
Enhanced piezo-phototronic effect in carbon nitride nanosheets via oxidative exfoliation for high-efficiency piezo-photocatalysis†
Xinyue
Yan
a,
Jiandong
Zhu
a,
Yonghui
Liu
 b,
Yazi
Liu
*ac,
Huan
He
a,
Chenmin
Xu
a,
Shaogui
Yang
*a,
Qiuyi
Ji
a,
Kai
Wang
*c and
Shaomin
Liu
d
b,
Yazi
Liu
*ac,
Huan
He
a,
Chenmin
Xu
a,
Shaogui
Yang
*a,
Qiuyi
Ji
a,
Kai
Wang
*c and
Shaomin
Liu
d
aSchool of Environment, Nanjing Normal University, Jiangsu Engineering Lab of Water and Soil Eco-Remediation, Nanjing 210023, China. E-mail: yazi.liu@njnu.edu.cn; yangsg@njnu.edu.cn
bSchool of Information Engineering, East China University of Technology, Nanchang 330013, China
cWA School of Mines: Minerals, Energy and Chemical Engineering, Curtin University, Perth, WA 6102, Australia. E-mail: kai.wang2@curtin.edu.au
dSchool of Engineering, Great Bay University, Dongguan 523000, China
First published on 2nd January 2025
Abstract
The application of piezo-photocatalysis in wastewater treatment by harnessing both solar and mechanical energy simultaneously offers a promising strategy for sustainable pollutant degradation. Nevertheless, integrating robust piezoelectric and semiconducting properties effectively within a single-component catalyst to achieve optimal synergy remains challenging due to their inherent trade-off. To tackle this limitation, a series of graphitic carbon nitride (g-C3N4) nanosheets with a controlled degree of exfoliation have been fabricated through a facile oxidative exfoliation strategy in this work. The exfoliated nanosheets demonstrate substantial improvements in both piezoelectricity (36 pm V−1) and semiconducting properties, significantly enhancing their piezo-photocatalytic performance for organic pollutant degradation. Combined experimental and theoretical analysis reveals that the oxidative exfoliation process not only enables the easy domain deformation feature of the nanosheets, but also introduces surface polar functional groups, thus accelerating polarization and the piezoelectric response. Meanwhile, the charge carrier migration pathway to surface-active sites has been largely shortened, benefiting from the nanosheet structure. Consequently, the optimized material E500-CN (g-C3N4 exfoliated at 500 °C) demonstrates superior piezo-photocatalytic degradation performance, achieving a degradation rate of 97% within 20 min and a first-order kinetic rate constant of 0.152 min−1, which is 5.1-fold faster than that of B-CN. The mechanism study highlights the critical role of the enhanced piezo-phototronic effect, where the vibration-induced in-plane piezopotential effectively manipulates the separation and transportation of photogenerated electron–hole pairs. This study offers a feasible design strategy for efficient 2D piezo-photocatalysts in water remediation, enabling a sustainable water treatment approach.
1. Introduction
With rapid urbanization and industrialization, water contamination has become a pressing global issue, attracting growing interest in sustainable remediation technologies. Photocatalysis is widely recognized as an eco-friendly and sustainable approach for utilizing sunlight to tackle these issues,1,2 which enables the direct conversion of renewable solar energy to chemical energy for organic pollutant degradation. However, the efficiency of photocatalysis remains unsatisfactory due to the rapid recombination of charge carriers and inefficient interfacial redox reaction in particulate photocatalysts.3 Therefore, manipulating the charge separation and transfer (CST) behavior in photocatalysts has become an effective strategy to enhance photocatalytic performance. In this context, some advanced methods have been proposed, including heterojunction photocatalyst fabrication, co-catalyst utilization, and defect engineering, aiming to improve the separation and transfer efficiency of electron–hole pairs, thereby reducing unwanted charge recombination.4–6Recently, the integration of piezoelectric polarization with photocatalysis has provided an alternative way to regulate the CST process. The development of this strategy is inspired by the piezo-phototronic effect.7 Specifically, it demonstrates that utilizing piezoelectric potential (piezopotential) generated by applying strain to a piezoelectric semiconductor can effectively control the generation, transport, separation, and recombination behavior of charge carriers within the material. Based on this, piezo-photocatalysis has been developed.8 Notably, piezo-photocatalysis generates a real-time electric field throughout the entire reaction, enhancing catalytic activity and providing greater adaptability to external conditions. This adaptability may lead to more robust catalytic performance under varying environmental conditions. Typically, when stress is applied to a piezoelectric semiconductor, the internal polarization field generated within the material by the piezoelectric effect provides a strong driving force to separate the photoexcited electron–hole pairs and drive them to migrate in opposite directions, which significantly enhances the photocatalytic performance.9,10 This strategy allows harvesting solar energy and mechanical energy simultaneously for sustainable applications. However, the core of this technology relies on the design of piezoelectric semiconductors. Since Wang's group first proposed piezo-photonics, numerous piezoelectric semiconductor materials have been developed, including ZnO, BiFeO3, PbTiO3, and others.7,11–15 These materials are employed as piezo-photocatalysts for water remediation, H2O2 production, and hydrogen evolution.16–19 Nevertheless, these conventional piezoelectric materials are specifically designed for electronics. Despite their notable piezoelectric properties, their semiconducting properties and catalytic features are sacrificed, rendering them unsuitable for piezo-photocatalysis. Consequently, the development of novel piezo-photocatalysts with well-balanced piezoelectric and semiconducting properties is of great importance.
Graphitic carbon nitride (g-C3N4) serves as a non-metallic photocatalyst with suitable bandgap structures, stable physicochemical properties, low-cost and environmentally friendly characteristics. Recently, the discovery of its inherent piezoelectricity has led to the development of g-C3N4-based piezo-photocatalysts.20–25 For example, Liang et al. fabricated porous-structured g-C3N4 nanosheets with significant piezo-photocatalytic degradation of rhodamine B (RhB).23 It was demonstrated that the porous structure exhibited enhanced mechanical vibration responsiveness and distinctive physiochemical properties, thereby enhancing the piezo-photocatalytic performance.24,26 However, the intrinsic weak piezoelectricity of g-C3N4 limits its overall piezo-photocatalytic performance. This limitation is primarily due to the inferior piezo-phototronic effect, characterized by poor synergy between its piezoelectric and semiconducting properties. Additionally, the bulk morphology of g-C3N4 hinders domain deformation, resulting in insignificant piezoelectric polarization that fails to ensure efficient charge separation and transfer. Therefore, it is highly desirable to overcome these bottlenecks and optimize the piezo-phototronic effect in g-C3N4 for a more efficient piezo-photocatalysis system.
In this study, we employed a facile oxidation exfoliation strategy with post-calcination treatment to fabricate g-C3N4 nanosheets and applied them for highly efficient piezo-photocatalysis. It is revealed that the controlled exfoliation serves dual functions in enhancing the piezo-photocatalytic performance of g-C3N4 nanosheets. Firstly, the nanosheet morphology enhances the sensitivity of the materials to external stress, thereby facilitating effective piezoelectric responses under catalytic conditions. In the meantime, extra polar groups introduced on the material surface significantly improved its piezoelectric properties. Secondly, appropriate exfoliation increases the specific surface area and reduces charge carrier recombination, benefiting from the shortened diffusion pathway. As a result, the piezo-phototronic effect dramatically improved with the overall piezo-photocatalytic performance enhanced in the highly exfoliated g-C3N4 nanosheets. The reaction mechanism has been fully elucidated through systematic experimental and theoretical analysis. This study thoroughly addresses the long-standing trade-off between piezoelectric and semiconducting properties in piezo-photocatalysts, paving the way for the design and fabrication of highly efficient piezo-photocatalytic materials for environmental remediation.
