
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Introducing phenol-yne chemistry for the design of lignin-based vitrimers: towards sustainable and recyclable materials†
Lisa
Sougrati
a,
Antoine
Duval
 *ab and
Luc
Avérous
*a
*ab and
Luc
Avérous
*a
aBioTeam/ICPEES-ECPM, UMR CNRS 7515, Université de Strasbourg, 25 Rue Becquerel, 67087 Strasbourg, Cedex 2, France. E-mail: antoine.duval@unistra.fr; luc.averous@unistra.fr; Fax: +33 368852716; Tel: +33 368852784
bSoprema, 15 Rue de Saint Nazaire, 67100, Strasbourg, France
First published on 7th January 2025
Abstract
Lignins are the main source of renewable aromatic compounds on the Earth. These polyphenols of high functionality are biobased building blocks of great interest for the development of aromatic polymer materials. However, one shortcoming of these thermosets is their inefficient recyclability, which increases their environmental impact in a situation where the need for high-performance materials with controlled end-of-life is paramount. Conceptualization of lignin-based vitrimers is an attractive solution to address this global challenge. In this work, we report the synthesis, evaluation and recycling of lignin-based phenol-yne vitrimers adhering to the principles of circular bioeconomy. Herein, the potential of phenols substituted in ortho position with one or two methoxy groups (G and S units) to undergo vinyl ether bond exchanges was examined with model compounds. Then, the addition kinetics of aliphatic hydroxyls and di-substituted phenols was investigated on model molecules representatives of lignin heterogeneous reactive groups. Subsequently, a series of materials from different lignin sources was prepared via a one-step click-addition under solvent-free conditions and achieved full atom-economy in accordance with different green chemistry principles. These high lignin-content (39 to 49 wt%) vitrimers were duly characterized. The thermal, morphological, rheological, and mechanical properties were comprehensively assessed. It was observed that the vitrimers underwent rearrangement through vinyl ether bond exchanges. Relaxation times in the range of 74 to 476 s were obtained at 200 °C. The determined activation energies were in the range of 72 to 89 kJ mol−1. Network structures and properties were analyzed through successive physical recycling steps, and the chemical recycling potential was surveyed via acid-catalyzed hydrolysis. Results clearly showed a proof-of-concept for the closed-loop recycling of lignin-based phenol-yne vitrimers, offering further upcycling opportunities for the development of sustainable aromatic materials for a greener future and towards a cradle-to-cradle approach.
1. Introduction
Lignocellulosic biomass is the main available source of aromatic renewable compounds on the Earth. Lignins are one of their major components and represent up to 40% of their dry weight. It is an attractive source of phenols for the design of sustainable and performant aromatic polymers. Lignin is an amorphous polyphenol with a complex structure, which is produced from the polymerization of three main aromatic precursors (monolignols), namely, coniferyl alcohol, sinapyl alcohol, and p-coumaryl alcohol, often referred to as G, S and H units, respectively. The complex lignin architecture mainly varies according to the feedstock's botanical origin and extraction process. According to the extraction and fractionation process, different lignins can be obtained with (sulfite and Kraft processes) or without sulfur (soda and organosolv processes).1Lignin has a high functionality in aliphatic and phenolic (mainly G and S) hydroxyls (OHs), enabling their utilization as building blocks for the preparation of high-performance thermosets. Aromatic structures present strong advantages, such as high mechanical properties and fire resistance. Nonetheless, the highly crosslinked structures of the ensuing polymer networks result in limited recycling opportunities due to their inability to melt. It is a profound global environmental concern.
For addressing the end-of-life issues of thermosets, the development of covalent adaptable networks (CANs) is an effective method. CANs are an ingenious novel class of polymers benefiting from the most sophisticated features of thermosets and thermoplastics that enables reprocessing (as for thermoplastics) without compromising the mechanical properties (as for thermosets).2–4 Thanks to the incorporation of stimuli-responsive bonds that are sensitive to temperature, light or pH variations for instance, these networks can undergo bond rearrangements to induce a macroscopic flow. CANs are generally divided in two main categories according to their bond exchange mechanisms. Dissociative CANs are subjected to the dissociation of the initial bond prior to the formation of a new one, resulting in decreased viscosity, and in some instances, to depolymerization. Conversely, associative CANs, also classified as vitrimers, rely on the formation of a new bond without bond cleavage, maintaining a constant crosslinking density.5–7 Because of the wide range of dynamic covalent chemistries, the development of recyclable alternatives to conventional thermoset polymers has been spread to different polymer types, such as polyolefins, polyesters, polyimines, and polyurethanes.8–12
The design of vitrimers derived from biomass is an elegant answer to address both the reduction of fossil chemicals and the end-of-life recyclability in a circular bioeconomy approach.13–15 Lignin has emerged as an attractive structure because of its compatibility with a large array of potential covalent dynamic chemistries. Recently, they have gained particular interest in the field of CANs.16–19 The valorization of lignin within CANs was successfully reported using various dynamic chemistries in polyester, polyurethane, vinylogous urethane, disulfide, and Diels–Alder networks.20–29 The design of these materials necessitates the chemical modification of lignin to enhance its reactivity, to homogenize its reactive end groups, or to graft new functions. This usually requires multi-step syntheses, consuming significant amounts of reagents and solvents, and generating substantial wastes. In a green chemistry approach, the limitation of the number of chemical modifications is paramount,30 and is key to facilitate the eventual scaling-up and industrialization.
In this global context, the emerging and still under-exploited phenol-yne dynamic chemistry is an elegant approach for the valorization of lignin into high-performance, recyclable and sustainable polymer materials. This strategy relies on the click-addition of phenols with activated alkynes under basic catalysis, which can undergo bond exchanges through addition/elimination pathways, inducing dynamicity.31–35 Phenol-yne should have the potential to enable the rapid preparation of lignin-based materials in a single atom-efficient step, without the prior chemical modification of lignin.
In this study, we report for the first time the synthesis of innovative phenol-yne vitrimers with high lignin content, up to around 50 wt%. Different lignins from various extraction and fractionation processes have been used, characterized and compared. In a first step, the impact of phenol ortho-substitution by methoxy groups (as in G and S units) on the bond exchange process was carefully analyzed using different model molecules. The click-addition of aliphatic and phenolic OHs (Al-OH and Ph-OH) with activated alkynes was then investigated on model compound representatives of the lignin heterogeneous structure. A polyethylene glycol (PEG)-derived activated dialkyne crosslinker was synthesized by esterification with propiolic acid. A series of vitrimers were then prepared using different lignin sources to evaluate the impact of their chemical structures on the material properties. Lignins were reacted via one-step click-addition with the PEG crosslinker in solvent-free conditions, leading to 100% atom-economy (Scheme 1). The structures and the thermal, rheological, and mechanical behaviors of these materials were duly determined. The end-of-life of these materials was investigated via several cycles of physical recycling. Alternatively, the chemical recycling potential was studied through the acid-catalyzed hydrolysis of vinyl ether and ester bonds, paving the way to a closed-loop recycling approach that would enable the recovery of both pristine lignin and PEG towards a cradle-to-cradle approach.
 | ||
| Scheme 1 Synthetic pathway for the preparation of (a) PEG dialkyne crosslinker (PEG-DA), and (b) phenol-yne (PY) networks from KL, OSL, and DOSL. | ||
2. Experimental section
2.1. Materials
BioPiva™100 softwood kraft lignin (KL) was supplied by UPM Biochemicals (Helsinki, Finland). Organosolv lignin (OSL) was isolated at pilot scale from beech wood using the aqueous acetone Fabiola™ process (TNO, Petten, Netherland). Detailed characterizations of this lignin has been largely described and previously published (formerly named “P-BEC-3”).36 A partially depolymerized lignin (DOSL) was obtained by catalytic depolymerization of OSL (TNO, Petten, Netherland). The steps of this reductive depolymerization and details of the DOSL characterizations have been previously described.37 All lignins were dried overnight at 50 °C prior to utilization. The main chemical and physicochemical characterizations of KL, OSL, and DOSL are reported in Table 1. The corresponding 31P NMR spectra and SEC distributions are available in Fig. S1 and S2 in ESI,† respectively.| Lignin | Al-OH content (mmol g−1) | Ph-OH content (mmol g−1) | S and condensed units (mmol g−1) | G units (mmol g−1) | H units (mmol g−1) | M n (g mol−1) | M w (g mol−1) | Đ |
|---|---|---|---|---|---|---|---|---|
| KL | 2.48 | 4.33 | 1.95 | 2.17 | 0.24 | 1580 | 5270 | 3.3 |
| OSL | 1.74 | 2.89 | 2.19 | 0.60 | 0.08 | 1340 | 3310 | 2.5 |
| DOSL | 2.17 | 3.27 | 2.39 | 0.78 | 0.09 | 1280 | 2310 | 1.8 |
Potassium carbonate anhydrous (K2CO3, ≥99%), benzyl alcohol (99%), 3,4,5-trichloropyridine (TCP, 98%), ethyl acetate (99%), and magnesium sulfate were obtained from Thermo Fisher Scientific. Polyethylene glycol with a number-average molar mass of 400 g mol−1 (PEG400), p-toluenesulfonic acid monohydrate (pTsOH, 99%), sodium bicarbonate (NaHCO3, 99.7%), and dimethyl sulfoxide anhydrous (DMSO, ≥99.7%) were obtained from Acros Organics. Ethyl propiolate (EP, 99%), propiolic acid (95%), 2-methoxyphenol, 2,6-dimethoxyphenol (99%), 4-methyl-2,6-dimethoxyphenol (S, ≥97%), 2-methoxy-4-methylphenol (G, ≥98%), 1-butanol, chromium(III) acetylacetonate (Cr(acac)3, 97%), cholesterol (>99%), deuterated chloroform (CDCl3), hydrochloric acid (37% in water), 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (Cl-TMDP, 95%), N,N-dimethylformamide anhydrous (DMF, 99.8%), and dichloromethane (CH2Cl2, ≥99.8%), were purchased from Sigma-Aldrich. Acetone was obtained from Carlo Erba Reagents. Deuterated dimethyl sulfoxide (DMSO-d6, 99.8%) was purchased from Eurisotop.
2.2. Synthesis of aromatic model compounds
Ethyl-3-(2-methoxy-4-methylphenoxy)acrylate (G-VE) and ethyl-3-(2,6-dimethoxy-4-methylphenoxy)acrylate (S-VE) have ben synthesized as aromatic models (Fig. S3†). For the synthesis of G-VE, 3 g of 2-methoxy-4-methylphenol (G) (21.71 mmol), 2.201 mL of ethyl propiolate (1 equivalent, 21.71 mmol) and 0.15 g of K2CO3 (0.05 equivalent, 1.09 mmol) were added to a 25 mL round bottom flask. The reactive mixture was stirred vigorously under argon flow, and heated at 80 °C for 3 h. After cooling, the mixture was diluted in 25 mL of ethyl acetate, and washed consecutively by 25 mL of 0.01 M NaOH, brine and water. The product was dried with anhydrous magnesium sulfate, filtered, concentrated under reduced pressure, and dried overnight at 50 °C in a vacuum oven. G-VE was obtained as a viscous liquid in 96% yield.1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 12.2 Hz, 1H), 6.92 (d, J = 7.9 Hz, 1H), 6.78 (s, 2H), 5.38 (d, J = 12.3 Hz, 1H), 4.28–4.08 (m, 2H), 3.84 (s, 3H), 2.34 (s, 3H), 1.26 (t, J = 7.2 Hz, 3H).
The same protocol was reproduced using 4-methyl-2,6-dimethoxyphenol (S), producing ethyl-3-(2,6-dimethoxy-4-methylphenoxy)acrylate (S-VE) in 89% yield.
1H NMR (400 MHz, CDCl3) δ 7.65 (d, J = 12.2 Hz, 1H), 6.55 (d, J = 7.0 Hz, 1H), 5.26 (d, J = 12.2 Hz, 1H), 4.98 (d, J = 7.0 Hz, 1H), 4.21 (q, J = 7.2 Hz, 3H), 4.15 (q, J = 7.2 Hz, 2H), 1.27 (d, J = 16.5 Hz, 4H).
