
Untangling the potential of non-entangled bottlebrush block copolymers as separator coating materials for high-rate and long-life sodium metal batteries†
Daun
Jeong
ab,
Gyu Hwang
Shin
c,
Hyojun
Lim
a,
Seunghyeon
Kim
d,
Da-Sol
Kwon
ae,
Jinsol
Yook
f,
Sang-Ok
Kim
 ag,
Kyung Yoon
Chung
ag,
Kyeounghak
Kim
*c and
Jimin
Shim
*d
ag,
Kyung Yoon
Chung
ag,
Kyeounghak
Kim
*c and
Jimin
Shim
*d
aEnergy Storage Research Center, Korea Institute of Science and Technology (KIST), 14 Gil 5 Hwarang-ro, Seongbuk-gu, Seoul 02792, Republic of Korea
bDepartment of Chemical Engineering and Materials Science, University of Minnesota, Minneapolis, Minnesota 55455, USA
cDepartment of Chemical Engineering, Hanyang University, 222 Wangsimni-ro, Seongdong-gu, Seoul 04763, Republic of Korea. E-mail: chemekim@hanyang.ac.kr
dDepartment of Chemistry Education, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Republic of Korea. E-mail: jiminshim@snu.ac.kr
eDepartment of Chemical and Biological Engineering, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
fDepartment of Chemistry, University of Minnesota, Minneapolis, Minnesota 55455, USA
gDivision of Energy & Environment Technology, KIST School, University of Science and Technology, Seoul 02792, Republic of Korea
First published on 12th December 2024
Abstract
Sodium (Na) metal batteries are of great interest as next-generation battery systems due to the high energy density, natural abundance, and cost advantages of Na metal. However, Na metal systems face significant challenges, primarily due to uncontrolled Na electrodeposition and unstable electrolyte–electrode interphases, leading to collateral cell failures. Herein, we introduce a bottlebrush block copolymer (BBP), consisting of oligomeric poly(ethylene oxide) (PEO) and polystyrene blocks, as a coating material for conventional glass fiber (GF) separators. The BBP exhibits a controlled hexagonal cylindrical morphology and non-entangled topology, which significantly enhances ion-conducting properties and mechanical robustness through well-defined microphase separation. Additionally, the non-entangled topology of the BBP, facilitated by its bottlebrush structure, promotes effective PEO–Na+ coordination, thereby effectively regulating Na+ flux at the electrolyte–electrode interphases. This uniform Na+ flux achieved by the BBP coating ultimately leads to dendrite-free Na deposition and stable electrode interphases, resulting in high-rate and long-lasting Na metal batteries. This study highlights the potential of non-entangled bottlebrush-structured polymers as viable separator coating materials for Na metal battery systems.
Introduction
Na metal batteries have been highlighted as promising next-generation battery systems primarily due to the high theoretical specific capacity (1166 mA h g−1) and low redox potential (−2.71 V vs. standard hydrogen electrode) of the Na metal anode.1–3 While lithium (Li) metal batteries previously held the spotlight, recent concerns regarding Li depletion and the escalating cost of raw materials underscore the importance of exploring Na as a viable alternative owing to its natural abundance and cost advantages.4,5 However, a persistent challenge in utilizing Na metal anodes is the occurrence of catastrophic cell failures accompanied by metallic dendrite growth and uncontrolled solid electrolyte interphase (SEI) formation during cycling.6 These undesired side reactions significantly impact the electrolyte composition, consequently deteriorating the uniform formation of the cathode–electrolyte interphase (CEI).7 Recent research efforts have made strides in mitigating the uncontrolled formation of dendrites and interphase layers through sophisticated engineering of electrolytes,7,8 artificial SEI layers,9,10 electrode structures,11–13 and separators.14,15Among these efforts, developing optimized separators for Na metal batteries holds significant importance because separators play a critical role in guiding Na+ flux between electrodes by regulating ion transport.16 Ensuring a spatially homogeneous Na+ distribution enabled by separators leads to uniform Na deposition during cycling, effectively suppressing dendrite growth and irregular SEI and CEI layers.17 Until now, GF membranes have predominantly been used as separators for Na metal batteries due to their high ionic conductivity and mechanical robustness.18 However, GF lacks the ability to regulate Na+ flux, primarily due to the absence of molecular-level interactions with Na+.19
One of the most accessible sodiophilic polymers is poly(ethylene oxide) (PEO), particularly since its alkali metal ion-coordinative capabilities were established in the 1970s.20 Compared to other polymer candidates, such as polyvinylidene fluoride (PVDF) and polytetrafluoroethylene (PTFE),18,21,22 PEO demonstrates stronger molecular-level interactions with alkali metal ions by forming pseudo-crown ether complexes, wherein a certain number of ether oxygens in PEO coordinate with the metal ions.23 However, it should be noted that PEO derivatives exhibit a semi-crystalline structure due to the partial packing of PEO chains, where the crystallized domains restrict facile coordination with Na+.24 To address this, extensive research efforts have been devoted to suppressing crystallization through structural variations of PEO to promote Na+-coordinating ability.25–30 However, significantly less is known about how polymer architectures and the resulting molecular-level interactions impact dendrite growth and electrode–electrolyte interphases in Na metal batteries.
Herein, we have developed a bottlebrush block copolymer (BBP)-coated GF as separators for Na metal batteries, where the BBP consists of oligomeric PEO and polystyrene bottlebrushes. The special non-entangled topology of the BBP, enabled by its bottlebrush structure, effectively facilitates PEO–Na+ interactions, thus regulating Na+ flux at the electrolyte–electrode interphases. The uniform Na+ flux achieved by the BBP coating ultimately leads to dendrite-free, high-rate, and long-life Na metal batteries. Through comprehensive analyses using X-ray scattering, rheology, and computational simulation, we revealed the potential of non-entangled BBP as separator coating materials for high-performance Na metal batteries.
Results and discussion
Design rationale of the GF-NaBBP separator