2. Experimental section
2.1. Reagents
Melamine (C3H6N6, 99%) and TC (C22H24N2O8·HCl, USP) were purchased from Macklin Biochemical Technology Co., Ltd (Shanghai, China). MB (C16H18ClN3S·3H2O, 98.5%), RhB (C28H31ClN2O3) and MO (C14H14N3NaO3S) were bought from Chemical Reagent Research Institute Co., Ltd (Tianjin, China). BPA (C15H16O2, 99%) was obtained from J&K Scientific Co., Ltd (Beijing, China). All chemical reagents were used without further purification. All experiments were conducted using distilled water.2.2. Synthesis of catalysts
Bulk g-C3N4 (B-CN) was synthesized via thermal condensation of melamine. Specifically, 8 g of melamine was placed in an alumina crucible, heated to 550 °C at a rate of 5 °C min−1 in static air, and held at this temperature for 4 h. The resulting yellow bulk g-C3N4 was subsequently ground to fine powder (3.8 g). For the exfoliation process, 0.5 g of B-CN was placed in an uncovered alumina crucible and heated at a rate of 5 °C min−1 in static air to temperatures of 460 °C, 480 °C, 500 °C, and 540 °C, with each heating treatment maintained for 2 hours. These samples were denoted as E460-CN, E480-CN, E500-CN and E540-CN, corresponding to different degrees of exfoliation.2.3. Characterization of catalysts
The crystal structures of samples were recorded using X-ray powder diffraction (XRD, Bruker D8 Advance, Germany). High-resolution scanning electron microscopy (SEM, Apreo 2S, USA), transmission electron microscopy (TEM, JEOL JEM-2100F, Japan) and atomic force microscopy (AFM, Bruker Dimension Icon, Germany) were used to confirm the morphology and thickness of the samples. Fourier transform infrared spectra (FTIR) were collected on a Nicolet IS5 spectrophotometer with samples embedded in potassium bromide (KBr) pellets. Brunauer–Emmett–Teller (BET) specific surface area was measured by the nitrogen adsorption–desorption method using a surface area analyzer (Micromeritics ASAP 2460). Chemical state analysis was performed by X-ray photoelectron spectroscopy (XPS, ESCALAB Xi+, USA). Kelvin probe force microscopy (KPFM, Bruker Dimension Icon) and piezoresponse force microscopy (PFM, Bruker Dimension Icon) were utilized to monitor the piezoelectric response of the synthesized samples. UV-vis spectra were acquired on a UV-vis spectrophotometer (UV-vis DRS, UV-3600i Plus, Japan). Photoelectrochemical measurements and Mott–Schottky (M–S) measurement were performed in a standard three-electrode system on a CHI660E electrochemical station (Text S1†). Photoluminescence (PL) with an excitation wavelength of 330 nm and time-resolved photoluminescence (TRPL) measurements were performed on a steady-state and lifetime fluorescence spectrofluorometer (LS-50B, PerkinElmer, USA). The electron paramagnetic resonance (EPR) signals, spin-trapped by the reagents 5,5-dimethyl-L-pyrroline N-oxide (DMPO), 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and 2,2,6,6-tetramethyl-4-piperidone (TEMP) were recorded with an EPR spectrometer (Bruker A300, Germany) to reconfirm the generated reactive species during the catalytic reactions.2.4. Piezocatalytic, photocatalytic and piezo-photocatalytic performance
The catalytic activities of the samples towards methylene blue (MB) degradation were investigated under light irradiation (300 W xenon lamp, CEL-HXF300-T3, Beijing China Education Au-Light Technology Co., Ltd), ultrasonic excitation (40 kHz, 120 W, KQ5200DE, Kunshan) or co-irradiation. A cut-off filter (1.5G) was equipped on the lamp, with the light intensity centered around 350 mW cm−2. A solution of MB (50 mL, 10 mg L−1) and catalyst (30 mg) was mixed in the dark for 60 min to reach adsorption–desorption equilibrium before performing the catalytic experiments. During the experiments, the temperature of the suspension was maintained with circulating cooling water. 1.5 mL of solution was collected at a predetermined time, followed by centrifugation at 9000 rpm to remove the catalyst. Finally, 1 mL of supernatant was taken to record the absorption spectrum of MB solution at 664 nm using a UV-visible spectrophotometer (UV723S, APL Instruments, China). The reaction equipment for piezo-photocatalysis is shown in Fig. S1.† The catalysts employed in the study were retrieved through centrifugal separation and subsequently washed with deionized water and ethanol for cycling tests.2.5. DFT calculation
The calculations were performed using the projector augmented-wave method implemented in the Vienna Ab initio Simulation Package (VASP)27–29 with a plane wave energy cutoff of 550 eV. We adopted the generalized gradient approximation (GGA) given by Perdew–Burke–Ernzerhof (PBE) for the exchange–correlation functional.30 Because van der Waals interactions play an important role in vdW heterostructure systems, the DFT-D3 method with Becke–Johnson damping was used to describe these vdW interactions.31 The thickness of the vacuum slab was set to no less than 20 Å in the perpendicular direction to avoid artificial interaction between two neighboring monolayers. The convergence threshold of total energy was 10−5 eV, and the convergence criteria of atomic force were set to 10−3 eV Å−1. A Monkhorst–Pack k-point grid of 2 × 1 × 1 was adopted for energy minimization of the system.3. Results and discussion
3.1. Material characterization
Exfoliated g-C3N4 nanosheets with varying degrees of exfoliation were prepared through direct thermal oxidation etching of bulk g-C3N4 (B-CN) under an air atmosphere (Fig. 1a). As the exfoliation temperature increased from 460 to 540 °C, the sample color gradually changed from yellow to light yellow, eventually becoming nearly white (Fig. S2†). This color change is attributed to the high-temperature-driven exfoliation process, wherein the layered structure of bulk g-C3N4 is exfoliated into thinner sheets.32 This structural modification alters the light absorption properties of the material, resulting in the observed visible color shift.33 Scanning electron microscopy (SEM) images shown in Fig. S3† revealed that the exfoliated g-C3N4 nanosheets became fluffy and exhibited a thin sheet-like morphology, in contrast to bulk g-C3N4, which was composed of blocks stacked with numerous layers and had a size of several micrometers. This transformation is attributed to the weakening of van der Waals forces between layers at elevated temperatures; meanwhile the oxidation process occurs, which facilitates the exfoliation of bulk g-C3N4 into thinner layers.34 Additionally, transmission electron microscopy (TEM) images in Fig. 1b–d show dimensional shrinkage in the material after heat treatment at 540 °C, compared to the smooth surface of the pristine carbon nitride particles. This shrinkage may be caused by the instability of the in-plane hydrogen bond network between polymeric tri-s-triazine units under high-temperature oxidation by O2, partially breaking the hydrogen bonds between melon strands.35 Atomic force microscopy (AFM) further demonstrated a reduction in thickness from 128 nm to approximately 2.5 nm as the etching temperature increased (Fig. 1e′–i′). This ultrathin sheet-like structure of the exfoliated g-C3N4 is expected to enhance mechanical energy harvesting due to its easy domain deformation under mechanical vibration. | ||
| Fig. 1 (a) A schematic illustration of the bulk g-C3N4 exfoliation process; TEM images of (b) B-CN, (c) E500-CN and (d) E540-CN; (e–i) AFM images of B-CN, E460-CN, E480-CN, E500-CN and E540-CN, respectively, where (e′–i′) are the corresponding height profiles. | ||
The microstructures of the different samples were characterized using X-ray diffraction (XRD) patterns and Fourier-transform infrared (FT-IR) spectra. As illustrated in Fig. 2a, the XRD patterns of all samples exhibited two distinct peaks: the (100) peak at 12.98°, corresponding to the in-plane stacking of tri-s-triazine units, and the (002) peak at 27.85°, attributed to interlayer stacking.36 However, the intensities of both peaks decreased after exfoliation, indicating a reduction in the thickness and size of the nanosheets after thermal oxidation etching. The (100) peak, representing the in-plane structural stacking motif, was significantly weakened after calcination at 540 °C, indicating that the long-range ordering of the in-plane atomic arrangement in E540-CN was disrupted.37 Furthermore, the (002) peak of E540-CN was shifted to a higher angle by approximately 0.2° compared to B-CN, implying a decreased interlayer stacking distance within the nanosheets.38 The FT-IR spectra of catalysts are presented in Fig. 2b. All samples exhibited the characteristic bands of g-C3N4. A prominent peak at 810 cm−1 signifies the out-of-plane bending vibration of heptazine rings,39 while a broad band from 3070 to 3280 cm−1 corresponds to O–H or N–H stretching vibrations.40 Several strong peaks within the range of 1200–1700 cm−1 are associated with the stretching vibration modes of aromatic CN heterocycles.41 Notably, a new peak at 2340 cm−1 in E540-CN is attributed to the stretching vibration of the cyano group (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N).42–44 This characteristic peak demonstrates that the cyano group is successfully introduced into the g-C3N4 structure. This finding aligns with the observation that the disruption of the long-range ordering structure within the in-plane motifs leads to the breaking of some hydrogen bonds, resulting in the formation of cyano groups.45 The specific surface area of the catalysts was determined by measuring nitrogen (N2) adsorption–desorption isotherms (Fig. 2c). The specific surface area of E500-CN was 123.26 m2 g−1, significantly larger than the 9.02 m2 g−1 of B-CN, as determined by the Brunauer–Emmett–Teller (BET) method.22 This notable increase in specific surface area after exfoliation (Fig. S4†) suggests a higher density of active sites in the exfoliated g-C3N4, which may enhance its photocatalytic performance.
