
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design for enhanced mechanical and kinetic properties of SnSb-based yolk–shell heterostructure as long cycle-life, high-rate Na-ion battery anode†
Jong Min
Im‡
ab,
Hyojun
Lim‡
 ac,
Hyunjin
Kim
ad,
Yun Chan
Kang
b,
Yoon
Hwa
e and
Sang-Ok
Kim
*ad
ac,
Hyunjin
Kim
ad,
Yun Chan
Kang
b,
Yoon
Hwa
e and
Sang-Ok
Kim
*ad
aEnergy Storage Research Center, Korea Institute of Science and Technology, 5, Hwarang-ro 14-gil, Seongbuk-gu, Seoul 02792, Republic of Korea
bDepartment of Materials Science and Engineering, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
cDepartment of Nuclear Science and Engineering and Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
dDivision of Energy & Environment Technology, KIST School, Korea University of Science and Technology, 5, Hwarang-ro 14-gil, Seongbuk-gu, Seoul 02792, Republic of Korea. E-mail: kimsok82@kist.re.kr
eSchool of Electrical, Computer and Energy Engineering, Arizona State University, Tempe, Arizona 85287, USA
First published on 21st January 2025
Abstract
Bimetallic SnSb has significantly attracted attention as a Na-ion battery (SIB) anode owing to its higher theoretical capacity of 752 mA h g−1 compared to conventional hard carbon anodes. However, practical applications are hindered by substantial volume changes during sodiation/desodiation. Herein, a SnSb-based heterostructured anode (SnSb@C-SiOC) with high SnSb content (∼85%) is developed via two-step pyrolysis using SnSbOx@polydopamine precursors dispersed in silicone oil. The resulting SnSb yolk nanoparticles, encapsulated within a multi-functional C–SiOC bi-layered shell, facilitate rapid Na-ion transport and provide effective volume buffering during cycling for efficient electrochemical reactions and enhanced structural integrity. Post-mortem analyses reveal reversible crystalline phase transformations of SnSb with uniform elemental distributions, demonstrating the effectiveness of bi-layered shells. With superior mechanical robustness of the heterostructure confirmed by nanoindentation, the SnSb@C-SiOC anode delivers a high capacity of 445.6 mA h g−1 after 250 cycles at 2 A g−1, retaining 87.9% of its initial capacity and greatly outperforming pure SnSb. Additionally, a full cell combining the anode with a Na3V2(PO4)3 cathode shows promising cycle and rate performances, suggesting potential for practical applications. This study presents a viable approach for developing durable and efficient anode materials to advance SIBs and provide next-generation energy storage systems.
1 Introduction
In the pursuit of cost-effective Na-ion batteries (SIBs) with high energy density, alloy-type anode materials including Sn, Sb, Ge, and Se have garnered considerable interest. Such materials exhibit significantly high specific capacities owing to alloying and dealloying reactions that involve the formation and breaking of alloys with Na during battery operation. Moreover, such elements exhibit relatively high sodiation potential compared to hard carbon, effectively mitigating the growth of Na-dendrites and enhancing the safety of SIBs. Furthermore, environment-friendly characteristics make the elements particularly advantageous for large-scale energy storage applications.1,2Among various alloy-type anodes, metallic Sn and Sb have emerged as attractive candidates. The elements offer high theoretical capacities of 847 and 660 mA h g−1 for Sn and Sb, respectively, suitable redox potentials (<1.0 V vs. Na+/Na), and minimal toxicity.3–5 Despite such advantages, Sn and Sb face significant scientific challenges including substantial volume expansion (420 and 390% for Na15Sn5 and Na3Sb, respectively) that occurs upon full sodiation. A volume change causes particle pulverization and detachment from current collectors during repeated cycling, thereby compromising the mechanical integrity and electrochemical performance of anodes. Additionally, the formation of an undesirable solid electrolyte interface (SEI) on the electrode surface restricts efficient Na-ion storage and negatively affects battery stability over multiple charge and discharge cycles.6
To overcome existing obstacles, researchers have explored applications of intermetallic compounds, particularly binary alloys, as a promising approach for enhancing the physicochemical and electrochemical characteristics of alloy-based anode materials.7–9 For example, SnSb alloys have demonstrated the ability to alleviate structural degradations of anodes and improve the electrochemical performance of SIBs compared to individual metallic elements such as Sn or Sb. However, significant volume variations and sluggish sodiation kinetics during alloying and dealloying reactions hinder the long-term cycling stability and high-rate capability of the alloys, limiting their practical application to SIBs.9
A widely recognized strategy to address existing issues includes controlling particle size (e.g., micro and nanoscale) and developing unique nanostructures (e.g., hollow and yolk–shell configurations) in alloy-type anodes. Structural modifications facilitate electron transfer and ionic diffusion while mitigating mechanical strain caused by volume changes, thereby enhancing battery performance.4,10–13 Another promising approach involves the creation of heterostructured anode materials comprising metal–semiconductor or semiconductor–semiconductor junctions with different energy bandgaps. The heterostructures generate an internal electric field at the interfaces to enhance electronic conductivity, boost charge transport capability, and demonstrate great potential for high-power storage.14,15 The most common and efficient method for achieving heterostructures incorporates carbonaceous materials (e.g., graphene, carbon nanotubes, and porous carbons) into composites, which serve as electronic conduction networks and stress buffers to enhance the overall Na storage performance of host materials.16,17 Despite such advantages, carbon-based materials exhibit several drawbacks such as insufficient mechanical robustness and large irreversible capacity, leading to capacity degradation during cycling. Therefore, additional structural reinforcement agents are required to overcome the limitations.
Among various coating agents that provide a buffering effect, SiOC is a suitable material for alleviating substantial mechanical stress from volume variations in anode materials for Li-ion batteries (LIBs) and SIBs. SiOC exhibits exceptional mechanical and electrochemical properties including elasticity, robustness, and superior electrical conductivity when compared to carbon-based materials.15 Nevertheless, conventional methods for synthesizing SiOC ceramic composites often require complex processes, high costs, and polysiloxane precursor usage. As an alternative source of SiOC, silicone oil has been investigated owing to its cost-effectiveness, facile pyrolysis, and eco-friendliness compared to polysiloxane precursors.3,15 The approach demonstrates enhanced anode performance by leveraging mechanical effectiveness through controlled surface modifications, such as encapsulation.
