
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature- and creep-resistant Diels–Alder salogels for shape stabilization of salt hydrate phase change materials†
Kartik Kumar Rajagopalan and
Svetlana A. Sukhishvili*
and
Svetlana A. Sukhishvili*
Department of Materials Science & Engineering, Texas A&M University, College Station, TX 77843, USA. E-mail: svetlana@tamu.edu
First published on 10th February 2025
Abstract
Shape stabilization of inorganic salt hydrate phase change materials (PCMs) above their melting point is required to overcome the leakage problem in the molten state. However, previous strategies in designing PCM-stabilizing polymer networks, e.g. salogels, resulted in a limited range of temperature stability (up to 50 °C) and thus could not be applied to PCMs with widely ranging melting temperatures. In this work, we introduce a polymer salogel system that was not only able to retain PCMs over a wide temperature range (up to 120 °C) but also provide robust mechanical stabilization allowing creation of leakage-free, creep-resistant thermal energy storage materials. This salogel system consisted of a polymer gel formed by the Diels–Alder (DA) reaction between furan-modified polyvinyl alcohol (PVA) and a bismaleimide crosslinker with an entrapped salt hydrate PCM – either lithium nitrate trihydrate (LiNO3·3H2O, LNH) or magnesium nitrate hexahydrate (Mg(NO3)2·6H2O, MgNH), which were chosen for their widely different melting temperatures of 29 and 89 °C, respectively. Rheological studies of DA salogels were compared side-by-side with the salogels stabilized by another type of dynamic covalent chemistry based on boronate ester bonds. Compared to the viscoelastic boronate ester salogels which showed poor and dramatically decreasing strain recovery with temperature in creep experiments, DA salogels exhibited high elasticity, high gel-to-sol transition temperature (Tgel > 120 °C) and superior creep resistance at elevated temperatures (≥80% strain recovery at T ≤ 95 °C). Moreover, the DA crosslinking preserved the temperature response of gelation above 120 °C, important for end-of-life material removal in thermal management applications. Finally, DA salogels demonstrated high retention of latent heat characteristics of neat PCMs due to the high (>90%) content of inorganic salt hydrates and preserved their thermal properties over 50 melting/crystallization cycles.
Introduction
Thermal energy storage (TES) technologies are gaining importance due to their ability to store and release large amounts of energy to meet the gap between energy demand and supply brought about by economic growth and climate change. Compared to sensible heat and thermochemical energy storage techniques, latent heat thermal energy storage based on solid-to-liquid phase change materials (PCMs) is preferred due to their compact volume, isothermal operation, and low cost per unit of energy stored.1,2 Among different types of PCMs, inorganic salt hydrates (MxNy·nH2O) stand out due to their high volumetric energy density, low cost, and non-flammability.1,3–5 However, there is a significant challenge related to the leakage of inorganic salt hydrates from the thermal storage modules during multiple melting and crystallization cycles due to the low viscosity of these PCMs.3,6 Therefore, shape stabilization is pursued to prevent the leakage and flow of the liquid PCM away from heat transfer surfaces during the solid–liquid phase transition by trapping the PCM in an appropriate matrix which can mechanically stabilize the PCM without altering the thermal properties significantly. Several strategies using inorganic matrices and polymer networks were developed to provide shape stabilization of the salt hydrates in their liquid state.3,6 However, these efforts were mostly focused on developing materials that were permanently crosslinked,7–12 which makes them difficult to remove from heat exchange modules at the end of their life cycle.Our group previously introduced a different class of PCM-stabilizing polymer matrices which are temperature-responsive and enable removal of the PCM from the thermal storage module. The approach is based on the ability of these polymer matrices to transition from gel to sol above a gel transition temperature (Tgel).13–16 These polymer-inorganic salt hydrate systems, called salogels, were constructed using hydrogen bonding polymers (such as poly(vinyl alcohol), PVA) dissolved in the water-scarce environment of the molten salt hydrates (e.g. 30% saturation of the primary hydration shells in LiNO3·3H2O (LNH) and 33% saturation in Ca(NO3)2·4H2O (CNH)). Due to the scarcity of water, hydrogen-bonding polymers remained strongly dehydrated and could form weak gels due to enhanced intermolecular hydrogen bonding and polymer-ion interactions.3,15 While incorporation of hydrogen-bonding crosslinkers, entanglements, and dynamic boronate ester bonds could improve salogel thermomechanical properties, these systems were not mechanically robust even at room temperature and their viscoelastic temperature response could only provide moderate shape stabilization of low-melting-point salt hydrate PCMs over a limited range of temperatures (<50 °C).13–16 The insufficient elasticity of these salogels makes them susceptible to creep even at physiological temperatures and suitable only for limited applications. However, for many other thermal energy storage applications such as wearable devices, personal thermal management, and application as patches, materials which are mechanically robust are required.10–12,17–20 Moreover, the limited temperature range of mechanical stability makes the previously explored dynamic crosslinking strategies unsuitable for shape stabilization of medium to high melting temperature salt hydrate PCMs (melting temperature > 50 °C) such as sodium acetate trihydrate (CH3COONa·3H2O) and magnesium nitrate hexahydrate (Mg(NO3)2·6H2O). These PCMs are relevant for industrial waste heat recovery such as operation of fuel cells, space and water heating in buildings, and solar thermal energy storage applications where temperatures in the range of 60–100 °C are reached.2,6,21–27 To overcome both these limitations of current salogel systems, here we explore the use of another dynamic covalent chemistry which provides a wide-temperature-range shape stabilization of PCM materials while preserving their temperature response necessary for end-of-life removal from a thermal energy storage module.
In this work we employ furan–maleimide Diels–Alder (DA) dynamic covalent chemistry for constructing highly creep-resistant salogels for shape stabilization of salt hydrate PCMs with widely varied melting temperatures. The furan–maleimide DA reactions are “click” in nature, occur without side products or a catalyst under ambient conditions,28–30 and can be reversed by heating above a retro-DA reaction temperature (TrDA ∼ 120 °C).30–32 The dynamic nature of the DA bonds was exploited to develop self-healing and thermo-responsive materials where they were included within solvent-free polymer networks or hydrogels for applications in soft robotics, shape morphing, and biomedical applications such as cell encapsulation and tissue engineering.31–42 However, the use of DA polymer networks in thermal energy storage applications has been limited to flammable organic PCMs (e.g. polyethylene glycol (PEG)38,39,42 and beeswax37) which typically yield TES materials with relatively low (40–83%) PCM content.37,39,42 In contrast, this work explores, to the best of our knowledge for the first time, the use of furan–maleimide dynamic covalent DA bonds to create salogels in an inflammable PCM which are based on inorganic salt hydrates. We demonstrate the construction of a novel salogel network using DA bonds and its ability to shape stabilize salt hydrate PCMs over a wide melting temperature range at a high PCM loading of 90 wt%. The salt hydrates chosen for this work were LNH and MgNH, which have melting points of ∼29 °C and 89 °C and high thermal energy storage capacities of 280 J g−1 and 162 J g−1, respectively.13,21,23,24,26,43 The large difference in melting points of LNH and MgNH, along with the low viscosity of their melts (only 5 mPa s at 35 °C for LNH and 0.18 mPa s at 95 °C for MgNH)3 makes these PCMs ideal candidates to explore the shape stabilizing capability of DA chemistry. We study the ability of DA crosslinks to yield mechanically robust salogels during PCM melting-solidification cycles while supporting the gel-to-sol transition above Tgel (Tgel = TrDA) for the end-of-life disposal and benchmark the performance of DA salogels against that of boronate ester salogels.