N).42–44 This characteristic peak demonstrates that the cyano group is successfully introduced into the g-C3N4 structure. This finding aligns with the observation that the disruption of the long-range ordering structure within the in-plane motifs leads to the breaking of some hydrogen bonds, resulting in the formation of cyano groups.45 The specific surface area of the catalysts was determined by measuring nitrogen (N2) adsorption–desorption isotherms (Fig. 2c). The specific surface area of E500-CN was 123.26 m2 g−1, significantly larger than the 9.02 m2 g−1 of B-CN, as determined by the Brunauer–Emmett–Teller (BET) method.22 This notable increase in specific surface area after exfoliation (Fig. S4†) suggests a higher density of active sites in the exfoliated g-C3N4, which may enhance its photocatalytic performance.
 | ||
| Fig. 2 (a) XRD patterns; (b) FT-IR spectra of all samples; (c) nitrogen adsorption–desorption isotherms of B-CN and E500-CN; (d) N 1s, (e) C 1s and (f) O 1s XPS spectra of B-CN and E500-CN. | ||
X-ray photoelectron spectroscopy (XPS) was conducted to investigate the chemical states and bonding structures of samples. The C 1s and N 1s peaks of E500-CN exhibit a shift to lower binding energies compared to B-CN, which can be attributed to the changes in the chemical environment of the C and N atoms caused by thermal treatment.46 The N 1s spectrum displays three sub-peaks at 398.8, 400.3, and 401.4 eV, attributed to the sp2-hybridized nitrogen (C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), N–(C)3 and amino groups (C–N–H), respectively (Fig. 2d).47 The peak area ratio of N–(C)3 to C–N–H in E500-CN and B-CN was calculated to be 6.42 and 5.26, respectively. The increased N–(C)3 to C–N–H area ratio in E500-CN suggests a higher proportion of orderly bonded tri-s-triazine units within the layers compared to B-CN.25 These units act as polar groups contributing to enhanced in-plane piezoelectricity. In the C 1s spectrum, peaks at binding energies of 284.8 and 288.2 eV primarily correspond to graphitic carbon (CC) and sp2-bonded carbon (NC–N), respectively (Fig. 2e).48,49 In the O 1s spectra (Fig. 2f), the characteristic peak at 532.5 eV is attributed to the adsorbed H2O.50
C), N–(C)3 and amino groups (C–N–H), respectively (Fig. 2d).47 The peak area ratio of N–(C)3 to C–N–H in E500-CN and B-CN was calculated to be 6.42 and 5.26, respectively. The increased N–(C)3 to C–N–H area ratio in E500-CN suggests a higher proportion of orderly bonded tri-s-triazine units within the layers compared to B-CN.25 These units act as polar groups contributing to enhanced in-plane piezoelectricity. In the C 1s spectrum, peaks at binding energies of 284.8 and 288.2 eV primarily correspond to graphitic carbon (CC) and sp2-bonded carbon (NC–N), respectively (Fig. 2e).48,49 In the O 1s spectra (Fig. 2f), the characteristic peak at 532.5 eV is attributed to the adsorbed H2O.50
3.2. Catalytic performance evaluation
Based on the above morphological and structural characterization, the exfoliated materials with significantly altered structures and properties are expected to have drastic effects on their piezocatalytic, photocatalytic, and piezo-photocatalytic activities. Here, methylene blue (MB) was used as a prototype organic pollutant for the degradation experiments. Initially, we conducted blank experiments without catalysts to examine the effect of ultrasonication and photo illumination alone on organic pollutant removal (Fig. S5†). It is evident that both processes have a minor contribution to degradation, ruling out the chemical effects of ultrasonication and light irradiation.With the simultaneous presence of ultrasonic vibration and catalyst, i.e., piezocatalysis (Fig. 3a), E540-CN exhibited a removal efficiency of 30.5% within 20 minutes, with a first-order kinetic rate constant of 0.011 min−1, surpassing B-CN, E460-CN, E480-CN, and E500-CN. In this process, the piezoelectric material subjected to mechanical stress from ultrasonication induces in-plane piezoelectric polarization, driving the migration of free charge carriers. The separated charge carriers then participated in the redox reactions. Among the bulk and exfoliated materials, E540-CN demonstrated the highest piezoelectric degradation efficiency. This can be attributed to the thinner nature of E540-CN, which enhances its susceptibility to deformation and consequently generates greater piezoelectric polarization under mechanical stress. Additionally, new cyano defects have been introduced into E540-CN as described in FT-IR. These defects not only enhance the material structural asymmetry, thereby improving its piezoelectric properties, but also provide active sites that enhance its catalytic performance.