This study develops a heterostructured anode comprising SnSb yolk and C–SiOC bilayer shell. SnSb@C-SiOC nanohybrid is synthesized by the simple pyrolysis of silicone oil suspension containing SnSbOx@polydopamine (SnSbOx@PDA). The process facilitates the formation of nanosized metallic SnSb yolk particles encapsulated within a multi-functional C–SiOC shell. Encapsulation is crucial for effectively managing volume changes in SnSb during alloying and dealloying reactions. The crystal structure, morphology, and surface chemical states of the SnSb@C-SiOC nanohybrid are systematically characterized. Furthermore, the influence of the yolk–shell heterostructure on the electrochemical performance and mechanical properties of SnSb-based anode materials is evaluated using various electrochemical tests and nanoindentation techniques. The SnSb@C-SiOC nanohybrid addresses critical challenges such as large volume expansion, sluggish Na-ion transport, structural instability, and undesirable SEI formation in SnSb-based alloy anodes, representing a significant advancement in SIB technology. The innovative heterostructure approach not only demonstrates viability as a practical anode material but also offers a scalable and environmentally friendly solution for large-scale energy storage applications.
2 Results and discussion
2.1 Materials characterization
Fig. 1a illustrates the overall synthesis of SnSb-based anode materials using a SnSbOx nanoparticle precursor, which was successfully prepared via solvothermal reaction. First, an in situ uniform PDA coating was coated on the SnSbOx precursors via adhesion and self-polymerization of dopamine hydrochloride in a basic aqueous solution. Fourier transform infrared spectroscopy (FT-IR) indicated the presence of functional groups related to PDA (C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, N–H, –OH, and –NH) (Fig. S1, ESI†). During two-step pyrolysis, the PDA layer was transformed into N-doped carbon structures owing to the N-containing functional groups, resulting in the formation of SnSb@C. The presence of N-doped carbon ensured high electrical conductivity and structural stability of the active materials.10,18 However, the realization of SnSb composites with carbon coatings was unsatisfactory considering long-term cycling stability and high-rate capability due to insufficient mechanical properties such as elasticity and hardness, which often resulted in cracking of the coating layer and electrode failure.7,12,17 Accordingly, a dual coating strategy based on N-doped carbon with SiOC shell (C–SiOC) was employed to further reinforce the aforementioned aspects. According to the literature,15,19 silicone oil-derived SiOC exhibits superior mechanical performance in accommodating the substantial stress caused by volume variations in alloy-based anode materials during alloying/dealloying reactions. Additionally, the mesoporous characteristics of SiOC were expected to accelerate ionic diffusion from bulk electrolyte to SnSb, thereby enhancing rate capability. To prepare the SnSb@C-SiOC nanohybrid, SnSbOx@PDA precursors were homogeneously dispersed in silicone oil, and the suspension was subsequently pyrolyzed to induce SiOC ceramization under an Ar atmosphere.20 Excess silicone oil, which was not used in the formation of SiOC, was volatilized during heat treatment. Meanwhile, SnSbOx was immediately reduced to SnSb during heat treatment, and the functional groups of the PDA coating layer (Fig. S1, ESI†) formed hydrogen bonds with the silicone oil, which serves as a driving force for forming a yolk–shell hybrid structure comprising SnSb nanoparticles encapsulated within the C–SiOC bilayer.21
C, N–H, –OH, and –NH) (Fig. S1, ESI†). During two-step pyrolysis, the PDA layer was transformed into N-doped carbon structures owing to the N-containing functional groups, resulting in the formation of SnSb@C. The presence of N-doped carbon ensured high electrical conductivity and structural stability of the active materials.10,18 However, the realization of SnSb composites with carbon coatings was unsatisfactory considering long-term cycling stability and high-rate capability due to insufficient mechanical properties such as elasticity and hardness, which often resulted in cracking of the coating layer and electrode failure.7,12,17 Accordingly, a dual coating strategy based on