The bottlebrush block copolymer, poly(PPG-b-PSt) (BBP), was synthesized by sequential ring-opening metathesis polymerization (ROMP) of the synthesized macromonomers, PPG and PSt (Fig. 1a), where the PPG block regulates Na+ flux and the PSt block imparts mechanical robustness and coating stability. The PPG and PSt macromonomers were synthesized via atom transfer radical polymerization (ATRP) to precisely control their degree of polymerization and dispersity (Scheme S1†). The volume fractions of the PPG and PSt blocks in BBP were determined to be 82% and 18%, respectively, to induce cylindrical phase separation,31 where the PPG block can act as a preferentially oriented ion conducting path while the insulating PSt block with high glass transition temperature provides mechanical integrity.32–35 Approximately 6.3 mol% of glycidyl moiety was statistically incorporated into the PPG block, serving as cross-linking sites for the subsequent dip-coating process. The molecular characteristics of the macromonomers and BBP confirmed by 1H NMR and size exclusion chromatography (SEC) are summarized in Fig. S1–S5 and Table S1.† With molecular weights deliberately targeted around 5 kDa for the macromonomers, which is significantly lower than the entanglement molecular weight (Me) of polystyrene (17 kDa),36 a bottlebrush structure was introduced to decrease topological constraints associated with inter-chain entanglement.37–39 As previously reported, side chains in bottlebrush graft polymers suppress the chain entanglement, as steric repulsion hinders the interpenetration between side chains belonging to neighboring polymers.38 The non-entangled and highly mobile oligomeric PEO chains facilitate dipole–dipole interactions with Na+ rather than engaging in inter- or intra-molecular interactions, effectively regulating Na+ flux at the electrolyte–electrode interphases. The synthesized BBP was coated on GF with NaTFSI via a straightforward dip-coating method to prepare the GF-NaBBP separator, as illustrated in Fig. 1b. After immersing GF in a solution that contains BBP and NaTFSI ([Na+]/[EO] = 0.13), the resulting GF membrane was collected and subjected to heat treatment at 120 °C under vacuum conditions for 15 h to induce a cross-linking reaction between glycidyl moieties of BBP and surface hydroxyl groups of GF (Fig. S6†). Furthermore, this thermal annealing also facilitates microphase separation between the PPG and PSt blocks by promoting self-assembly into the equilibrium morphology.40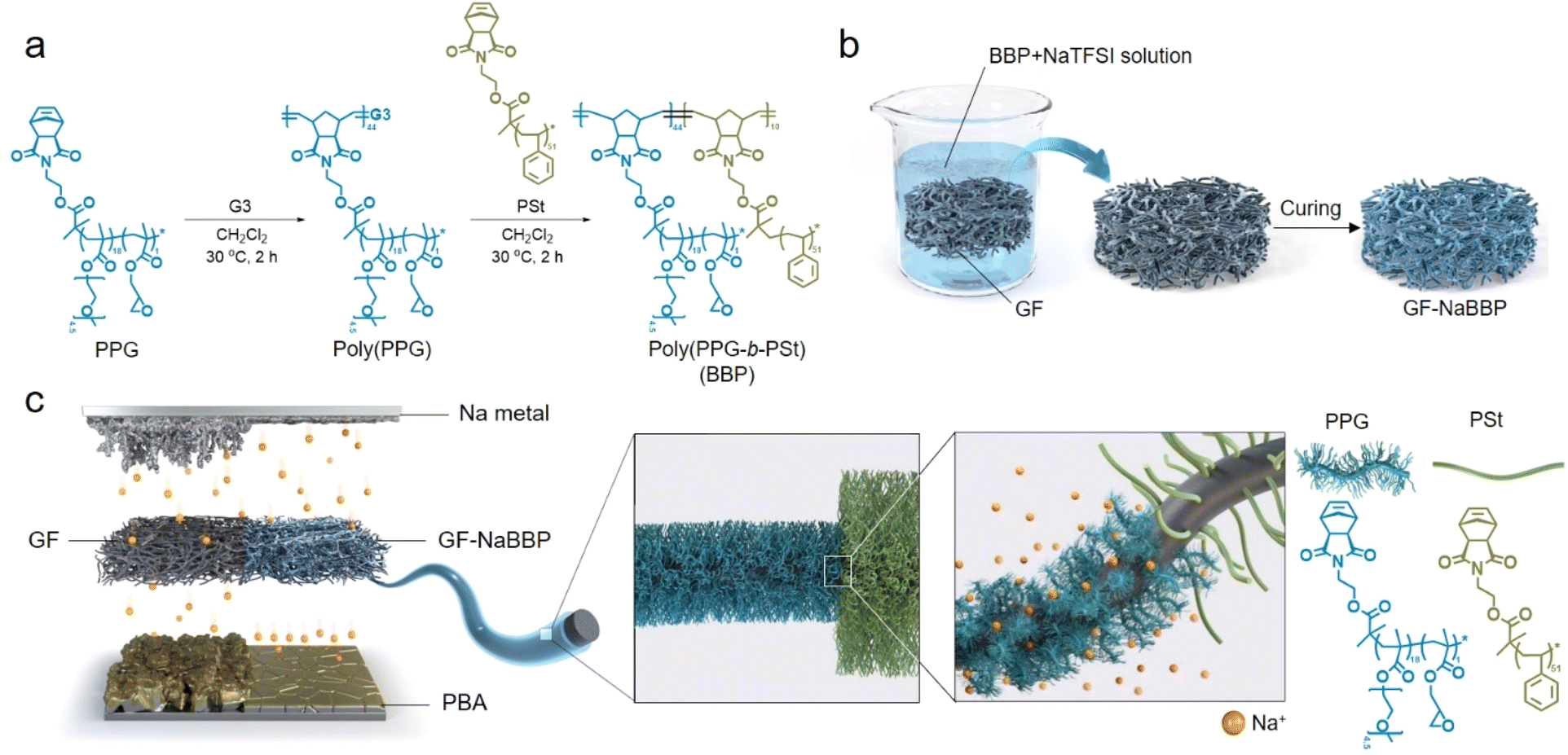 | ||
| Fig. 1 Synthetic strategy and functional contributions of the GF-NaBBP separator. (a) Synthetic route to the bottlebrush block copolymer (BBP) by sequential ROMP. (b) Preparation of GF coated with NaTFSI-doped BBP (GF-NaBBP). (c) Schematic illustration highlighting the functional contributions of GF-NaBBP to regulating Na+ flux and building stable interphases on the electrodes. | ||
Fig. 1c highlights the overall functional contributions of GF-NaBBP when applied to Na metal batteries. By introducing the NaTFSI-doped BBP (NaBBP) coating layer into the GF separator, the Na+ flux-regulating capability is significantly improved. As previously discussed, the non-entangled oligomeric PEO chains within the PPG block primarily coordinate with Na+, resulting in homogeneous Na+ flux over the surfaces of the Na metal anode and Prussian blue analogue (PBA) cathode during cycling. We speculate that this uniform Na+ flux minimizes the localized current density, effectively stabilizing the electrode interphases as well as suppressing Na dendrite growth.41 Furthermore, the microphase separation of the BBP into a cylindrical morphology improves both ionic conductivity and mechanical strength, leading to enhanced electrochemical performance as will be discussed later.32,33,42
Structural characteristics of BBP and NaBBP
To investigate the unique self-assembly behavior of BBP and NaBBP, a series of analyses were conducted. The DSC thermograms in Fig. 2a demonstrate the phase separation of BBP by detecting two distinct Tg values, derived from PPG (−48.1 °C) and PSt (81.3 °C) blocks. Interestingly, upon NaTFSI doping ([Na+]/[EO] = 0.13), the Tg of the PPG block significantly increased from −48.1 to 16.7 °C, while the increase in the Tg of the PSt block was much less pronounced. This finding provides evidence that NaTFSI is exclusively present in the PPG domain through molecular-level interactions between Na+ and the ether oxygens in PPG, leading to the formation of physical cross-links.43 Furthermore, the mechanical properties were also affected by NaTFSI doping, as shown in the representative loading–displacement curves of BBP and NaBBP obtained by nano-indentation (Fig. 2b). It was found that NaBBP required a higher indentation load than BBP to reach an equal displacement, possibly due to the enhanced phase separation upon NaTFSI doping.44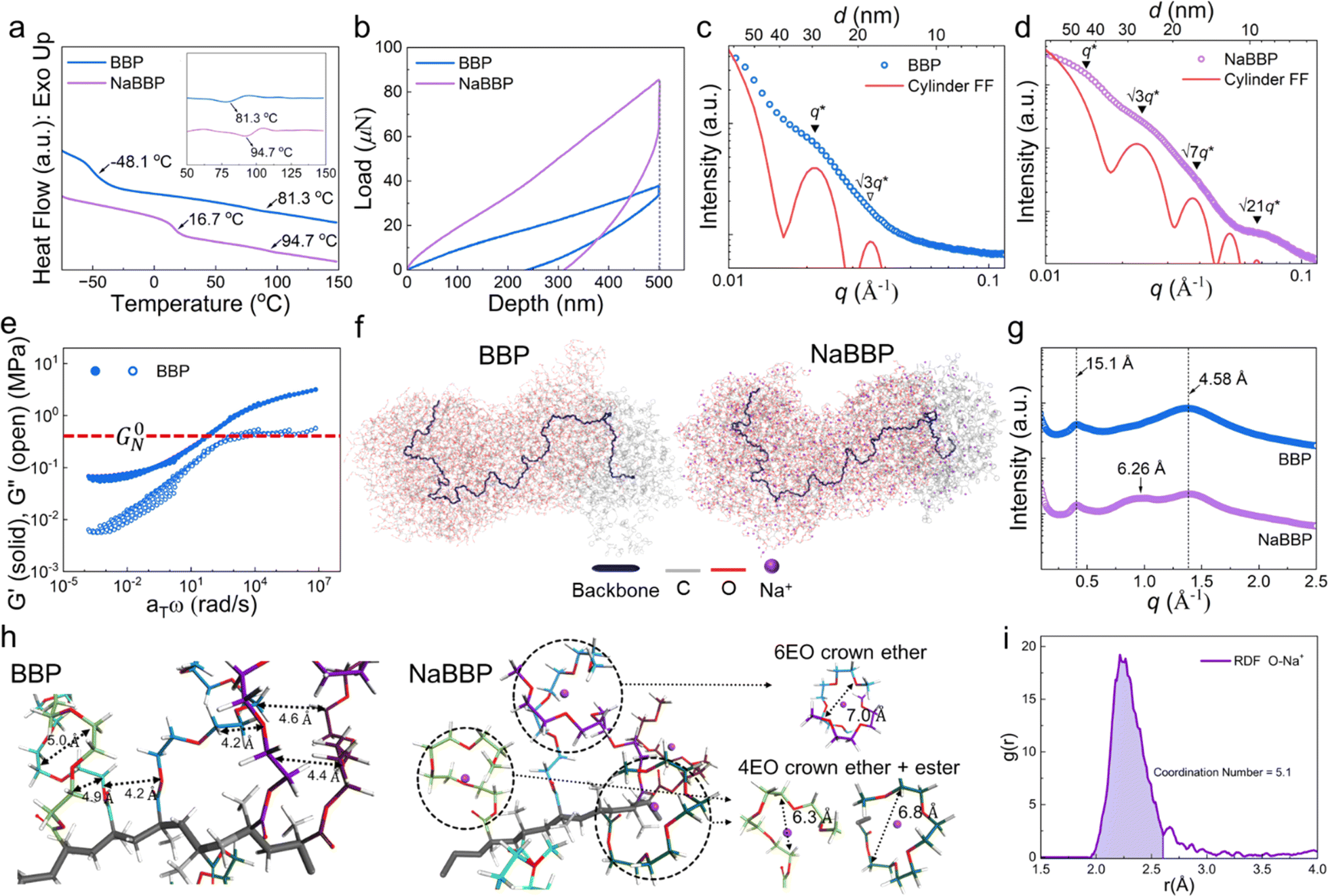 | ||
| Fig. 2 Self-assembly and simulated topologies of BBP and NaBBP. (a) DSC thermograms of BBP and NaBBP, where the inset shows the corresponding 1st derivative profiles. (b) Load–displacement curves of BBP and NaBBP obtained by nano-indentation. SAXS profiles of (c) BBP and (d) NaBBP fitted with the cylindrical form factor (FF) (red line). Solid (▼) and open (▽) triangle symbols denote cylindrical reflections that are present and absent, respectively. (e) Time–temperature superposition (TTS) master curve of BBP, where the red-dashed horizontal line indicates the plateau modulus (G0N). (f) Optimized molecular dynamics (MD) simulation snapshots of BBP and NaBBP. The gray and red colors indicate carbon and oxygen, respectively, and the dark blue line represents the norbornene imide backbone. (g) WAXS profiles of BBP and NaBBP. (h) Detailed images of selected regions from MD simulations of BBP and NaBBP. (i) Radial distribution function (RDF) of O–Na+ at NaBBP. | ||
The equilibrium morphologies of BBP and NaBBP resulting from phase separation were identified by SAXS analysis (Fig. 2c and d). Note that both BBP and NaBBP were heated above their order–disorder transition temperatures (TODT) (Fig. S7†) and then slowly cooled to room temperature to achieve their equilibrium morphologies. Both BBP and NaBBP exhibited a series of broad scattering peaks with a lack of higher-order reflections, presumably because the high molecular weight of BBP (322 kDa) hinders fast equilibrium kinetics of long-range ordering.45 Despite this, BBP and NaBBP are phase-separated into a hexagonal cylindrical structure, as their SAXS patterns correlate well with the cylindrical form factor (FF) curves represented by the following eqn (1):