 | ||
| Fig. 3 MB degradation performance of all the samples via (a) piezocatalysis, (b) photocatalysis, and (c) piezo-photocatalysis; (d) comparison of first-order kinetic rate constants for MB degradation of all the samples under different conditions; (e) degradation of various organic pollutants using E500-CN in piezo-photocatalysis; (f) stability test of E500-CN. Experimental conditions: [catalyst]0 = 0.6 g L−1, [organic pollutants]0 = 10 mg L−1. | ||
In terms of photocatalysis, under light irradiation alone (Fig. 3b), E500-CN exhibited the highest degradation efficiency of MB, achieving approximately 88% degradation within 20 minutes, with a first-order kinetic rate constant of 0.083 min−1. In comparison, B-CN, E460-CN, E480-CN, and E540-CN showed degradation efficiencies of 27%, 50%, 71%, and 79%, respectively, with corresponding rate constants of 0.014, 0.028, 0.050, and 0.062 min−1. Interestingly, the photocatalytic performance of E500-CN surpassed that of E540-CN. This is because the long-range ordering structure of E540-CN was destroyed and too many defects were formed, leading to a decrease in the in-plane driving force and electron–hole recombination sites, subsequently damaging its photocatalytic activity.35,45
Notably, under simultaneous light and ultrasound irradiation for piezo-photocatalysis, the degradation efficiency of the system is significantly enhanced across all samples (Fig. 3c), suggesting a strong synergy between piezocatalysis and photocatalysis. B-CN, E460-CN, E480-CN, E500-CN, and E540-CN exhibited degradation rates of 47%, 60%, 88%, 97%, and 86% within 20 min, respectively. The corresponding rate constants were determined to be 0.030, 0.039, 0.094, 0.152, and 0.083 min−1, respectively. This improvement can be attributed to the synergistic effect between the piezoelectric effect and the photocatalytic process. The ultrasonication-induced piezoelectric potential field within g-C3N4 materials acts as an internal driving force for the separation and migration of photoexcited electron–hole pairs, effectively preventing their recombination and thereby providing more available charge carriers for the reaction.51 Remarkably, E500-CN exhibited the highest piezo-photocatalytic activity among all materials, demonstrating a significant enhancement with a reaction rate constant 5.1 times greater than that of B-CN. Interestingly, despite E540-CN offering superior piezocatalytic performance, its piezo-photocatalytic activity was lower than that of E500-CN, indicating that E500-CN achieves the highest synergy between piezoelectric and photocatalytic effects. These results imply that the exfoliation degree of g-C3N4 plays a critical role in piezo-photocatalytic activity.
For an intuitive comparison, Fig. 3d presents the first-order kinetic rate constants of piezocatalysis, photocatalysis, and piezo-photocatalysis for each material. Across the three experimental conditions, the rate constants for piezocatalytic degradation were noticeably lower, likely due to the limited number of free charge carriers. In contrast, the efficiency of photocatalysis is significantly higher than that of piezocatalysis. When both individual processes are coupled, the piezo-photocatalytic activities of all the materials are dramatically improved. Piezo-photocatalytic performance is significantly greater than the combined effects of piezocatalysis and photocatalysis for all catalysts. Particularly, the E500-CN catalyst exhibited the optimal synergy and highest efficiency (first-order apparent rate constant k = 0.152 min−1), surpassing that of sole photocatalysis and piezocatalysis by 1.82 and 22.8 times, respectively.
To examine the viability of the system, the system based on E500-CN has been employed for diverse recalcitrant pollutants degradation via piezo-photocatalysis, including Rhodamine B (RhB), Methyl Orange (MO), Tetracycline (TC), and Bisphenol A (BPA). As illustrated in Fig. 3e, the system exhibited remarkable efficiency for organic pollutant removal. Thus, E500-CN emerges as a versatile piezo-photocatalyst that effectively degrades a wide range of environmental contaminants. Compared to other similar catalytic systems, E500-CN also demonstrated outstanding MB removal capability (Table S1†). Additionally, cyclic experiments of E500-CN demonstrated sustained activity even after four cycles, highlighting its good stability (Fig. 3f). Moreover, XRD of E500-CN after the piezo-photocatalytic reaction revealed no impurities or additional phases (Fig. S6a†). Furthermore, the morphology of E500-CN after the reaction was preserved, as determined through SEM characterization, thus validating its recyclability and stability (Fig. S6b†). After the reaction, the solution was slightly acidified (Fig. S7†). In addition, as shown in Fig. S8,† the TOC of the reaction system decreased to 65.38% within 60 min, confirming the partial mineralization of MB. This also suggests that the intermediates underwent continuous oxidation and decomposition, driven by the production of active species.
3.3. Mechanism study
To investigate the involved reactive species in the piezo-photocatalytic process of E500-CN, quenching experiments were performed with the introduction of various scavengers into the reaction system. EDTA–2Na (2 mmol L−1), histidine (L-His, 2 mmol L−1), 1,4-benzoquinone (BQ, 2 mmol L−1), and tert-butanol (TBA, 2 mmol L−1) were respectively employed as scavengers for h+, singlet oxygen (1O2), superoxide radicals (˙O2−), and hydroxyl radicals (˙OH).52,53 The results revealed that the addition of EDTA–2Na caused a significant decrease in the piezo-photocatalytic degradation rate of MB, indicating the pivotal role of holes in the degradation process. On the other hand, the introduction of L-His and BQ led to only a slight reduction in catalytic efficiency, suggesting that 1O2 and ˙O2− played a relatively minor role in this process. Overall, the contribution of each species follows the sequence of h+ > 1O2 > ˙O2− > ˙OH (Fig. 4a and b). | ||
| Fig. 4 (a and b) Degradation of MB in the presence of various scavengers using E500-CN in piezo-photocatalysis; EPR spectra of (c) DMPO–˙O2−, (d) TEMP–1O2, and (e) TEMPO–h+ in piezocatalysis, photocatalysis and piezo-photocatalysis of E500-CN; (f) EPR spectra of TEMPO–h+ in piezo-photocatalysis of B-CN, E500-CN and E540-CN. Experimental conditions: [catalyst]0 = 0.6 g L−1, [MB]0 = 10 mg L−1. | ||
Electron paramagnetic resonance (EPR) was used to further investigate the role of reactive species in the system. The results in Fig. 4c–e depict that ˙O2−, 1O2, and h+ are successfully detected in the system, in line with the quenching experimental results. Importantly, a strong synergistic effect between piezocatalysis and photocatalysis has been observed, where the production of superoxide radicals and singlet oxygen species is dramatically improved fin piezo-photocatalysis, while individual photocatalysis and piezocatalysis result in weak signals. In addition, the remarkable weakening of the TEMPO–h+ signal in piezo-photocatalysis suggests that more holes were produced, thus consuming the TEMPO. This observation further evidences that the piezoelectric effect can effectively assist photocatalysis. The built-in piezopotential field within the material not only serves as the direct driving force to regulate the free charge dynamics, but also induces band tilting, favorable for charge separation and transportation processes.50,54 However, the signal of DMPO–˙OH disappeared after joint exposure to light and ultrasound, likely due to band tilting under piezoelectric polarization (Fig. S9†). Moreover, as depicted in Fig. 4f, to substantiate the superior performance of E500-CN, EPR tests were carried out during the piezo-photocatalytic processes over B-CN, E500-CN, and E540-CN, respectively. The dominant active species, i.e., holes, was monitored. It was evident that under simultaneous ultrasound and light exposure, E500-CN generated a significantly higher amount of available photoinduced holes compared to B-CN and E540-CN. These holes could facilitate oxidation reactions and lead to more efficient degradation of MB.