N-doped carbon with SiOC shell (C–SiOC) was employed to further reinforce the aforementioned aspects. According to the literature,15,19 silicone oil-derived SiOC exhibits superior mechanical performance in accommodating the substantial stress caused by volume variations in alloy-based anode materials during alloying/dealloying reactions. Additionally, the mesoporous characteristics of SiOC were expected to accelerate ionic diffusion from bulk electrolyte to SnSb, thereby enhancing rate capability. To prepare the SnSb@C-SiOC nanohybrid, SnSbOx@PDA precursors were homogeneously dispersed in silicone oil, and the suspension was subsequently pyrolyzed to induce SiOC ceramization under an Ar atmosphere.20 Excess silicone oil, which was not used in the formation of SiOC, was volatilized during heat treatment. Meanwhile, SnSbOx was immediately reduced to SnSb during heat treatment, and the functional groups of the PDA coating layer (Fig. S1, ESI†) formed hydrogen bonds with the silicone oil, which serves as a driving force for forming a yolk–shell hybrid structure comprising SnSb nanoparticles encapsulated within the C–SiOC bilayer.21
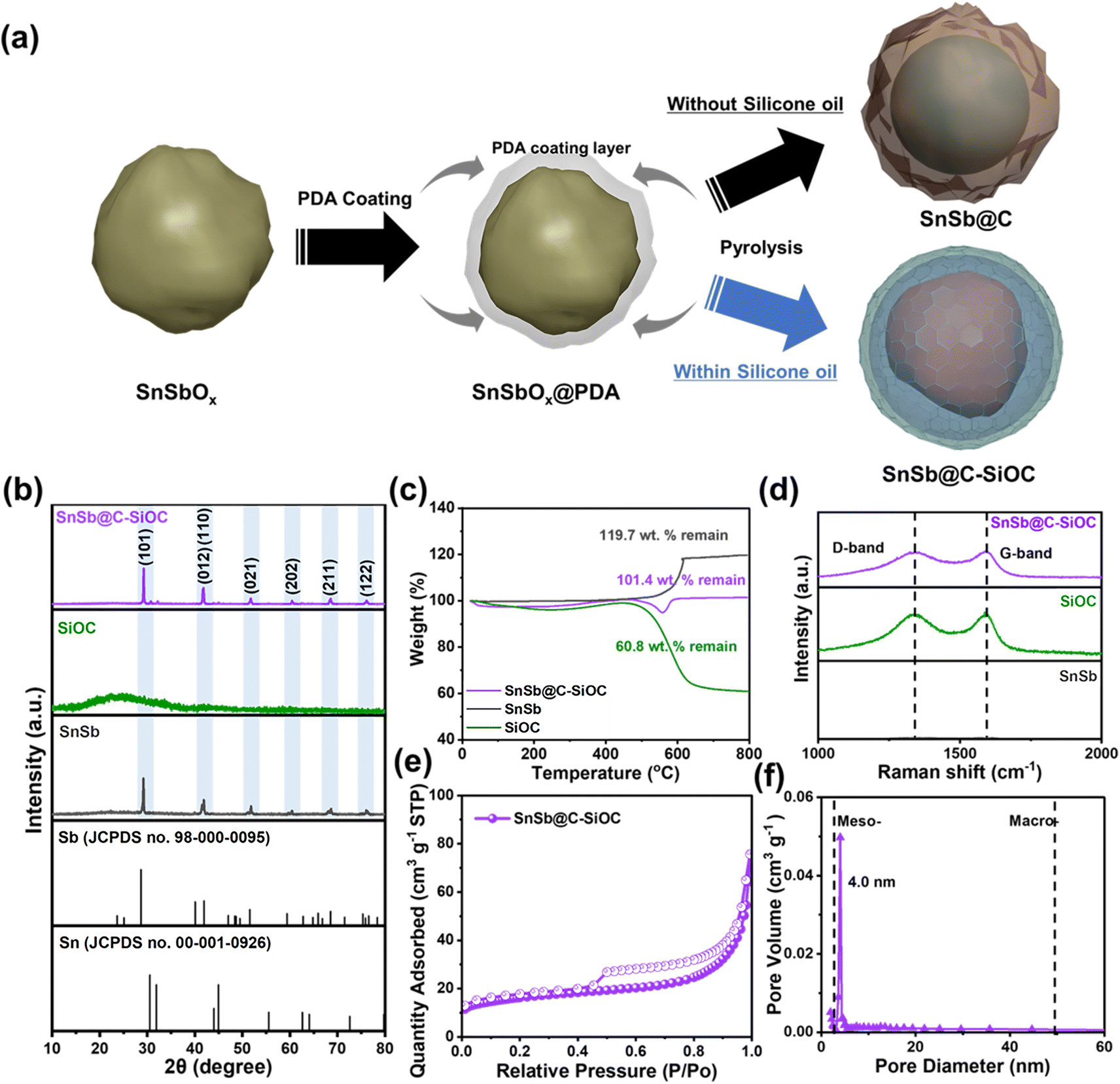 | ||
| Fig. 1 (a) Schematic of SnSb@C and SnSb@C-SiOC nanohybrid preparation. (b) XRD patterns, (c) TGA, and (d) Raman spectra of SnSb, SiOC, and SnSb@C-SiOC. (e) N2 adsorption–desorption isotherm curve and (f) corresponding pore size distribution of SnSb@C-SiOC. | ||
The X-ray diffraction (XRD) patterns of pure SnSb, SiOC, and SnSb@C-SiOC nanohybrids were compared (Fig. 1b). Pure SiOC exhibited a broad peak at approximately 23°, indicating the characteristic features of an amorphous structure, consistent with previous reports.19,20,22 During pyrolysis, SnSbOx was surrounded by amorphous SiOC and reduced to SnSb; therefore, distinct peaks corresponding to SnSb (JCPDS no. 33-0118) were observed in the XRD pattern of the final material.23,24 Two additional insignificant peaks related to metallic Sn in the 30–45° range were detected compared to those of pure SnSb, owing to different reduction kinetics between Sn and Sb.9 To investigate the composition ratio in SnSb@C-SiOC nanohybrid, thermogravimetric analysis (TGA) was conducted between 30 and 800 °C under air conditions (Fig. 1c). Above 500 °C, carbon and SnSb reacted with oxygen and oxidized to CO2 (C (s) → CO2 (g)) and metal oxide (SnSb (s) → SnSbOx (s)), respectively. From the TGA results, SnSb@C-SiOC contained approximately 84.7 and 15.3 wt% SnSb and C–SiOC bilayers, respectively. Due to the low coating layer content in the composite material, the XRD pattern of the final product in Fig. 1b shows distinct SnSb peaks without a reduction in peak intensity. Such a high active material content could be beneficial for improving the energy density of SIBs.
The Raman spectra of pure SnSb, SiOC, and SnSb@C-SiOC were collected to characterize the carbon structures of the anode materials (Fig. 1d). The G-band at 1345 cm−1 corresponded to ordered carbon (sp2-carbon), whereas the D-band at 1595 cm−1 was attributable to disordered carbon with defects (sp3-hybridized carbon).23,25,26 The high-intensity ratio of D to G bands (ID/IG) within the deconvoluted Raman peaks suggested superior electrical conductivity.26 The intensity ratios (ID/IG) of SiOC and SnSb@C-SiOC nanohybrids were 0.96 and 0.97, respectively, suggesting the contribution of SiOC with free carbon domains to improve the electrical conductivity of SnSb@C-SiOC, facilitating electrode kinetics. Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda methods were employed to assess the specific surface area and pore size distribution of SnSb@C-SiOC (Fig. 1e and f). The N2 adsorption–desorption isotherm showed type IV curves for SnSb@C-SiOC nanohybrid, indicating mesoporous characteristics with specific surface area and total pore volume of 57.3 m2 g−1 and 0.12 cm3 g−1, respectively (Fig. 1e). Additionally, the pore size distribution of SnSb@C-SiOC (Fig. 1f) revealed the largest pore size (∼4.0 nm, which was classified as mesoporous). The mesoporous structure was primarily due to the high surface area and mesoporous nature of SiOC (Fig. S2 and Table S1, ESI†). The large specific surface area and mesoporous properties facilitated fast Na-ion transport by creating abundant redox-active sites, ultimately improving the rate capability.