Experimental
Materials
Polyvinyl alcohol (PVA) with a molecular weight of 90 kDa and degree of hydrolysis of 98% and sodium tetraborate decahydrate (ACS, >99%) were purchased from Alfa Aesar. Anhydrous lithium nitrate (ACS, >99.0%) purchased from Alfa Aesar was used for preparation of lithium nitrate trihydrate (LNH) by adding a stoichiometric amount of deionized (DI) water. Furfuryl isocyanate (FIC) (97% purity) was purchased from Thermo Fisher. Anhydrous dimethyl sulfoxide (DMSO), isopropyl alcohol, triethylamine (TEA), magnesium nitrate hexahydrate (Mg(NO3)2·6H2O, MgNH), deuterium oxide (D2O) with 99.9 D atom%, and deuterated dimethyl sulfoxide (d6-DMSO) were obtained from Sigma-Aldrich. A bismaleimide crosslinker with a polyethylene glycol backbone (BM-PEG) of molecular weight 2 kDa was purchased from JenKem Technology, USA. All reagents except anhydrous lithium nitrate used for preparation of LNH were used as received.Furan modification of PVA
DA salogels were made by attaching furan groups to PVA followed by crosslinking with a BM-PEG crosslinker in liquid salt hydrate (LNH or MgNH) as a solvent. Furan modification of PVA was performed using the well-known hydroxyl–isocyanate reaction between PVA and FIC which has been reported for small-molecule hydroxyl compounds and another hydroxyl polymer, dextran.44,45 Specifically, PVA (1 g) was dissolved in anhydrous DMSO (20 mL) with stirring at 80 °C and then the solution was cooled to room temperature. FIC (5.56 mmol) and TEA (5.56 mmol) were then added, and the hydroxyl–isocyanate reaction was carried out at 70 °C for 6 hours. After allowing the mixture to cool down to room temperature, the polymer was precipitated in isopropyl alcohol and washed several times. The polymer was then dried in vacuum at room temperature. The furan-modified PVA obtained from this procedure was denoted as Fx-PVA, where x = 2.5, 5, and 10 denote the mol% of furan groups on PVA. The covalent attachment of furan groups to the PVA was also confirmed by ATR-FTIR31 and quantified by 1H NMR (Fig. 1). The degrees of PVA modification above 10 mol% were not pursued due to deteriorated solubility of the modified polymers in LNH and MgNH as the hydroxyl groups of PVA were replaced with the furan moiety.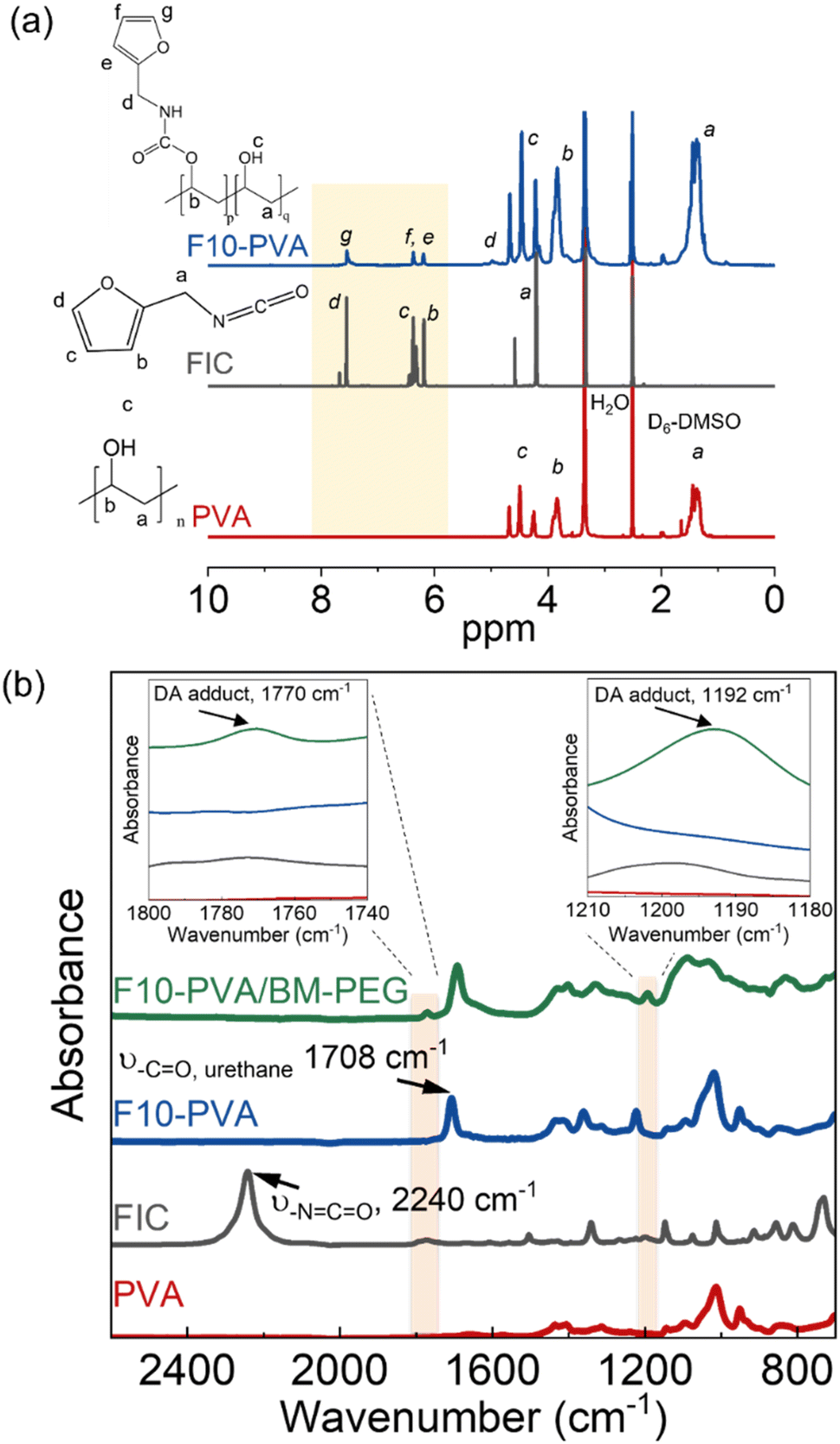 | ||
| Fig. 1 (a) 1H NMR of PVA, FIC, and F10-PVA and (b) ATR-FTIR spectra of PVA, FIC, F10-PVA, and F10-PVA/BM-PEG showing successful attachment of furan groups on PVA and formation of DA bonds. Insets in (b) show the DA adduct peaks at 1770 cm−1 (left) and 1190 cm−1 (right). | ||
Preparation of salogels and hydrogels
DA salogels were prepared by adding 5 wt% Fx-PVA to LNH while stirring at 70 °C to form a homogeneous solution and adding the BM-PEG crosslinker at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 furan-to-maleimide molar ratio to crosslink the furan groups on the Fx-PVA backbone (Scheme 1). The BM-PEG crosslinker dissolved readily upon heating to 70 °C and gelation occurred within a few minutes of gentle stirring due to the click nature of the DA reaction. Mixing was stopped and the samples were annealed for 3 hours at 70 °C to ensure completion of the crosslinking reaction. DA hydrogels were made using the same procedure as described for the salogels, except that the maximum furan modification was 2.5 mol% since PVA with a higher percentage of furan groups became insoluble in water. Comparison of DA hydrogels with DA salogels was done using PVA with the matched degree of furan modification (F2.5-PVA). The DA salogel in magnesium nitrate hexahydrate (MgNH) was prepared at 100 °C using F10-PVA following the same steps as those described for the salogel in LNH. Note that DA salogels were formed in PCMs above their melting points, i.e. at 70 °C in the case of LNH and 100 °C in the case of MgNH and did not contain residual salt hydrate crystals.
:1 furan-to-maleimide molar ratio to crosslink the furan groups on the Fx-PVA backbone (Scheme 1). The BM-PEG crosslinker dissolved readily upon heating to 70 °C and gelation occurred within a few minutes of gentle stirring due to the click nature of the DA reaction. Mixing was stopped and the samples were annealed for 3 hours at 70 °C to ensure completion of the crosslinking reaction. DA hydrogels were made using the same procedure as described for the salogels, except that the maximum furan modification was 2.5 mol% since PVA with a higher percentage of furan groups became insoluble in water. Comparison of DA hydrogels with DA salogels was done using PVA with the matched degree of furan modification (F2.5-PVA). The DA salogel in magnesium nitrate hexahydrate (MgNH) was prepared at 100 °C using F10-PVA following the same steps as those described for the salogel in LNH. Note that DA salogels were formed in PCMs above their melting points, i.e. at 70 °C in the case of LNH and 100 °C in the case of MgNH and did not contain residual salt hydrate crystals.
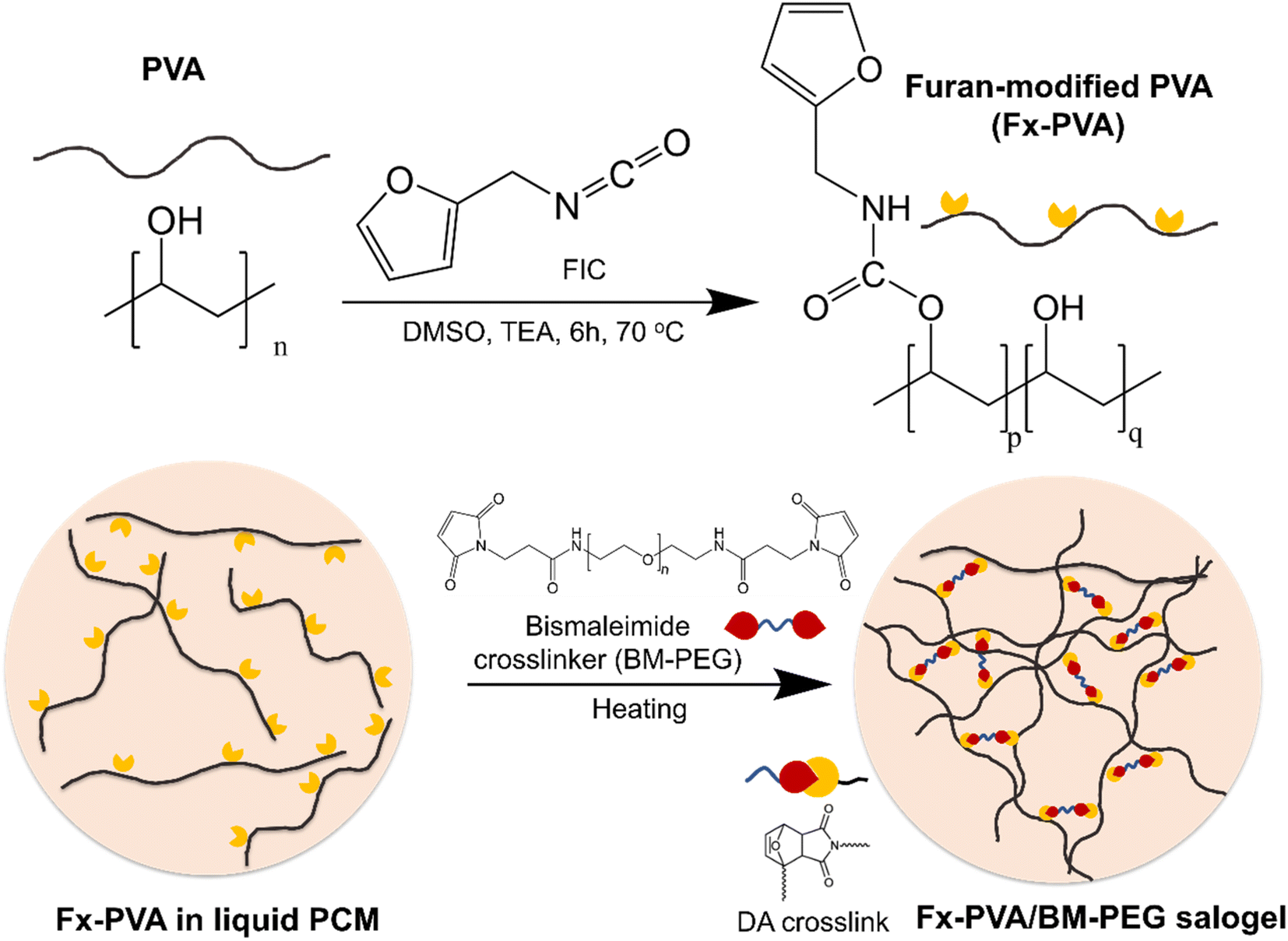 | ||
| Scheme 1 Furan modification of PVA using furfuryl isocyanate (FIC) and crosslinking of furan-modified PVA with BM-PEG to form DA salogels in salt hydrate PCMs. | ||
Boronate ester salogels (in LNH and MgNH) and hydrogels (in water) with matched mol% of hydroxyl groups crosslinked were compared with DA salogels and hydrogels. The salogels and hydrogels were prepared by varying the borax concentration (0.125, 0.25, and 0.5 wt% borax) using a procedure described previously.15 These gels are represented as PVA/borax-x, where x = 2.5, 5, and 10, respectively to indicate the mol% of –OH groups crosslinked. Boronate ester salogels were prepared under the same conditions as DA salogels (5 wt% PVA dissolved in LNH at 70 °C) but without additional annealing after addition of borax. The samples were then cooled down to room temperature before adding the borax crosslinker. PVA/borax salogels were prepared by adding borax followed by heating the mixture to 80 °C while stirring to facilitate the dissolution of borax and cooling down to room temperature to induce gelation. PVA/borax-10 salogels in MgNH were prepared using the same steps as described above for the salogels in LNH, except that all the mixing was done at 100 °C. PVA/borax hydrogels were prepared the same way as the salogels, except that the mixing was done at room temperature since borax is easily soluble in water compared to LNH. Mixing was stopped after the formation of the gel and the samples were allowed to homogenize at room temperature for 24 hours. Because of the known supercooling effect that is significant in the case of LNH,46 no crystallization occurred in both salogel systems (DA and boronate ester) at room temperature, and freezing temperatures (−18 °C) were used to induce crystallization of LNH for testing the performance of the matrix during multiple crystallization/melting cycles. Supercooling also exists in MgNH, but the high melting temperature results in crystallization well above room temperature (62 °C). Due to the crystallized state of MgNH at room temperature, all room temperature experiments (time sweep for gelation and creep–recovery at room temperature) were performed using the DA salogel in LNH.