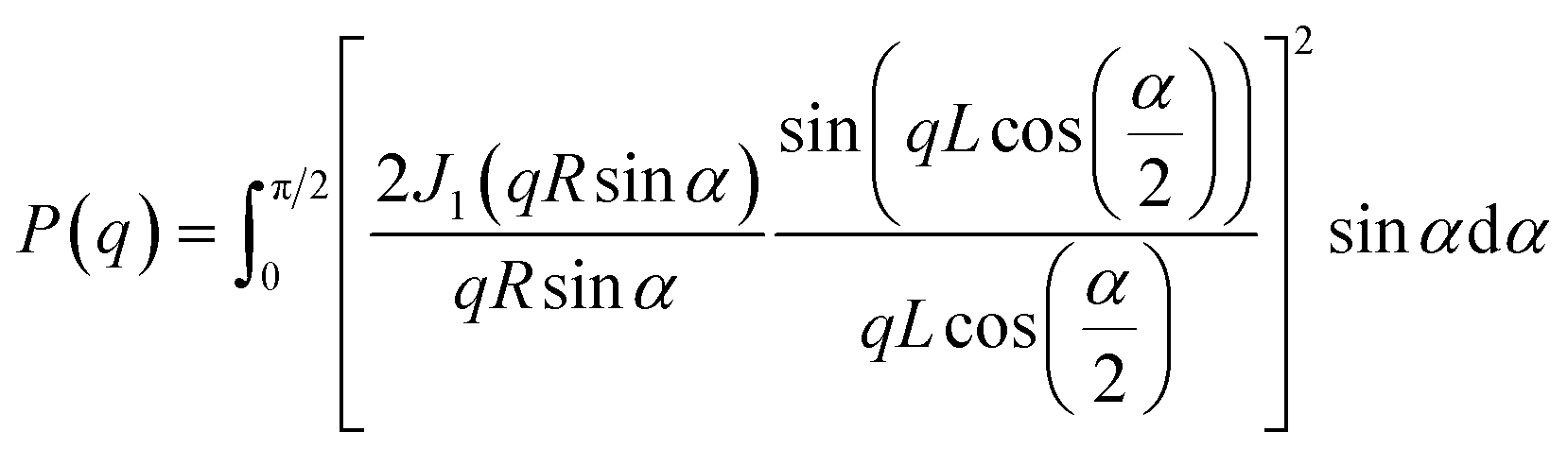 | (1) |
To investigate the special chain entanglement behavior of BBP, rheological characterization was conducted. Fig. 2e exhibits a time–temperature superposition (TTS) master curve of BBP, where each frequency sweep was conducted within a linear viscoelastic regime determined by dynamic strain sweeps. Notably, the absence of relaxation behavior in the high-frequency region indicates the presence of non-entangled side chains.52 Additionally, the Me value of BBP was roughly estimated using eqn (2) as reported by Fetters et al.:
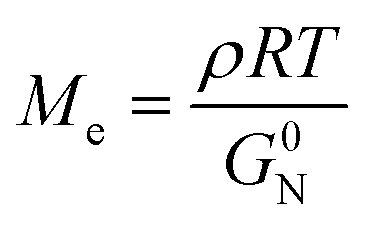 | (2) |
To better expose the molecular-level interactions between Na+ and the bottlebrush-structured PPG block, WAXS analysis was conducted. As shown in Fig. 2g, both BBP and NaBBP exhibited two distinct scattering peaks at q ≈ 0.4 and 1.3 Å−1, corresponding to the average distances between the backbones (15.1 Å) and side chains (4.58 Å), respectively.55 Interestingly, an additional peak appeared only in the case of NaBBP at q ≈ 1.0 Å−1 corresponding to 6.26 Å, which is quantitatively coincident with the average distance between the Na+-doped side chains of the PPG block, as revealed by MD calculations (Fig. 2h). It was found that the ethylene oxide (EO) repeating units in the non-entangled PPG domain stabilize the Na+-doped state by forming pseudo-crown ether complexes with larger inter- and intra-chain distances than the neutral state.56,57 For example, crown ethers consisting of six EO groups exhibited an inter-chain distance of 7.0 Å, while those with four EO and ester groups demonstrated intra-chain distances of 6.3–6.8 Å, matching well with the WAXS results. The radial distribution function (RDF) for Na+ and oxygen (O) observed in NaBBP revealed a strong peak corresponding to the distance between Na+ ions and O atoms in the PPG domains (Fig. 2i). This peak reflects a coordination number (CN) of 5.1, which is consistent with previous observations, as Na+ ions are typically known to exhibit a CN ranging from 5 to 7 with PEO.58–61 Interestingly, NaBBP exhibited the lowest CN (5.1) within this range, particularly noteworthy because a reduced CN implies a lower energy barrier for ion diffusion, thereby enhancing ion conducting properties.62 Furthermore, after the first peak, the g(r) value rapidly decreased, indicating minimal long-range interactions between Na+ ions and O atoms in NaBBP. This suggests that particles behave independently at distances longer than approximately 3.0 Å, facilitating Na+ ion diffusion.63 This intriguing molecular-level interaction between Na+ ions and the PPG domain evidenced by the RDF analysis further demonstrates optimal Na+ ion mobility in NaBBP. Based on a series of computational simulation results, we speculate that at long distances, facile ion conduction is ensured due to minimal long-range interactions between Na+ and the PPG domains, while at shorter distances, NaBBP effectively regulates Na+ flux by forming pseudo-crown ether structures that strongly interact with Na+ ions.
Electrochemical and physical properties of GF and GF-NaBBP
The non-entangled NaBBP with cylindrical morphology was coated onto a GF separator by the dip-coating method as previously described. The resulting GF-NaBBP exhibited a slightly more brownish color compared to the pristine GF, as shown in the optical photographs (Fig. 3a). Furthermore, the surface SEM images of GF and GF-NaBBP show that the NaBBP coating layer surrounds every single fiber of the GF given the apparent decrease in pore size. To quantitatively investigate the change in pore sizes upon introducing the NaBBP coating, mercury (Hg) intrusion porosimetry was conducted. As summarized in Fig. 3b, porosity as well as median and average pore diameters decreased upon NaBBP coating. The more pronounced decrease in the median pore diameter compared to the average pore diameter and porosity indicates that the NaBBP coating primarily fills the large pores of the GF, leading to a uniform pore size distribution. Indeed, the population of large-sized pores significantly decreased in the pore size distribution profiles, while the average pore size did not change significantly (Fig. 3c). This finding demonstrates that a more uniformly distributed porous structure is achieved by NaBBP coating. As revealed by TGA profiles (Fig. 3d), approximately 36 wt% of NaBBP was coated on the GF through the dip-coating method. The FT-IR spectra of GF, NaBBP, and GF-NaBBP further corroborate the presence of the NaBBP coating on the GF (Fig. 3e). The peaks originating from NaBBP, including aliphatic C–H stretching at 2870 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1725 cm−1, C–H aliphatic bending at 1450 cm−1, and C–O vibration at 1098 cm−1, are observed in GF-NaBBP, indicating the successful inclusion of NaBBP on the GF.64,65
O stretching at 1725 cm−1, C–H aliphatic bending at 1450 cm−1, and C–O vibration at 1098 cm−1, are observed in GF-NaBBP, indicating the successful inclusion of NaBBP on the GF.64,65
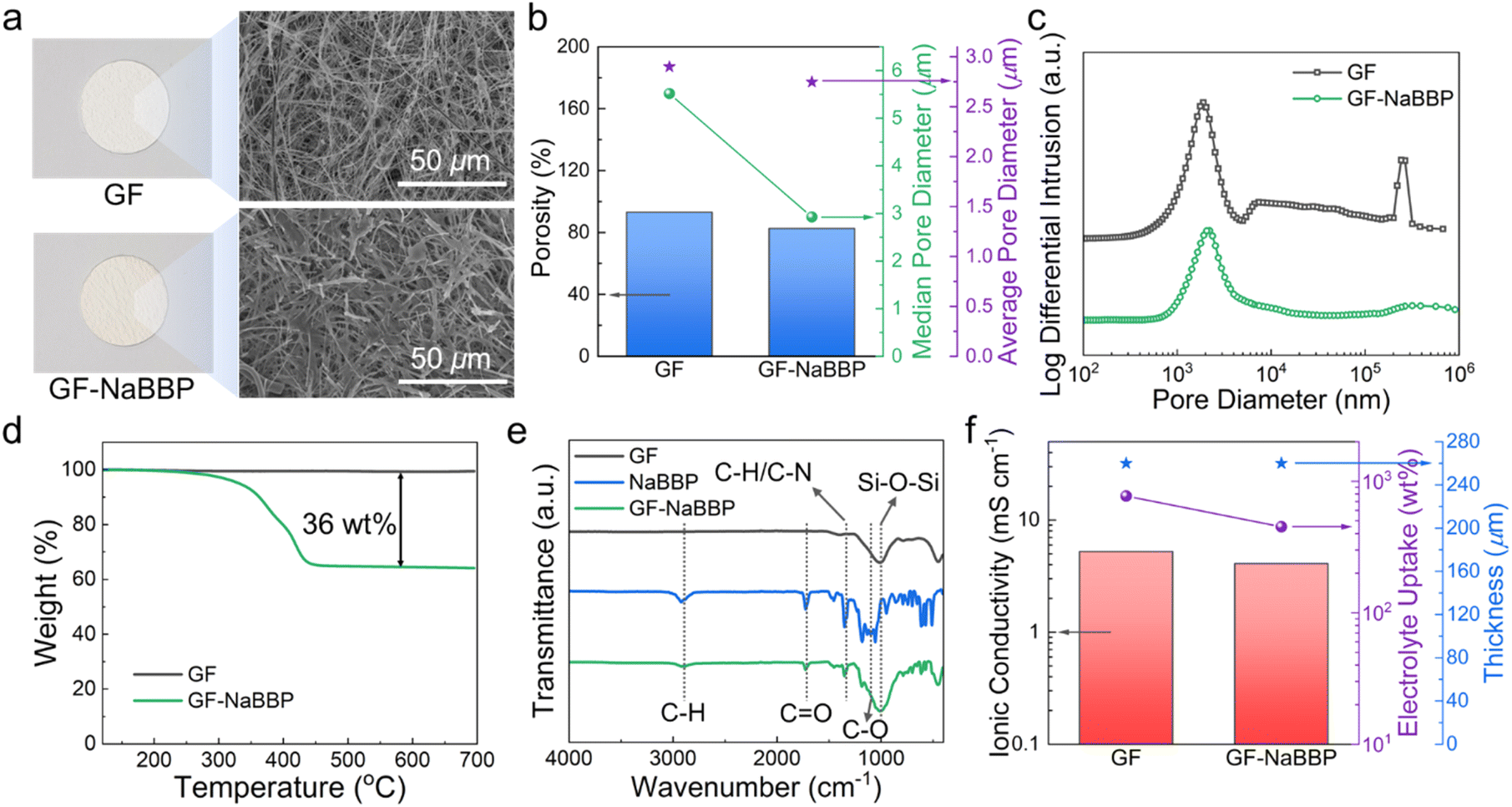 | ||
Fig. 3 Electrochemical and physical properties of GF and GF-NaBBP. (a) Optical photographs and SEM images of GF and GF-NaBBP. (b) Summary of porosities, median pore diameters, and average pore diameters of GF and GF-NaBBP determined by Hg intrusion porosimetry and the corresponding (c) pore size distribution profiles. (d) TGA profiles of GF and GF-NaBBP (heating rate: 10 °C min−1). (e) FT-IR spectra of GF, NaBBP, and GF-NaBBP. (f) Ionic conductivities, electrolyte uptakes, and thicknesses of GF and GF-NaBBP at 30 °C, where the electrolyte is 1 M NaPF6 in EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC:DEC (1:1:1 vol%) with 2 wt% FEC. :PC:DEC (1:1:1 vol%) with 2 wt% FEC. | ||