Field-emission scanning electron microscopy (FE-SEM), dynamic light scattering (DLS), transmission electron microscopy (TEM) coupled with energy dispersive X-ray spectroscopy (EDS), and X-ray photoelectron spectroscopy (XPS) depth profiles were employed to thoroughly investigate the morphology and structure of SnSb-based materials. The SnSbOx precursor, which served as the starting material, exhibited an average particle diameter of approximately 234.1 nm (Fig. S3a, ESI†). After thermal treatment, the SEM image of pure SnSb displayed large micro-sized clusters up to 5.5 μm with rough surfaces (Fig. S3b, ESI†). Particle agglomeration was mainly attributed to the low melting points of Sn (231 °C) and Sb (630 °C) that resulted in the growth of active particles during pyrolysis. Contrarily, SnSb-based heterostructured materials comprised nano-sized particles (Fig. 2a and S3c, ESI†) and maintained nearly unchanged average particle sizes of 527 and 371.8 nm for SnSb@C and SnSb@C-SiOC (Fig. 2b and S3c, ESI†), respectively. The realization of heterostructure with coating agents effectively prevented the formation of aggregated particles throughout thermal reduction. The selected area electron diffraction (SAED) pattern (Fig. 2c) revealed lattice fringes with an interplanar spacing of 0.31 and 0.22 nm, corresponding to the (101) and (012)/(110) crystal planes of rhombohedral SnSb.23,24 TEM-EDS elemental mapping (Fig. 2d), line scan profile (Fig. S4, ESI†), and XRD and SAED results confirmed the uniform distribution of Sn and Sb in the yolk region, indicating the presence of SnSb within the SnSb@C-SiOC nanohybrid.
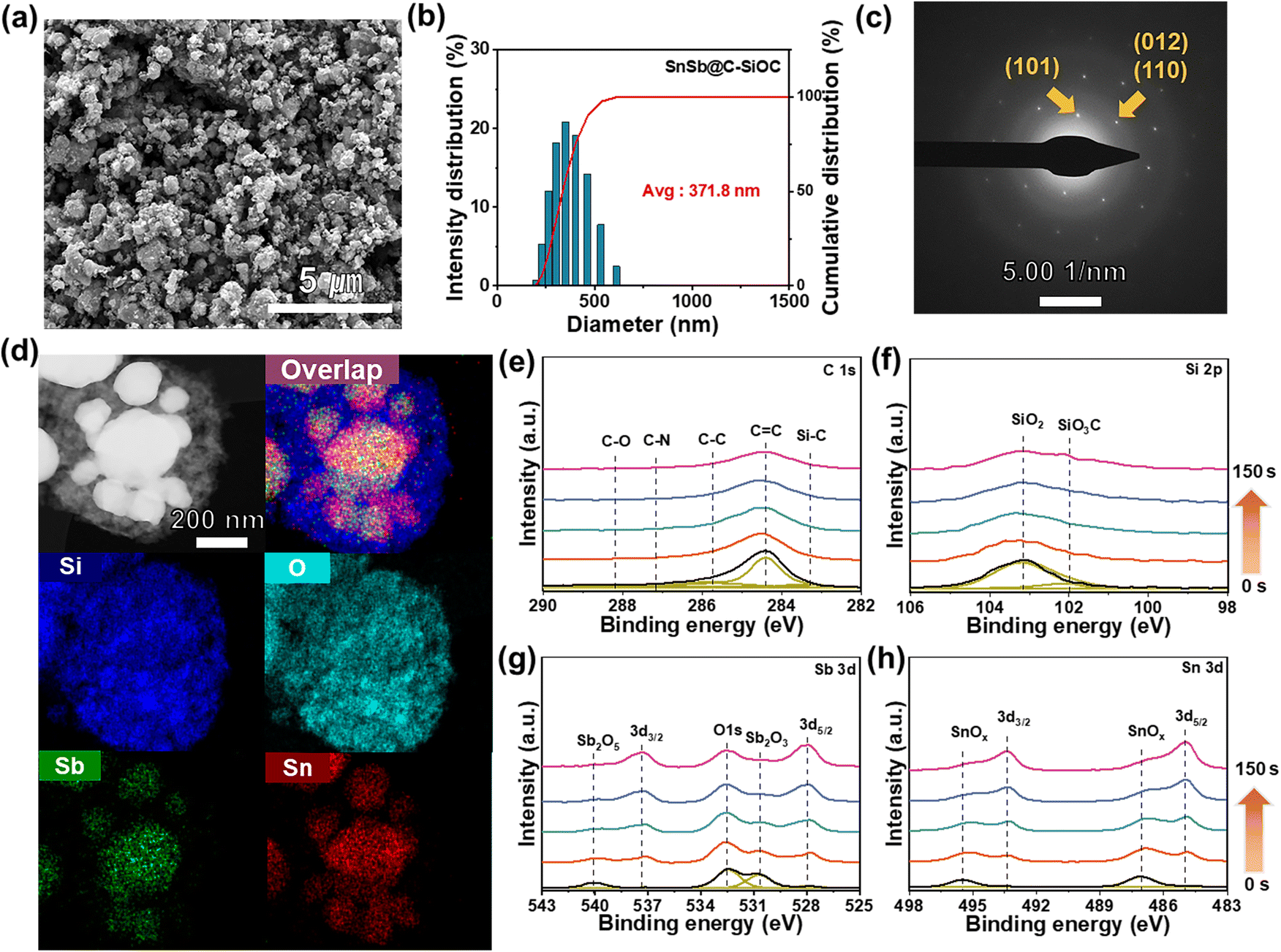 | ||
| Fig. 2 Morphology and structural characterization. (a) FE-SEM, (b) particle-size distribution, (c) SAED pattern, and (d) TEM/EDS mapping images of SnSb@C-SiOC (Si, Sb, and Sn). (e) XPS depth profiles of SnSb@C-SiOC in the regions of C 1s, Si 2p, Sb 3d, and Sn 3d (sputtering rate: 0.5 nm s−1). | ||
The XPS survey spectrum of the SnSb@C-SiOC nanohybrid was obtained to determine its surface composition and bond valence states (Fig. S5, ESI†). To observe the detailed chemical composition, XPS depth profiles were collected via Ar+ sputtering at intervals of 0, 25, 50, 100, and 150 s (Fig. 2e). The C 1s spectrum revealed a C–N peak (287.1 eV) attributable to the formation of PDA-derived carbon.21 The Si 2p spectrum displayed two distinctive peaks associated with the SiOC shell, identified as SiO2 (103.1 eV) and SiO3C (102 eV).27 In the initial surface region, SiOC-related chemical bonding peaks were predominantly observed, providing strong evidence of SiOC generation. Considering the Sn 3d and Sb 3d spectra, insignificant peaks corresponding to Sn oxide (495.5 and 487 eV) and Sb oxide (540 and 530.7 eV) were detected before sputtering, indicating the presence of minor oxidized substances at the surface during pyrolysis.28,29 Nano-sized metal particles underwent partial oxidation reactions at the surface, attributable to their large surface area with high surface reactivity. During XPS depth profiling, the SiOC-related peaks gradually decreased. The primary constituents of Sn (493.4 and 485 eV at Sn 3d) and Sb (537.4 and 528 eV at Sb 3d) peaks were prominently observed with high intensity during sputtering.28–30 The obtained results confirmed that the proposed yolk–shell type SnSb@C-SiOC heterostructure was achieved via simple pyrolysis.