Materials characterization
where Ag is the area of the furan proton peak g at 7.54 ppm, and Aa is the area of the CH2 proton peak a on the PVA backbone (Fig. 1, S1, and S2†).
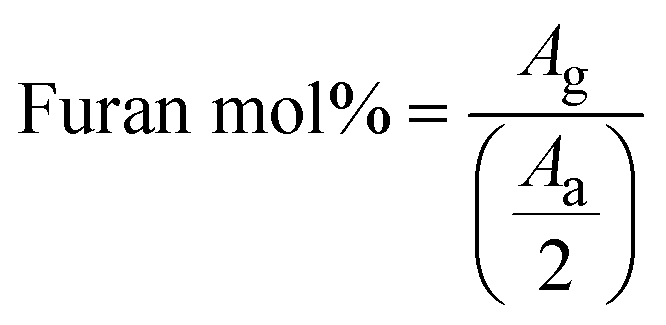
Thermal cycling, shape stabilization, and self-healing capabilities of DA salogels
Retention of thermal properties in the DA salogel in LNH and MgNH over multiple melting and crystallization cycles was explored by subjecting salogel samples (F10-PVA/BM-PEG) sealed in a 20 mL vial to appropriate temperatures to induce melting (50 °C for LNH and 95 °C for MgNH) and crystallization (−18 °C). DSC scans were performed after 50 cycles to check for retention of thermal properties (heat of fusion and melting temperature).Shape stabilization of LNH and MgNH in DA salogels was studied using hexagon shaped and rectangle shaped DA salogels by exposing them to temperatures of 70 °C and 95 °C (above the melting point of LNH and MgNH), respectively in an air-tight container and observing signs of leakage and retention of shape over a period of time. A boronate ester salogel hexagon in LNH was also employed as a control. Leakage prevention of PCM from the DA salogel was also confirmed by heating small pieces of the salogel in LNH (75 mg) and MgNH (60 mg) at 70 °C and 95 °C, respectively, for 12 h in a closed container and measuring their weights again after the heat treatment. Additionally, to simulate the creep resistance of the DA salogels we applied stress by placing three 50 gram weights (total of 150 grams) on the DA salogels in LNH and MgNH at room temperature when the salogels were in the crystallized state. The DA salogels with the weights placed on them were then heated to 70 °C and 95 °C, respectively for LNH and MgNH and held at this temperature for 10 minutes and then changes in dimensions due to creep were measured. The ability of the DA network to prevent leakage of the PCM under load was also tested from weight measurements of 376 mg and 562 mg of the LNH and MgNH salogels after 10 minutes of exposure to 70 and 95 °C, respectively.
The self-healing capability of the DA salogel was demonstrated by heating two pieces of the salogel in LNH to a temperature close to but below the TrDA to partially break DA bonds in the network for a short duration of time (∼2 min). During the experiment the samples were sandwiched between the 40 mm parallel plates of a rheometer, but no stress or strain was applied. The gap between the plates was set at a value where the plates would hold the sample stationary. In this state the samples were heated for two minutes and then cooled to room temperature. This was followed by visual evaluation of the self-healing capability by handling the salogels with tweezers and optical microscopy imaging of the healed area using a Nikon TI2-U inverted microscope with a 10× objective. The mechanical properties of the healed sample were compared with those of the original sample by performing a time sweep oscillatory rheology experiment at a strain of 1% and frequency of 10 rad s−1.
Results and discussion
DA salogels were prepared by furan modification of PVA followed by dissolution of this polymer in the highly ionic environment of a salt hydrate and crosslinking to form the gel network (Scheme 1). The procedure involved modification of PVA with furfuryl isocyanate (FIC), followed by the formation of a salogel network in liquid PCM using a bismaleimide crosslinker, BM-PEG. Modification of PVA was performed to obtain polymers with different percentages of furan-modified units; these polymers are abbreviated as Fx-PVA, where x is 2.5, 5 or 10.Crosslinking of Fx-PVA polymers with BM-PEG resulted in Fx-PVA/BM-PEG networks in which crosslinking density was controlled by the degree of PVA modification. Fig. 1a and b show the characterization of furan-modified PVA with the highest content of furan groups, i.e. F10-PVA, using 1H NMR and ATR-FTIR, respectively. The appearance of the peaks of protons from the furan ring at 6.19, 6.36, and 7.54 ppm in the 1H NMR spectrum of F10-PVA in Fig. 1a indicates successful furan modification. The extent of furan modification was determined from a ratio of furan proton peak g at 7.54 ppm to the methylene proton peak a of the PVA backbone at 1.4 ppm (see the Materials and methods for details). 1H NMR spectra of F2.5-PVA and F5-PVA are shown in Fig. S1 and S2.† The emergence of the peak at 1708 cm−1 associated with the formation of urethane bonds47 in the FTIR spectra of the F-PVA (Fig. 1b) provides evidence of successful furan modification. The salogels were prepared using a 5 wt% solution of Fx-PVA polymers in LNH or MgNH, which all gelled within several minutes after the addition of BM-PEG due to the formation of DA crosslinks. This concentration was chosen as it is above the critical overlap concentration of 3.5 wt% as was determined for PVA of the same molecular weight (90 kDa) in salt hydrate solvents in a previous paper from our group.14 Gelation and other room temperature experiments were explored in greater detail using LNH rather than MgNH due to the low melting point of this PCM. The low melting point enables salogel preparation at ambient temperature and the existence of the gel state with a liquid-phase PCM at this temperature. Fig. S4† shows that in the F10-PVA/LNH system, the “click” nature of the DA reaction resulted in gelation after ∼8.5 minutes after adding the crosslinker as observed from the crossover of storage (G′) and loss (G′′) moduli. Fig. S5† shows that the moduli of the DA salogel did not change between 3 and 12 hours of exposure at 70 °C indicating that the crosslinking was complete after 3 hours. These conditions were used to prepare all the DA salogels in LNH (DA salogel in MgNH was prepared at 100 °C, see the Materials and methods for details). The DA hydrogels which were used as a control to compare with the DA salogel were prepared using the same procedure as the salogel. The gelation time for the DA salogel and the hydrogel was approximately the same from visual observation, suggesting that the highly ionic environment of the salt hydrate did not affect the kinetics of the DA reaction. The formation of DA crosslinks could be observed in ATR-FTIR spectra with the appearance of furan–maleimide DA adduct peaks at 1192 and 1770 cm−1 (insets of Fig. 1b, see the Materials and methods for a procedure of preparing salt-free DA matrices used for ATR-FTIR measurements).
The viscoelastic properties of the DA salogels were first studied at room temperature using oscillatory rheology and compared with those of boronate ester salogels. Note that all rheological measurements were made in the crystal-free, gel state of the salogels where the salt hydrate was in the liquid state. To enable these measurements, the low-melting-point LNH was used in these experiments as a PCM since MgNH was in a crystalline state at room temperature. The boronate ester salogel used as a control dynamic covalent salogel could not be formed in MgNH due to its Tgel being lower than the melting point of MgNH. Fig. S6† shows room-temperature frequency sweep rheology experiments for DA and boronate ester salogels and hydrogels used for comparison. The results suggest that both the DA gels containing liquid LNH and water exhibited elastic behavior throughout the 0.1–110 rad s−1 frequency range showing that these were true gels (Fig. S6a and b†). In contrast, the boronate ester gels showed viscoelastic behavior with a G′–G′′ crossover in the 1–10 rad s−1 range, with the salogel being both stronger and having a higher relaxation time compared to the hydrogel (Fig. S6c and d†). These results are consistent with those reported in our previous work in a different salt hydrate (CNH).15 It was also interesting to note that both the boronate ester and DA salogels were stronger than their corresponding hydrogels. These differences can be understood from the nature of the solvent (LNH vs. H2O) used in the salogel and hydrogel. The high ionic content (18 M) and low water concentration in LNH led to a dehydrated state of PVA and ions triggering a competition between ions and polymer for the scarce water resulting in a blue shift of the –OH stretching peak of F2.5-PVA dissolved in deuterated LNH (LND) as compared to D2O (Fig. S7†).
Further differences in viscoelastic properties between the DA and boronate ester salogels were revealed in the temperature dependence experiments (Fig. 2 and S8†). First, the G′ values were higher and G′′ values much smaller for the DA salogels (Fig. 2a and b) when compared with the corresponding values for the boronate ester salogels (Fig. 2c and d). The higher elasticity endowed to the polymer gel with DA crosslinks as compared to boronate ester bonds is illustrated by the extremely low values of tanδ 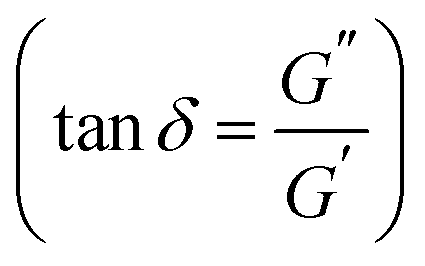 of DA salogel systems of both PCMs (LNH and MgNH) across a wide temperature range (Fig. S8†). Specifically, temperature sweep experiments shown in Fig. 2a, b and S8† indicate that the elastic DA salogels exhibit essentially unchanged values of G′ and G′′ between room temperature and 120 °C indicating that DA bonds remained largely intact. In the case of MgNH, the DA salogel showed a gel-like behavior between 95 and 120 °C (Fig. 2b) related to melting of MgNH at 89 °C and successful trapping of liquid MgNH in the DA network. Beyond 120 °C, the retro-DA reaction started to occur, and further heating resulted in gel-to-sol transition at ∼145 °C in LNH accompanied by two orders of magnitude reduction in viscosity (Fig. S9a†). A temperature ramp DSC experiment showed a slightly lower TrDA temperature of ∼135 °C (Fig. S10a†). The differences in Tgel from rheology and TrDA from DSC could be due to water evaporation from the sample in the rheometer due to lack of hermetic sealing. During the gel–sol transition the breaking of the thermo-responsive DA bonds results in the entire system (salt hydrate and polymer) converting to a low viscosity liquid (Fig. S9a†) due to the solubility of the polymer and crosslinker in the salt hydrate. The thermal response of the DA crosslinks resulting in the formation of a low viscosity liquid (Fig. S9a†) after gel–sol transition facilitates the removal of the spent salt hydrate PCM at the end-of-life cycle which is demonstrated using a simple experiment where the DA salogel flame-sealed in an ampoule was heated using a heat gun until the gel turned into a liquid and started flowing (Video S1†). Heating the DA salogel above the TrDA and then cooling did not affect the melting transition of LNH as observed in the second heating cycle indicating that in a hermetically sealed environment the water content in the salt hydrate did not change (Fig. S10a†). Note that in MgNH, the gel-to-sol transition of the DA polymer network was higher than that of LNH and could not be observed till the maximum tested temperature of 150 °C, which was also confirmed from DSC. However, a similar reduction in viscosity was also observed in this system in the retro-DA reaction temperature region (Fig. S9b†). At the same time, melting transition of the PCM (MgNH) was not affected by heating to 150 °C as observed in the second heating cycle (Fig. S10b†). In both salt hydrate PCMs, the retro-DA reaction temperature was higher than the previously reported value of 120–130 °C for furan–maleimide DA networks.31 This increase can be attributed to the effect of the salt hydrate solvent as well as the flexibility of the PEG-based bismaleimide crosslinker which was shown to result in a higher retro-DA reaction temperature compared to that of a stiff crosslinker.48 In contrast to the thermomechanical behavior of the DA salogel, the boronate ester salogel showed a sharp decrease in storage modulus (Fig. 2c) and increase in tanδ (Fig. S8†) with increasing temperature and underwent gel-to-sol transition at Tgel = 65 °C in LNH, which was 55–65 °C lower than the gel-to-sol transition temperature of the DA salogel. Note that boronate ester could not form a gel network in MgNH above its melting point (Fig. 2d and S8†) due to the thermo-reversible nature of the crosslinks.15,49,50 We believe that the persistence of DA elasticity between room temperature and 120 °C is the key for future applications of DA networks for hosting PCM materials with widely different melting temperatures while the temperature responsiveness allows removal of the PCM at the end-of-life cycle.
