. Sato, T. Ishihara and J. Kilner, Interaction of SrO-terminated SrTiO3 surface with oxygen, carbon dioxide, and water, J. Mater. Chem. A, 2018, 6, 22662–22672, 10.1039/c8ta05177a.
- J. Yang, J. M. Polfus, Z. Li, H. L. Tuller and B. Yildiz, Role of Adsorbate Coverage on the Oxygen Dissociation Rate on Sr-Doped LaMnO3Surfaces in the Presence of H2O and CO2, Chem. Mater., 2020, 32, 5483–5492, DOI:10.1021/acs.chemmater.9b05243.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07063a |
| This journal is © The Royal Society of Chemistry 2025 |