2.2 Electrochemical performance
Various cell tests and post-mortem analyses were performed to evaluate the effects of the SiOC coating layer and heterostructure on the electrochemical performance of the SnSb-based materials. First, the electrochemical behavior of the SnSb-based electrodes was investigated using differential capacity (dQ/dV) plots (Fig. 3a and S6a, ESI†). In the initial dQ/dV plot, the hump-shaped peak at 0.44 V upon sodiation indicated an overlap between Sb sodiation and SEI formation on the surface.31 In the 2nd cycle scan, two pairs of sodiation peaks were observed at 0.6, 0.51 (Na–Sb), 0.33, and 0.03 V (Na–Sn), while desodiation peaks were located at 0.18, 0.36 (Na–Sn), 0.54, and 0.88 V (Na–Sb), respectively.31–33 The cathodic/anodic peaks of pure SnSb electrode were observed at slightly delayed voltages compared to those of C–SiOC anode due to the slower sodiation/desodiation kinetics of pure SnSb, attributable to the lack of an electrically conductive bilayer shell.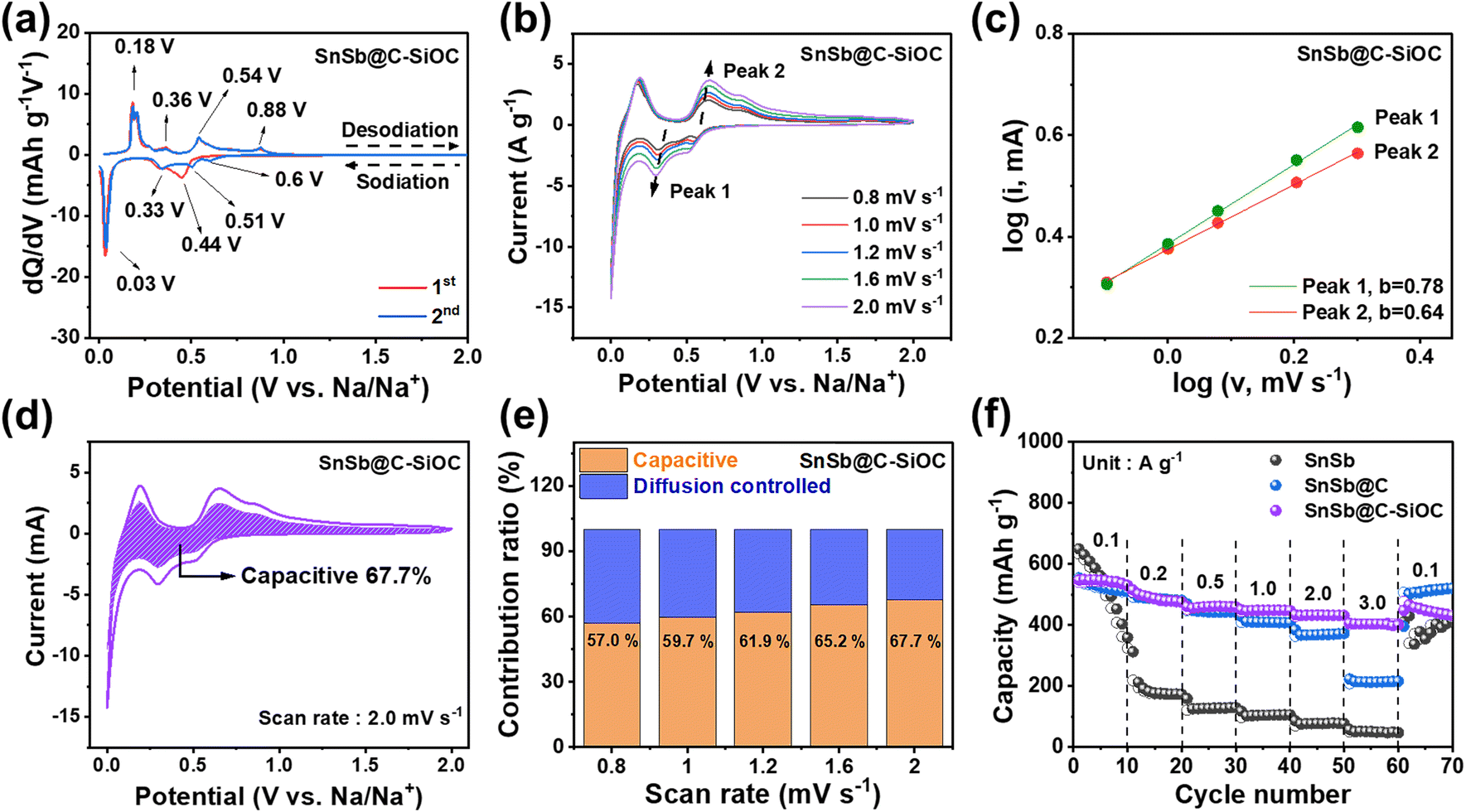 | ||
Fig. 3 (a) dQ/dV plots of SnSb@C-SiOC at 0.1 A g−1 for the 1st and 2nd cycles. (b) CV curves of the SnSb@C-SiOC electrode at different scan rates from 0.8 to 2.0 mV s−1 in the voltage range of 0.001–2.0 V. (c) log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) i versus logv plots and (d) capacitive contribution of SnSb@C-SiOC at 2.0 mV s−1. (e) Comparative contribution ratio of the SnSb@C-SiOC electrode (diffusion-controlled vs. capacitive) at various scan rates. (f) Rate performance of the SnSb-based electrodes at various current densities from 0.1 to 3.0 A g−1. i versus logv plots and (d) capacitive contribution of SnSb@C-SiOC at 2.0 mV s−1. (e) Comparative contribution ratio of the SnSb@C-SiOC electrode (diffusion-controlled vs. capacitive) at various scan rates. (f) Rate performance of the SnSb-based electrodes at various current densities from 0.1 to 3.0 A g−1. | ||
For a deeper understanding of the charge storage mechanism, cyclic voltammetry (CV) was performed for SnSb-based electrodes at various scan rates from 0.8 to 2.0 mV s−1, measured within a voltage window from 0.001 to 2.0 V (Fig. 3b and S6b, ESI†). Compared with pure SnSb, the SnSb@C-SiOC nanohybrid maintained a consistent CV shape during oxidation and reduction, even at high scan rates, indicating highly stable redox reactivity of the composite electrode with Na ions. The relationship between capacitive and diffusion-controlled mechanisms was estimated using eqn (1).34,35
| logi = loga + blogv | (1) |
| i = k1v + k2v1/2 | (2) |
The capacitive behavior, which corresponds to fast reactions, exhibits a higher contribution rate in electrochemical reactions when the material has a larger surface area and mesoporous structure because abundant active sites for Na-ion storage are provided on the surface. Additionally, such structures facilitate electrolyte penetration, promoting the facile movement of Na-ions, and further contributing to the improvement of the electrochemical rate characteristics of the active material.3,20 To investigate the rate capability of the SnSb-based anodes, changes in specific capacity at various current densities from 0.1 to 3.0 A g−1 were compared (Fig. 3f). Pure SnSb showed a high specific capacity at a low current density of 0.1 A g−1 but underwent rapid capacity decay as the current density increased. While the SnSb@C electrode revealed better rate capability up to 1.0 A g−1, its capacity decreased at higher current densities (>2.0 A g−1). Contrarily, the SnSb@C-SiOC electrode exhibited the highest reversible capacities of 547, 487.6, 461.6, 448.9, 432.7, and 403.1 mA h g−1 at 0.1, 0.2, 0.5, 1.0, 2.0, and 3.0 A g−1, respectively. The obtained results suggested that the porous SiOC shell not only promoted Na-ion transport but also stored additional energy at high current densities through surface-based capacitive reactions.