of DA salogel systems of both PCMs (LNH and MgNH) across a wide temperature range (Fig. S8†). Specifically, temperature sweep experiments shown in Fig. 2a, b and S8† indicate that the elastic DA salogels exhibit essentially unchanged values of G′ and G′′ between room temperature and 120 °C indicating that DA bonds remained largely intact. In the case of MgNH, the DA salogel showed a gel-like behavior between 95 and 120 °C (Fig. 2b) related to melting of MgNH at 89 °C and successful trapping of liquid MgNH in the DA network. Beyond 120 °C, the retro-DA reaction started to occur, and further heating resulted in gel-to-sol transition at ∼145 °C in LNH accompanied by two orders of magnitude reduction in viscosity (Fig. S9a†). A temperature ramp DSC experiment showed a slightly lower TrDA temperature of ∼135 °C (Fig. S10a†). The differences in Tgel from rheology and TrDA from DSC could be due to water evaporation from the sample in the rheometer due to lack of hermetic sealing. During the gel–sol transition the breaking of the thermo-responsive DA bonds results in the entire system (salt hydrate and polymer) converting to a low viscosity liquid (Fig. S9a†) due to the solubility of the polymer and crosslinker in the salt hydrate. The thermal response of the DA crosslinks resulting in the formation of a low viscosity liquid (Fig. S9a†) after gel–sol transition facilitates the removal of the spent salt hydrate PCM at the end-of-life cycle which is demonstrated using a simple experiment where the DA salogel flame-sealed in an ampoule was heated using a heat gun until the gel turned into a liquid and started flowing (Video S1†). Heating the DA salogel above the TrDA and then cooling did not affect the melting transition of LNH as observed in the second heating cycle indicating that in a hermetically sealed environment the water content in the salt hydrate did not change (Fig. S10a†). Note that in MgNH, the gel-to-sol transition of the DA polymer network was higher than that of LNH and could not be observed till the maximum tested temperature of 150 °C, which was also confirmed from DSC. However, a similar reduction in viscosity was also observed in this system in the retro-DA reaction temperature region (Fig. S9b†). At the same time, melting transition of the PCM (MgNH) was not affected by heating to 150 °C as observed in the second heating cycle (Fig. S10b†). In both salt hydrate PCMs, the retro-DA reaction temperature was higher than the previously reported value of 120–130 °C for furan–maleimide DA networks.31 This increase can be attributed to the effect of the salt hydrate solvent as well as the flexibility of the PEG-based bismaleimide crosslinker which was shown to result in a higher retro-DA reaction temperature compared to that of a stiff crosslinker.48 In contrast to the thermomechanical behavior of the DA salogel, the boronate ester salogel showed a sharp decrease in storage modulus (Fig. 2c) and increase in tanδ (Fig. S8†) with increasing temperature and underwent gel-to-sol transition at Tgel = 65 °C in LNH, which was 55–65 °C lower than the gel-to-sol transition temperature of the DA salogel. Note that boronate ester could not form a gel network in MgNH above its melting point (Fig. 2d and S8†) due to the thermo-reversible nature of the crosslinks.15,49,50 We believe that the persistence of DA elasticity between room temperature and 120 °C is the key for future applications of DA networks for hosting PCM materials with widely different melting temperatures while the temperature responsiveness allows removal of the PCM at the end-of-life cycle.
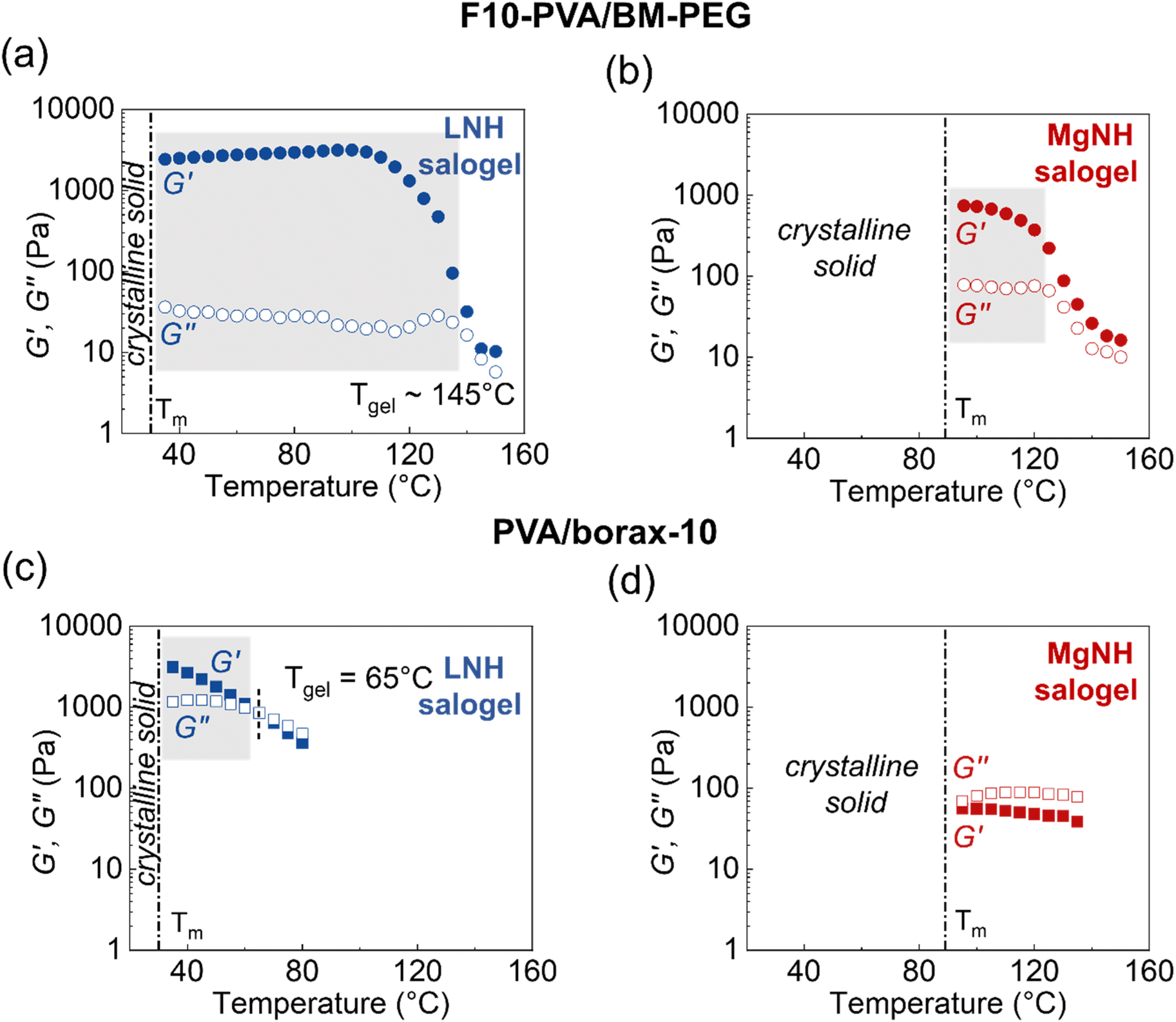 | ||
| Fig. 2 Temperature sweep oscillatory rheology experiments performed at a frequency of 10 rad s−1 and 1% strain comparing DA salogels in (a) LNH and (b) MgNH, and boronate ester salogels in (c) LNH and (d) MgNH. Crosslink density was matched in both salogel systems with 10 mol% of –OH groups consumed in DA and boronate ester crosslinks. The grey boxes in (a)–(c) indicate the temperature ranges over which the DA and boronate ester salogels can operate as a shape stabilizing matrix. Polymer (F10-PVA or PVA) concentration was 5 wt%. | ||
Quantitative insight into DA salogel shape stability is also important since the PCMs are typically subjected to multiple melting and crystallization cycles and exposed to temperatures above the melting point of the PCM over long periods of time. Loss of shape due to creep during thermal cycling may lead to leakage of PCM and inefficient heat transfer resulting in reduced thermal energy storage capability. This feature is especially important in applications where the shape stabilized PCM is used as a free-standing thermal energy storage material.10–12,17–19 Thus, we studied the shape stability characteristics of DA salogels and compared them with those of boronate-ester-based salogels in rheological creep–recovery experiments. The creep resistance experiments were first performed using the salogels with the highest crosslink density (Fig. 3a and b) at room temperature. A stress of 100 Pa was applied for 5 minutes to induce creep after which the stress was removed, and the gels were allowed to recover for 10 minutes. The more elastic DA salogel reached a strain of about 8% after 5 min of stress whereas the boronate ester salogel reached a strain of 20% and did not plateau, indicating a continued creep behavior (Fig. 3a and b). The behavior of the two salogels was also drastically different upon removal of stress: while the boronate ester salogel showed viscoelastic behavior recovering only 46% of strain, DA salogels demonstrated nearly complete strain recovery of ∼96% after 10 minutes (Fig. 3a and b). These data show that DA crosslinks resulted in superior creep resistance and excellent recovery behavior of the salogels compared to the boronate–ester-crosslinked salogels. This was further highlighted when DA salogels with lower crosslink density (2.5 and 5 mol% of –OH groups consumed in crosslinking) were compared with the boronate ester salogels. Fig. 3c, S11, and S12† show that even the lowest-crosslink-density DA salogel outperformed the strongest boronate ester salogel showing a strain recovery of 71% compared to the 46% achieved with the boronate–ester-based salogels. Furthermore, even when the DA salogel was subjected to higher stresses of 500 and 1000 Pa, high strain recovery percentages of 95% and 92%, respectively, were achieved due to the elastic nature of the DA salogels (Fig. 3d).