The 1H NMR spectra of both compounds are available in Fig. S4 and S5, in ESI.†
2.3. Vinyl ether model exchange reactions
In a 10 mL round bottom flask were added 0.2 g of G-VE (0.85 mmol), 93 μL of 2-methoxyphenol (G′, 1 equivalent, 0.85 mmol) and 5.9 mg of K2CO3 (0.05 equivalent, 0.042 mmol). The mixture was heated at 120 °C for 24 h. The exchange reaction between the GG′ units was monitored by 1H NMR. The protocol was adapted to study the exchange kinetics for the GS′, SG′, and SS′ units. 2-Methoxyphenol (G′) is used as a G-type exchangeable unit, 2,6-dimethoxyphenol (S′) as an S-type exchangeable unit, and benzyl alcohol as an aliphatic OH unit. All the reaction conditions are summarized in Table S1 in ESI.† The reactions are designated as follows: XY, with X = G-VE or S-VE model compounds, and Y = G′ or S′.2.4. Click-addition kinetics study on aliphatic and phenolic models
To study the reactivity of the phenolic model, 0.5 g of 4-methoxyl-2,6-dimethoxyphenol (2.97 mmol), 0.301 mL of ethyl propiolate (1 equivalent, 2.97 mmol), and 0.0205 g of K2CO3 (0.05 equivalent, 0.15 mmol) were added to a 25 mL round bottom flask. The mixture was heated at 60, 80, or 120 °C for 6 h. The reaction kinetics were monitored by 1H NMR. The same procedure was adopted with 1-butanol as a model for aliphatic OHs.2.5. Lignin modification with ethyl propiolate
In a 25 mL round bottom flask equipped with a condenser, 1 g of lignin was dissolved in 4 mL DMF or DMSO. Ethyl propiolate (1 equivalent with respect to the phenol groups of lignin) and K2CO3 (0.05 equivalent with respect to the phenol groups) were then added. The reactive mixture was stirred vigorously under an argon flow, and heated at 80 or 120 °C. The reaction was stopped after complete consumption of the alkyne groups, as evaluated by 1H NMR. The product was precipitated in acidified water (pH = 2), filtered, and washed with water. The powder was dried overnight at 50 °C in a vacuum oven. Detailed reaction conditions including quantities and time are available in Table S2 in ESI.† The 2D HSQC spectra are available in Fig. S6 in ESI.†31P NMR spectra are displayed in Fig. S7–S10, in ESI.†2.6. Synthesis of PEG dialkyne crosslinker (PEG-DA)
In a 250 mL round-bottom flask equipped with a distillation apparatus connected to a trap filled with a saturated NaHCO3 aqueous solution were added 50 g of PEG400 (0.273 mol), 33.65 mL of propiolic acid (2 equivalent, 0.547 mol), and 2.60 g of p-toluenesulfonic acid monohydrate (0.05 equivalent, 0.014 mol). The mixture was heated at 85 °C under 550 mbar with vigorous stirring for 24 h. After cooling to room temperature, the mixture was diluted in 200 mL of dichloromethane, and the product was washed consecutively with 200 mL of saturated NaHCO3, brine, and water. The product was dried with anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure. PEG-DA was dried 48 h at 60 °C in a vacuum oven. The 1H NMR characterization is available in Fig. S11 in ESI.† A yield of 58% and 82% conversion were obtained.2.7. Synthesis of lignin-based phenol-yne materials
5.50 g of KL (4.33 mmol g−1 of phenol groups), 8.569 g of PED-DA (0.8 equivalent with respect to phenol groups), and 0.658 g of K2CO3 (0.2 equivalent with respect to phenol groups) were added in a mortar. Using a pestle, the blend was mixed until complete visible homogenization. The resulting paste was poured into a Teflon mold, and cured in an oven at 120 °C for 2.5 h. The material was compression-molded for 30 minutes at 160 °C under a constant applied force of 16 MPa to obtain a homogeneous film (10 cm × 10 cm × 1 mm). The same procedure was also applied to OSL and DOSL. The detailed experimental conditions are reported in Table S3 in ESI.† The material designations were established as follows: X-PY (PY for “phenol-yne”), where X = KL, OSL, or DOSL.2.8. Physical and chemical recycling studies of phenol-yne materials
Physical recycling was performed as follows: the material was ground into a fine and homogenous powder using a 200 W Livoo DOD192 grinder. The powder was compression-molded into a new generation material for 30 minutes at 160 °C under a constant applied force of 16 MPa to obtain the films (10 cm × 10 cm × 1 mm).Chemical recycling was performed by acid-catalyzed hydrolysis. 1.2 g of KL-PY sample that had been previously ground was immersed in 18 mL 5 M HCl and 85 mL DMSO. The reaction was equipped with a cooler and heated at 70 °C for 72 h. The solid fraction was removed by centrifugation and washed with water for quantification. It represented only 0.5 wt% of the initial sample. The soluble fraction was precipitated in 400 mL of acidified water (pH around 2) and filtered. To ensure optimum purification, the product was resolubilized in a minimum of DMSO (3 mL) and reprecipitated in 100 mL of acidified water. The resulting powder was filtered and dried overnight at 50 °C in a vacuum oven.
2.9. Characterization techniques
1H and 31P NMR spectra were acquired on a Bruker 400 MHz spectrometer. 1H NMR calibration was performed based on the CDCl3 (δH = 7.26 ppm) chemical shift over 16 scans. Quantitative 1H NMR (15 s relaxation delay) was performed using TCP as the internal standard. Approximately 20 mg of sample was dissolved in 500 μL of solvent and 100 μL of a standard solution of TCP (0.5 M) was added. 31P NMR samples were prepared according to standard protocols, employing the reaction with a phosphylating reagent (Cl-TMDP).38–40 The spectra were acquired with 128 scans and 15 s relaxation delays. Eqn (1) was used for the calculation of the OH content (in mmol g−1), where Cs (M) is the concentration of the cholesterol standard, Vs (μL) is the volume of the cholesterol standard, A is the area of the OH groups peak obtained on the 31P NMR spectrum, and m (mg) is the sample weight: | (1) |
2D HSQC NMR spectra were recorded on a Bruker 500 MHz spectrometer at 25 °C. About 100 mg of sample was dissolved in DMSO-d6, and 32 scans were recorded (1024 × 256 increments, 1.5 s relaxation delay, 0.13 s acquisition time).
Fourier transform infrared (FT-IR) spectra were assessed at room temperature using a Nicolet 380 spectrometer from Thermo Electron Corporation, equipped with an attenuated total reflectance (ATR) diamond module. Each analysis required 32 scans in the range of 4000–500 cm−1.
Diffuse reflectance FT-IR (DRIFT) spectra were collected on a Bruker VERTEX 70 spectrometer equipped with a MCT (HgCdTe) detector with a 4 cm−1 spectral resolution in the range of 4000–500 cm−1. The viscous samples were mixed with KBr to obtain powders before introduction in a Praying Mantis™ low temperature reaction chamber from Harrick Scientific. The samples were heated under a 10 mL min−1 He flow rate with a ramp of 10 °C min−1 from room temperature to 80 or 120 °C, followed by an isotherm.
Size-exclusion chromatography (SEC) was carried out on a Acquity APC apparatus from Waters for the determination of the number-average molar mass (Mn), weight-average molar mass (Mw) and dispersity (Đ). THF was employed as an eluent (0.6 mL min−1) at 40 °C. Three 150 mm APC XT columns (a 45 Å, 1.7 μm column; a 200 Å, 2.5 μm column; and a 450 Å, 2.5 μm column) were connected, and the detection was performed with a UV or a refractive index detector. The apparatus was calibrated using polystyrene (PS) standards. To increase lignins solubility in THF, samples were acetylated prior to analysis, in accordance with a previously reported procedure.41
Thermogravimetric analysis (TGA) was recorded on a Mettler Toledo TGA 2 apparatus. Samples of around 3 to 5 mg were heated in a crucible from room temperature to 800 °C at a rate of 20 °C min−1 under an air atmosphere (25 mL min−1 flow rate).
Differential scanning calorimetry (DSC) was carried out on a TA Instruments Discovery DSC-25 apparatus under dry nitrogen flow of 50 mL min−1. Around 2 to 3 mg of sample were weighed in aluminum pans. Samples were first equilibrated at 160 °C for 3 minutes to erase their thermal history. They were then cooled down at −80 °C at a 5 °C min−1 rate, and equilibrated for 3 min. Finally, samples were heated at 160 °C at a 10 °C min−1 rate. The glass transition temperature (Tg) was measured on the last heating ramp as the change of slope.
Wide angle X-ray scattering (WAXS) analysis was performed on a Bruker D8 Advance diffractometer using Cu Kα radiation (λ = 1.5406 Å) at room temperature in the 2θ range of 3 to 60°. A scan step of 0.03° and a step time of 0.5 s were used. Samples of 1 mm thickness were analyzed. Eqn (2) and (3) were employed for the calculation of the scattering vectors (q) and the real-space distances (D), where 2θ (rad) is the scattering angle and λ is the X-rays wavelength (Å).
 | (2) |
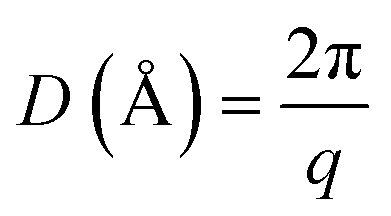 | (3) |
The peaks were deconvoluted with 4 Gaussian functions, using the Scipy library openly developed on GitHub, on Python 3.0. The proportions of the different interactions were calculated from the integrated areas under each Gaussian curves.
Dynamic mechanical analyses (DMA) were recorded on a TA Instruments Discovery Hybrid Rheometer HR-3 equipped with a rectangular torsion geometry. Rectangular samples of around 2 cm × 1 cm × 1 mm were heated at 3 °C min−1 from −60 to 180 °C, with a 0.01% strain at 1 Hz frequency. Eqn (4) was used for the calculation of the crosslinking densities (ν) in accordance with the theory of rubber elasticity for small deformations on the rubbery plateau, where G′ (Pa) is the storage modulus, and R the molar gas constant (8.314 J mol−1 K−1). Tα (K), the temperature of the α transition, is determined at the maximum of the tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ curve.42,43
δ curve.42,43
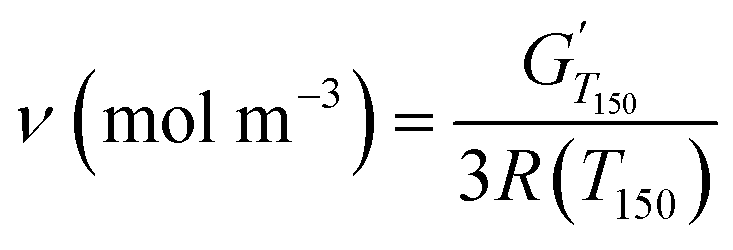 | (4) |
Stress relaxations were measured on a TA Instruments Discovery Hybrid Rheometer HR-3 equipped with ETC-steel 25 mm parallel plates geometry. Sample discs of 25 mm diameter and 1 mm thickness were analyzed. The series of experiments at different temperatures were carried out successively on the same sample after 10 min equilibration at temperature, with a fixed gap of around 1 mm and 1% strain. The curves were fitted with the Kohlrausch–Williams–Watts (KWW) stretched exponential decay using eqn (5), where G(t)/G0 is the normalized stress at relaxation time t, τ* is a characteristic relaxation time, and β (0 ≤ β ≤ 1) is the exponent representing the breadth of the relaxation times distribution.44–47 Activation energies (Ea) were determined according to an Arrhenius law (eqn (6)), by plotting lnτ* = f(1000/T).
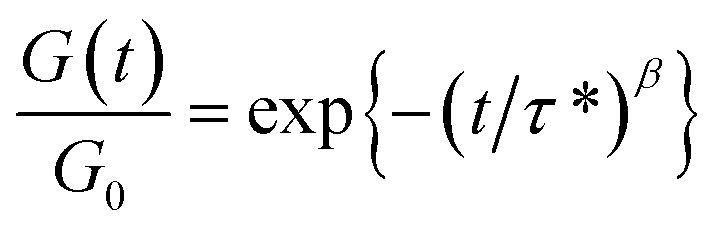 | (5) |
 | (6) |
Creep experiments were performed on a TA Instruments Discovery Hybrid Rheometer HR-3 equipped with a rectangular torsion geometry. Tests were carried out on rectangular samples of around 2 cm × 1 cm × 1 mm using a constant axial force of 2 N and a 0.2 MPa stress over 1000 s, followed by a recovery time of 1000 s. The stress was followed over time at constant temperature (20 and 70 °C).