After identifying the reactive species involved in the degradation process, we systematically examined the piezoelectric and semiconducting properties of the materials to gain deeper insights into their synergistic interactions. Piezoresponse force microscopy (PFM) was employed to assess the piezoelectric properties of B-CN, E500-CN, and E540-CN (Fig. S10a–c†). It is well established that the deformation of piezoelectric materials under an applied electric field is manifested by the amplitude signal in the PFM model.55 Fig. S10a′–c′† present clear amplitude butterfly loop curves of B-CN, E500-CN, and E540-CN, confirming the presence of piezoelectricity across all the materials. Phase analysis revealed polarization relaxation within the piezoelectric domains, with phase diagrams closely aligning with the morphological features, indicating a uniform polarization distribution throughout both the bulk and exfoliated nanosheets (Fig. S10a′′–c′′†). The robust piezoelectricity of these materials is further reflected by the calculated effective piezoelectric coefficients. E540-CN exhibited the highest piezoelectric coefficient of ∼36 pm V−1, followed by E500-CN (∼19 pm V−1) and B-CN (∼14 pm V−1) as shown in Fig. 5a–c. This observation underscores that a higher degree of exfoliation can significantly enhance the piezoelectric properties of the material. On the one hand, thinner nanosheets, which are more prone to deformation due to their reduced thickness, exhibit higher sensitivity to externally applied mechanical vibrations. The increased flexibility of these nanosheets allows them to more effectively respond to external mechanical stimuli, generating stronger piezoelectric effects. As a result, the thinner 2D nanosheets facilitate the conversion of mechanical energy into piezoelectric energy.56,57 On the other hand, the increased exposure of orderly arranged tri-s-triazine units and non-centrosymmetric triangular holes intrinsically contributes to the enhanced piezoelectric properties. These units and non-centrosymmetric holes serve as polar groups and enhance the material asymmetry.58 The accumulation of polar tri-s-triazine units along the a-axis is identified as a key contributor to the robust in-plane polarization observed in g-C3N4. And as the treatment temperature reaches 540 °C, the in-plane polarity continues to increase due to the presence of cyano defects. Consequently, when subjected to ultrasonic excitation, the exfoliated g-C3N4, with its higher deformability and enhanced in-plane polarization, is capable of converting more external mechanical energy than B-CN. This leads to a more significant piezoelectric potential in exfoliated g-C3N4.
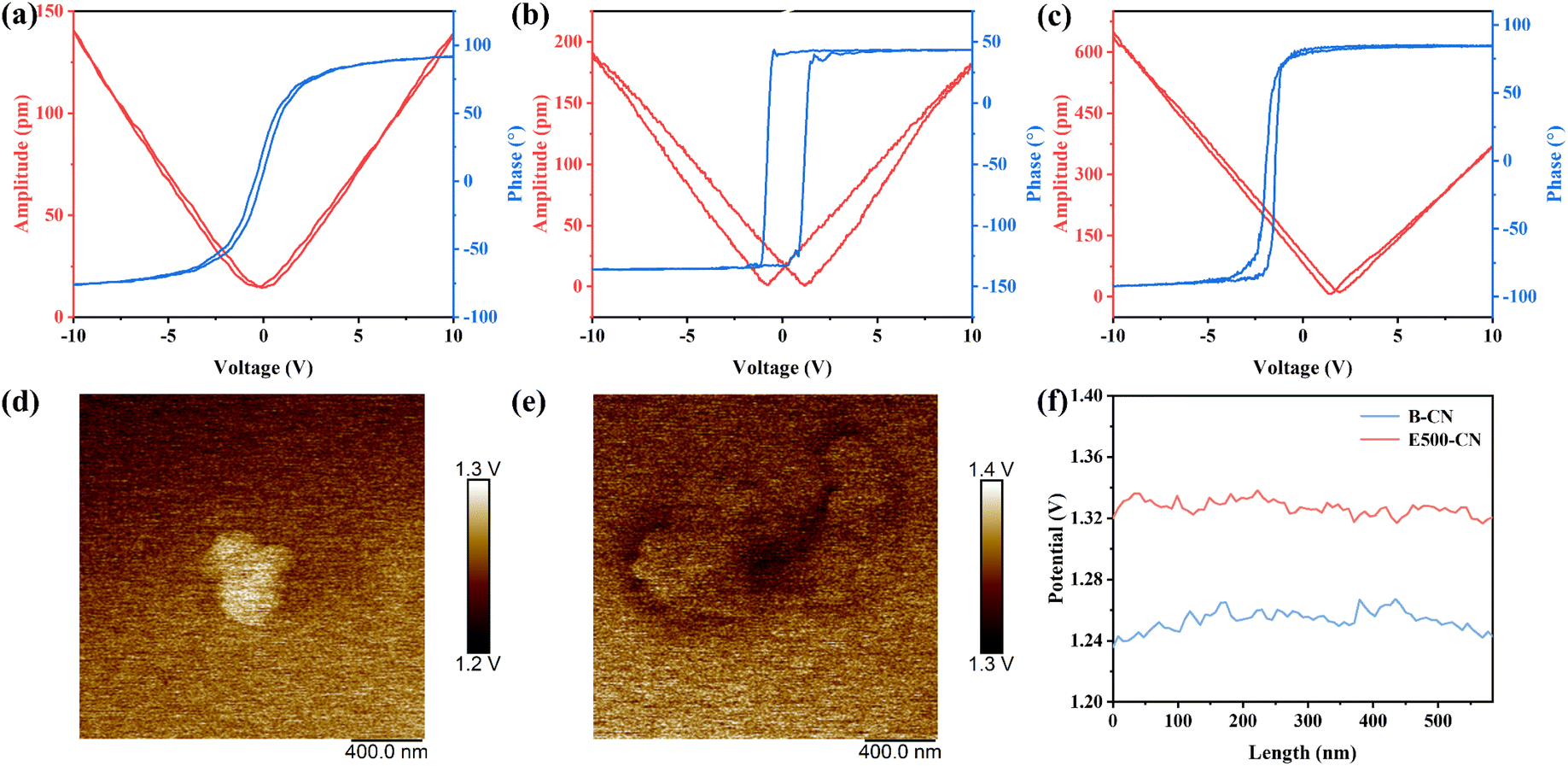 | ||
| Fig. 5 (a–c) PFM amplitude–voltage butterfly loops and phase–voltage hysteresis loops of B-CN, E500-CN and E540-CN; (d and e) KPFM images, and (f) the corresponding surface potential of B-CN and E500-CN. | ||
Moreover, Kelvin probe force microscopy (KPFM) was used to examine the surface potential of B-CN and E500-CN, further highlighting the relationship between catalyst thickness and the strength of the piezoelectric effect. As shown in Fig. 5d–f, when subjected to the stress from the KPFM probe, the average surface potential of B-CN was recorded at 1.25 V, whereas E500-CN exhibited a slightly higher surface potential of 1.33 V. This result suggests that as the thickness of g-C3N4 decreases, the surface potential increases. The increase in surface potential with thinner nanosheets is indicative of a stronger piezoelectric effect.26
The optical properties and band structures of all samples were also evaluated. The UV-vis diffuse reflectance spectra (DRS) exhibited a blue shift in the absorption edge of thermally etched g-C3N4 compared to bulk g-C3N4 (Fig. 6a). This phenomenon is attributed to the quantum confinement effect induced by its ultrathin structure.33 Using the Kubelka–Munk method, the band gaps of B-CN, E460-CN, E480-CN, E500-CN, and E540-CN were determined to be 2.81, 2.89, 2.92, 2.95, and 3.00 eV, respectively (Fig. 6b).59 The Mott–Schottky curves of samples revealed that the flat-band potentials of B-CN, E460-CN, E480-CN, E500-CN, and E540-CN were −0.593, −0.468, −0.505, −0.537 and −0.476 V vs. Ag/AgCl, respectively (Fig. 6c). The positive slope in the Mott–Schottky plots revealed the n-type semiconductor characteristics of all samples.60 The equation E(NHE) = E(Ag/AgCl) + 0.197 is used, along with the theory that the flat-band potential of n-type semiconductors is 0.2 eV larger than the conduction band.61 Consequently, the band structures of all samples were delineated, satisfying the thermodynamic requisites for redox processes (Fig. 6d).