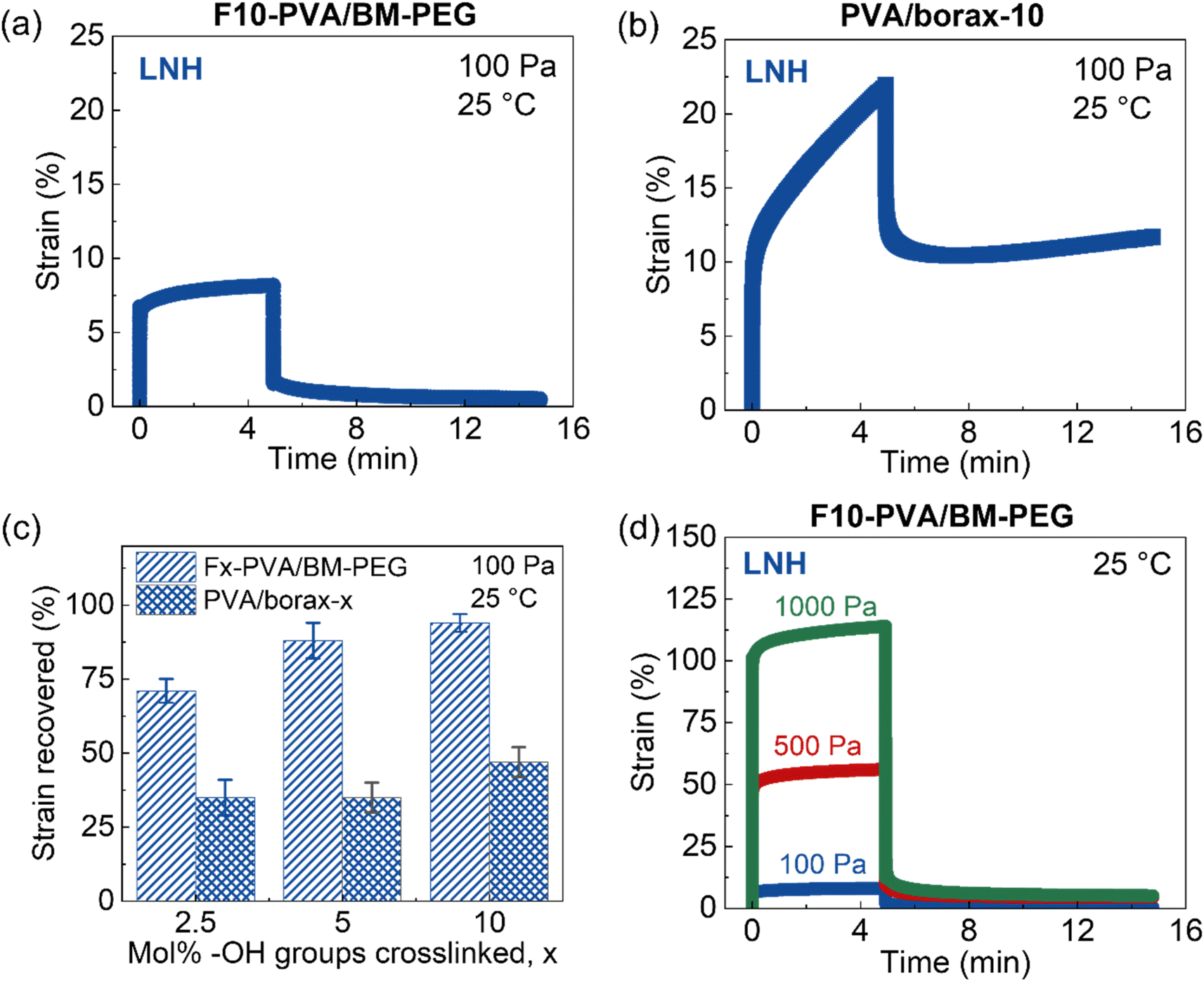 | ||
| Fig. 3 Creep–recovery experiments performed at 25 °C with (a) F10-PVA/BM-PEG and (b) PVA/borax-10 (salogels containing 10 mol% of –OH groups consumed by crosslinks). (c) Comparison of strain recovered after 10 minutes by DA and boronate ester salogels after creep with 100 Pa stress applied as a function of mol% of –OH groups crosslinked. (d) Creep–recovery experiments at different stress levels for the DA salogel where 10 mol% of –OH groups are crosslinked. Note that the DA salogel in MgNH is not shown here since it is in a crystallized state at room temperature. Polymer (F10-PVA or PVA) concentration was 5 wt%. | ||
The potential use of DA chemistry in salogels for shape stabilization of salt hydrates with a wide range of melting points above room temperature requires understanding creep performance as a function of temperature. Therefore, creep–recovery experiments were performed at increased temperature with the stress, creep, and recovery durations kept the same as those at 25 °C. Note that these experiments were performed with only the highest crosslink density DA and boronate ester salogels (10 mol%). Fig. 4a shows that the strain in the DA salogel did not increase with temperature indicating that the DA salogel did not exhibit significant creep due to the DA bonds being intact below TrDA. The strain recovery of the DA salogels was greater than 90% at 35 and 50 °C and only slightly reduced to 85% at 70 °C, still being ∼ three-fold higher than that for the boronate ester salogel at 50 °C (Fig. 4a–c). Note that the boronate ester salogel was not tested at 70 °C since this temperature was above the Tgel of 65 °C. The creep–recovery behavior can be understood from the temperature sweep experiments in Fig. 2 where a decrease in moduli for the boronate ester salogel and no change in moduli for the DA salogel with an increase in temperature up to 120 °C were observed. The ability of DA bonds to shape stabilize a high-melting-temperature salt hydrate PCM was tested by performing creep–recovery experiments on the DA salogel in MgNH. Creep–recovery experiments at 95 °C showed a strain of 15% and recovery of 80% (Fig. 4d) indicating that the DA salogel is a good candidate for shape stabilization of MgNH even at this high temperature.
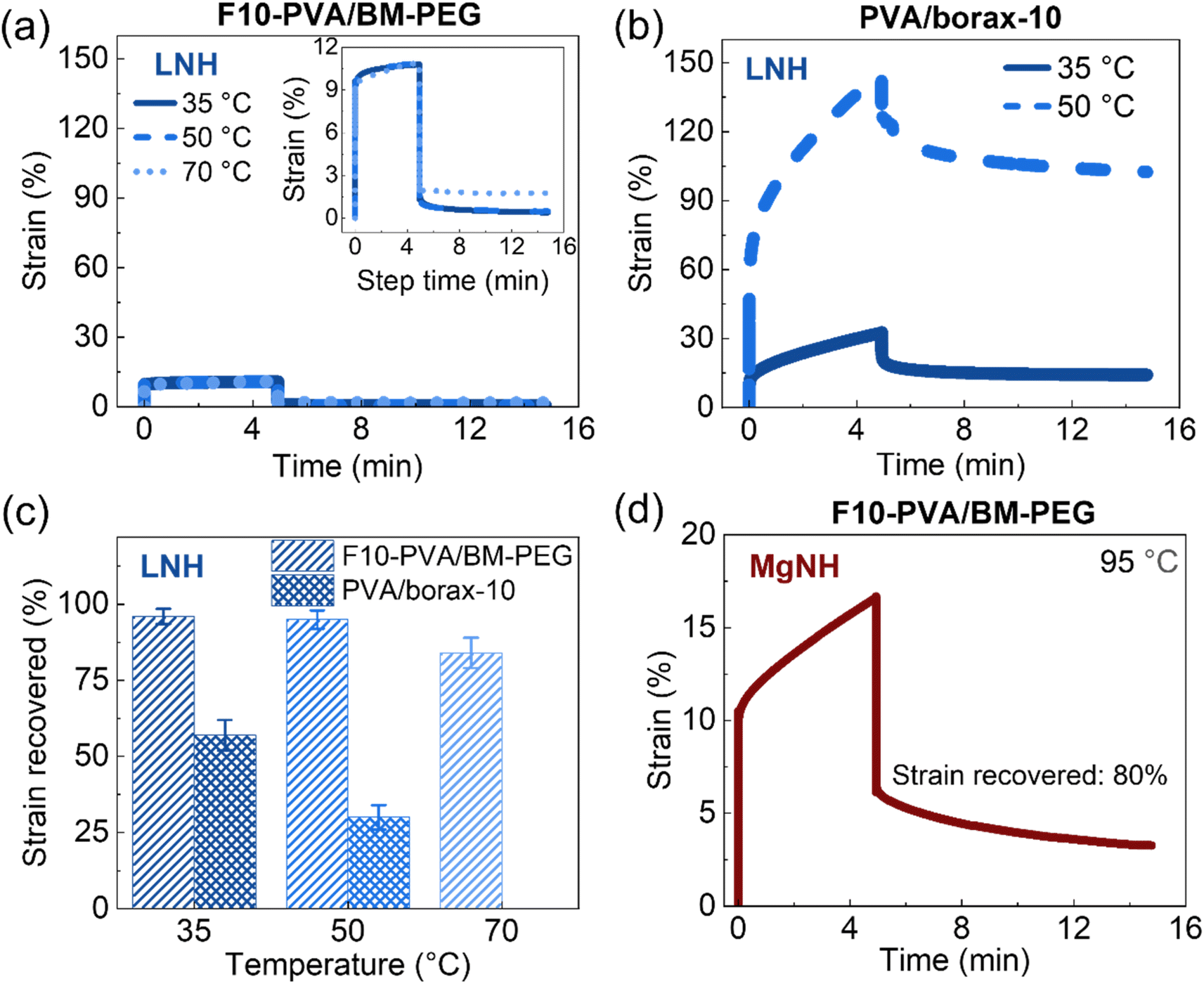 | ||
| Fig. 4 Creep–recovery experiments at different temperatures for (a) F10-PVA/BM-PEG and (b) PVA/borax-10 salogels. (c) Strain recovered after 10 minutes as a function of temperature for the creep–recovery data shown in (a) and (b). (d) Creep–recovery experiment for the F10-PVA/BM-PEG salogel in MgNH at 95 °C. Polymer (F10-PVA or PVA) concentration was 5 wt%. A stress of 100 Pa was applied during the creep phase of the experiment. | ||
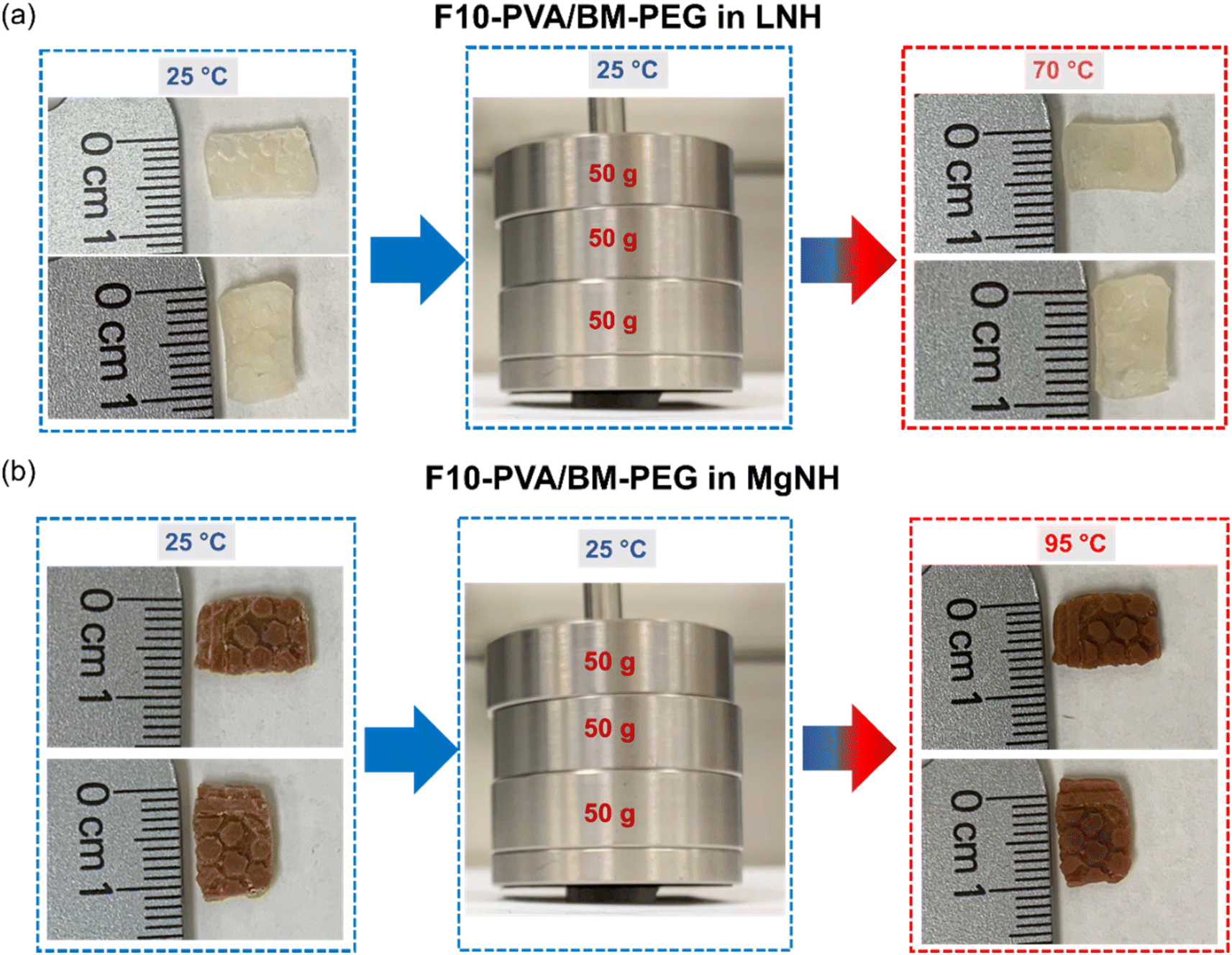
Shape stabilization and leakage prevention capability of LNH and MgNH within the DA salogels could also be observed visually and compared with the boronate ester salogel as a control (only in LNH). For this purpose, bulk hexagon-shaped and rectangular-shaped DA salogels were prepared in LNH and MgNH, respectively, by reacting F10-PVA solution in a molten PCM with BM-PEG in the corresponding mold and crystallizing once the gelation was completed. The boronate ester salogel hexagon was prepared by heating the salogel prepared in a vial to 80 °C, a temperature above the Tgel (65 °C), poured into the hexagon mold and then cooled to room temperature to allow gelation to occur and then crystallized. The LNH-crystallized DA and boronate ester salogels were heated to 70 °C, a temperature well above the melting point of LNH (29 °C) (Fig. 5a and b). While the DA salogel (Fig. 5a) showed no leakage of LNH and no loss of shape even after 1 h (Fig. S13†) due to the superior creep resistance of the DA salogel as demonstrated in Fig. 4a, the boronate ester salogel (Fig. 5b) could not retain its shape above its Tgel of 65 °C. Similarly, heating the MgNH-crystallized salogel to 95 °C (above the melting point of MgNH) did not result in loss of shape of the salogel due to elasticity endowed by DA bonds and no leakage of MgNH was observed (Fig. 5b). The ability of the DA network to prevent the leakage of PCM over an extended period of time was also confirmed from gravimetric experiments. In these experiments, no weight loss was observed when 75 and 60 mg pieces of LNH and MgNH salogels were heated in a closed container at 70 and 95 °C, respectively, for 12 hours. These results also indicate that the DA salogels were able to accommodate the volume change of the salt hydrate PCMs due to density changes during phase transitions. Further, we studied the creep resistance of the bulk DA salogel in LNH and MgNH at 70 and 95 °C, respectively, by performing an experiment where we applied pressure on rectangle shaped DA salogels by placing three 50 gram weights on the salogel for 10 minutes (Fig. 6a and b). Both