Uniaxial tensile tests were carried out on an Instron 5567H dynamometer (USA) equipped with a 10 kN load cell. Tests were performed on a set of 5 dumbbell-shaped samples of dimension 45 × 5 × 1 mm3 with a constant crosshead speed of 20 mm min−1 in a room set at 23 °C. Young's modulus (E), stress at break (σ), and elongation at break (ε) were measured. Averages and standard deviations were determined. Representative curves were selected to be displayed.
The swelling ratio (SR) and gel fraction (GF) of the series of materials were investigated by immersion of previously dried samples of around 100 mg in both acetone and water for 48 h. The mass of the material after swelling (m1) was then measured. The materials were finally dried in a vacuum oven for 24 h at 50 °C, and their final mass (mf) was recorded. The test was conducted in triplicate. SR and GF were calculated according to eqn (7) and (8), respectively.
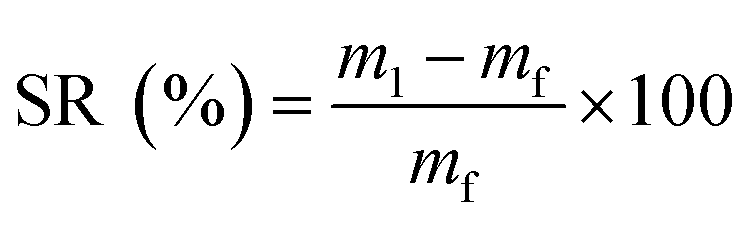 | (7) |
 | (8) |
The apparent water contact angles (WCA) of the final materials were measured by sessile test using a TRACKER™ goniometer from Teclis Scientific (France). 3 droplets of distilled water with volumes of 8 μL were dropped onto the recently dried materials. The WCA are given as average values with their standard deviations.
3. Results and discussion
3.1. Analysis of the vinyl ether rearrangements: a study on lignin model compounds
The emerging phenol-yne chemistry has been reported to undergo exchange reactions on small organic molecules,32 and in polymer networks based on bisphenol A.34 The bond exchange was suggested to occur through an associative pathway in the presence of free phenols: (i) the deprotonation of phenol under basic catalysis and subsequent nucleophilic addition of the phenolate anion to the vinyl ether bond, forming a negatively charged acetal enolate intermediate, and (ii) the elimination of the substituted phenol, generating a new vinyl ether (Scheme 2).34 Previous studies reported on the facile exchange at low temperature of vinyl ether bonds with phenols that are unsubstituted in ortho-positions. To transpose these previous results to the case of lignins, it is necessary to evaluate whether such rearrangements are also possible with mono- or di-ortho-substituted phenols, as in lignin G and S units. | ||
| Scheme 2 Mechanistic pathway for the associative exchange of phenyl vinyl ether dynamic bonds. | ||
Two model compounds containing vinyl ether bonds were thus synthesized from the lignin G and S models, by reaction with ethyl propiolate (G-VE and S-VE, respectively, Fig. 1 and Scheme S1 in ESI†). Their 1H NMR spectra are available in Fig. S3 and S4 in ESI.† The reaction proceeds in an anti-Markovnikov fashion, as confirmed by the absence of the Markovnikov product, revealing good regioselectivity (Scheme S2 in ESI†).33,48,49G-VE and S-VE show the formation of the E and Z-isomers, which are readily assigned due to their distinct coupling constants. G-VE resonances at 5.36 and 7.68 were assigned to E-isomer vinylene protons (JHH = 12.2 Hz) while doublets at 5.04 and 6.96 were attributed to Z-isomer (JHH = 7 Hz).50,51 Similarly, S-VE resonances at 5.25 and 7.64 ppm were assigned to the E-isomer, while doublets at 4.97 and 6.54 ppm were attributed to the Z-isomer. The proportion of E/Z-isomers in G-VE and S-VE were found to be 88/12 and 42/58, respectively, whereas Santos et al. observed only the E-isomer with unsubstituted phenols.32 The results herein suggest that the substituents in ortho-position tend to increase the proportion of the Z-isomer.
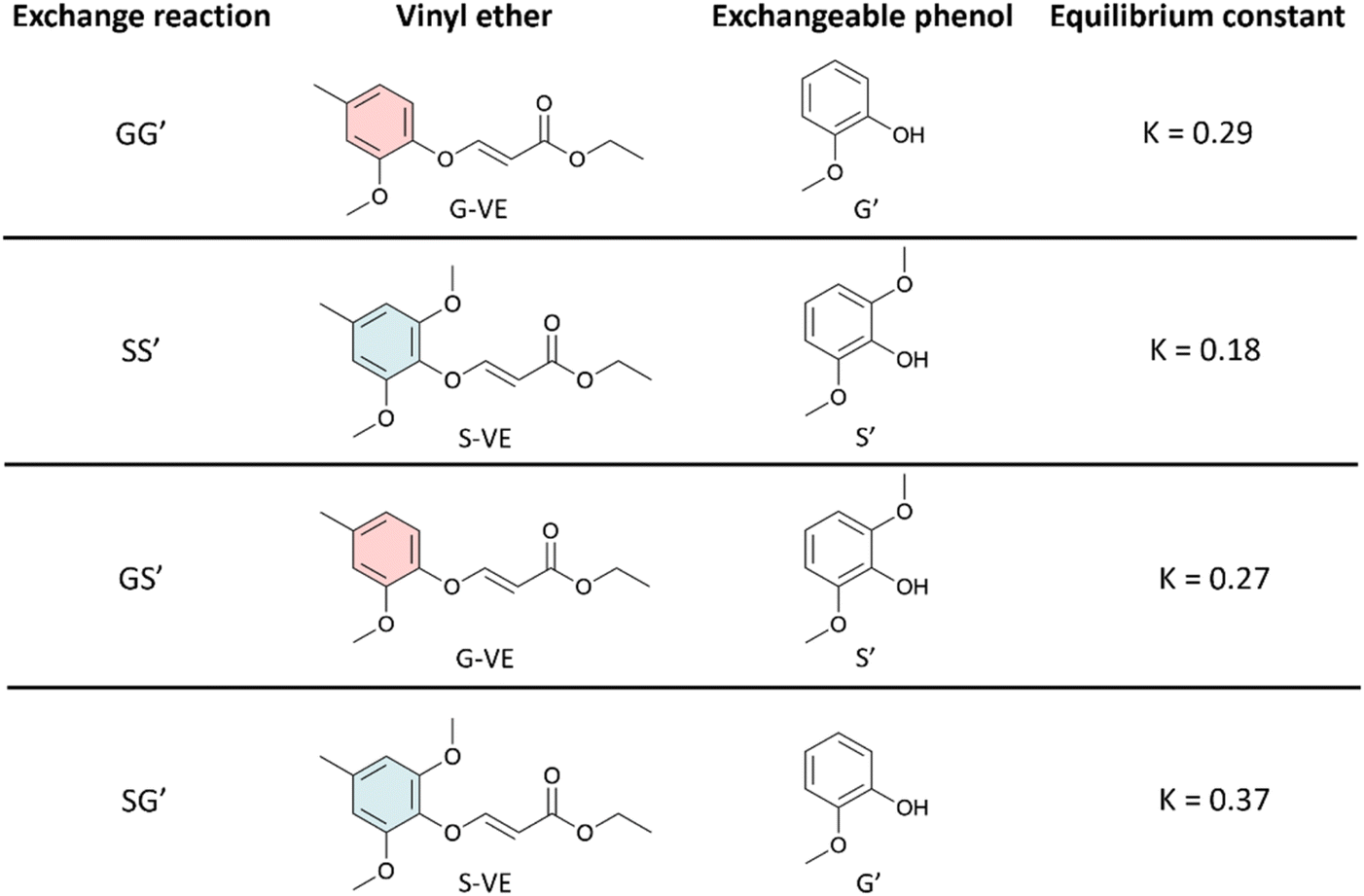 | ||
| Fig. 1 Exchange reaction conditions and equilibrium constants for GG′, SS′, GS′, and SG′ reactions. For clarity, only the E-isomers are presented. | ||
The dynamicity of the phenol-yne reaction between the various units was studied (GG′, GS′, SG′, and SS′) by reacting the G-VE and S-VE models with G′ or S′ (Fig. 1). The reactions were monitored by 1H NMR over 24 h. The spectra and corresponding plotted kinetics are available in ESI (Fig. S5–S11).†G-VE and S-VE were demonstrated to undergo exchange reactions when mixed with equimolar amounts of the G′ and S′ compounds (Fig. 2).
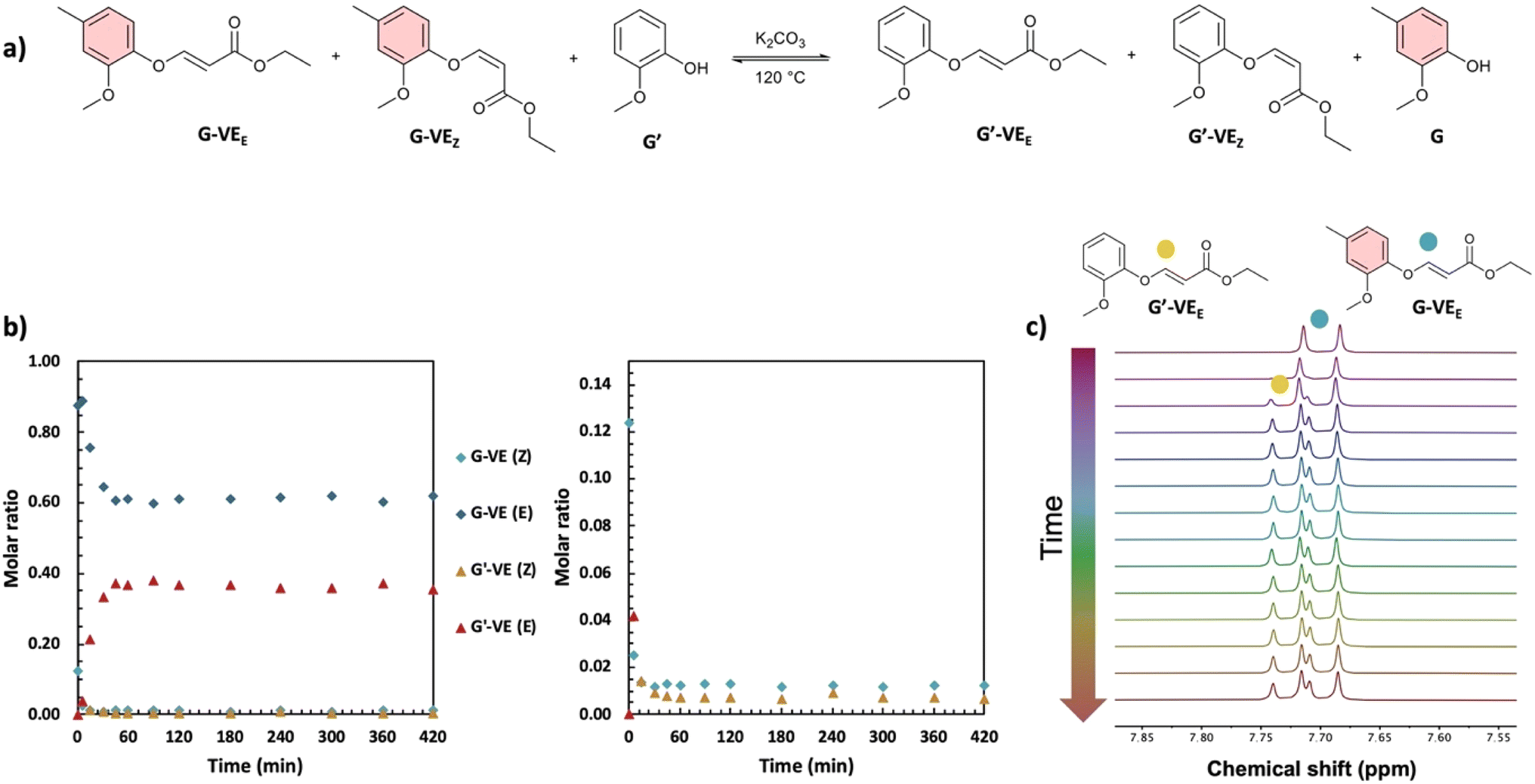 | ||
| Fig. 2 (a) Scheme for the dynamic exchange of substituted phenols, (b) kinetics of the exchange reaction with K2CO3 at 120 °C, with a closer view of the molar ratios from 0 to 0.15, and (c) detail of the stacked 1H NMR spectra of the exchange reaction at different times, showing the characteristic doublet of the vinylene protons in the reactant and product. | ||
Equilibration constants (K) were determined (Fig. 1), as detailed from Tables S2–S5, in ESI.† For all reactions, K < 1 is obtained. This is associated with reactions favoring the formation of G-VE and S-VE structures over G′-VE and S′-VE compounds. Two influencing parameters were observed: (i) the ability of mono- or di-substituted compounds to react on existing vinyl ether bonds, and (ii) the ability of the mono- or di-substituted bonds to undergo addition/elimination reactions. The higher K values of 0.37 and 0.29 obtained for the SG′ and GG′ reactions suggest that G′ undergoes smoother addition onto G-VE and S-VE model compounds than S′. Interestingly, the addition of G′ on S-VE is favored compared to G-VE, despite the larger steric hindrance induced by the two methoxy substituents. This is suspected to result from the larger proportion of Z-isomer (58%) in S-VE making the addition site more accessible compared to an E-isomer, which is the main configuration of G-VE. The S-VEZ-configuration appears to be more prone to the addition of a new phenol, as indicated by the SG′ and SS′ kinetics (Fig. S7 and S11 in ESI†). For instance, in the case of SS′, S-VEZ is predominantly consumed in a first kinetic step, generating the S′-VEE compound and free S. Then, in a second step, both S-VEZ and the newly generated S′-VEZ are consumed to preferably reform S-VEE and produce S′-VEE structures. Eventually, all the Z-isomers will be consumed into the more stable E-compounds, leading to the reaction equilibrium. The same kinetic profile is observed with the SG′ reaction.