Fig. 3f summarizes the ionic conductivities, electrolyte uptakes, and thicknesses of GF and GF-NaBBP. Although the electrolyte uptake of GF-NaBBP (451%) decreased compared to that of GF (773%), presumably due to the decreased porosity, the decrease in the ionic conductivity (22%) was much less than that of electrolyte uptake (42%). GF-NaBBP exhibited an ionic conductivity of 4.1 mS cm−1 at 30 °C, comparable to that of GF (5.2 mS cm−1). This result is attributed to the ion-conducting nature of oligomeric PEO in the PPG block, which should contribute to facile ion conduction, as demonstrated by RDF analysis. Interestingly, a nontrivial difference in the ionic conductivity was observed depending on the thermal annealing process during the dip-coating method (Fig. S9†). GF-NaBBP without thermal annealing showed a slightly decreased ionic conductivity (2.9 mS cm−1) and electrolyte uptake (422%) compared to the annealed GF-NaBBP. We speculate that the microphase separation of NaBBP into a cylindrical nanostructure induced by thermal annealing constructs a well-defined ion-conducting path, while the disordered state of non-annealed GF-NaBBP does not provide any preferential conducting path for Na+ (Fig. S10†).32,34,35,66
Na deposition behavior
The Na deposition behavior of Na/Cu cells with GF and GF-NaBBP was compared to evaluate the effectiveness of GF-NaBBP in promoting uniform Na deposition (Fig. 4a). Interestingly, the GF-NaBBP cell exhibited a notably higher areal capacity (0.50 mA h cm−2) than the GF cell (0.33 mA h cm−2) during the initial Na deposition cycle, where the initial capacity was determined by the point where a sudden voltage drop occurs. The lower capacity with GF can be attributed to the formation of an unstable SEI layer due to non-uniform Na deposition, which led to severe side reactions between Na+ and the liquid electrolyte, as well as the formation of dead Na. Furthermore, as revealed by the surface SEM images in Fig. 4b, the deposited Na metal in the GF-NaBBP cell exhibited a surprisingly smooth surface without any dendrites, while that in the GF cell showed a rough surface covered by ill-defined morphologies of Na agglomerates with numerous fibers detached from GF. The cross-sectional SEM images of the Na metal further revealed even more pronounced morphological disparities between the GF and GF-NaBBP cells (Fig. 4c). The deposited Na metal in the GF-NaBBP cell displayed a remarkably clean cross-section without noticeable defects, while that in the GF cell exhibited many microcracks and pinholes, indicative of an inhomogeneous Na deposition. It is striking that the simple NaBBP coating on the GF brings about a significant difference in Na deposition behavior even in the first cycle. We speculate that the homogeneous Na deposition achieved by NaBBP is associated with effectively regulated Na+ flux, primarily enabled by strong molecular interactions between Na+ and non-entangled oligomeric PEO chains in the PPG block. Additionally, achieving uniform pore size distributions upon NaBBP coating further contributes to regulating spatially homogeneous Na+ flux across Na metal surfaces.14 As a result, the GF-NaBBP cell demonstrated significantly prolonged long-term cycling performance, retaining high coulombic efficiency over 100 cycles (Fig. 4d). The GF cell, however, experienced early cell failure accompanied by an abrupt decrease in coulombic efficiency, presumably due to the inhomogeneous Na electrodeposition.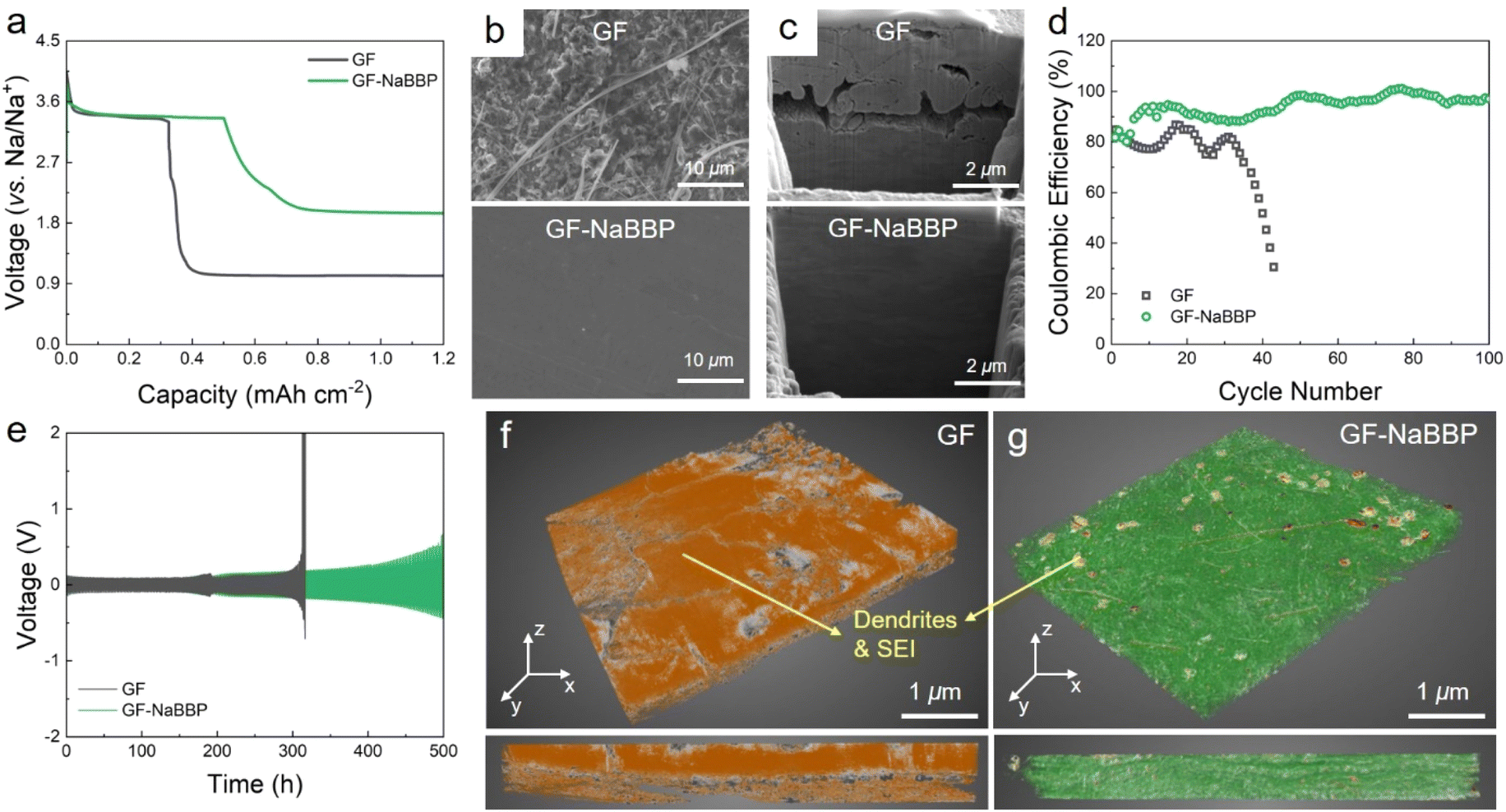 | ||
| Fig. 4 Na deposition behavior. (a) Capacity–voltage profiles corresponding to the 1st Na deposition cycle of the Na/Cu cells with GF and GF-NaBBP at a current density of 0.5 mA cm−2 and a total capacity of 0.5 mA h cm−2 at 30 °C. (b) Surface and (c) cross-sectional SEM images of the deposited Na metal in the GF and GF-NaBBP cells after one cycle. (d) Coulombic efficiency profiles of the Na/Cu cells with GF and GF-NaBBP. (e) Galvanostatic cycling profiles of symmetric Na/Na cells with GF and GF-NaBBP cycled under a current density of 0.5 mA cm−2 and a total capacity of 0.5 mA h cm−2 at 30 °C. 3D XRM images of (f) GF and (g) GF-NaBBP collected from the Na/Na cells after 200 h of cycling, where the orange color indicates Na deposits and SEI layers. The bottom images correspond to the cross-sections of GF and GF-NaBBP. | ||
To further probe the dendrite growth behavior, galvanostatic Na plating–stripping tests were conducted on symmetric Na/Na cells with GF and GF-NaBBP (Fig. 4e). The GF cell exhibited a drastic voltage increase after 300 h, associated with dendrite growth and electrolyte depletion caused by dead Na.67 Although GF exhibited substantial electrolyte uptake and ionic conductivity, the uneven distribution of large pores makes it vulnerable to internal short-circuiting caused by dendrite penetration.17 In contrast, the GF-NaBBP cell demonstrated an extended lifespan of over 500 h without any evidence of short-circuiting, although a gradual increase in overpotential was observed. This result is consistent with the ex situ EIS results obtained during the galvanostatic cycling (Fig. S11†). The GF cell showed large fluctuations in the SEI layer resistance (RSEI) over 300 h, followed by a substantial increase in the overall resistance prior to short-circuiting. This indicates that unstable SEI formation during cycling eventually accelerates dendrite growth and the resulting short-circuit.6 In contrast, the RSEI of the GF-NaBBP cell increased gradually and consistently over 500 h, suggesting that a relatively stable SEI layer is constructed during cycling through uniform Na deposition. As demonstrated by SEM images (Fig. S12†), the cycled Na metal in the GF cell exhibited mossy dendrites that covered the entire surface, while that in the GF-NaBBP cell showed surprisingly homogeneous Na plating. The 3D X-ray microscopy (XRM) images of the cycled GF and GF-NaBBP further highlight the effect of NaBBP coating on dendrite growth and SEI layer formation. As shown in Fig. 4f, the cycled GF was completely covered with Na deposits and SEI layers as indicated by the orange color, and the original pores were almost filled with them. Furthermore, it was difficult to detach the cycled GF from the Na metal presumably, due to Na dendrites penetrating through the pores of the GF (Fig. S13†).68 In sharp contrast, GF-NaBBP was easily separated from the Na metal, revealing only a few noticeable SEI and Na deposit layers in the XRM image (Fig. 4g), while both the fibers and the open pores were well preserved. The cross-sectional XRM images also showed that the thick SEI and Na plating are only detected from the GF.