DA salogel samples showed no loss of shape due to creep (Fig. 6a and b) and no leakage of the salt hydrate from the polymer network as determined from weight measurements on separate samples which showed only a small (∼0.5%) weight decrease. These experiments demonstrate the ability of the DA salogels to work as truly shape stable, leakage-proof thermal energy storage materials. Thus, the DA salogels showed the potential to efficiently entrap salt hydrate PCMs with a wide range of melting temperatures at a low polymer concentration of 5 wt% without loss of shape. This behavior was supported by the elasticity of the DA network between room temperature and the high temperature of the retro-DA reaction – a feature not available with the boronate ester salogel due to its low gel-to-sol transition temperature.
 | ||
| Fig. 5 Optical images of (a) the hexagon shaped DA salogel showing shape retention and leakage prevention of LNH after 5 minutes at 70 °C, (b) PVA/borax-10 salogel lacks shape retention at 70 °C and the sol wets the paper underneath resulting in a heterogeneous appearance, and (c) rectangular shaped DA salogels showing shape retention and leakage prevention of MgNH at 95 °C. The DA salogels in LNH and MgNH are F10-PVA/BM-PEG. | ||
 | ||
| Fig. 6 Optical images showing creep demonstration experiments on DA salogels (F10-PVA/BM-PEG) with no loss of shape or leakage of (a) LNH and (b) MgNH when three 50 gram weights are applied on the salogel for 10 minutes. Polymer (F10-PVA) concentration was 5 wt%. | ||
In addition, DA salogels demonstrated self-healing ability which was supported by the dynamic nature of DA bonds. The self-healing behavior of DA salogels was demonstrated by heating two pieces of the DA salogel in LNH sandwiched between the plates of a rheometer with the solvent trap to ensure no loss of water during the experiment. The salogel pieces were heated to 140 °C, a temperature where the retro-DA reaction is occurring but below the TrDA (∼145 °C) so as not to completely melt the polymer network and held at this temperature for two minutes followed by cooling to room temperature. Fig. S14a† shows that the two pieces of the salogel healed and bonded to each other during this treatment indicating that the thermo-responsive nature of the DA reaction can enable self-healing of the salogel. Moreover, these healed pieces could be handled with tweezers immediately upon cooling to room temperature since the click nature of the DA reaction resulted in the formation of the crosslinks restoring the material's elasticity. The time sweep oscillatory rheology experiment comparing the healed sample to the original sample in Fig. S14b† shows ∼33% recovery of storage modulus (G′) during this short heat treatment.
Evaluation of thermal energy storage materials for potential applications also involves assessment of the materials' thermal energy storage capability. The relatively low (5%) content of polymers in the creep-resistant salogels reported here is an important feature useful for the preservation of the high volumetric energy density of a polymer-entrapped PCM. Melting and crystallization cycles were performed on a DSC with neat LNH and neat MgNH, along with the DA and boronate ester salogels (10 mol% of –OH groups crosslinked) (Fig. 7a and b and S15†). Table 1 shows the heat of fusion and melting temperatures obtained from the endothermic melting peak, and the % heat of fusion of neat PCM retained for the DA salogels. The data for the boronate ester salogel is shown in Table S1.† Note that the enthalpies of crystallization do not represent an equilibrium thermodynamic property due to the large undercooling that exists in this material. Both DA and boronate ester salogels retained >85% of the heat of fusion of the PCM in the respective salogel causing only a 3–6 °C reduction in melting temperature (Tables 1 and S1†). Overall, the polymer and crosslinker added as part of the DA salogel did not affect the melting behavior and thermal properties of LNH and MgNH in the salogel. However, the undercooling during thermal cycling, which is a significant unsolved issue for salt hydrate PCMs21,43 and also present in LNH (∼60 °C) (Fig. 7a) and MgNH (∼25 °C) (Fig. 7b), was reduced in the DA salogels (Fig. S16†). The crystallization onset temperature was even lower in the boronate ester salogel (Fig. S15 and Table S1†) but was increased to about −6 °C (a more than 25 °C increase compared to that of neat LNH) in the DA salogel (Fig. S16† and Table 1). A smaller but yet significant increase of about 10 °C was also observed in the DA salogel in MgNH with crystallization occurring at 74 °C. The data illustrate enhanced heterogeneous nucleation of inorganic salt hydrate crystals on the DA salogel network and demonstrate that the network can not only work as a shape stabilizing matrix for salt hydrates over a wide temperature range but could potentially reduce supercooling as well.