The bond exchange mechanism was postulated by Santos et al.32 to be associative in the presence of free phenols (Scheme 2). However, the postulated acetal enolate intermediate was not observed; therefore, other potential mechanisms were not ruled out. In closely related thiol-yne systems, two equivalents of thiols can react with one equivalent of alkyne, forming a stable thioacetal linkage, which can further exchange with free thiols. The mechanism is dissociative in this case, indicating a retro-Michael addition to the vinylogous thioether.52 To evaluate whether a dissociative mechanism could also occur in the phenol-yne case, G-VE was reacted with an excess of methyl propiolate (Fig. S12 in ESI†). A dissociative mechanism would indicate the rupture of the phenyl vinyl ether bond into phenol and alkyne, and the reaction of the phenol with the excess methyl propiolate. However, no trace of the products was observed, therefore excluding the possibility of a dissociative exchange mechanism.
Aliphatic OHs can also react with activated alkynes,53 forming alkyl vinyl ethers, which were reported to be non-dynamic.32 This point was confirmed by the synthesis and study of a benzyl alcohol vinyl ether model (Benz-VE) (Fig. S13 in ESI†). Benz-VE was reacted with G, but bond exchange was not observed (Fig. S14 in ESI†). The inverse reaction of benzyl alcohol with G-VE was also investigated and revealed that bond exchange occurred, leading to the formation of an alkyl vinyl ether and the release of the G compound (Fig. S15 in ESI†). It means that in the presence of Al-OH groups, non-dynamic alkyl vinyl ether bonds can be generated from dynamic phenyl vinyl ether linkages, leading to a loss in dynamicity. This might cause limitations when transposed to lignin, which also contain Al-OH groups.
3.2. Analysis of vinyl ether formations onto aliphatic and phenolic OH: from models to lignins
The formation of vinyl ether bonds on aliphatic and aromatic OH was first investigated and analyzed with the selected model molecules. 1-Butanol and S were chosen to model Al-OH and the S unit Ph-OH, respectively. They were reacted with ethyl propiolate. The conversion was followed by integration of the distinct doublet peaks corresponding to vinylene protons in 1H NMR spectra (Fig. 3). The Ph-OH model shows the formation of E and Z-isomers, as previously observed (Fig. S16 in ESI†). Resonances at 5.24 and 7.63 ppm were assigned to the E-isomer vinylene protons, while doublets at 4.98 and 6.55 ppm were attributed to the Z-isomer. The E/Z ratio was found to be 41/59, 37/63, and 52/48 at 120, 80, and 60 °C, respectively, showing no clear trend with the temperature variation. The E/Z stereochemistry is reported to be dependent on the temperature, catalyst and solvent conditions, enabling the tuning of the target isomeric configuration.53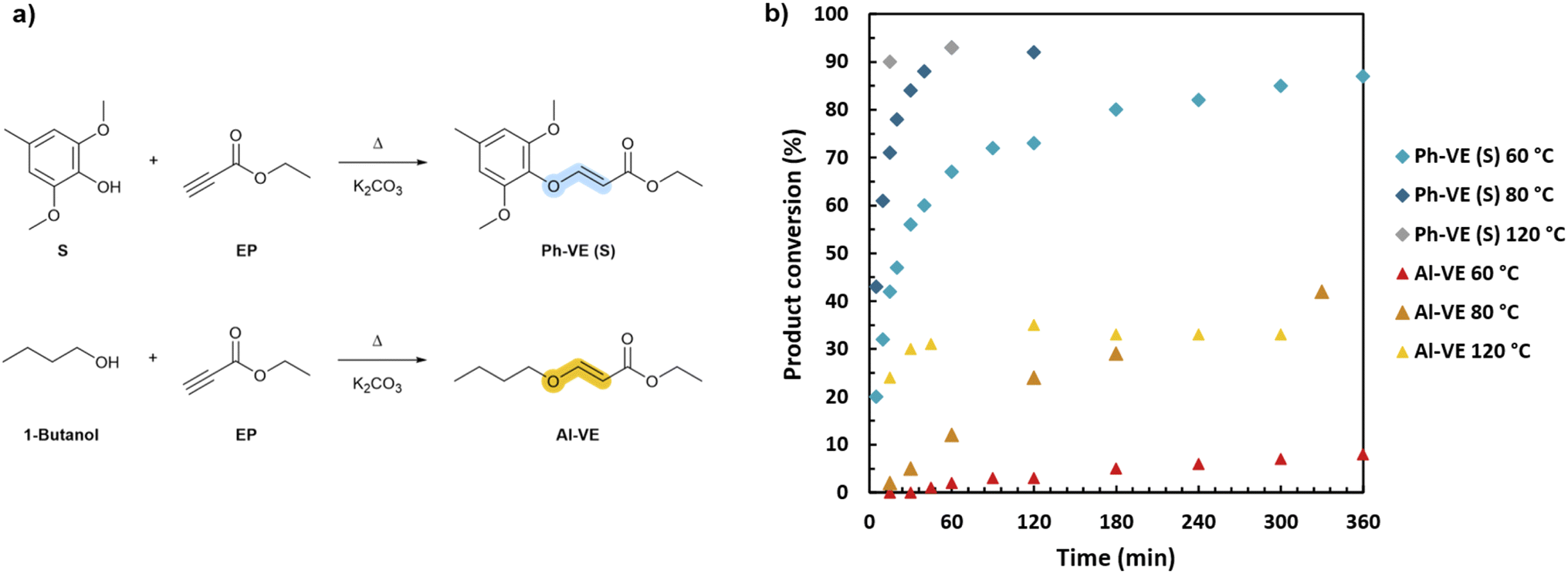 | ||
| Fig. 3 (a) Formation of phenyl and alkyl vinyl ethers from the reaction of models S and 1-butanol with ethyl propiolate. (b) Kinetic monitoring of the formation of alkyl (Al-VE) and phenyl vinyl ether (Ph-VE) adducts at different temperatures. | ||
Similarly, the Al-OH model shows the formation of E/Z-isomers. According to their coupling constants, resonances at 4.82 and 7.56 ppm were attributed to E-isomer vinylene protons and signals at 4.81 and 6.49 ppm were attributed to Z-isomer protons. Additionally, the spectra indicate that some transesterification occurs between butanol and esters, forming ethanol. The different potential products and their corresponding vinylene protons signals are given in Fig. S17, in ESI.† Transesterification products were observed irrespective of the tested conditions, but could not be quantified because of signal overlap in 1H NMR.
Overall, Ph-OH exhibit enhanced reactivity towards activated alkynes compared to Al-OH. At 120 °C, the conversion plateau around 90% is obtained in 15 minutes for Ph-OH, whereas the product conversion does not exceed 33% after 5 h of reaction for Al-OH. This discrepancy in reactivity is observed regardless of the temperature. This should be attributed to the relatively low pKa of K2CO3, which limits the deprotonation of aliphatic alcohols, while successfully deprotonating the phenols. In the literature, nucleophilic organic bases such as 4-(dimethylamino)pyridine (DMAP) and 1,4-diazabicyclo[2.2.2]octane (DABCO) or strong organic bases such as phosphazenes (t-BuP4) were employed to catalyze the reaction between activated alkynes and aliphatic alcohols.54–56
Furthermore, the temperature is found to strongly influence the reaction kinetics by accelerating the formation of vinyl ether adducts. For Ph-OH, the adduct conversion reaches 87% in 6 h at 60 °C, and 90% in just 15 minutes at 120 °C. The same trend is observed for Al-OH, although significantly lower conversion rates are obtained. The reaction of Al-OH forming non-dynamic alkyl vinyl ethers is thus limited in these conditions, as opposed to Ph-OH generating dynamic phenyl vinyl ethers, which is positive for the target elaboration of dynamic systems.
In the continuity of this large study based on model molecules, different lignins with various G/S units ratios were then selected. Table 1 shows that KL extracted from softwood is rich in G units, whereas OSL extracted from hardwood presents a high S content. Their reactivity towards click-addition with ethyl propiolate was investigated (Scheme S3 in ESI†). Two reaction temperatures (80 and 120 °C) were analyzed to evaluate the impact of the curing temperature on the reactivity of Al-OH and Ph-OH. The modified lignins were analyzed by 31P NMR to quantify the amount of Al-OH and Ph-OH that have reacted with the monoalkyne. The spectra are available in Fig. S18 and S19, in ESI.† The conversion of Al-OH and Ph-OH was thus evaluated to determine the content in phenyl and alkyl vinyl ethers, as well as the ratio between dynamic and non-dynamic bonds. The results are reported in Table 2, and detailed data are available in Table S6, in ESI.† Overall, between 73% and 78% of dynamic bonds were obtained in the different experiments. The lignin architecture and source (KL or OSL) did not significantly influence this content, with values varying from 73% to 75%. Increasing the temperature from 80 to 120 °C significantly reduced the reaction time (Table S6 in ESI†), but the proportion of dynamic bonds remained constant (73%). The influence of the solvent was evaluated by replacing DMF by DMSO, which can be biobased,57 and a similar reactivity was observed.
| Lignin | Temperature (°C) | Solvent | Alkyl vinyl ether dynamic bonds (%) | Phenyl vinyl ether non-dynamic bonds (%) |
|---|---|---|---|---|
| OSL | 80 | DMF | 75 | 25 |
| KL | 80 | DMF | 73 | 27 |
| KL | 120 | DMF | 73 | 27 |
| OSL | 80 | DMSO | 78 | 22 |
2D HSQC NMR, displayed in Fig. S20 in ESI,† confirmed the formation of vinyl ether moieties with the apparition of signals labelled A, associated with vinylene protons, and B, attributed to the newly grafted ester groups. It also shows a good preservation of the lignin structure, since ether inter-unit linkages are still visible after the modification.