The galvanostatic charge discharge (GCD) profiles of the SnSb-based electrodes were compared to examine the variations in their reversible capacity and coulombic efficiency (CE) for up to the 15th cycle (Fig. 4a and S7, ESI†). The initial charge/discharge capacities of the SnSb@C-SiOC electrode were 543.0/644.1 mA h g−1, with an initial CE of 84.3%. The first irreversible capacity loss was primarily attributed to SEI formation and organic electrolyte decomposition, commonly observed in various host anode materials.36,37 As the cycles progressed, the reversible capacity of the SnSb@C-SiOC nanohybrid stabilized at approximately 500 mA h g−1, maintaining high CE values above 99% (Fig. 4a). Conversely, pure SnSb exhibited a rapid decline in capacity, retaining only approximately 100 mA h g−1 by the 15th cycle. Thus, pure SnSb possessed significantly poorer structural stability than the SnSb@C-SiOC nanohybrid composite over multiple cycles.
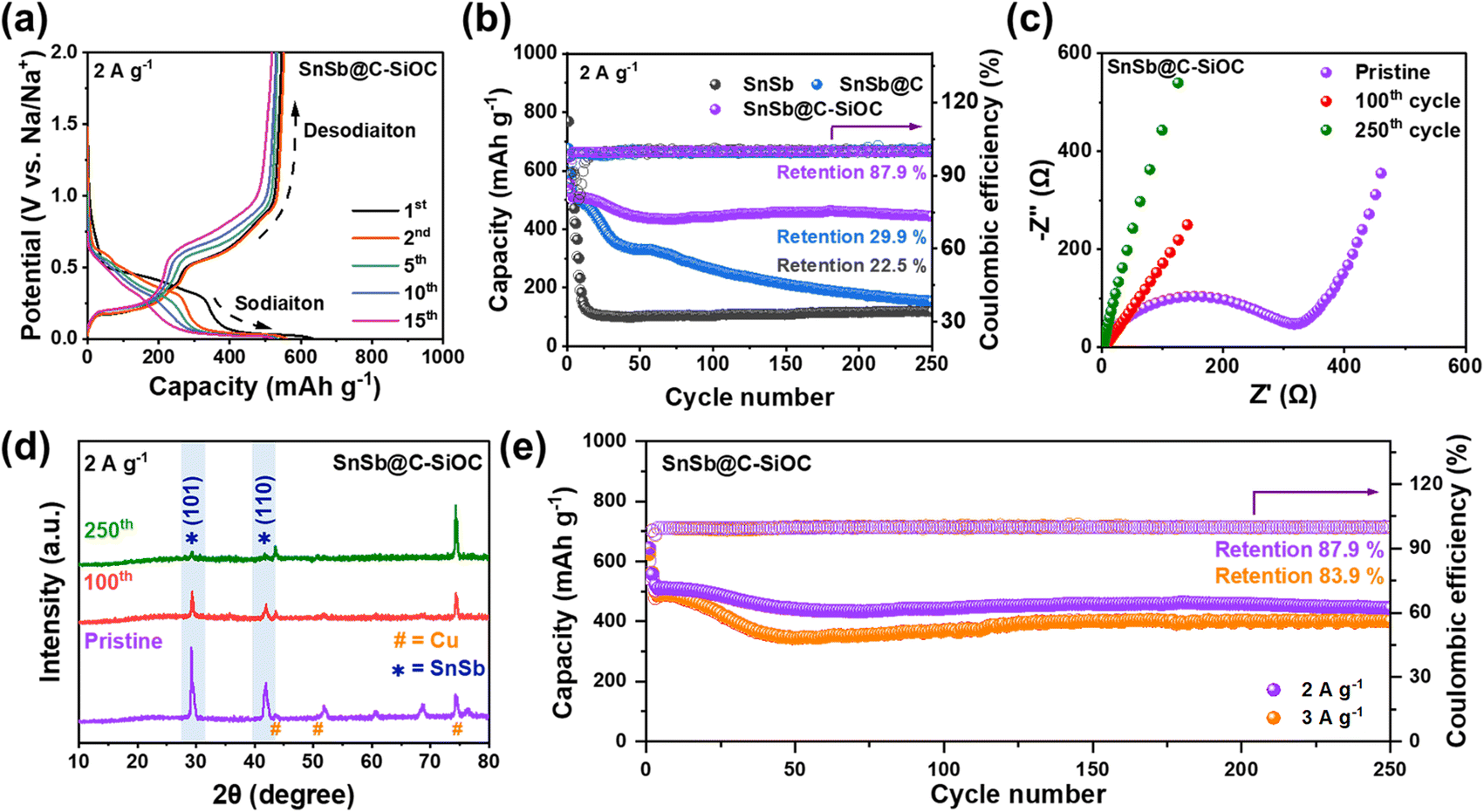 | ||
| Fig. 4 (a) GCD profiles of the SnSb@C-SiOC nanohybrid cell after several cycles (1st, 2nd, 5th, 10th, and 15th). (b) Cycling performance of SnSb, SnSb@C, and SnSb@C-SiOC at 2.0 A g−1, (c) Nyquist plots, and (d) ex situ XRD patterns of the SnSb@C-SiOC electrode obtained before cycling and after the 100th and 250th cycles. (e) High rate cycling performance of the SnSb@C-SiOC electrode at current densities of 2.0 and 3.0 A g−1. | ||
Fig. 4b displays the long-term cycling performance of SnSb-based electrodes at 2.0 A g−1 after the initial formation and two cycles at 0.2 A g−1. As anticipated from the GCD curves, pure SnSb electrodes experienced a rapid capacity decline, exhibiting specific charge and discharge capacities of 118.7 and 119.2 mA h g−1, respectively, with a capacity retention of approximately 22.5% after the 250th cycle. Conversely, the SnSb@C electrode demonstrated enhanced cycling stability compared with pure SnSb; however, its capacity sharply declined after the 50th cycle, retaining only approximately 29.9% of its initial capacity by the 250th cycle. In sharp contrast, the SnSb@C-SiOC electrode maintained high reversible charge and discharge capacities of 444.0 and 444.6 mA h g−1, respectively, even after 250 cycles, achieving a capacity retention of 87.9%. The enhanced performance suggested that the SiOC shell in the heterostructure effectively accommodated significant volume changes in the SnSb yolk, thereby preserving the structural stability of the yolk and contributing to excellent long-term battery stability.