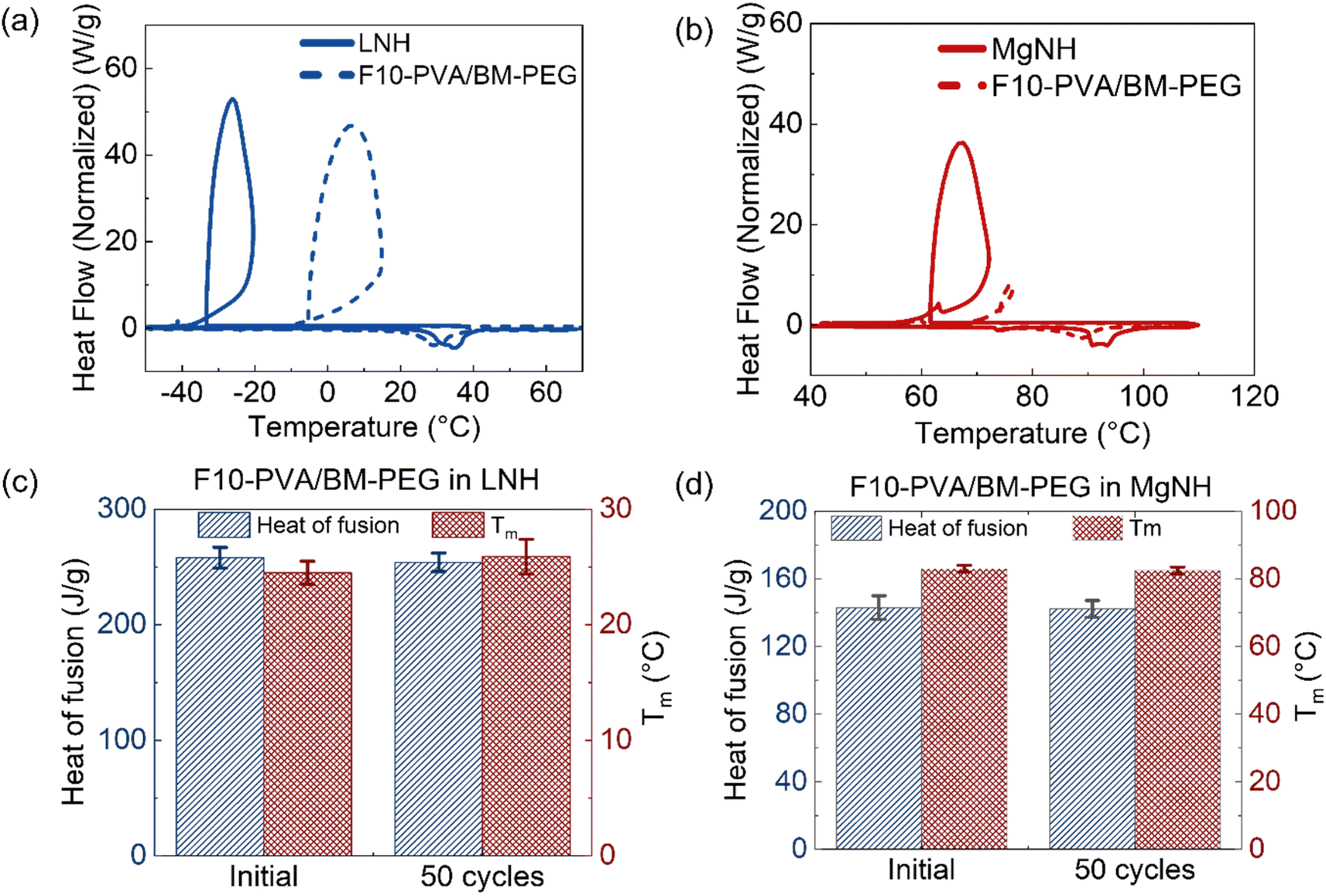 | ||
| Fig. 7 DSC thermograms showing melting and crystallization transitions in (a) neat LNH and DA salogel, and (b) neat MgNH and DA salogels. Comparison of heat of fusion and melting temperature (Tm) in the DA salogel in (c) LNH and (d) MgNH before and after thermal cycling. These experiments were performed with a few grams of DA salogel sealed in a glass vial. | ||
| Sample | Heat of fusion (J g−1) | Melting temperature (°C) | Crystallization temperature (°C) | % heat of fusion retained in the salogel | wt% PCM in the salogel |
|---|---|---|---|---|---|
| a These values were used as reference to calculate the % heat of fusion retention of the salt hydrate in the salogel. | |||||
| LNH | 280 ± 4 | 29 ± 1 | −32 ± 6 | —a | N/A |
| DA salogel in LNH | 258 ± 9 | 25 ± 1 | −6.5 ± 3 | 91 ± 1 | 90 |
| MgNH | 163 ± 3 | 89 ± 1 | 62 ± 2 | —a | N/A |
| DA salogel in MgNH | 143 ± 7 | 83 ± 1 | 72 ± 4 | 88 ± 3 | 90 |
Fig. S17† presents a comparison of the percentage of heat of fusion retention in the salogels as a function of the amount of PCM (LNH and MgNH) in the shape stabilizing matrices found by others and reported in this work. Compared to other DA networks, which were demonstrated with organic PCMs,37–39,42 and the polymer systems containing LNH13,14 and MgNH,18,21–23 the DA salogels reported here show both high PCM loading and heat of fusion retention, which is crucial in their application as thermal energy storage materials.
Finally, Fig. 7c and d and S18† illustrate the ability of the DA salogels to retain thermal characteristics that are important for thermal energy storage applications, over multiple melting and crystallization cycles in both LNH and MgNH. After 50 cycles, the melting and crystallization transitions were not altered (Fig. S18†), and the thermal properties (heat of fusion and melting temperature) remained unaffected in both LNH and MgNH salogels (Fig. 7c and d).
The effect of repeated melting and crystallization on mechanical properties was evaluated by performing the creep–recovery experiment at 70 °C on the salogel in LNH subjected to 50 cycles. Fig. S19† shows that there was a ∼2× reduction in strain after creep and a 2% improvement in strain recovery. These results indicate that the polymer network constructed from just 5% polymer and strengthened using DA crosslinks (Fig. 2a and 4a) can withstand the stress from repeated cycles of crystal formation and melting (Fig. S19†). The gel network not only prevented leakage of liquid salt hydrate from its network but also retained the thermal properties of the salt hydrate both of which are crucial for long-term thermal energy storage applications. These data further confirm that construction of DA salogels is a viable strategy for shape stabilization of salt hydrate PCMs.
Conclusions
In this work, DA crosslinks were used in the highly ionic environment of an inorganic salt hydrate PCM to create a mechanically robust, creep-resistant shape stabilizing matrix. The ability of the DA salogel to achieve shape stabilization at elevated temperatures was demonstrated using low-viscosity salt hydrate PCMs with melting temperatures varying from room temperature (LNH) to 89 °C (MgNH). In comparison to the viscoelastic behavior of boronate ester salogels, the elastic nature of the DA crosslinks resulted in robust salogels which could maintain their moduli over a broad temperature range up to the onset of retro-DA reaction temperature (120 °C) and showed excellent creep resistance and strain recovery. Importantly, this work demonstrated the ability of the DA salogel to shape stabilize a salt hydrate PCM with a high melting temperature – a property not achievable with previously developed salogel materials. In addition, the low polymer concentration of 5 wt% in DA salogels had effective energy storage capabilities, showing heat of fusion retention of the neat PCM during multiple melting and crystallization cycles. We believe that the DA crosslinking strategy can be broadly applied to other salt hydrate PCM families such as chlorides, phosphates, acetates, etc. and even organic PCMs via a careful choice of soluble polymer and furan modification strategy. When used with high-melting point PCMs, the DA salogels can be useful as a shape stabilizing matrix for energy harvesting in industrial processes which involve high temperatures in the range of 60–90 °C. At the same time integration of DA networks with PCMs having close-to-physiological melting temperatures yields robust creep-resistant TES materials for applications in personal thermal management and wearable devices.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy (EERE) under the Buildings and Technologies Award Number DE-EE0009155. Use of the Texas A&M University Soft Matter Facility (RRID: SCR_022482) is acknowledged.References
- S. A. Mohamed, F. A. Al-Sulaiman, N. I. Ibrahim, M. H. Zahir, A. Al-Ahmed, R. Saidur, B. S. Yılbaş and A. Z. Sahin, A review on current status and challenges of inorganic phase change materials for thermal energy storage systems, Renewable Sustainable Energy Rev., 2017, 70, 1072–1089 CrossRef CAS.
- P. Dixit, V. J. Reddy, S. Parvate, A. Balwani, J. Singh, T. K. Maiti, A. Dasari and S. Chattopadhyay, Salt hydrate phase change materials: Current state of art and the road ahead, J Energy Storage, 2022, 51, 104360 CrossRef.
- K. K. Rajagopalan, P. Karimineghlani, X. Zhu, P. J. Shamberger and S. A. Sukhishvili, Polymers in molten inorganic salt hydrate phase change materials: solubility and gelation, J. Mater. Chem. A, 2021, 9, 25892–25913 RSC.
- G. Alva, Y. Lin and G. Fang, An overview of thermal energy storage systems, Energy, 2018, 144, 341–378 CrossRef.
- M. Telkes, Thermal energy storage in salt hydrates, Sol. Energy Mater., 1980, 2(4), 381–393 CrossRef CAS.
- K. Yu, Y. Liu and Y. Yang, Review on form-stable inorganic hydrated salt phase change materials: Preparation, characterization and effect on the thermophysical properties, Appl. Energy, 2021, 292, 116845 CrossRef CAS.
- T. Wang, N. Wu, H. Li, Q.-L. Lu and Y. Jiang, Preparation and properties of a form-stable phase-change hydrogel for thermal energy storage, J. Appl. Polym. Sci., 2016, 133(34), 43836 CrossRef.
- X. Qi, T. Zhu, W. Hu, W. Jiang, J. Yang, Q. Lin and Y. Wang, Multifunctional polyacrylamide/hydrated salt/MXene phase change hydrogels with high thermal energy storage, photothermal conversion capability and strain sensitivity for personal healthcare, Compos. Sci. Technol., 2023, 234, 109947 CrossRef CAS.
- K. Yu, Y. Liu, F. Sun, M. Jia and Y. Yang, Graphene-Modified Hydrate Salt/UV-Curable Resin Form-Stable Phase Change Materials: Continuously Adjustable Phase Change Temperature and Ultrafast Solar-to-Thermal Conversion, Energy Fuels, 2019, 33(8), 7634–7644 CrossRef CAS.
- Y. Luo, W. Yu, J. Qiao, X. Zhao, H. Wu, X. Sheng, Y. Chen and P. Lin, Self-healing inorganic hydrated salt gels for personal thermal management in the static and dynamic modes, Chem. Eng. J., 2022, 440, 135632 CrossRef CAS.
- C. Yin, J. Lan, X. Wang, Y. Zhang, R. Ran and L.-Y. Shi, Shape-Stable Hydrated Salts/Polyacrylamide Phase-Change Organohydrogels for Smart Temperature Management, ACS Appl. Mater. Interfaces, 2021, 13(18), 21810–21821 CrossRef CAS.
- M. Song, L. Wang, F. Shao, H. Xie, H. Xu and W. Yu, Thermally induced flexible phase change hydrogels for solar thermal storage and human thermal management, Chem. Eng. J., 2023, 464, 142682 CrossRef CAS.
- P. Karimineghlani, E. Emmons, M. J. Green, P. Shamberger and S. A. Sukhishvili, A temperature-responsive poly(vinyl alcohol) gel for controlling fluidity of an inorganic phase change material, J. Mater. Chem. A, 2017, 5(24), 12474–12482 RSC.