 | ||
| Fig. 6 (a) UV-vis diffuse reflectance spectra, (b) Tauc's plots of (ahν)2vs. photon energy, (c) Mott–Schottky plots, and (d) schematic diagram of the band structure of B-CN, E460-CN, E480-CN, E500-CN and E540-CN; (e) steady-state photoluminescence (PL) spectroscopy, (f) time-resolved photoluminescence (TR-PL) spectroscopy, (g) EIS spectra and (h) transient photocurrent spectra of B-CN, E460-CN, E480-CN, E500-CN and E540-CN; (i) transient photocurrent spectra of E500-CN under different conditions in 0.5 mol L−1 Na2SO4 solution. | ||
In addition to bandgap and light absorption, photocatalytic activity is closely related to the separation efficiency of photogenerated charge carriers. Therefore, steady-state photoluminescence (PL) spectroscopy and time-resolved photoluminescence (TR-PL) spectroscopy were utilized to examine the charge separation efficiency of the samples. As illustrated in Fig. 6e, the fluorescence intensity of exfoliated g-C3N4 under 330 nm excitation decreases significantly with the increasing exfoliation degree, suggesting enhanced suppression of electron–hole radiative recombination.62 This can be attributed to the nanosheet structure and the enhanced internal spontaneous polarization, both of which promote more efficient charge separation and transfer. However, after treatment at 540 °C, charge recombination worsens due to the aggregation of cyano defects, which act as recombination centers. This finding is further supported by TR-PL spectra, where the charge carrier lifetime is extended by exfoliation, with the longest lifetime observed for E500-CN (Fig. 6f).63 This indicates that E500-CN has the best charge separation efficiency among the tested samples, accounting for its superior photocatalytic performance. Electrochemical impedance spectroscopy (EIS) measurements confirmed that E500-CN exhibited the smallest arc radius (Fig. 6g), indicating lower resistance and enhanced interfacial charge carrier transfer. Additionally, transient photocurrent measurements provided further insights into charge transfer efficiency of the samples. The photocurrent responses followed the trend of E500-CN > E540-CN > E480-CN > E460-CN > B-CN (Fig. 6h). For E500-CN, current responses under different conditions, i.e., light, ultrasound, and combined ultrasound and light irradiation, were measured. Notably, the highest current density was achieved under simultaneous ultrasound and light irradiation (Fig. 6i). This observation firmly proves the role of piezoelectric polarization in promoting charge separation and transfer, underscoring the significant synergy between E500-CN's semiconducting and piezoelectric properties. The combination of these properties enhances the overall piezo-photocatalytic performance. Overall, the thinner layer thickness, which reduces the diffusion distance, along with the internal piezo-potential of the material, significantly facilitates the photogenerated electron–hole pairs separation and diffusion, thus successfully suppressing their recombination. However, if the layers become excessively thin due to extreme etching temperatures (540 °C), the structure of tri-s-triazine units could be compromised, and an excess of cyano defects can be introduced, which can negatively impact its semiconducting properties and photocatalytic reaction. As a result, E500-CN shows a balance between piezoelectric and semiconducting properties, demonstrating the best piezo-photocatalytic performance.
DFT calculations were conducted to gain insights into the internal mechanism. Structural models were simulated for both (Fig. S11†), with E500-CN exhibiting a long-range ordered structure connected by hydrogen bonds. In contrast, the elevated temperature treatment in E540-CN led to the disruption of these hydrogen bonds, accompanied by the formation of cyano defects. This structural disruption resulted in a significant increase in the material's polarity, as confirmed by electrostatic potential calculations (Fig. 7a and b). This increase in polarity aligns with experimental results that the material shows enhanced piezoelectricity. Furthermore, theoretical calculations have revealed that cyano groups can act as electron acceptors (Fig. 7c) and the cyano defects could trap photoexcited electrons, leading to severe recombination. As a result, despite showing superior piezoelectricity, the presence of cyano defects in E540-CN hinders its semiconducting properties due to electron recombination, explaining its lower piezo-photocatalytic performance compared to E500-CN (Fig. 7d).
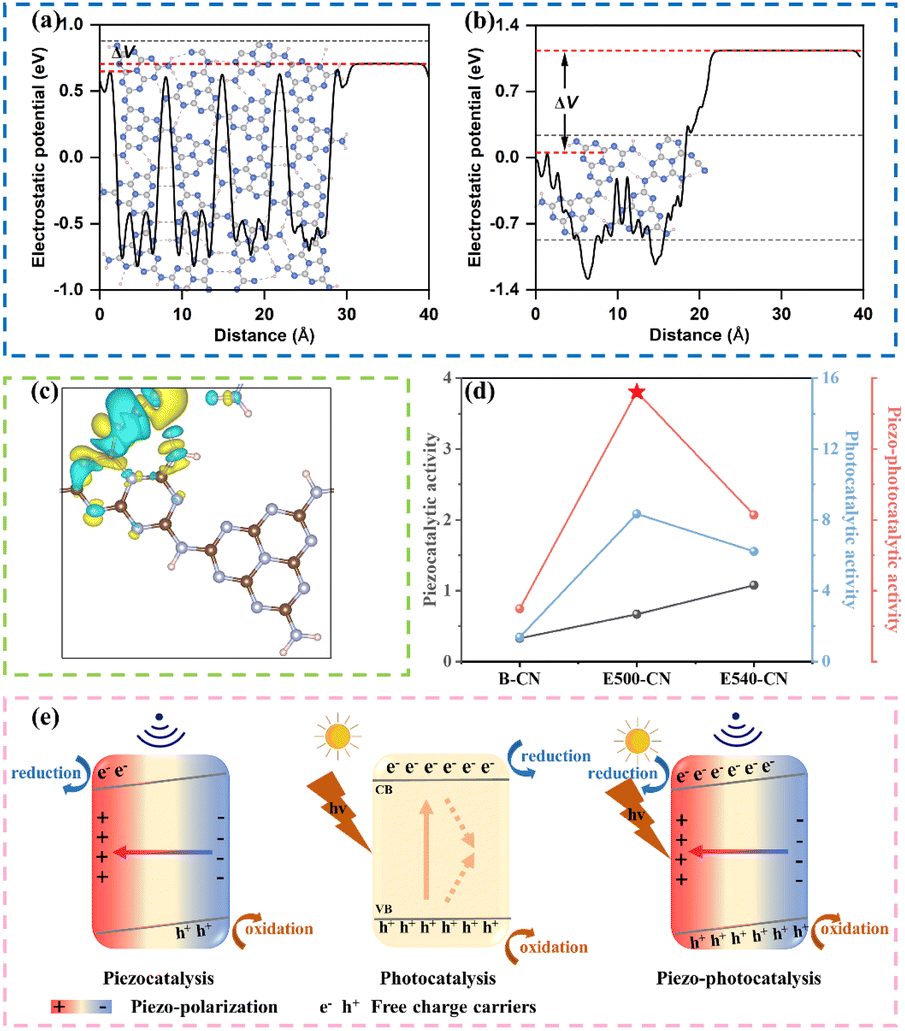 | ||
| Fig. 7 The calculated electrostatic potentials of (a) E500-CN with hydrogen bonds and (b) E540-CN with cyano groups; (c) the charge difference density in E540-CN; blue and yellow regions represent electron deficiency and electron enrichment, respectively. (d) Comparison of the piezocatalytic, photocatalytic and piezo-photocatalytic activities of B-CN, E500-CN, and E540-CN; (e) schematic illustration of the charge transfer process of the piezocatalysis, photocatalysis, and piezo-photocatalysis using E500-CN. | ||
Based on the analysis presented above, a plausible mechanism for the enhancement of photocatalytic performance through piezoelectric polarization of E500-CN is proposed in Fig. 7e. When E500-CN is subjected to ultrasonic vibration, the collapse of cavitation bubbles induces considerable pressure (up to approximately 108 Pa).64 This extreme pressure leads to notable deformations in the nanosheets, thereby generating an internal polarized electric field. This field separates negative and positive charges on opposite surfaces, triggering the piezocatalytic redox reaction. Upon light irradiation, photoinduced h+ and e− are generated within E500-CN; however, a substantial portion of these charge carriers typically recombine. In contrast, under simultaneous light and ultrasonic stimulation, the built-in piezoelectric potential tilts the conduction band (CB) and valence band (VB) of E500-CN, leading to more effective separation of the photoinduced electron–hole pairs. The internal polarization electric field also drives these charge carriers to migrate toward the material's surface in opposite directions, subsequently inducing redox reaction to form reactive species. Consequently, the generated h+, 1O2 and ˙O2− facilitate rapid decomposition of pollutants, demonstrating the strong synergy between piezocatalysis and photocatalysis.