3.3. Analysis of the lignin-based vitrimers based on phenol-yne
The reaction kinetics of lignin with PEG-DA was monitored by DRIFT spectroscopy at 80 and 120 °C. 3D plots of the IR spectra over time are available in Fig. S23, in ESI.† The evolution of the normalized absorbance of the alkyne signal at 2111 cm−1 was plotted as a function of time in Fig. 4. At 120 °C, the absorbance of the characteristic alkyne band at 2111 cm−1 decreased to less than 0.15 in 4 h, indicating a conversion of around 85%. On the other hand, the experiment performed at 80 °C exhibited an absorbance of 0.66, revealing a lower conversion of 34% after 4 h of reaction. This is suspected to originate from a restricted segmental mobility of lignin moieties at 80 °C, preventing reactive groups from interacting and forming covalent vinyl ether bonds. It is noteworthy to mention that because the samples are dispersed in KBr and the temperature is induced by IR light only at the surface of the materials, the reaction time is herein overestimated compared to a reaction performed in an oven. The curing temperature was then set at 120 °C for the rest of the study.
 | ||
| Fig. 4 Evolution of the normalized absorbance of the alkyne signal at 2111 cm−1 as a function of time, at 80 and 120 °C. The corresponding FT-IR spectra are available in ESI (Fig. S23).† | ||
An optimization of the reaction conditions of PEG-DA with lignin phenols was performed under basic catalysis, including (i) the amount of excess Ph-OH available to induce addition/elimination rearrangements, and (ii) the molar equivalents of catalyst (K2CO3) on lignin-based materials relaxation times. Following this preliminary study detailed in Fig. S24 and Table S9 in ESI,† a series of materials were synthesized by the click-addition of KL, OSL and DOSL with PEG-DA (Scheme 1b, the detailed compositions are provided in Table S8 in ESI†).
The chemical structures of the KL-PY, OSL-PY, and DOSL-PY materials were characterized by FT-IR, and are displayed in Fig. 5. The disappearance of peaks at 3211 and 2111 cm−1 characteristic of alkyne C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H and CC stretching vibrations, respectively, and the diminution of the absorption peak at 3390 cm−1 associated with OH stretching indicated the full conversion of PEG-DA alkyne groups and partial conversion of lignin OHs. The appearance of a new absorption band at 1640 cm−1 attributed to the C
C–H and CC stretching vibrations, respectively, and the diminution of the absorption peak at 3390 cm−1 associated with OH stretching indicated the full conversion of PEG-DA alkyne groups and partial conversion of lignin OHs. The appearance of a new absorption band at 1640 cm−1 attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration confirmed the formation of vinyl ether moieties.62,63
C stretching vibration confirmed the formation of vinyl ether moieties.62,63
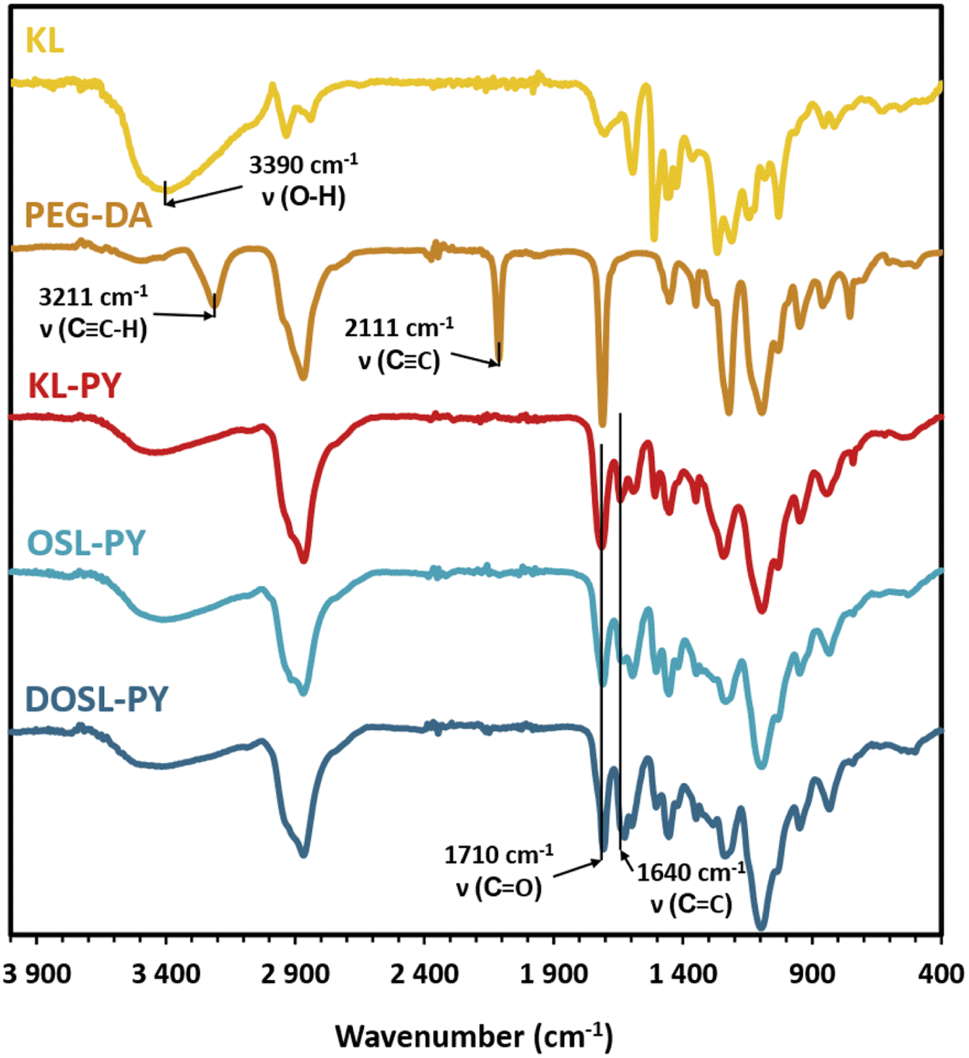 | ||
| Fig. 5 FT-IR spectra of KL, PEG-DA, and the series of lignin-based vitrimers. | ||
Swelling ratio (SR) and gel fraction (GF) experiments were performed after immersion of the materials in acetone or water for 48 h to analyze the corresponding networks. Table 3 reports the resulting SR and GF values. Interestingly, KL-PY and OSL-PY prepared from non-depolymerized lignins exhibit similar low swelling ability in acetone at around 30%, while DOSL-PY derived from depolymerized organosolv lignin shows a higher (2-fold) value approaching 60%. This is the first suggestion that KL and OSL, despite their distinct extraction processes, have a similar affinity with acetone when integrated into networks. On the other hand, catalytic treatments used to obtain DOSL from OSL enhance the affinity of the resulting structure with acetone, inducing greater swelling ability of the corresponding PY-based material. Overall, smaller variations of SR are observed between each material in water, as compared to acetone. The evolution of SR could be correlated with the Al-OH content, which is available in the network (Fig. S25 in ESI†), since increased swellings are observed for the higher Al-OH contents through hydrogen bonding with water. GF were subsequently measured in both solvents. In acetone, excellent GF values of between 90.6% and 100.0% were obtained. As previously discussed with SR, KL and OSL exhibit comparable behaviors in acetone, resulting in similar GF, while the DOSL structure induces a small reduction in GF. In water, GF slightly decreased around 90%, but remained rather high. This is believed to result from a difference of affinity of the materials with water and acetone due to the polar/apolar balance and the polymer–solvent interactions. Moreover, the results are in good accordance with the reported GF in water for ester containing vitrimers.23,64 Globally, very high gel fractions were obtained, indicating a good solvent resistance in both acetone and water. The developed lignin-based PY vitrimers herein showed better solvent resistance compared to previously reported lignin-based vitrimers also containing PEG chains.27 The results confirm that the materials were indeed highly crosslinked, and that the soluble fractions remain marginal.
| PY-based networks | Lignin content (wt%) | SRa (%) | SRw (%) | GFa (%) | GFw (%) | T d5% (°C) | T d (°C) | T g (°C) | T α (°C) | ν (mol m−3) |
|---|---|---|---|---|---|---|---|---|---|---|
| KL-PY | 39 | 30.6 ± 0.3 | 64.8 ± 0.8 | 98.0 ± 0.1 | 91.1 ± 0.1 | 274 | 400 | 2 | 46 | 3321 |
| OSL-PY | 49 | 29.5 ± 0.4 | 57.6 ± 2.0 | 100.0 ± 0.1 | 92.2 ± 0.4 | 266 | 388 | 19 | 76 | 1207 |
| DOSL-PY | 46 | 57.8 ± 0.7 | 61.4 ± 0.3 | 90.6 ± 0.3 | 90.1 ± 0.1 | 263 | 384 | −4 | 10 | 1563 |
The organization of the series of materials was analyzed by WAXS, as displayed in Fig. 6. The materials exhibit patterns with broad background scattering signals, which are specific to amorphous polymers comprising regions with a certain degree of ordering. The organization is associated with inter- and intra-molecular physical interactions occurring between the polymer segments. Scattering vectors q (also known as momentum transfer in reciprocal-space) and the associated real-space distances D (also known as Bragg spacings) were determined using eqn (2) and (3) (Table 4). The proportions of physical interactions were identified through the scattering signal deconvolution using four Gaussian functions, as previously reported for lignins and lignin-based materials.65,66 The first two contributions (q1 and q2), corresponding to interplanar distances of around 6.1–6.9 Å and 4.2–4.3 Å, respectively, correspond well to the reported distances for the π–π interactions in lignins, when the aromatic rings are parallel (“sandwich” π–π interactions) or perpendicular to each other (T-shaped π–π interactions).67–71 Microstructure organization of the materials was not found to significantly vary between KL and OSL. This suggests that both lignins, despite their distinct macromolecular architectures, have similar abilities towards the generation of physical interactions when integrated into polymer networks. Material prepared from DOSL has a significantly different organization, with a predominant q1 contribution. It could indicate that lignin–lignin interactions are more important than in the other systems. The lower molar mass of DOSL might facilitate these interactions, resulting in a higher structuration of lignin-rich domains in the materials. Finally, the materials display narrow peaks in the regions from q ≈ 2.0 to 3.0 Å−1. Those peaks indicate a higher organization of some domains. DOSL-PY shows better network organization characterized by greater amounts of narrow peaks.
 | ||
| Fig. 6 WAXS curves and deconvolutions of (a) KL-PY, (b) OSL-PY, and (c) DOSL-PY. | ||
| PY (designation) | KL-PY | OSL-PY | DOSL-PY |
|---|---|---|---|
| q 1 (Å−1) | 1.02 | 1.01 | 0.91 |
| q 2 (Å−1) | 1.47 | 1.47 | 1.50 |
| q 3 (Å−1) | 1.85 | 1.86 | 1.70 |
| q 4 (Å−1) | 2.85 | 2.85 | 2.53 |
| D 1 (Å) | 6.14 | 6.21 | 6.89 |
| D 2 (Å) | 4.29 | 4.26 | 4.19 |
| D 3 (Å) | 3.40 | 3.38 | 3.70 |
| D 4 (Å) | 2.21 | 2.20 | 2.49 |
| q 1 content (%) | 7 | 7 | 36 |
| q 2 content (%) | 33 | 34 | 17 |
| q 3 content (%) | 20 | 19 | 16 |
| q 4 content (%) | 40 | 40 | 31 |
Rheological properties of the series of networks were investigated by DMA. The main rheological parameters are reported in Table 3, and the curves are available in Fig. S29 in ESI.† Only OSL-PY exhibits a well-marked α-transition at 76 °C. KL-PY exhibited a broader signal originating around −30 °C and terminating around 140 °C, with a maximum at 46 °C. DOSL-PY demonstrated a main peak at 10 °C and a less intense second signal around 120 °C. The two maxima of DOSL-PY are suspected to be associated with phase separation. This partial phase separation is suggested to occur at the sub-micro scale, owing to partially aggregated DOSL structures and homogeneous phase, in which lignin is dispersed in the flexible PEG chains.28,73 This is in good agreement with DSC and WAXS observations on DOSL-PY, which respectively indicated the presence of two Tg's and interactions potentially originating from the microphase separation.