To further examine the resistance of the SnSb-based electrodes, electrochemical impedance spectroscopy (EIS) was performed (Fig. 4c and S8, ESI†). Table S2 (ESI)† summarizes parameters such as electrolyte resistance (Rele), SEI resistance (RSEI), and charge transfer resistance (Rct) according to the equivalent circuit.16,26 In all states, the SnSb@C-SiOC electrode showed lower resistance than the pure SnSb electrode. Particularly, when comparing the resistances after the 100th and 250th cycles, the nanohybrid electrode consistently demonstrated a lower resistance than the pure electrode. Electrodes typically undergo a stabilization step during formation cycling, resulting in a decrease in the resistance of the electrode. However, despite the decrease in resistance through stabilization, degradation of the morphology and structure of the material after repeated cycling will lead to an increase in resistance. Therefore, despite the decrease in resistance after 100 cycles for the SnSb and SnSb@C-SiOC electrodes, the greater reduction in the SnSb@C-SiOC electrode and significantly lower resistance after 250 cycles indicate that the well-designed yolk–shell structure of the SnSb composite material not only contributed to the stable formation of the SEI layer on the particle surface but also helped maintain the microstructure and morphology of the active particles.
Fig. 4d shows the ex situ XRD patterns of the electrodes before and after cycling (pristine, 100th, and 250th cycles) to confirm the crystalline phase stability of the SnSb electrodes. Even after the 100th and 250th cycles, the SnSb@C-SiOC electrode exhibited crystalline peaks at (101) and (110), corresponding to the hexagonal SnSb phase. Contrarily, the XRD pattern of the pure SnSb electrode showed only the Cu current collector peak without any SnSb-related peaks, indicating that the crystal structure of pure SnSb deteriorated during battery operation (Fig. S9 and S10, ESI†).8 The obtained results demonstrated that the SnSb@C-SiOC electrodes recovered their crystal structure even after several cycles, suggesting that the presence of the SiOC shell provided enhanced structural stability compared to the pure SnSb sample. As illustrated in the high-rate cycling performance (Fig. 4e), the SnSb@C-SiOC electrode maintained high charge/discharge capacities of 401.0/401.7 mA h g−1 (capacity retention: 83.9%) after the 250th cycle, with a high CE of 99.8%, even under a high current density of 3.0 A g−1, demonstrating enhanced Na-ion storage behavior compared to that reported in previous literature on SnSb-based anodes for SIBs (Table S3†).11,13,23,26,31,38–40
In general, alloy-type anodes undergo significant volume variations during sodiation and desodiation, resulting in electrode pulverization and cracking, which can profoundly influence cycling stability.41 Therefore, to investigate morphological changes in the electrodes, coin cells in their first sodiated and desodiated states were disassembled, and the thickness and morphological changes in the electrodes during the charge and discharge processes were observed using cross-sectional and top-view FE-SEM. Before sodiation, both electrodes exhibited a thickness of approximately 12 μm with no significant differences on their surfaces (Fig. 5a and b). During the first sodiation step, the thickness of the pure SnSb electrode expanded significantly, reaching up to 18.4 μm (148.4% of the initial value), and the electrode surface became non-uniform (Fig. 5a and S11a, ESI†). During the desodiation step, the thickness of the pure SnSb electrode did not return to its initial state and remained at 14.4 μm (116.1%), with microcracks appearing on the surface. Contrarily, the SnSb@C-SiOC electrode exhibited a smaller expansion (16.9 μm, 138.5%) compared to the pure SnSb electrode (148.4%) during the sodiation step, and the electrode surface remained uniform without cracks (Fig. 5b and S11b, ESI†). After the desodiation step, the thickness of the SnSb@C-SiOC electrode recovered to almost its initial state and remained at 12.5 μm (102.5%), with no microcracks developing on the surface. Notably, the thickness and surface states of the composite electrodes nearly returned to their initial states after desodiation, with no microcracks or electrode pulverization. Thus, the mechanically stable C–SiOC shell effectively buffered the volume changes of the SnSb alloy during the initial charge and discharge processes, enabling the nanohybrid electrode to exhibit superior long-term cycling stability compared to electrodes without the C–SiOC shell.