4. Conclusion
In conclusion, this study successfully unveils the inherent tradeoff between piezoelectric and semiconducting properties in piezo-photocatalysts through the development of graphitic carbon nitride (g-C3N4) nanosheets via a thermal-driven oxidative exfoliation approach. The enhanced piezoelectric and semiconducting properties of the exfoliated nanosheets significantly improve their piezo-phototronic effect, leading to an impressive degradation rate of 97% for organic pollutants within only 20 minutes. The mechanism insights reveal that the optimized material, E500-CN, leverages the vibration-induced in-plane piezopotential to facilitate effective separation and transport of charge carriers, enhancing the generation of reactive species. The remarkable performance underscores a practical design strategy for developing efficient 2D piezo-photocatalysts with an optimized piezo-phototronic effect to advance sustainable water treatment technologies. The insights gained from the enhanced piezoelectric and semiconducting properties of the materials pave the way for future innovations in environmental remediation.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Special Science and Technology Innovation Program for Carbon Peak and Carbon Neutralization of Jiangsu Province (BE2022612), the Natural Science Research Major Project of Jiangsu Higher Education Institutions of ChinaReferences
- Q. Zhang, J. Chen, H. N. Che, B. Liu and Y. H. Ao, Small, 2023, 19, 2302510 CrossRef CAS PubMed.
- S. J. Li, C. C. Wang, Y. P. Liu, Y. Z. Liu, M. J. Cai, W. Zhao and X. G. Duan, Chem. Eng. J., 2023, 455, 140943 CrossRef CAS.
- Z. B. Luo, T. Wang, J. J. Zhang, C. C. Li, H. M. Li and J. L. Gong, Angew. Chem., Int. Ed., 2017, 56, 12878–12882 CrossRef CAS PubMed.
- Y. M. Ma and S. Y. Li, RSC Adv., 2019, 9, 33519–33524 RSC.
- Q. Y. Ji, K. X. Du, J. D. Zhu, X. Q. Ye, H. J. Li, X. Y. Cheng, Y. Z. Liu, Z. Xu, G. C. Zuo, S. Y. Li, S. G. Yang, L. M. Zhang and H. He, Chem. Eng. J., 2023, 462, 142116 CrossRef CAS.
- Z. R. Liu, L. W. Wang, X. Yu, J. Zhang, R. Q. Yang, X. D. Zhang, Y. C. Ji, M. Q. Wu, L. Deng, L. L. Li and Z. L. Wang, Adv. Funct. Mater., 2019, 29, 1807279 CrossRef CAS.
- X. Y. Xue, W. L. Zang, P. Deng, Q. Wang, L. L. Xing, Y. Zhang and Z. L. Wang, Nano Energy, 2015, 13, 414–422 CrossRef CAS.
- S. C. Tu, Y. X. Guo, Y. H. Zhang, C. Hu, T. R. Zhang, T. Y. Ma and H. W. Huang, Adv. Funct. Mater., 2020, 30, 2005158 CrossRef CAS.
- C. Y. Wang, C. Hu, F. Chen, T. Y. Ma, Y. H. Zhang and H. W. Huang, Nano Energy, 2023, 107, 108093 CrossRef CAS.
- J. H. Liu, W. L. Qi, M. M. Xu, T. Thomas, S. Q. Liu and M. H. Yang, Angew. Chem., Int. Ed., 2023, 62, e202213927 CrossRef CAS PubMed.
- W. X. Zheng, Y. F. Tang, Z. W. Liu, G. X. Xing and K. Zhao, J. Mater. Chem. A, 2022, 10, 13544–13555 RSC.
- Y. Zhang, C. H. Liu, G. L. Zhu, X. Huang, W. Liu, W. G. Hu, M. Song, W. D. He, J. Liu and J. Y. Zhai, RSC Adv., 2017, 7, 48176–48183 RSC.
- M. L. Xu, M. Lu, G. Y. Qin, X. M. Wu, T. Yu, L. N. Zhang, K. Li, X. Cheng and Y. Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202210700 CrossRef CAS PubMed.
- W. Amdouni, M. Fricaudet, M. Otoničar, V. Garcia, S. Fusil, J. Kreisel, H. Maghraoui-Meherzi and B. Dkhil, Adv. Mater., 2023, 35, 2301841 CrossRef CAS PubMed.
- X. Y. Huang, R. Lei, J. Yuan, F. Gao, C. K. Jiang, W. H. Feng, J. D. Zhuang and P. Liu, Appl. Catal., B, 2021, 282, 119586 CrossRef CAS.
- C. Sun, Y. M. Fu, Q. Wang, L. L. Xing, B. D. Liu and X. Y. Xue, RSC Adv., 2016, 6, 87446–87453 RSC.
- Z. J. Wang, T. C. Hu, H. X. He, Y. M. Fu, X. Zhang, J. Sun, L. L. Xing, B. D. Liu, Y. Zhang and X. Y. Xue, ACS Sustainable Chem. Eng., 2018, 6, 10162–10172 CrossRef CAS.
- S. Y. Lan, C. Yu, F. Sun, Y. X. Chen, D. Y. Chen, W. J. Mai and M. S. Zhu, Nano Energy, 2022, 93, 106792 CrossRef CAS.
- K. T. Wong, C. E. Choong, I. W. Nah, S. H. Kim, B. H. Jeon, Y. Yoon, E. H. Choi and M. Jang, Appl. Catal., B, 2022, 315, 121581 CrossRef CAS.
- Z. Li, Y. Y. Zhou, Y. T. Zhou, K. Wang, Y. Yun, S. Y. Chen, W. T. Jiao, L. Chen, B. Zou and M. S. Zhu, Nat. Commun., 2023, 14, 5742 CrossRef CAS PubMed.
- P. L. Wang, S. Y. Fan, X. Y. Li, J. Duan and D. K. Zhang, ACS Catal., 2023, 13, 9515–9523 CrossRef CAS.
- H. Lei, Q. S. He, M. X. Wu, Y. Y. Xu, P. F. Sun and X. P. Dong, J. Hazard. Mater., 2022, 421, 126696 CrossRef CAS PubMed.
- F. J. Liang, Z. W. Chen, Z. Y. Lu and X. Wang, J. Colloid Interface Sci., 2023, 630, 191–203 CrossRef CAS PubMed.
- T. Wu, Z. F. Liu, B. B. Shao, Q. Y. He, Y. Pan, X. S. Zhang, J. W. Sun, M. He, L. Ge, C. Y. Cheng and T. J. Hu, Nano Energy, 2024, 120, 109137 CrossRef CAS.
- C. Hu, F. Chen, Y. G. Wang, N. Tian, T. Y. Ma, Y. H. Zhang and H. W. Huang, Adv. Mater., 2021, 33, e2101751 CrossRef PubMed.
- J. Y. Hu, C. Yu, C. Li, S. Y. Lan, L. X. Zeng and M. S. Zhu, Nano Energy, 2022, 101, 107583 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- H. Lamkaouane, H. Ftouhi, M. Richard-Plouet, N. Gautier, N. Stephant, M. Zazoui, M. Addou, L. Cattin, J. C. Bernède, Y. Mir and G. Louarn, Nanomaterials, 2022, 12, 3171 CrossRef CAS PubMed.