Crosslinking densities (ν) were determined on the rubbery plateau (at 150 °C) according to eqn (4). ν between 1207 and 3321 mol m−3 were obtained. Unlike previously reported values, ν was found to decrease with increasing lignin content.27 However, it increases with the lignin content in Ph-OH groups, which varies significantly between lignin samples (Fig. S30 in ESI†). The high functionality of lignin is the only source for crosslinking in the designed materials. Thus, lignins with high Ph-OH content lead to the formation of a greater number of crosslinks within the networks, therefore increasing the crosslink density.
The dynamicity of the lignin-based networks was investigated through stress relaxation experiments. For all materials, full stress relaxation is observed (Fig. 7). It confirms that networks are mainly composed of dynamic phenyl vinyl ether bonds, and that the formation of non-dynamic alkyl vinyl ether bonds was low. Experimental curves were fitted using the KWW stretched exponential decay (eqn (5)), displayed as dashed lines in Fig. 7a–c. This model allowed the determination of the characteristic relaxation time τ* and an exponent β (0 < β < 1), which characterizes the distribution of relaxation times for materials dissipating energy through various relaxation modes. The main relaxation parameters are reported in Table 5. Relaxation times ranging from 74 to 476 s were obtained at 200 °C. The relaxation times were found to decrease with the molar mass of lignin, as illustrated in Fig. S31, in ESI.† The fastest relaxation was observed for DOSL exhibiting the lowest molar mass. It is suggested that bond rearrangements are highly dependent on the network segmental mobility, which is improved by employing low molar mass lignins.
 | ||
| Fig. 7 Normalized stress relaxation curves of (a) KL-PY, (b) OSL-PY and (c) DOSL-PY. The data were fitted with a stretched exponential decay (eqn (5)), which is represented as dashed lines. (d) Arrhenius plots of the characteristic relaxation times. | ||
| PY (designations) |
|
β | E a (kJ mol−1) | T v (°C) |
|---|---|---|---|---|
| KL-PY | 476 | 0.49 ± 0.01 | 72 ± 1 | 114 |
| OSL-PY | 252 | 0.56 ± 0.02 | 89 ± 9 | 107 |
| DOSL-PY | 74 | 0.44 ± 0.09 | 80 ± 5 | 81 |
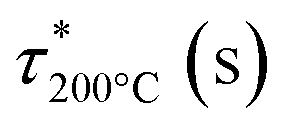
The dynamicity is expected to originate from vinyl ether addition/elimination bond exchange (Scheme 2), although transesterification reactions involving lignin free Al-OH groups might also occur in the presence of the K2CO3 catalyst.74 Based on the small molecules model study, transesterification in the networks is expected to be marginal because of the low reactivity of the Al-OH groups in these conditions, but it cannot be ruled out. Further experiments are required to better evidence its potential impact.
The β-parameter was found to vary between 0.44 and 0.56. These values are characteristic of heterogenous distributions of relaxation times originating from the different chemical environments of the phenyl vinyl ether groups. The different ortho-substituents of phenols (H, G and S units), which showed distinctive bond exchange kinetics, are suspected be the source of this discrepancy in relaxation times.
Activation energy (Ea) values of 72, 89, and 80 kJ mol−1 were obtained for KL-PY, OSL-PY, and DOSL-PY, respectively (Table 5 and Fig. S32 in ESI†). These values are in perfect agreement with the only reported Ea for phenol-yne vitrimers of 73.7 and 89.8 kJ mol−1.34Ea was found to increase with the lignin content (Fig. S33 in ESI†), as previously reported for lignin-based vitrimers.27 Theoretical topology freezing temperatures (Tv), associated with temperatures above which bond exchange induce a macroscopic flow (elastic solid to viscoelastic liquid), were calculated by extrapolation from Arrhenius plots for a viscosity conventionally set at 1012 Pa s (Table 5).75,76 Parameters employed for the calculations are reported in Table S10, in ESI.†Tv of KL-PY, OSL-PY, and DOSL-PY were determined to be 114, 107, and 81 °C, respectively. Each vitrimer exhibits Tv > Tg which is characteristic of a viscoelastic flow that is mainly controlled by the bond exchange kinetics.77 At ambient temperature (25 °C), KL-PY and DOSL-PY (Tg < Tamb < Tv) behave like elastomers in the rubbery state with fixed chemical structures because of their high Tv. OSL-PY (Tg ≈ Tamb < Tv) has limited segmental movements because (i) the rubbery plateau has not been completely reached, and (ii) the rigidity and structure of the chemical network induce such a behavior.
Frequency sweep measurements were also performed on the materials. The data are reported in Fig. S34 in ESI.† The materials show a plateau modulus that slightly increases with temperature over the studied temperature range, which is consistent with an associative bond exchange mechanism.9,78 It corroborates the results of the small molecules model study, and confirms that the materials can be classified as vitrimers.
Creep behavior of the vitrimers was evaluated under 0.2 MPa stress over 1000 s, followed by a recovery time of 1000 s (Fig. 8). Creep recovery experiments were performed at 20 and 70 °C. At 20 °C, greater strain can be observed for vitrimers with lower Tg (DOSL-PY > KL-PY > OSL-PY). After 1000 s of recovery, 77%, 85%, and 79% of the creep is recovered for KL-PY, OSL-PY and DOSL-PY, respectively. At 70 °C, higher strains are observed because the temperature is much greater than the vitrimers Tg. After 1000 s of recovery time, 79%, 87%, and 83% of the creep was recovered for KL-PY, OSL-PY, and DOSL-PY, respectively. Creep is not significantly impacted by the variation between 20 and 70 °C, indicating that the phenyl vinyl ether bond exchanges are limited in those conditions, which is in good agreement with Tv values. This reveals the good creep resistance of these lignin-based vitrimers.
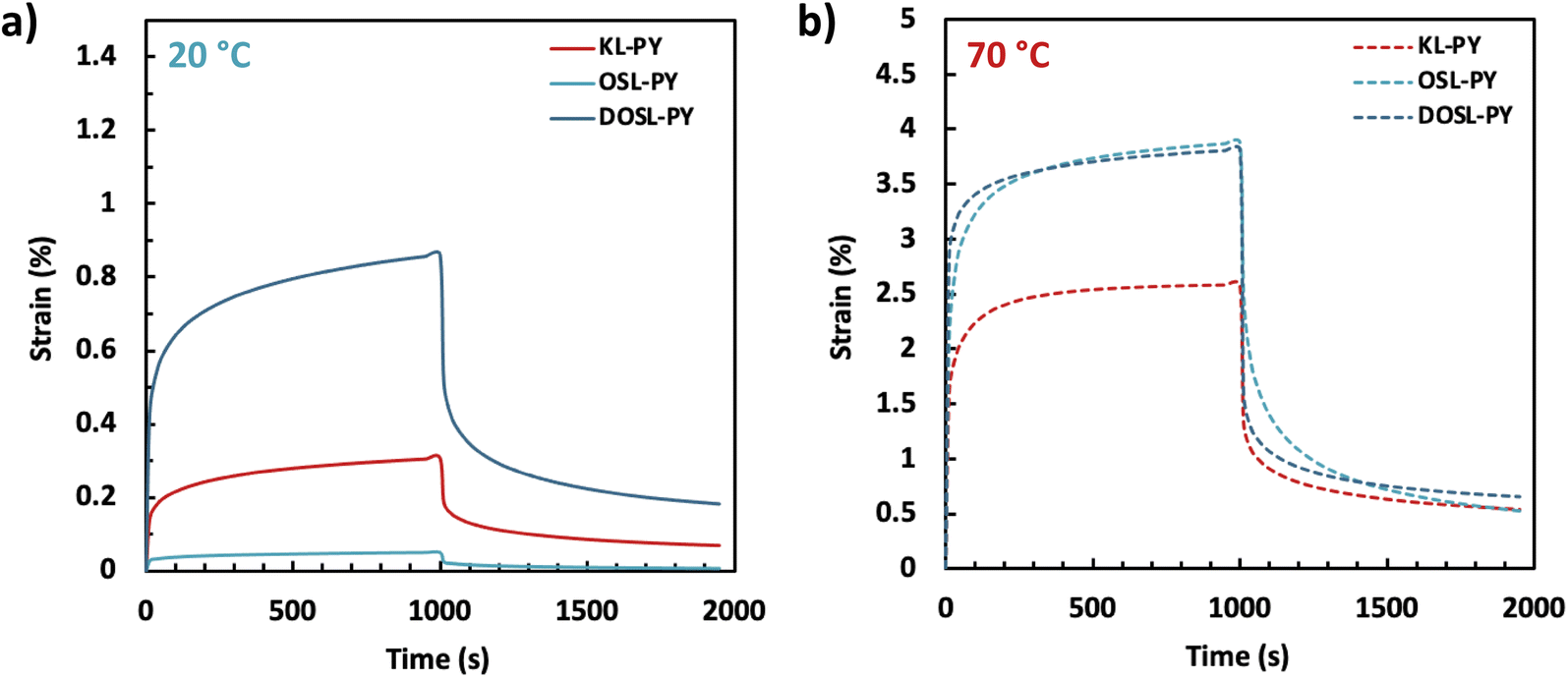 | ||
| Fig. 8 Creep recovery plots of the vitrimers at (a) 20 °C and (b) 70 °C. | ||
Static mechanical properties were assessed by uniaxial tensile tests. The resulting stress–strain curves displayed in Fig. 9 show that the materials globally have a typical thermoset behavior, with a rather steep slope in the initial linear region associated with the high modulus, and limited elongation at break. The corresponding values of the Young's modulus (E), stress at break (σ), and elongation at break (ε) are reported in Table 6. Two distinct mechanical behaviors are observed, since Tg of OSL-PY is close to the room temperature (Tg of 19 °C). This material exhibits the greatest Young's modulus and maximal stress of 899 MPa and 24.6 MPa, respectively, while achieving an elongation of 4.0%. KL-PY and DOSL-PY, whose Tg are far below testing temperature, are in their rubbery state. They respectively show E values of 263 and 42 MPa, σ of 9.6 and 2.0 MPa, and ε of 10.0 and 13.1%. E and σ values show a similar evolution to that of the dynamic storage modulus determined at 23 °C  and reported in Table 6, with OSL-PY > KL-PY > DOSL-PY.
and reported in Table 6, with OSL-PY > KL-PY > DOSL-PY.
 | ||
| Fig. 9 Representative stress–strain curves of the series of lignin-based vitrimers. | ||
| PY (designations) | E (MPa) | σ (MPa) | ε (%) |
|
|---|---|---|---|---|
| a Values labelled with different capital letters (A, B, C) are significatively different (Student's t-test, α = 0.05). | ||||
| KL-PY | 263 ± 21A | 9.6 ± 0.2A | 10.0 ± 1.0A | 586 |
| OSL-PY | 899 ± 41B | 24.6 ± 2.8B | 4.0 ± 1.2B | 915 |
| DOSL-PY | 42 ± 3C | 2.0 ± 0.2C | 13.1 ± 2.6A | 211 |
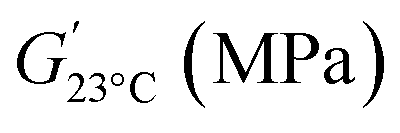 C region shows no variation.
C region shows no variation.