Cycling performance
The cycling performance of the Na/PBA cells prepared with GF and GF-NaBBP was evaluated. Note that the cathode active material, PBA, was prepared by a precipitation method, and its morphological characteristics, including the crystal structure, were confirmed by SEM and XRD analyses (Fig. S14†). The electrochemical stability of GF and GF-NaBBP within the operation voltage range was preliminarily confirmed by LSV and CV (Fig. S15†). The optimum NaBBP content of GF-NaBBP was determined by comparing the long-term cycling performance of a series of GF-NaBBP with varying NaBBP contents (Fig. S16†). A coating amount of 80 mg of NaBBP was identified as optimum; therefore, all subsequent electrochemical analyses were conducted using this GF-NaBBP. As shown in Fig. 5a, the GF-NaBBP cell demonstrated superior rate capability compared to the GF cell, even up to an extremely high current density of 20C (corresponding to 2.4 A g−1). This enhanced rate performance is attributable to the effective suppression of Na dendrites and the formation of stable electrolyte–electrode interphases facilitated by a uniform Na+ flux through GF-NaBBP.69 Indeed, as shown in Fig. 5b and c, significantly smaller overpotentials in the voltage–capacity curves were observed from the GF-NaBBP cell compared to the GF cell throughout the entire cycle. Furthermore, the GF-NaBBP cell exhibited a noticeable charge/discharge plateau even above 10C, indicating efficient electrochemical kinetics,70,71 while the corresponding plateau in the GF cell became much less distinguishable at high current densities.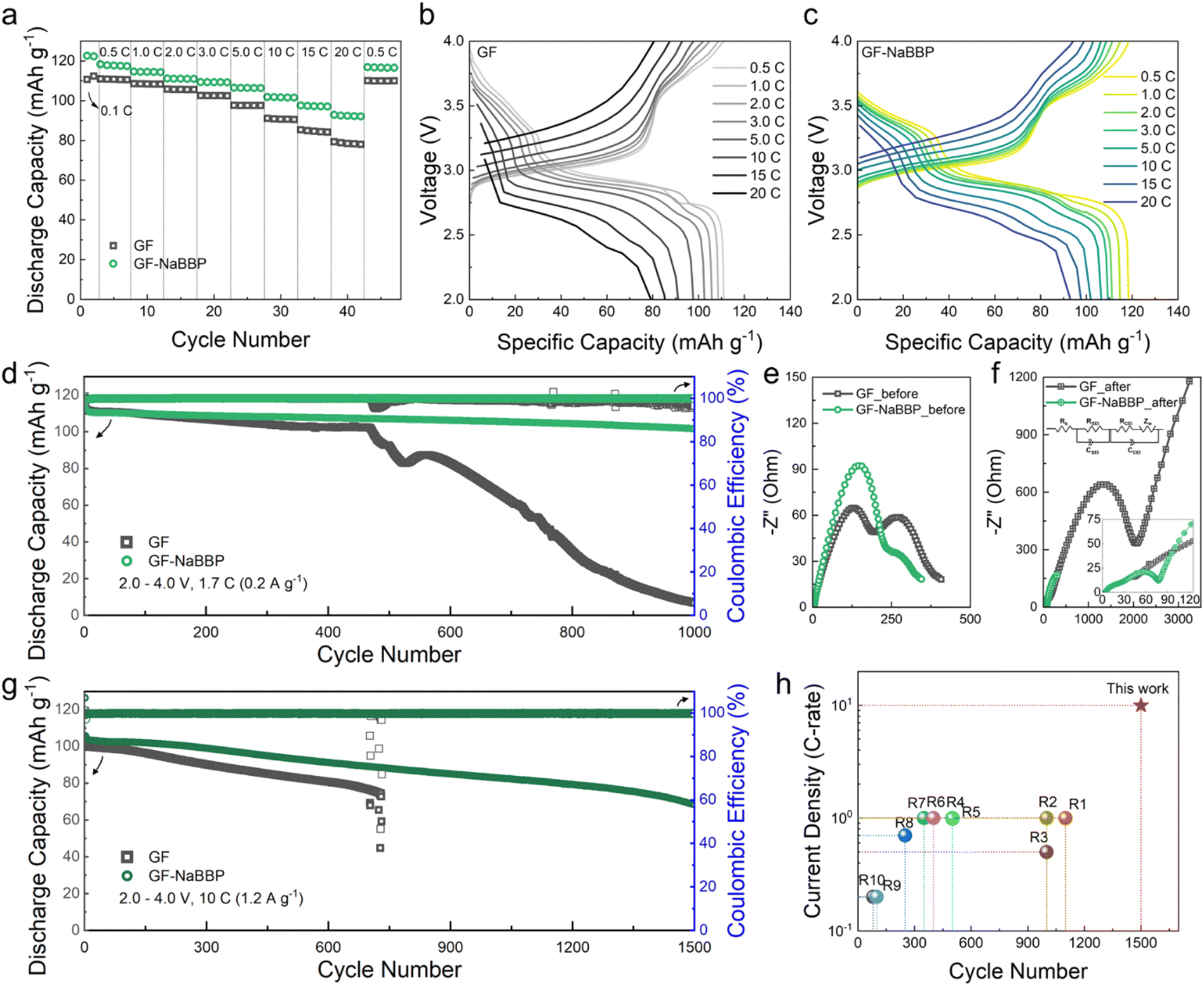 | ||
| Fig. 5 Cycling performance of Na/PBA cells prepared with GF and GF-NaBBP within the voltage range of 2.0–4.0 V at 30 °C. (a) Rate capability of the cells with GF and GF-NaBBP. Voltage–capacity profiles of the cells with (b) GF and (c) GF-NaBBP cycled at 0.5–20C. (d) Long-term cycling performance of the cells with GF and GF-NaBBP cycled at 1.7C (0.2 A g−1). Nyquist plots of the cells with GF and GF-NaBBP (e) before and (f) after 1000 cycles. (g) Long-term cycling performance of the cells with GF and GF-NaBBP cycled at 10C (1.2 A g−1). (h) Comparison of the current density and cycle number of the GF-NaBBP cell compared with other studies. | ||
Fig. 5d shows the long-term cycling performance of the GF and GF-NaBBP cells cycled at 1.7C (corresponding to 0.2 A g−1) (see Fig. S17† for the voltage–capacity curves). The GF-NaBBP cell exhibited a remarkably high capacity retention of 90% after 1000 cycles (approximately 0.0096% decay per cycle), surpassing the GF cell, which experienced a drastic capacity decay after 500 cycles. The cell failure observed from the GF cell should be primarily associated with dendrite growth.6 Furthermore, the coulombic efficiency of the GF-NaBBP cell remained strikingly stable compared to the GF cell (Fig. S18†). This superior cycling performance of the GF-NaBBP cell over the GF counterpart primarily stems from the formation of stable SEI and CEI layers,72 as corroborated by the EIS results (Fig. 5e and f). After 1000 cycles, the GF-NaBBP cell showed substantially smaller charge transfer resistances, representing SEI and CEI layer resistances, compared to the GF cell.73 These results indicate the formation of much more stable electrolyte–electrode interphases in the GF-NaBBP cell, presumably enabled by Na+ flux regulation.17,74 We further evaluated the long-term cycling performance of the GF-NaBBP cell under an extremely high current density of 10C (1.2 A g−1, Fig. 5g) (see Fig. S19† for the voltage–capacity profiles). Notably, the GF-NaBBP cell retained sufficient discharge capacities even after 1500 cycles, while the GF cell failed before 800 cycles. As summarized in Fig. 5h and Table S2,† GF-NaBBP exhibited superior cycling performance compared to many other previous reports, surpassing them in terms of rate property and long-term cycling.