- B. Feng, Y. N. Liu, K. Wan, S. J. Zu, Y. Pei, X. X. Zhang, M. H. Qiao, H. X. Li and B. N. Zong, Angew. Chem., Int. Ed., 2024, 63, e202401884 CrossRef CAS PubMed.
- P. Niu, L. Zhang, G. Liu and H. M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- Y. Kang, Y. Yang, L. C. Yin, X. Kang, L. Wang, G. Liu and H. M. Cheng, Adv. Mater., 2016, 28, 6471–6477 CrossRef CAS PubMed.
- Q. Gu, Y. S. Liao, L. S. Yin, J. L. Long, X. X. Wang and C. Xue, Appl. Catal., B, 2015, 165, 503–510 CrossRef CAS.
- Y. Kang, Y. Yang, L. C. Yin, X. Kang, G. Liu and H. M. Cheng, Adv. Mater., 2015, 27, 4572–4577 CrossRef CAS PubMed.
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS.
- C. Y. Feng, L. Tang, Y. C. Deng, J. J. Wang, J. Luo, Y. N. Liu, X. L. Ouyang, H. R. Yang, J. F. Yu and J. J. Wang, Adv. Funct. Mater., 2020, 30, 2001922 CrossRef CAS.
- C. Y. Zhou, P. Xu, C. Lai, C. Zhang, G. M. Zeng, D. L. Huang, M. Cheng, L. Hu, W. P. Xiong, X. F. Wen, L. Qin, J. L. Yuan and W. J. Wang, Chem. Eng. J., 2019, 359, 186–196 CrossRef CAS.
- Y. Yang, G. M. Zeng, D. L. Huang, C. Zhang, D. H. He, C. Y. Zhou, W. J. Wang, W. P. Xiong, X. P. Li, B. S. Li, W. Y. Dong and Y. Zhou, Appl. Catal., B, 2020, 272, 118970 CrossRef CAS.
- X. L. Hu, P. Lu, R. Pan, Y. X. Li, J. W. Bai, Y. Z. He, C. H. Zhang, F. Y. Jia and M. Fu, Chem. Eng. J., 2021, 423, 130278 CrossRef CAS.
- C. Z. Li, J. L. Liu, H. Li, K. F. Wu, J. H. Wang and Q. H. Yang, Nat. Commun., 2022, 13, 2357 CrossRef CAS PubMed.
- C. C. Chu, D. C. Yao, Z. Chen, X. R. Liu, Q. S. Huang, Q. J. Li and S. Mao, Small, 2023, 19, 2303796 CrossRef CAS PubMed.
- P. Niu, M. Qiao, Y. F. Li, L. Huang and T. Y. Zhai, Nano Energy, 2018, 44, 73–81 CrossRef CAS.
- Y. Z. Zhang, Z. X. Huang, C.-L. Dong, J. W. Shi, C. Cheng, X. J. Guan, S. C. Zong, B. Luo, Z. N. Cheng, D. X. Wei, Y.-C. Huang, S. H. Shen and L. J. Guo, Chem. Eng. J., 2022, 431, 134101 CrossRef CAS.
- J. J. Jiang, Z. Q. Zhao, J. Y. Gao, T. R. Li, M. Y. Li, D. D. Zhou and S. S. Dong, Environ. Sci. Technol., 2022, 56, 5611–5619 CrossRef CAS PubMed.
- B. Jiang, H. Huang, W. B. Gong, X. Q. Gu, T. Liu, J. C. Zhang, W. Qin, H. Chen, Y. C. Jin, Z. Q. Liang and L. Jiang, Adv. Funct. Mater., 2021, 31, 2105045 CrossRef CAS.
- K. Li and W. D. Zhang, Small, 2018, 14, 1703599 CrossRef PubMed.
- Y. J. Sun, Z. Y. Fang, X. T. Huang, C. W. Bai, K. A. Zhu, X. J. Chen, B. B. Zhang, Y. S. Zhang, Q. Yang, J. X. Zheng and F. Chen, Appl. Catal., B, 2023, 337, 122994 CrossRef CAS.
- J. Shi, M. B. Starr and X. Wang, Adv. Mater., 2012, 24, 4683–4691 CrossRef CAS PubMed.
- Y. Zheng, X. Y. Wu, Y. C. Zhang, Y. Q. Li, W. Q. Shao, J. Fu, Q. Lin, J. S. Tan, S. W. Gao, W. N. Ye and H. T. Huang, Chem. Eng. J., 2023, 453, 139919 CrossRef CAS.
- J. D. Zhu, X. Y. Yan, L. L. Wu, Q. W. Yu, W. D. Zhou, Q. Y. Ji, Q. Zhong, Y. Z. Liu, G. C. Zuo, Z. Xu, S. G. Yang, L. M. Zhang and H. He, Sep. Purif. Technol., 2024, 333, 125950 CrossRef CAS.
- H. R. Wang, X. L. Zhang, C. Hu, H. Cai, S. C. Tu and H. W. Huang, Appl. Surf. Sci., 2024, 650, 159214 CrossRef CAS.
- Y. M. Du, T. Lu, X. N. Li, Y. Liu, W. P. Sun, S. J. Zhang and Z. X. Cheng, Nano Energy, 2022, 104, 107919 CrossRef CAS.
- D. F. Yu, Z. H. Liu, J. M. Zhang, S. Li, Z. C. Zhao, L. F. Zhu, W. S. Liu, Y. H. Lin, H. Liu and Z. T. Zhang, Nano Energy, 2019, 58, 695–705 CrossRef CAS.
- W. H. Feng, J. Yuan, L. L. Zhang, W. T. Hu, Z. H. Wu, X. L. Wang, X. Y. Huang, P. Liu and S. Y. Zhang, Appl. Catal., B, 2020, 277, 119250 CrossRef CAS.
- M. Zelisko, Y. Hanlumyuang, S. B. Yang, Y. M. Liu, C. H. Lei, J. Y. Li, P. M. Ajayan and P. Sharma, Nat. Commun., 2014, 5, 4284 CrossRef CAS PubMed.
- A. X. Deng, E. Zhao, Q. Li, Y. Sun, Y. Z. Liu, S. G. Yang, H. He, Y. Xu, W. Zhao, H. O. Song, Z. Xu and Z. P. Chen, ACS Nano, 2023, 17, 11869–11881 CrossRef CAS PubMed.
- Y. F. Li, R. X. Jin, Y. Xing, J. Q. Li, S. Y. Song, X. C. Liu, M. Li and R. C. Jin, Adv. Energy Mater., 2016, 6, 1601273 CrossRef.
- J. H. Zheng and Z. Lei, Appl. Catal., B, 2018, 237, 1–8 CrossRef CAS.
- Y. Z. Liu, Y. Sun, E. Zhao, W. W. Yang, J. K. Lin, Q. Zhong, H. F. Qi, A. X. Deng, S. G. Yang, H. Y. Zhang, H. He, S. M. Liu, Z. P. Chen and S. B. Wang, Adv. Funct. Mater., 2023, 33, 2301840 CrossRef CAS.
- B. Q. Xia, B. W. He, J. J. Zhang, L. Q. Li, Y. Z. Zhang, J. G. Yu, J. R. Ran and S.-Z. Qiao, Adv. Energy Mater., 2022, 12, 2201449 CrossRef CAS.
- J. Wu, N. Qin and D. H. Bao, Nano Energy, 2018, 45, 44–51 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07713j |
| This journal is © The Royal Society of Chemistry 2025 |