KL-PY was ground and thermo-mechanically remolded at 160 °C over the duration of three cycles. The potential evolution of the chemical structure was also followed by FT-IR, as illustrated in Fig. 10 (full FT-IR spectra are given in Fig. S37 in ESI†). The FT-IR spectra display a change in the chemical structure after the first recycling with the slight intensity decrease of the ester band et 1710 cm−1 and the increase of the OH vibration band at 3390 cm−1. The shape of the ester peak is subtly modified with the appearance of a small shoulder at 1720 cm−1. Moreover, the region between 1640 and 1580 cm−1 evidences some alterations with the appearance of a new band around 1619 cm−1. This new band can be attributed to alkyl vinyl ethers generated by the reaction of lignin free Al-OH onto the existing phenyl vinyl ethers, as observed on model compounds. This hypothesis is supported by the FT-IR spectra of model molecules, displayed in Fig. S38 in ESI,† which indicate that vinyl ether CC bands wavenumber is shifted depending on its chemical environment and stereoisomer configuration.79 While the broadness of the peaks might result from the different isomeric configurations, the maximum of the peaks tends to reach 1620 cm−1 for alkyl vinyl ether (Benz-VE and Al-VE model molecules), whereas phenyl vinyl ethers (S-VE and G-VE) maximum is shifted towards 1640 cm−1. It is thus suspected that the alteration in the networks may be partially induced by thermal treatment (linked to the esters bonds). However, it is possible that it mostly arises from the high mechanical constraints involved in the remolding process, favoring side reactions. This hypothesis is supported by stress relaxation experiments, which indicated that heating the samples under low strain did not result in the formation of non-dynamic alkyl vinyl ether crosslinks. The side reaction generating alkyl vinyl ether might severely hamper the dynamic properties of the material over consecutive physical recycling cycles.
 | ||
| Fig. 10 Detailed FT-IR spectra of the pristine and recycled KL-PY. Corresponding full spectra are available in ESI (Fig. S37).† | ||
The evolution of the mechanical behavior was followed by uniaxial tensile tests after various physical recycling occurrences. The resulting stress–strain curves are available in Fig. 11a. The corresponding values are reported in Table 7. A considerable loss of mechanical performance can be observed over successive cycles, resulting in a substantial reduction in the Young's modulus, maximum stress at break, and elongation at break from 263 MPa, 9.6 MPa, and 10.0% to 198 MPa, 3.2 MPa, and 2.9% for the pristine and three times recycled materials, respectively. DMA (Fig. S39 in ESI†) shows a decrease in storage modulus leading to the collapse of crosslinking density from 3321 to 350 mol m−3 after the last recycling. Those results are indicative of a significant loss of network integrity during the remolding cycles. A suggested explanation for this crosslinking density loss is the successive cleavage of non-dynamic alkyl vinyl ether bonds during the grinding step that are no longer able to create new crosslinks. The materials might lose their dynamic properties during the successive physical recycling cycles. In addition to some chemical degradation of the structure observed by FT-IR, it is suspected that the physical recycling temperature of 160 °C for 30 minutes is too low and too short, respectively, to induce sufficient bond exchange in KL-PY. These remolding conditions (temperature and time) were initially chosen to limit the thermal degradation during the physical recycling steps. However, according to the KL-PY stress relaxation experiments, a relaxation time of 1677 s (28 minutes) is observed at 170 °C, suggesting that more than 30 minutes at 160 °C could be needed to achieve complete network rearrangement. It is noteworthy to mention that this extrapolation is only indicative.
 | ||
| Fig. 11 Representative stress–strain curves of (a) the pristine and recycled KL-PY, and (b) the pristine and recycled DOSL-PY. | ||
| Designations | E (MPa) | σ (MPa) | ε (%) | T α (°C) | ν (mol m−3) |
|---|---|---|---|---|---|
| a Values labelled with different capital letters (A, B, C, D) are significatively different (Student's t-test, α = 0.05). | |||||
| Original KL-PY | 263 ± 21A | 9.6 ± 0.3A | 10.0 ± 1.0A | 46 | 3321 |
| KL-PY 1st cycle | 159 ± 7B | 4.2 ± 0.2B,C | 4.6 ± 0.4B | 53 | 1430 |
| KL-PY 2nd cycle | 208 ± 4C | 5.9 ± 0.1B | 5.9 ± 0.1B | 49 | 790 |
| KL-PY 3rd cycle | 198 ± 20C | 3.2 ± 0.7C | 2.9 ± 0.1B | 52 | 350 |
| Original DOSL-PY | 42 ± 3D | 2.0 ± 0.2D | 13.1 ± 2.6C | 10 | 1563 |
| DOSL-PY 1st cycle | 52 ± 6D | 2.4 ± 0.1D | 23.4 ± 5.3D | 23 | 455 |
To support this suggestion, DOSL-PY, which is the vitrimer displaying the fastest relaxation at temperature, was physically recycled once. As previously discussed for KL-PY, the FT-IR spectra of the pristine and recycled DOSL-PY (Fig. S40 in ESI†) exhibit diminished ester and alkene bands but a stronger OH band, indicating chemical alteration of the network. Crosslinking densities obtained by DMA (Fig. S41 in ESI†) once again indicate a loss of network integrity with ν values that collapse from 1563 to 455 mol m−3 after remolding. Surprisingly, the mechanical properties are maintained after the first reprocessing cycle, despite the decrease in crosslink density (Fig. 11b and Table 7).
Vinyl ether bonds are known to be susceptible to hydrolysis in acidic conditions.33 Moreover, ester bonds are known to break under acidic condition in the presence of water.81 Then, acid-catalyzed hydrolytic degradation of the PY materials could theoretically lead to the recovery of both pristine lignin and PEG400 by the cleavage of vinyl ethers and esters moieties, as illustrated in Scheme 3.
 | ||
| Scheme 3 Acid-catalyzed hydrolysis of the KL-PY vinyl ether and ester groups. | ||
Various degradation conditions were investigated. Tested conditions are reported in Table S11 in ESI,† and displayed in Fig. S42, in ESI.† It was found that aqueous 2 M HCl and solutions of acetone/aqueous 2 M HCl (7:3) could not degrade the network. This was probably due to the poor solubilization ability of the products of the depolymerization. The addition of two drops of concentrated HCl (37%) within the solution of acetone/aqueous 2 M HCl (7:3) helped to disperse the material into small, but insoluble particles in the mixture. DMSO/aqueous 1 M HCl (8:2) was found to completely solubilize the materials after 48 h at 70 °C. The depolymerization was performed on 1.2 g of KL-PY (0.012 g L−1) using the latter conditions for 72 h.
Lignin was then precipitated from the DMSO/HCl solution. The recovered brown powder was analyzed by 31P NMR, as displayed in Fig. S43, in ESI.† The content of free phenols resulting from the cleavage of vinyl ether bonds was determined by integrating the Ph-OH area in the spectrum of the recovered KL (between 137.6 and 144.0 ppm). A resulting Ph-OH content of 1.62 mmol g−1 was obtained (Table S12 in ESI†). Only 21% of the initial Ph-OH were recovered during this acid-catalyzed depolymerization. The 31P NMR spectrum also indicates the presence of OH end chains at 147.1 ppm corresponding to the chemical shift of PEG400 (Fig. S43 in ESI†), as well as an increase in signal intensity in the carboxylic acid region from 133.6 to 136.0.82 This suggests that the applied conditions allowed for partial ester hydrolysis, but were not sufficient to cleave all vinyl ether bonds. Finally, because PEG400 is highly soluble in water which was used for the purification of the recovered powder, it is suspected that a large part of PEG moieties is still grafted to the lignin via vinyl ether linkages.
FT-IR analysis was performed on the recovered product. The spectra show the presence of the vinyl ether CC band at 1640 cm−1 and ester band at 1710 cm−1 (Fig. S44 in ESI†). The simultaneous increase in the OH band around 3400 cm−1, and the decrease of the C–H stretching (2863 cm−1) and C–O–C ether stretching (1086 cm−1) indicate that considerable PEG400 amounts were removed during degradation, but some moieties are still linked by ester bonds to lignin.83
To further analyze the depolymerized structure obtained, the product was analyzed by 2D HSQC NMR. The spectra of pristine KL and the recovered KL are displayed in Fig. 12. 2D HSQC indicates that the main inter-linkages and end chains of KL remain unchanged before and after click-addition. Characteristic signals associated with the B and C structures are also observed on recovered KL. Signals A are less intense, suggesting that some β-O-4 linkages might have been cleaved during the material processing or during the depolymerization. Overall, these results indicate that the hydrolysis conditions do not completely alter the KL structure. However, the presence of PEG moieties in the recovered compound is evidenced by the characteristic signals of methylene protons in the α and β positions from the ester. Vinylene protons are also visible, confirming incomplete depolymerization, in agreement with the 31P NMR and FT-IR results.
 | ||
| Fig. 12 2D HSQC of (a) KL and (b) the recovered KL. | ||
Finally, the depolymerized product was acetylated, and the soluble fractions were analyzed by SEC. The resulting curves are displayed in Fig. S45† and detailed values are reported in Table S12, in ESI.† The soluble fraction of recovered KL showed a Mn of 1520 g mol−1 indicating that the most depolymerized structures approach Mn of pristine KL. The presence of insoluble fractions after acetylation suggests the presence of structures with Mn > 1520 g mol−1. The Đ value reached 2.7 and 3.3 for the recovered and pristine KL, respectively.
At this level and for further applied development, the hydrolysis potential of the materials prepared with OSL and DOSL should be assessed to compare the lignin structure influence on acid-catalyzed degradation. Hydrophilicity might be a determinant parameter in the degradation rates of the vitrimers.80,84
4. Conclusion
The phenol-yne click-addition has been clearly demonstrated to be a very promising dynamic covalent chemistry for the development of performing, aromatic and sustainable vitrimers from lignins.The dynamicity of the substituted phenyl vinyl ether was investigated on model compounds that were representative of the lignin heterogenous reactive groups. In this context, ortho-substitution of the G and S units was found to strongly influence the bond exchange potential of vinyl ethers originating from different steric hindrance and Z/E isomerization. While the G units are more easily subjected to addition on vinyl ether bonds owing to reduced steric hindrance, the S units favoring Z-vinyl ethers undergo easier rearrangements due to their improved accessibility. The gradual replacement of Z-configurations into more stable E-linkages was found to eventually hamper bond exchanges. Kinetics studies on model molecules and on KL and OSL lignins showed the undeniably superior reactivity of phenols with activated alkynes compared to aliphatic OH, indicating the favored formation of dynamic phenyl vinyl ether bonds over non-dynamic alkyl vinyl ethers.
The valorization of different technical lignins from sulfur and sulfur-free processes was successfully achieved by click phenol-yne addition with a PEG-dialkyne crosslinker to develop a series of sustainable aromatic vitrimers. The type of lignin significantly influenced the thermal and mechanical properties. A detailed study of the material structure revealed a potential phase separation in the case of the materials prepared from DOSL, which would require deeper characterization. The high lignin-content vitrimers (up to 49 wt% lignin) could undergo rearrangement through vinyl ether bond exchanges. Network rearrangements were found to be favored with lignins of lower molar mass, facilitating the segmental mobility needed for bond exchanges.
To consider different potential recycling processes at the end of life of these materials, physical and chemical recycling were analyzed. Physical recycling was found to be hampered by the gradual formation over successive cycles of non-dynamic alkyl vinyl ether bonds, resulting from the reaction of free aliphatic OHs onto dynamic phenyl vinyl ethers. Finally, chemical recycling investigated through acid-catalyzed hydrolysis showed that ester and vinyl ethers were cleavable, enabling the recovery of a lignin-derived product. Although the tested conditions allowed only partial hydrolysis of vinyl ether moieties with a recovery of 21% of the phenols, the results showed proofs-of-concepts for the recovery of both pristine lignin and PEG. The closed-loop recycling of the lignin-based phenol-yne vitrimer could also offer further upcycling opportunities for the development of sustainable aromatic materials in the context of a circular bioeconomy.
This work clearly opens several new perspectives in the exploration of lignin-based vitrimer materials, for which the phenol-yne click reaction presents strong advantages, since it does not require any chemical modification of the lignin, unlike most other pathways towards lignin-based CANs. Moreover, the click addition reaction yields 100% atom economy, which satisfies the green chemistry principles. Dynamicity occurs as a result of the presence of free Ph-OH groups in the networks, leading to exchanges with the vinyl ether bonds. However, the presence of Al-OH was shown to negatively affect the dynamicity because of the formation of non-dynamic alkyl vinyl ethers during reprocessing. In this context, masking Al-OH groups could be a potential future strategy to further enhance the dynamicity of these adaptable and sustainable materials.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Lisa Sougrati: investigation, data curation, conceptualization, methodology, writing – original draft, writing – review & editing. Antoine Duval: investigation, data curation, conceptualization, methodology, supervision, validation, writing – review & editing. Luc Avérous: conceptualization, supervision, funding acquisition, validation, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors are grateful to the Foundation Jean Marie Lehn (Strasbourg-France) for their financial support, with the project “LigniCAN”. We want to thank Dr Arjan T. Smit (TNO, Petten, Netherland) for kindly supplying the organosolv lignin used in this study. We are grateful for the help of Thierry Dintzer with WAXS acquisitions, Rodolphe Migneret with the WAXS deconvolution, Sécou Sall with DRIFT acquisitions, Dr Emeric Wasielewski with 2D HSQC NMR acquisitions, and Nathan Wybo with the SEC experiments.References
- S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed.