Post-mortem analyses of the electrodes
To demonstrate our speculation that a stable interphase layer is constructed by introducing GF-NaBBP, a comprehensive series of post-mortem analyses were conducted. Fig. 6a presents the surface SEM images of the cycled Na metal from the GF and GF-NaBBP cells. On the surface of Na metal from the GF cell, numerous mossy dendrites and detached GF fibers were observed. In contrast, Na deposits in the GF-NaBBP cell exhibited much smoother and more compact morphologies, indicating the uniform Na deposition regulated by GF-NaBBP.21 Cross-sectional SEM images further highlight significant differences in the morphologies of Na deposits between the two cases. As shown in Fig. 6b, the cycled Na metal in the GF-NaBBP cell exhibited a more compact and smooth cross-section compared to that in the GF cell. The surface morphology of the GF and GF-NaBBP separators after cycling also demonstrates the effectiveness of GF-NaBBP in stabilizing interphase and suppressing dendrite growth (Fig. S20†). Remarkably, GF and GF-NaBBP exhibited significantly different morphologies: the surface of cycled GF was completely covered by thick and rough Na deposits, while GF-NaBBP largely retained its pristine morphology without any deposited Na. High-magnification images further revealed that the pores of GF were almost entirely filled with Na deposits and dendrites, which penetrated the large pores during cycling. However, GF-NaBBP exhibited well preserved pristine morphology (Fig. 3a), verifying that GF-NaBBP effectively inhibited Na dendrite growth and stabilized the SEI layers by promoting uniform Na deposition during cycling. These findings strongly support the 3D-XRM results (Fig. 4f).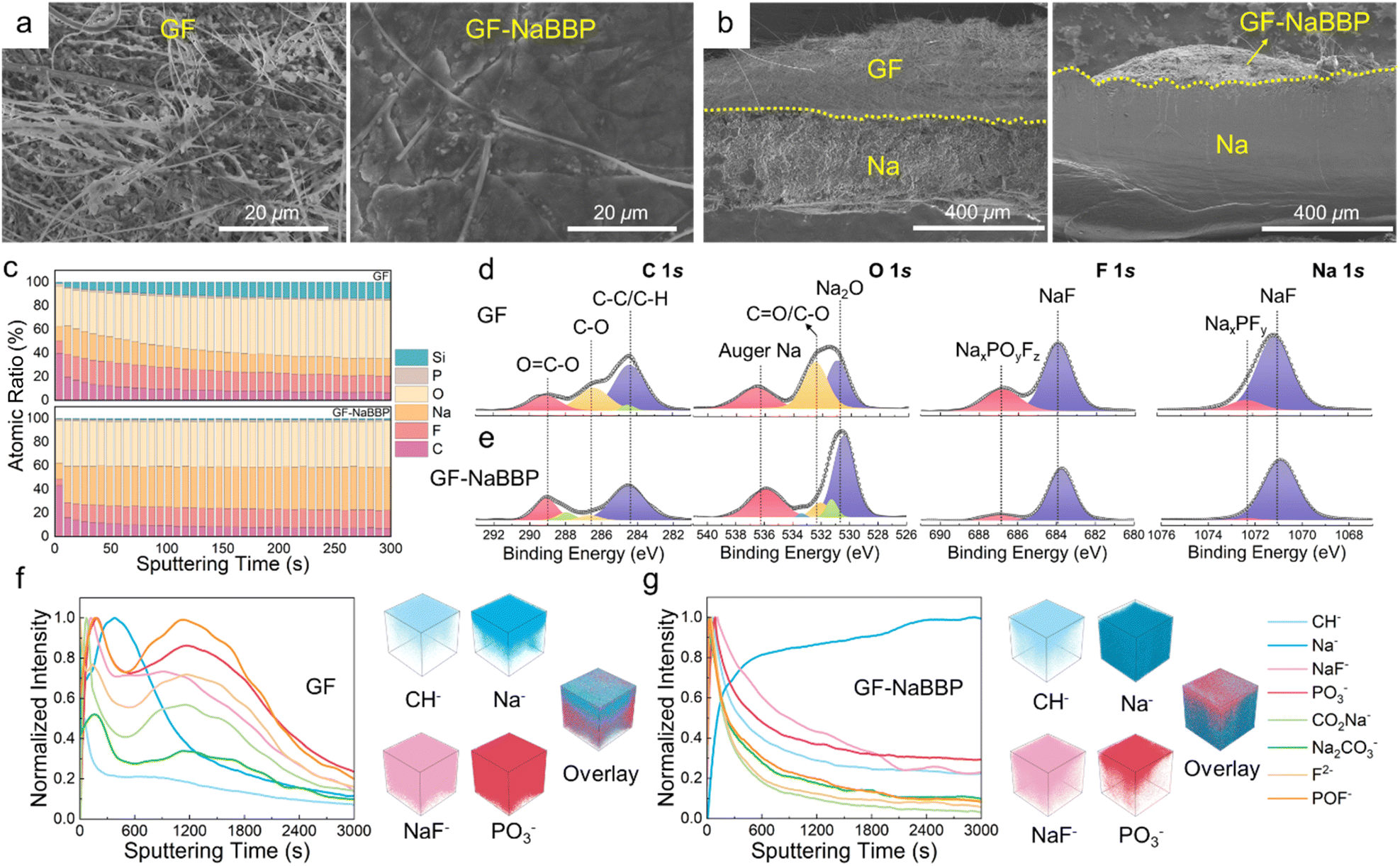 | ||
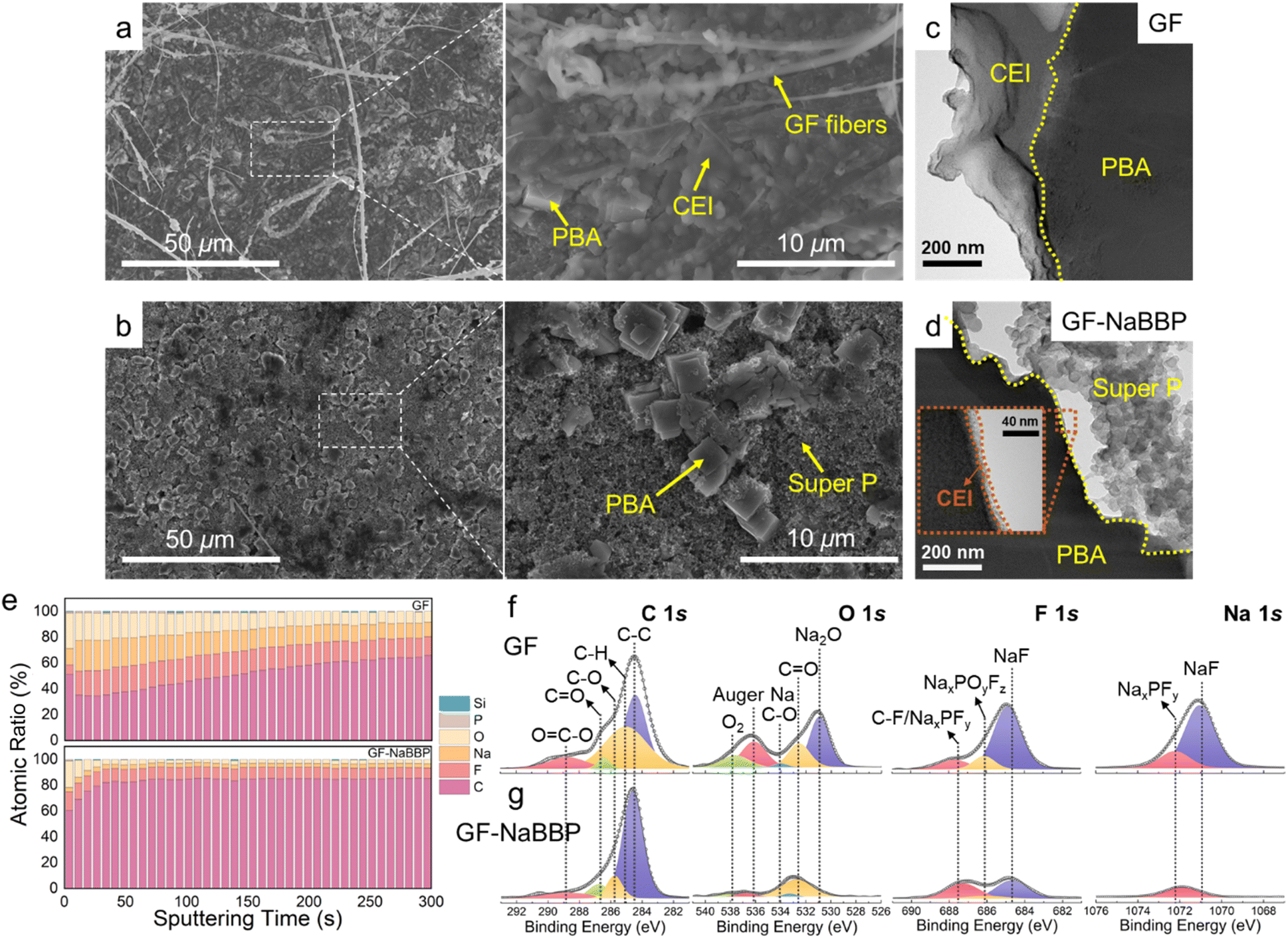
| Fig. 6 Post-mortem analyses of the cycled Na metal after 1000 cycles under 1.7C (0.2 A g−1) at 30 °C. (a) Surface and (b) cross-sectional SEM images of the Na anode from the GF and GF-NaBBP cells. The yellow dashed line indicates the demarcation between Na metal and the separators. (c) In-depth XPS profiling of the atomic ratio and the representative deconvoluted C 1s, O 1s, F 1s, and Na 1s XPS spectra of the SEI layers on the cycled Na metal in the (d) GF and (e) GF-NaBBP cells. Normalized TOF-SIMS depth profiles of the SEI layers on the cycled Na metal in the (f) GF and (g) GF-NaBBP cells with 3D maps of the selected species and overlay images. | ||
The quantitative chemical composition of the SEI layers on the cycled Na metal was probed by in-depth XPS analysis. Fig. 6c presents the XPS depth profiling obtained by Ar+ etching through the SEI layers of the cycled Na metal in the GF and GF-NaBBP cells. The GF-NaBBP cell exhibited smaller numbers of C and O atoms in the SEI layer compared to the GF cell, likely due to the effective suppression of electrolyte decomposition.14 Furthermore, the SEI layer in the GF cell showed a persistent Si content during sputtering, indicating the presence of detached GF fibers. It is very intriguing to note that the GF-NaBBP cell exhibited a remarkably constant atomic ratio in the SEI layer upon sputtering, demonstrating the formation of a uniform SEI layer.75 The chemical composition of the SEI layer was further evaluated using the representative deconvoluted XPS spectra (Fig. 6d and e). In the C 1s and O 1s spectra, significantly larger amounts of C–O and CO species were detected from the GF cell compared to those in the GF-NaBBP cell, indicating a greater quantity of carbonate-based electrolyte decomposition during cycling.76 In addition, in the F 1s and Na 1s spectra of the GF cell, the peaks corresponding to NaxPOyFz and NaxPFy exhibited stronger intensities than those in the GF-NaBBP cell, indicating a larger amount of NaPF6 decomposition.77 Consequently, the suppressed electrolyte decomposition in the GF-NaBBP cell leads to the formation of uniform and compact SEI layers.41 To further probe the spatial distribution of the SEI layers, TOF-SIMS analysis was conducted. Fig. 6f and g present the TOF-SIMS depth profiles with the corresponding 3D maps of SEI species of interest. The GF cell exhibited a substantially stronger normalized intensity of CH−, NaF−, and PO3− species than the GF-NaBBP cell, primarily originating from electrolyte decomposition.78,79 Furthermore, the intensity of such decomposed species strongly persisted upon sputtering, while that of Na− was relatively weak, indicating the formation of a thick SEI layer.79 In sharp contrast, the intensity of CH−, NaF−, and PO3− species in the GF-NaBBP cell rapidly diminished upon sputtering, while the Na− intensity became stronger even at the early stage, confirming the formation of a thin SEI layer. The corresponding 3D maps visually illustrate the spatial distribution of the SEI species, showing a good correlation with the aforementioned findings.