- P. Karimineghlani, A. Palanisamy and S. A. Sukhishvili, Self-Healing Phase Change Salogels with Tunable Gelation Temperature, ACS Appl. Mater. Interfaces, 2018, 10(17), 14786–14795 CrossRef CAS PubMed.
- K. K. Rajagopalan, X. Zhu and S. A. Sukhishvili, Strong, thermo-reversible salogels with boronate ester bonds as thermal energy storage materials, J. Mater. Chem. A, 2022, 10(40), 21622–21632 RSC.
- K. K. Rajagopalan, S. Haney, P. J. Shamberger and S. A. Sukhishvili, Hybrid Polymer Salogels for Reversible Entrapment of Salt-Hydrate-Based Thermal Energy Storage Materials, ACS Appl. Eng. Mater., 2024, 2(3), 553–562 CrossRef CAS.
- Y. Luo, H. Wu, J. Qiao, J. Zhang, K. Liu, L. Zou, Y. Chen and P. Lin, Self-repairing thermal energy storage gels demonstrating superior thermophysical properties and wearability towards personal thermal management in static and dynamic modes, Chem. Eng. J., 2023, 457, 141201 CrossRef CAS.
- S. N. Lak, C.-M. Hsieh, L. AlMahbobi, Y. Wang, A. Chakraborty, C. Yu and E. B. Pentzer, Printing Composites with Salt Hydrate Phase Change Materials for Thermal Energy Storage, ACS Appl. Eng. Mater., 2023, 1(8), 2279–2287 CrossRef CAS.
- Y. Fang, X. Xiong, L. Yang, W. Yang, H. Wang, Q. Wu, Q. Liu and J. Cui, Phase Change Hydrogels for Bio-Inspired Adhesion and Energy Exchange Applications, Adv. Funct. Mater., 2023, 33(27), 2301505 CrossRef CAS.
- C. Yin, J. Sun, C. Cui, K.-K. Yang, L.-Y. Shi and Y. Li, Chaotropic Ions Mediated Polymer Gelation for Thermal Management, Adv. Sci., 2024, 11(32), 2405077 CrossRef CAS.
- H. Wang, L. Guo, K. Liu, Z. Song, L. Wu, M. Fang and J. Li, Investigation of magnesium nitrate hexahydrate based phase change materials containing nanoparticles for thermal energy storage, Mater. Res. Express, 2019, 6(10), 105512 CrossRef CAS.
- Y. Zhang, J. Sun, G. Ma, Z. Wang, S. Xie, Y. Jing and Y. Jia, Hydrophilic expanded graphite–magnesium nitrate hexahydrate composite phase change materials: Understanding the effect of hydrophilic modification on thermophysical properties, Int. J. Energy Res., 2019, 43(3), 1121–1132 CrossRef CAS.
- Y.-F. Shih, Z.-T. Liao, N.-C. Tsai and Y.-H. Chen, The application of magnesium nitrate hexahydrate/hybrid carbon material phase change composites in solar thermal storage, J. Chin. Chem. Soc., 2023, 70(8), 1644–1655 Search PubMed.
- B. C. Zhao, T. X. Li, F. He, J. C. Gao and R. Z. Wang, Demonstration of Mg(NO3)2·6H2O-based composite phase change material for practical-scale medium-low temperature thermal energy storage, Energy, 2020, 201, 117711 CrossRef CAS.
- R. Naumann, T. Fanghänel and H. H. Emons, Thermoanalytical investigation of sodium acetate trihydrate for application as a latent heat thermal energy storage material, J. Therm. Anal., 1988, 33(3), 685–690 CrossRef.
- Y. Li, C. Wang, J. Zong, J. Ma and Y. Fang, Experimental Research of the Heat Storage Performance of a Magnesium Nitrate Hexahydrate-Based Phase Change Material for Building Heating, Energies, 2021, 14(21), 7108 CrossRef CAS.
- K. Nagano, K. Ogawa, T. Mochida, K. Hayashi and H. Ogoshi, Performance of heat charge/discharge of magnesium nitrate hexahydrate and magnesium chloride hexahydrate mixture to a single vertical tube for a latent heat storage system, Appl. Therm. Eng., 2004, 24(2), 209–220 CrossRef CAS.
- C. García-Astrain, A. Gandini, C. Peña, I. Algar, A. Eceiza, M. Corcuera and N. Gabilondo, Diels–Alder “click” chemistry for the cross-linking of furfuryl-gelatin-polyetheramine hydrogels, RSC Adv., 2014, 4(67), 35578–35587 RSC.
- H.-L. Wei, Z. Yang, L.-M. Zheng and Y.-M. Shen, Thermosensitive hydrogels synthesized by fast Diels–Alder reaction in water, Polymer, 2009, 50(13), 2836–2840 CrossRef CAS.
- R. C. Cioc, M. Crockatt, J. C. van der Waal and P. C. A. Bruijnincx, The Interplay between Kinetics and Thermodynamics in Furan Diels–Alder Chemistry for Sustainable Chemicals Production, Angew. Chem., Int. Ed., 2022, 61(17), e202114720 CrossRef CAS PubMed.
- Q. Zhou, F. Gardea, Z. Sang, S. Lee, M. Pharr and S. A. Sukhishvili, A Tailorable Family of Elastomeric-to-Rigid, 3D Printable, Interbonding Polymer Networks, Adv. Mater., 2020, 30(30), 2002374 Search PubMed.
- A. Gandini, The furan/maleimide Diels–Alder reaction: A versatile click–unclick tool in macromolecular synthesis, Prog. Polym. Sci., 2013, 38(1), 1–29 CrossRef CAS.
- S. Kirchhof, F. P. Brandl, N. Hammer and A. M. Goepferich, Investigation of the Diels–Alder reaction as a cross-linking mechanism for degradable poly(ethylene glycol) based hydrogels, J. Mater. Chem. B, 2013, 1(37), 4855–4864 RSC.
- L. J. Smith, S. M. Taimoory, R. Y. Tam, A. E. G. Baker, N. Binth Mohammad, J. F. Trant and M. S. Shoichet, Diels–Alder Click-Cross-Linked Hydrogels with Increased Reactivity Enable 3D Cell Encapsulation, Biomacromolecules, 2018, 19(3), 926–935 CrossRef CAS PubMed.
- C. M. Nimmo, S. C. Owen and M. S. Shoichet, Diels−Alder Click Cross-Linked Hyaluronic Acid Hydrogels for Tissue Engineering, Biomacromolecules, 2011, 12(3), 824–830 CrossRef CAS.
- C. M. Madl and S. C. Heilshorn, Rapid Diels–Alder Cross-linking of Cell Encapsulating Hydrogels, Chem. Mater., 2019, 31(19), 8035–8043 CrossRef CAS PubMed.
- Z. Liu, X. Zhu, Y. Tian, K. Zhou, J. Cheng and J. Zhang, Bio-based recyclable Form-Stable phase change material based on thermally reversible Diels–Alder reaction for sustainable thermal energy storage, Chem. Eng. J., 2022, 448, 137749 CrossRef CAS.
- B. Wu, Y. Wang, Z. Liu, Y. Liu, X. Fu, W. Kong, L. Jiang, Y. Yuan, X. Zhang and J. Lei, Thermally reliable, recyclable and malleable solid–solid phase-change materials through the classical Diels–Alder reaction for sustainable thermal energy storage, J. Mater. Chem. A, 2019, 7(38), 21802–21811 RSC.
- C. Lin, H. Ge, P. Ying, T. Wang, M. Huang, P. Zhang, T. Yang, J. Wu and V. Levchenko, Synthesis and Properties of Dynamic Crosslinking Polyurethane/PEG Shape-Stable Phase Change Materials Based on the Diels–Alder Reaction, ACS Appl. Polym. Mater., 2023, 5(6), 4190–4198 CrossRef CAS.
- G. Zhang, Q. Zhao, L. Yang, W. Zou, X. Xi and T. Xie, Exploring Dynamic Equilibrium of Diels–Alder Reaction for Solid State Plasticity in Remoldable Shape Memory Polymer Network, ACS Macro Lett., 2016, 5(7), 805–808 CrossRef CAS PubMed.
- A. M. Peterson, R. E. Jensen and G. R. Palmese, Room-Temperature Healing of a Thermosetting Polymer Using the Diels−Alder Reaction, ACS Appl. Mater. Interfaces, 2010, 2(4), 1141–1149 CrossRef CAS.
- S. Yang, X. Du, S. Deng, J. Qiu, Z. Du, X. Cheng and H. Wang, Recyclable and self-healing polyurethane composites based on Diels-Alder reaction for efficient solar-to-thermal energy storage, Chem. Eng. J., 2020, 398, 125654 CrossRef CAS.
- P. J. Shamberger and T. Reid, Thermophysical Properties of Lithium Nitrate Trihydrate from (253 to 353) K, J. Chem. Eng. Data, 2012, 57(5), 1404–1411 CrossRef CAS.
- M. N. Belgacem, J. Quillerou, A. Gandini, J. Rivero and G. Roux, Urethanes and polyurethanes bearing furan moieties—2. comparative kinetics and mechanism of the formation of furanic and other monourethanes, Eur. Polym. J., 1989, 25(11), 1125–1130 CrossRef CAS.
- T. Elschner, F. Obst, K. Stana-Kleinschek, R. Kargl and T. Heinze, Synthesis and film formation of furfuryl- and maleimido carbonic acid derivatives of dextran, Carbohydr. Polym., 2017, 161, 1–9 CrossRef CAS.
- A. Sharma, V. V. Tyagi, C. Chen and D. Buddhi, Review on thermal energy storage with phase change materials and applications, Renewable Sustainable Energy Rev., 2009, 13(2), 318–345 CrossRef CAS.
- L. Xu, C. Li and K. Y. S. Ng, In-Situ Monitoring of Urethane Formation by FTIR and Raman Spectroscopy, J. Phys. Chem. A, 2000, 104(17), 3952–3957 CrossRef CAS.
- Q. Zhou, Z. Sang, K. K. Rajagopalan, Y. Sliozberg, F. Gardea and S. A. Sukhishvili, Thermodynamics and Stereochemistry of Diels–Alder Polymer Networks: Role of Crosslinker Flexibility and Crosslinking Density, Macromolecules, 2021, 54(22), 10510–10519 CrossRef CAS.
- K. Koga, A. Takada and N. Nemoto, Dynamic Light Scattering and Dynamic Viscoelasticity of Poly (vinyl alcohol) in Aqueous Borax Solutions. 5. Temperature Effects, Macromolecules, 1999, 32(26), 8872–8879 Search PubMed.
- E. Pezron, L. Leibler, A. Ricard and R. Audebert, Reversible gel formation induced by ion complexation. 2. Phase diagrams, Macromolecules, 1988, 21(4), 1126–1131 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07157c |
| This journal is © The Royal Society of Chemistry 2025 |