- J. M. Winne, L. Leibler and F. E. D. Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- M. Ahmadi, A. Hanifpour, S. Ghiassinejad and E. van Ruymbeke, Chem. Mater., 2022, 34, 10249–10271 CrossRef CAS.
- A. Breuillac, A. Kassalias and R. Nicolaÿ, Macromolecules, 2019, 52, 7102–7113 CrossRef CAS.
- S. Dhers, G. Vantomme and L. Avérous, Green Chem., 2019, 21, 1596–1601 RSC.
- M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 175–198 CrossRef CAS PubMed.
- J. Zheng, Z. M. Png, S. H. Ng, G. X. Tham, E. Ye, S. S. Goh, X. J. Loh and Z. Li, Mater. Today, 2021, 51, 586–625 CrossRef CAS.
- M. A. Lucherelli, A. Duval and L. Avérous, Prog. Polym. Sci., 2022, 127, 101515 CrossRef CAS.
- X.-L. Zhao, P.-X. Tian, Y.-D. Li and J.-B. Zeng, Green Chem., 2022, 24, 4363–4387 RSC.
- T. Vidil and A. Llevot, Macromol. Chem. Phys., 2022, 223, 2100494 CrossRef CAS.
- L. Sougrati, A. Duval and L. Avérous, Mater. Sci. Eng., R, 2024, 161, 100882 CrossRef.
- D. B. Tiz, F. A. Vicente, A. Kroflič and B. Likozar, ACS Sustain. Chem. Eng., 2023, 11, 13836–13867 CrossRef CAS.
- P. K. Karoki, S. Zhang, Y. Pu and A. J. Ragauskas, Mater. Adv., 2024, 5, 7075–7096 RSC.
- A. Adjaoud, N. Brosse and P. Verge, Mater. Today Chem., 2024, 42, 102342 CrossRef CAS.
- P. Buono, A. Duval, L. Averous and Y. Habibi, Polymer, 2017, 133, 78–88 CrossRef CAS.
- A. Duval, W. Benali and L. Avérous, Green Chem., 2024, 26, 8414–8427 RSC.
- A. Duval, H. Lange, M. Lawoko and C. Crestini, Green Chem., 2015, 17, 4991–5000 RSC.
- S. Gao, Z. Cheng, X. Zhou, Y. Liu, J. Wang, C. Wang, F. Chu, F. Xu and D. Zhang, Chem. Eng. J., 2020, 394, 124896 CrossRef CAS.
- W. Liu, C. Fang, F. Chen and X. Qiu, ChemSusChem, 2020, 13, 4691–4701 CrossRef CAS PubMed.
- X. Ma, S. Li, F. Wang, J. Wu, Y. Chao, X. Chen, P. Chen, J. Zhu, N. Yan and J. Chen, ChemSusChem, 2023, 16, e202202071 CrossRef CAS PubMed.
- A. Moreno, M. Morsali and M. H. Sipponen, ACS Appl. Mater. Interfaces, 2021, 13, 57952–57961 CrossRef CAS PubMed.
- L. Sougrati, A. Duval and L. Avérous, ChemSusChem, 2023, 16, e202300792 CrossRef CAS PubMed.
- M. Thys, J. Brancart, G. Van Assche, R. Vendamme and N. Van den Brande, Macromolecules, 2021, 54, 9750–9760 CrossRef CAS.
- A. Duval, W. Benali and L. Avérous, ChemSusChem, 2024, e202401480 CrossRef PubMed.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- X. Fu, A. Qin and B. Z. Tang, J. Polym. Sci., 2024, 62, 787–798 CrossRef CAS.
- T. Santos, Y. Pérez-Pérez, D. S. Rivero, R. Diana-Rivero, F. García-Tellado, D. Tejedor and R. Carrillo, Org. Lett., 2022, 24, 8401–8405 CrossRef CAS PubMed.
- Y. Shi, T. Bai, W. Bai, Z. Wang, M. Chen, B. Yao, J. Z. Sun, A. Qin, J. Ling and B. Z. Tang, Chem.–Eur. J., 2017, 23, 10725–10731 CrossRef CAS PubMed.
- W. Zhang, F. Gao, L. Shen, Y. Chen and Y. Lin, ACS Mater. Lett., 2022, 4, 2090–2096 CrossRef CAS.
- B. He, J. Huang, X. Liu, J. Zhang, J. W. Y. Lam and B. Z. Tang, Prog. Polym. Sci., 2022, 126, 101503 CrossRef CAS.
- A. T. Smit, M. Verges, P. Schulze, A. van Zomeren and H. Lorenz, ACS Sustain. Chem. Eng., 2022, 10, 10503–10513 CrossRef CAS.
- A. T. Smit, T. Dezaire, L. A. Riddell and P. C. A. Bruijnincx, ACS Sustain. Chem. Eng., 2023, 11, 6070–6080 CrossRef CAS.
- Y. Archipov, D. S. Argyropoulos, H. I. Bolker and C. Heitner, J. Wood Chem. Technol., 1991, 11, 137–157 CrossRef.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- X. Meng, C. Crestini, H. Ben, N. Hao, Y. Pu, A. J. Ragauskas and D. S. Argyropoulos, Nat. Protoc., 2019, 14, 2627–2647 CrossRef CAS PubMed.
- A. Duval and L. Avérous, ACS Sustain. Chem. Eng., 2017, 5, 7334–7343 CrossRef CAS.
- J.-M. Chenal, L. Chazeau, L. Guy, Y. Bomal and C. Gauthier, Polymer, 2007, 48, 1042–1046 CrossRef CAS.
- G. R. Palmese and R. L. McCullough, J. Appl. Polym. Sci., 1992, 46, 1863–1873 CrossRef CAS.
- G. Williams and D. C. Watts, Trans. Faraday Soc., 1970, 66, 80–85 RSC.
- A. Dhinojwala, J. C. Hooker and J. M. Torkelson, J. Non-Cryst. Solids, 1994, 172–174, 286–296 CrossRef CAS.
- M. L. Martins, X. Zhao, Z. Demchuk, J. Luo, G. P. Carden, G. Toleutay and A. P. Sokolov, Macromolecules, 2023, 56, 8688–8696 CrossRef CAS.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- Q. Jiang, H. Zhang, Q. Jiang, S. Zhang, S. Guan, W. Huang, X. Xue, H. Yang, L. Jiang, B. Jiang and S. Komarneni, Polym. Chem., 2023, 14, 1232–1240 RSC.
- B. Yao, J. Mei, J. Li, J. Wang, H. Wu, J. Z. Sun, A. Qin and B. Z. Tang, Macromolecules, 2014, 47, 1325–1333 CrossRef CAS.
- Y. Chiang, A. J. Kresge and C. I. Young, Can. J. Chem., 1978, 56, 461–464 CrossRef CAS.
- B. Song, D. Lu, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2022, 144, 1672–1680 CrossRef CAS PubMed.
- N. V. Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. D. Prez, Angew. Chem., Int. Ed., 2020, 59, 3609–3617 CrossRef PubMed.
- J. C. Worch, C. J. Stubbs, M. J. Price and A. P. Dove, Chem. Rev., 2021, 121, 6744–6776 CrossRef CAS PubMed.
- B. Li, C. Xu, L. Liu, J. Yu and Y. Fan, Green Chem., 2021, 23, 479–489 RSC.
- H. Si, K. Wang, B. Song, A. Qin and B. Z. Tang, Polym. Chem., 2020, 11, 2568–2575 RSC.
- J. Wang, B. Li, D. Xin, R. Hu, Z. Zhao, A. Qin and B. Z. Tang, Polym. Chem., 2017, 8, 2713–2722 RSC.
- L. Marcos Celada, J. Martín, S. V. Dvinskikh and P. Olsén, ChemSusChem, 2024, 17, e202301233 CrossRef CAS PubMed.
- A. R. Katritzky, S. K. Singh, N. K. Meher, J. Doskocz, K. Suzuki, R. Jiang, G. L. Sommen, D. A. Ciaramitaro and P. J. Steel, Arkivoc, 2006, 2006, 43–62 Search PubMed.
- V. X. Truong and A. P. Dove, Angew. Chem., 2013, 125, 4226–4230 CrossRef.
- J. Huang and X. Jiang, ACS Appl. Mater. Interfaces, 2018, 10, 361–370 CrossRef CAS PubMed.
- A. Maity, J. Chen, N. Wilson-Faubert, A. Laventure and A. Nazemi, Macromolecules, 2024, 57, 305–316 CrossRef.
- C.-Y. Cui, W. Chen, H.-R. Wang, W.-M. Ren, X.-B. Lu and H. Zhou, Eur. Polym. J., 2024, 211, 112997 CrossRef CAS.
- H. Si, K. Wang, B. Song, A. Qin and B. Z. Tang, Polym. Chem., 2020, 11, 2568–2575 RSC.
- P. Song, L. Du, J. Pang, G. Jiang, J. Shen, Y. Ma, S. Ren and S. Li, Ind. Crops Prod., 2023, 197, 116498 CrossRef CAS.
- I. Ribca, B. Sochor, M. Betker, S. V. Roth, M. Lawoko, O. Sevastyanova, M. A. R. Meier and M. Johansson, Eur. Polym. J., 2023, 194, 112141 CrossRef CAS.
- I. Ribca, B. Sochor, S. V. Roth, M. Lawoko, M. A. R. Meier and M. Johansson, ACS Omega, 2023, 8, 25478–25486 CrossRef CAS PubMed.
- Y. Li and S. Sarkanen, Macromolecules, 2005, 38, 2296–2306 CrossRef CAS.
- Y.-Y. Wang, Y. Chen and S. Sarkanen, Green Chem., 2015, 17, 5069–5078 RSC.
- S. Sarkanen, Y. Chen and Y.-Y. Wang, ACS Sustain. Chem. Eng., 2016, 4, 5223–5229 CrossRef CAS.
- Y.-Y. Wang, Y. Chen and S. Sarkanen, Faraday Discuss., 2017, 202, 43–59 RSC.
- Y. Chen, S. Sarkanen and Y.-Y. Wang, Molecules, 2019, 24, 4611 CrossRef CAS PubMed.
- W. G. Glasser, Front. Chem., 2019, 7, 565 CrossRef CAS PubMed.
- C. Wang, S. S. Kelley and R. A. Venditti, ChemSusChem, 2016, 9, 770–783 CrossRef CAS PubMed.
- J. R. Ochoa-Gómez, O. Gómez-Jiménez-Aberasturi, B. Maestro-Madurga, A. Pesquera-Rodríguez, C. Ramírez-López, L. Lorenzo-Ibarreta, J. Torrecilla-Soria and M. C. Villarán-Velasco, Appl. Catal., A, 2009, 366, 315–324 CrossRef.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, ACS Macro Lett., 2014, 3, 607–610 CrossRef CAS PubMed.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. D. Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
- F. C. Meunier, L. Domokos, K. Seshan and J. A. Lercher, J. Catal., 2002, 211, 366–378 CrossRef CAS.
- Y. Liu, Z. Yu, B. Wang, P. Li, J. Zhu and S. Ma, Green Chem., 2022, 24, 5691–5708 RSC.
- M. D. Rowe, E. Eyiler and K. B. Walters, Polym. Test., 2016, 52, 192–199 CrossRef CAS.
- Y. Pu, S. Cao and A. J. Ragauskas, Energy Environ. Sci., 2011, 4, 3154–3166 RSC.
- K. Shameli, M. Bin Ahmad, S. D. Jazayeri, S. Sedaghat, P. Shabanzadeh, H. Jahangirian, M. Mahdavi and Y. Abdollahi, Int. J. Mol. Sci., 2012, 13, 6639–6650 CrossRef CAS PubMed.
- S. Zhou, K. Huang, X. Xu, B. Wang, W. Zhang, Y. Su, K. Hu, C. Zhang, J. Zhu, G. Weng and S. Ma, ACS Sustain. Chem. Eng., 2023, 11, 3466–3473 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta07880b |
| This journal is © The Royal Society of Chemistry 2025 |