To explore the morphological and compositional characteristics of the CEI layers on the cycled PBA cathodes, a series of post-mortem analyses were carried out. Fig. 7a and b show the surface SEM images of the cycled PBA cathodes in the GF and GF-NaBBP cells. Surprisingly, the cycled PBA cathode in the GF-NaBBP cell preserved its pristine state (Fig. S21†), while that in the GF cell was covered by a thick CEI layer and detached GF fibers. As shown in the TEM images in Fig. 7c and d, a significantly thinner CEI layer was formed on the PBA surface in the GF-NaBBP cell, while that in the GF cell showed a relatively thick and irregular passivating layer. These findings suggest that the uniform Na+ flux enabled by GF-NaBBP also affects the formation of stable CEI layers.41
 | ||
| Fig. 7 Post-mortem analyses of the cycled PBA cathode after 1000 cycles under 1.7C (0.2 A g−1) at 30 °C. Surface SEM images of the cycled PBA cathode in the (a) GF and (b) GF-NaBBP cells. TEM images of the cycled PBA cathode in the (c) GF and (d) GF-NaBBP cells. (e) In-depth XPS profiling of the atomic ratio and the representative deconvoluted C 1s, O 1s, F 1s, and Na 1s XPS spectra of the CEI layers on the cycled PBA cathodes in the (f) GF and (g) GF-NaBBP cells. | ||
The chemical composition of the CEI species was further investigated by in-depth XPS analysis. As shown in Fig. 7e, the CEI layer of the GF cell exhibited a significantly higher O and F ratio than that of the GF-NaBBP cell.80 This is likely due to the effective suppression of electrolyte decomposition, consistently observed from the SEI layers on Na metal.81 Furthermore, the CEI layer in the GF-NaBBP cell possessed a surprisingly uniform composition throughout its depth, compared to that in the GF cell. Interestingly, the XPS results for the CEI layer on the PBA cathodes were almost identical to those obtained for the SEI layer on Na metal. This implies that GF-NaBBP plays a critical role in forming stable electrode–electrolyte interphases on both the cathode and the metallic anode. Indeed, as shown in the representative deconvoluted C 1s, O 1s, F 1s, and Na 1s spectra (Fig. 7f and g), a large amount of oxygen-containing species, NaxPOyFz, and NaxPFy was detected in the GF cell as a consequence of electrolyte decomposition.80 In particular, the NaF peak of the CEI layer in the GF cell was significantly stronger than that in the GF-NaBBP cell, indicating greater amounts of FEC and PF6− decomposition. Given the poor Na+ diffusivity of NaF, the NaF-rich CEI layer should impede electrochemical kinetics,82 further corroborating the inferior rate capability and long-term cycling performance previously observed. Overall, it was demonstrated that the NaBBP coating on the GF not only stabilizes the SEI layer on the Na metal but also facilitates the formation of a uniform and thin CEI layer on the PBA cathode. This is primarily because the non-entangled bottlebrush structure of NaBBP effectively regulates Na+ flux through molecular-level coordination, thereby mitigating dendrite growth and forming uniform electrode–electrolyte interphases.
Conclusions
We have demonstrated that a bottlebrush block copolymer, phase-separated into a hexagonal cylindrical morphology with a non-entangled topology, effectively mitigates Na dendrite growth and stabilizes the SEI and CEI layers of Na metal batteries. The NaBBP coating on the GF separator not only uniformizes the pore size distribution but also phase-separates into a hexagonal cylindrical structure, creating ion-conducting pathways that facilitate ion conduction. Unlike conventional PEO derivatives, the oligomeric PEO bottlebrushes in the BBP exhibit a non-entangled dynamic nature primarily due to steric repulsion, which prevents interpenetration between neighboring PEO brushes. This results in the formation of efficient molecular-level PEO–Na+ complexes, as systematically demonstrated through X-ray scattering analyses, rheological characterization, and computational simulations. The regulated Na+ flux through the GF-NaBBP separator enables remarkably uniform Na deposition on the electrodes during cycling, significantly stabilizing both the SEI and CEI layers of the Na metal and the PBA cathode, respectively. Consequently, the resulting Na metal batteries exhibit outstanding long-term cycling performance over 1500 cycles, even at an extremely high current density of 10C. These findings highlight the critical role of polymer architecture in regulating Na+ flux and achieving dendrite-free Na metal batteries, providing a powerful platform for designing non-entangled polymer materials for metal battery systems.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
D. Jeong: conceptualization, investigation, methodology, and writing – original draft. G. H. Shin: investigation, methodology, and formal analysis. H. Lim: methodology and resources. S. Kim: validation. D.-S. Kwon: validation. J. Yook: methodology. S.-O. Kim: methodology and resources. K. Y. Chung: validation. K. Kim: conceptualization, supervision, and writing – review & editing. J. Shim: conceptualization, funding acquisition, supervision, writing – original draft, and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. RS-2023-00209631). This work was also supported by the Korea Basic Science Institute (National Research Facilities and Equipment Center) grant funded by the Ministry of Science and ICT (No. RS-2024-00400789). This research was also supported by the ‘Science Education in Infosphere (SEI)' funded by the four-stage BK21 and the Korea Institute of Science and Technology (KIST) Institutional Program (No. 2E33271).Notes and references
- Y. Wang, Y. Wang, Y.-X. Wang, X. Feng, W. Chen, X. Ai, H. Yang and Y. Cao, Chem, 2019, 5, 2547–2570 Search PubMed
.
- Y. Zhao, K. R. Adair and X. Sun, Energy Environ. Sci., 2018, 11, 2673–2695 RSC
- S. V. Venkatesan, A. Nandy, K. Karan, S. R. Larter and V. Thangadurai, Electrochem. Energy Rev., 2022, 5, 16 CrossRef
- B. Sun, P. Xiong, U. Maitra, D. Langsdorf, K. Yan, C. Wang, J. Janek, D. Schroder and G. Wang, Adv. Mater., 2020, 32, 1903891 CrossRef
- D. S. Kwon, H. J. Kim and J. Shim, Macromol. Rapid Commun., 2021, 42, 2100279 CrossRef
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Chem. Rev., 2019, 119, 5416–5460 CrossRef
- Y. Jin, Y. Xu, P. M. L. Le, T. D. Vo, Q. Zhou, X. Qi, M. H. Engelhard, B. E. Matthews, H. Jia, Z. Nie, C. Niu, C. Wang, Y. Hu, H. Pan and J.-G. Zhang, ACS Energy Lett., 2020, 5, 3212–3220 CrossRef
- X. Zheng, Z. Cao, Z. Gu, L. Huang, Z. Sun, T. Zhao, S. Yu, X.-L. Wu, W. Luo and Y. Huang, ACS Energy Lett., 2022, 7, 2032–2042 CrossRef CAS
- W. Luo, C. F. Lin, O. Zhao, M. Noked, Y. Zhang, G. W. Rubloff and L. Hu, Adv. Energy Mater., 2016, 7, 1601526 CrossRef
- Z. Xu, J. Yang, T. Zhang, L. Sun, Y. Nuli, J. Wang and S. i. Hirano, Adv. Funct. Mater., 2019, 29, 1901924 CrossRef
- Y. Gu, W. W. Wang, Y. J. Li, Q. H. Wu, S. Tang, J. W. Yan, M. S. Zheng, D. Y. Wu, C. H. Fan, W. Q. Hu, Z. B. Chen, Y. Fang, Q. H. Zhang, Q. F. Dong and B. W. Mao, Nat. Commun., 2018, 9, 1339 CrossRef PubMed
- X. Song, X. Li, H. Shan, J. Wang, W. Li, K. Xu, K. Zhang, H. M. K. Sari, L. Lei, W. Xiao, J. Qin, C. Xie and X. Sun, Adv. Funct. Mater., 2023, 34, 2303211 CrossRef
- J. Qin, Z. Yang, F. Xing, L. Zhang, H. Zhang and Z.-S. Wu, Electrochem. Energy Rev., 2023, 6, 9 CrossRef
- D.-S. Kwon, D. Jeong, H. B. Kang, W. Chang, J. Bang and J. Shim, J. Mater. Chem. A, 2024, 12, 1465–1475 RSC
- B. Tong and X. Li, Mater. Chem. Front., 2024, 8, 309–340 RSC
- H. Liu, X. Zheng, Y. Du, M. C. Borras, K. Wu, K. Konstantinov, W. K. Pang, S. Chou, H. Liu, S. Dou and C. Wu, Adv. Mater., 2024, 36, 2307645 CrossRef PubMed
- D.-S. Kwon, S. H. Gong, S. Yun, D. Jeong, J. Je, H. J. Kim, S.-O. Kim, H.-S. Kim and J. Shim, ACS Appl. Mater. Interfaces, 2022, 14, 47650–47658 CrossRef PubMed
- H. Gao, B. Guo, J. Song, K. Park and J. B. Goodenough, Adv. Energy Mater., 2015, 5, 1402235 CrossRef
- J. Wang, Y. Gao, D. Liu, G. Zou, L. Li, C. Fernandez, Q. Zhang and Q. Peng, Adv. Mater., 2024, 36, 2304942 CrossRef PubMed
- P. V. Wright, Br. Polym. J., 2007, 7, 319–327 Search PubMed
- Y. Liu, Z. Tai, I. Rozen, Z. Yu, Z. Lu, A. P. LaGrow, O. Bondarchuk, Q. Chen, G. Goobes, Y. Li and L. Liu, Adv. Energy Mater., 2023, 13, 2204420 CrossRef
- J. I. Kim, Y. Choi, K. Y. Chung and J. H. Park, Adv. Funct. Mater., 2017, 27, 1701768 CrossRef
- O. Borodin and G. D. Smith, Macromolecules, 2006, 39, 1620–1629 CrossRef
- D. R. Payne and P. V. Wright, Polymer, 1982, 23, 690–693 CrossRef
- J. Shim, D.-G. Kim, J. H. Lee, J. H. Baik and J.-C. Lee, Polym. Chem., 2014, 5, 3432–3442 RSC
- D.-G. Kim, J. Shim, J. H. Lee, S.-J. Kwon, J.-H. Baik and J.-C. Lee, Polymer, 2013, 54, 5812–5820 CrossRef
- S.-J. Kwon, D.-G. Kim, J. Shim, J. H. Lee, J.-H. Baik and J.-C. Lee, Polymer, 2014, 55, 2799–2808 CrossRef
- J. Shim, L. Kim, H. J. Kim, D. Jeong, J. H. Lee and J.-C. Lee, Polymer, 2017, 122, 222–231 CrossRef
- J. Shim, J. W. Lee, K. Y. Bae, H. J. Kim, W. Y. Yoon and J. C. Lee, ChemSusChem, 2017, 10, 2274–2283 CrossRef PubMed
- D. Jeong, J. Shim, H. Shin and J. C. Lee, ChemSusChem, 2020, 13, 2642–2649 CrossRef PubMed
- Y. Gai, D.-P. Song, B. M. Yavitt and J. J. Watkins, Macromolecules, 2017, 50, 1503–1511 CrossRef
- X. Wang, C. Zhang, M. Sawczyk, J. Sun, Q. Yuan, F. Chen, T. C. Mendes, P. C. Howlett, C. Fu, Y. Wang, X. Tan, D. J. Searles, P. Kral, C. J. Hawker, A. K. Whittaker and M. Forsyth, Nat. Mater., 2022, 21, 1057–1065 CrossRef
- T. Niitani, M. Amaike, H. Nakano, K. Dokko and K. Kanamura, J. Electrochem. Soc., 2009, 156, A577 CrossRef
- C. G. Arges, Y. Kambe, M. Dolejsi, G.-P. Wu, T. Segal-Pertz, J. Ren, C. Cao, G. S. W. Craig and P. F. Nealey, J. Mater. Chem. A, 2017, 5, 5619–5629 RSC
- L. Gwee, J.-H. Choi, K. I. Winey and Y. A. Elabd, Polymer, 2010, 51, 5516–5524 CrossRef
- S. Alexandris, K. Peponaki, P. Petropoulou, G. Sakellariou and D. Vlassopoulos, Macromolecules, 2020, 53, 3923–3932 CrossRef
- B. M. Yavitt, Y. Gai, D.-P. Song, H. H. Winter and J. J. Watkins, Macromolecules, 2016, 50, 396–405 CrossRef
- M. Abbasi, L. Faust and M. Wilhelm, Adv. Mater., 2019, 31, 1806484 CrossRef
- H. Liang, Z. Cao, Z. Wang, S. S. Sheiko and A. V. Dobrynin, Macromolecules, 2017, 50, 3430–3437 CrossRef
- I. P. Campbell, C. He and M. P. Stoykovich, ACS Macro Lett., 2013, 2, 918–923 CrossRef
- X. Xia, C. F. Du, S. Zhong, Y. Jiang, H. Yu, W. Sun, H. Pan, X. Rui and Y. Yu, Adv. Funct. Mater., 2022, 32, 2110280 CrossRef
- Y. Orimo and A. Hotta, Macromolecules, 2011, 44, 5310–5317 CrossRef
- J. Shim, F. S. Bates and T. P. Lodge, Nat. Commun., 2019, 10, 2108 CrossRef PubMed
- S. K. Siddique, H. Sadek, T.-L. Lee, G.-M. Manesi, A. Avgeropoulos, C.-W. Wang, C.-C. Lee, E. L. Thomas and R.-M. Ho, Giant, 2024, 17, 100205 CrossRef
- M. B. Runge and N. B. Bowden, J. Am. Chem. Soc., 2007, 129, 10551–10560 CrossRef
- J. S. Pedersen, J. Appl. Crystallogr., 2000, 33, 637–640 CrossRef
- H. J. Kim, K. Jin, J. Shim, W. Dean, M. A. Hillmyer and C. J. Ellison, ACS Sustain. Chem. Eng., 2020, 8, 12036–12044 CrossRef
- W.-S. Young and T. H. Epps, Macromolecules, 2009, 42, 2672–2678 CrossRef
- I. Nakamura, N. P. Balsara and Z. G. Wang, ACS Macro Lett., 2013, 2, 478–481 CrossRef PubMed
- A. N. Semenov, Sov. Phys. – JETP, 1985, 61, 733–742 Search PubMed
- W. S. Loo, G. K. Sethi, A. A. Teran, M. D. Galluzzo, J. A. Maslyn, H. J. Oh, K. I. Mongcopa and N. P. Balsara, Macromolecules, 2019, 52, 5590–5601 CrossRef
- R. Xie, I. Lapkriengkri, N. B. Pramanik, S. Mukherjee, J. R. Blankenship, K. Albanese, H. Wang, M. L. Chabinyc and C. M. Bates, Macromolecules, 2022, 55, 10513–10521 CrossRef
- L. J. Fetters, D. J. Lohse, C. A. García-Franco, P. Brant and D. Richter, Macromolecules, 2002, 35, 10096–10101 CrossRef
- C. M. Bates, A. B. Chang, N. Momčilović, S. C. Jones and R. H. Grubbs, Macromolecules, 2015, 48, 4967–4973 CrossRef
- L. Liberman, M. L. Coughlin, S. Weigand, J. Edmund, F. S. Bates and T. P. Lodge, Macromolecules, 2022, 55, 4947–4955 CrossRef
- Z. Xiao, B. Zhou, J. Wang, C. Zuo, D. He, X. Xie and Z. Xue, J. Membr. Sci., 2019, 576, 182–189 CrossRef
- T. Terashima, M. Kawabe, Y. Miyabara, H. Yoda and M. Sawamoto, Nat. Commun., 2013, 4, 2321 CrossRef PubMed
- R. Tatara, G. M. Leverick, S. Feng, S. Wan, S. Terada, K. Dokko, M. Watanabe and Y. Shao-Horn, J. Phys. Chem. C, 2018, 122, 18316–18328 CrossRef
- D. Monti, E. Jónsson, M. R. Palacín and P. Johansson, J. Power Sources, 2014, 245, 630–636 CrossRef
- Y. Ugata, K. Shigenobu, R. Tatara, K. Ueno, M. Watanabe and K. Dokko, Phys. Chem. Chem. Phys., 2021, 23, 21419–21436 RSC
- X. Zhang, F. Niu, D. Liu, S. Yang, Y. Yang and Z. Tong, RSC Adv., 2019, 9, 32085–32096 RSC
- M. Ozlek, E. Burgaz, I. Inanc and M. Andac, Ionics, 2022, 28, 3255–3268 CrossRef
- L. Zhao, Y. Du, C. Wang, D. Li, H. Li and Y. Zhao, Energy Storage Mater., 2024, 68, 103360 CrossRef
- M. Mecozzi, M. Pietroletti, M. Scarpiniti, R. Acquistucci and M. E. Conti, Environ. Monit. Assess., 2012, 184, 6025–6036 CrossRef PubMed
- L. J. M. R. Davis, Curr. Res. Technol. Educ. Top Appl. Microbiol. Microb. Biotechnol., 2010, 2, 1582–1594 Search PubMed
- M. D. Galluzzo, W. S. Loo, A. A. Wang, A. Walton, J. A. Maslyn and N. P. Balsara, J. Phys. Chem. B, 2020, 124, 921–935 CrossRef PubMed
- M. Moorthy, B. Moorthy, B. K. Ganesan, A. Saha, S. Yu, D. H. Kim, S. Hong, S. Park, K. Kang, R. Thangavel and Y. S. Lee, Adv. Funct. Mater., 2023, 33, 2300135 CrossRef
- X. Li, J. Zhang, X. Guo, C. Peng, K. Song, Z. Zhang, L. Ding, C. Liu, W. Chen and S. Dou, Adv. Mater., 2023, 35, 2203547 CrossRef
- S. Xia, J. Lopez, C. Liang, Z. Zhang, Z. Bao, Y. Cui and W. Liu, Adv. Sci., 2019, 6, 1802353 CrossRef PubMed
- Z. Li, F. Du, X. Bie, D. Zhang, Y. Cai, X. Cui, C. Wang, G. Chen and Y. Wei, J. Phys. Chem. C, 2010, 114, 22751–22757 CrossRef
- K. Wang, M. Hou, S. Yuan, H. Yu, Y. Wang, C. Wang and Y. Xia, Electrochem. Commun., 2015, 55, 6–9 CrossRef
- B. Ma, Y. Lee and P. Bai, Adv. Sci., 2021, 8, 2005006 CrossRef
- X. Li, W. Peng, R. Tian, D. Song, Z. Wang, H. Zhang, L. Zhu and L. Zhang, Electrochim. Acta, 2020, 363, 137185 CrossRef
- Y. Feng, C. Zhang, B. Li, S. Xiong and J. Song, J. Mater. Chem. A, 2019, 7, 6090–6098 RSC
- C. Gong, S. D. Pu, S. Zhang, Y. Yuan, Z. Ning, S. Yang, X. Gao, C. Chau, Z. Li, J. Liu, L. Pi, B. Liu, I. Capone, B. Hu, D. L. R. Melvin, M. Pasta, P. G. Bruce and A. W. Robertson, Energy Environ. Sci., 2023, 16, 535–545 RSC
- D. Lei, Y. B. He, H. Huang, Y. Yuan, G. Zhong, Q. Zhao, X. Hao, D. Zhang, C. Lai, S. Zhang, J. Ma, Y. Wei, Q. Yu, W. Lv, Y. Yu, B. Li, Q. H. Yang, Y. Yang, J. Lu and F. Kang, Nat. Commun., 2019, 10, 4244 CrossRef
- B. Qin, A. Schiele, Z. Jusys, A. Mariani, T. Diemant, X. Liu, T. Brezesinski, R. J. Behm, A. Varzi and S. Passerini, ACS Appl. Mater. Interfaces, 2020, 12, 3697–3708 CrossRef
- R. Rodriguez, K. E. Loeffler, S. S. Nathan, J. K. Sheavly, A. Dolocan, A. Heller and C. B. Mullins, ACS Energy Lett., 2017, 2, 2051–2057 CrossRef
- Y. Ji, J. Qiu, W. Zhao, T. Liu, Z. Dong, K. Yang, G. Zheng, G. Qian, M. Yang, Q. Chen, K. Amine, F. Pan and L. Yang, Chem, 2023, 9, 2943–2955 Search PubMed
- X. Xu, K. Lin, D. Zhou, Q. Liu, X. Qin, S. Wang, S. He, F. Kang, B. Li and G. Wang, Chem, 2020, 6, 902–918 Search PubMed
- S. Wan, K. Song, J. Chen, S. Zhao, W. Ma, W. Chen and S. Chen, J. Am. Chem. Soc., 2023, 145, 21661–21671 CrossRef
- L. Deng, K. Goh, F.-D. Yu, Y. Xia, Y.-S. Jiang, W. Ke, Y. Han, L.-F. Que, J. Zhou and Z.-B. Wang, Energy Storage Mater., 2022, 44, 82–92 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07347a |
| This journal is © The Royal Society of Chemistry 2025 |