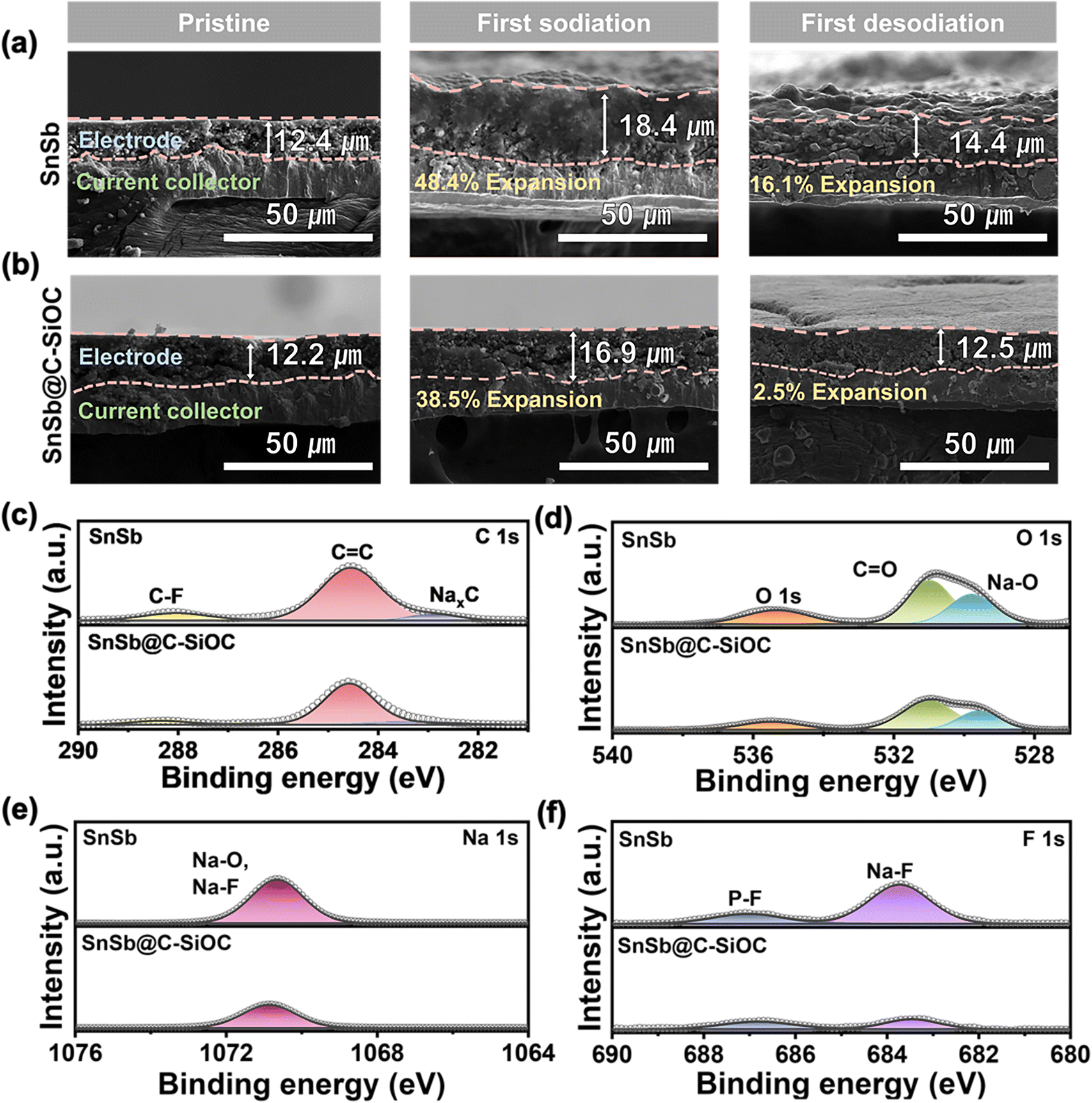 | ||
| Fig. 5 Cross-sectional SEM images of the (a) pure SnSb and (b) SnSb@C-SiOC electrodes before cycling, first sodiation, and first desodiation. (c–f) XPS spectra of C 1s, O 1s, Na 1s, and F 1s regions of the SnSb-based electrodes after the 100th cycle. | ||
To investigate the effect of the C–SiOC shell on the interfacial reaction with the electrolyte during sodiation/desodiation of the SnSb-based electrodes, post-mortem XPS analysis was performed on the electrodes after the 100th cycle. Overall, the electrode surface exhibited organic (RCH2ONa) and inorganic (Na2O and NaF) components related to SEI and NaPF6-based DME electrolytes (Fig. 5c–f).42,43 Compared to the pure SnSb electrode, the SnSb@C-SiOC electrode showed lower peak intensities for organic components C and O (Fig. 5c and d). Additionally, owing to the reduction of NaPF6, the peak intensities of O, Na, and F related to the Na2O and NaF components were lower for the SnSb@C-SiOC electrode (Fig. 5d–f). The reduced peak intensities associated with SEI formation indicated that the chemically and electrochemically stable C–SiOC shell protected the SnSb electrode from irreversible decomposition of the electrolyte. As shown in Fig. 5a, b and S11 (ESI),† the pure SnSb electrode generated microcracks on its surface during battery operation, creating new contact areas with the electrolyte. The new contact areas lead to additional SEI formation and electrolyte decomposition, which caused metal dissolution from the electrode, thereby accelerating surface-side reactions.44,45
To investigate the dissolution of active materials, inductively coupled plasma optical emission spectrometry (ICP-OES) was performed using glass fibers collected after the 100th cycle (Fig. S12, ESI†). Although the pure SnSb electrode exhibited 0.8 and 1.1% Sn and Sb dissolutions after the 100th cycle, respectively, the SnSb@C-SiOC electrode exhibited only 0.3 and 0.1% Sn and Sb dissolutions, respectively. The obtained results further demonstrated that the C–SiOC coating layer not only prevented electrode pulverization by enhancing the mechanical stability of the SnSb electrode but also suppressed surface-side reactions, such as electrolyte decomposition and metal dissolution, ensuring excellent long-term cycling performance.
To understand the morphological and structural changes in SnSb@C-SiOC during repeated charge/discharge processes, TEM-EDS and nanoindentation techniques were used on the SnSb@C-SiOC cell after the 100th cycle. Fig. 6a shows the elemental distribution of the nanohybrid powder collected from the SnSb@C-SiOC electrode. Even after the 100th cycle, the nanohybrid particles maintained their initial hierarchical yolk–shell architecture without structural degradation. The mechanical characteristics of the cycled electrodes were analyzed by nanoindentation. The SEM images of the nano-indented SnSb-based electrode after the 100th cycle (Fig. 6b, c and S13a, b, ESI†) revealed that the surface of the pure SnSb electrode was rough and swollen compared with that of the SnSb@C-SiOC electrode. The observation agreed well with the cross-sectional and top-view SEM images of the electrodes during the 1st sodiation/desodiation (Fig. 5a, b and S11, ESI†).
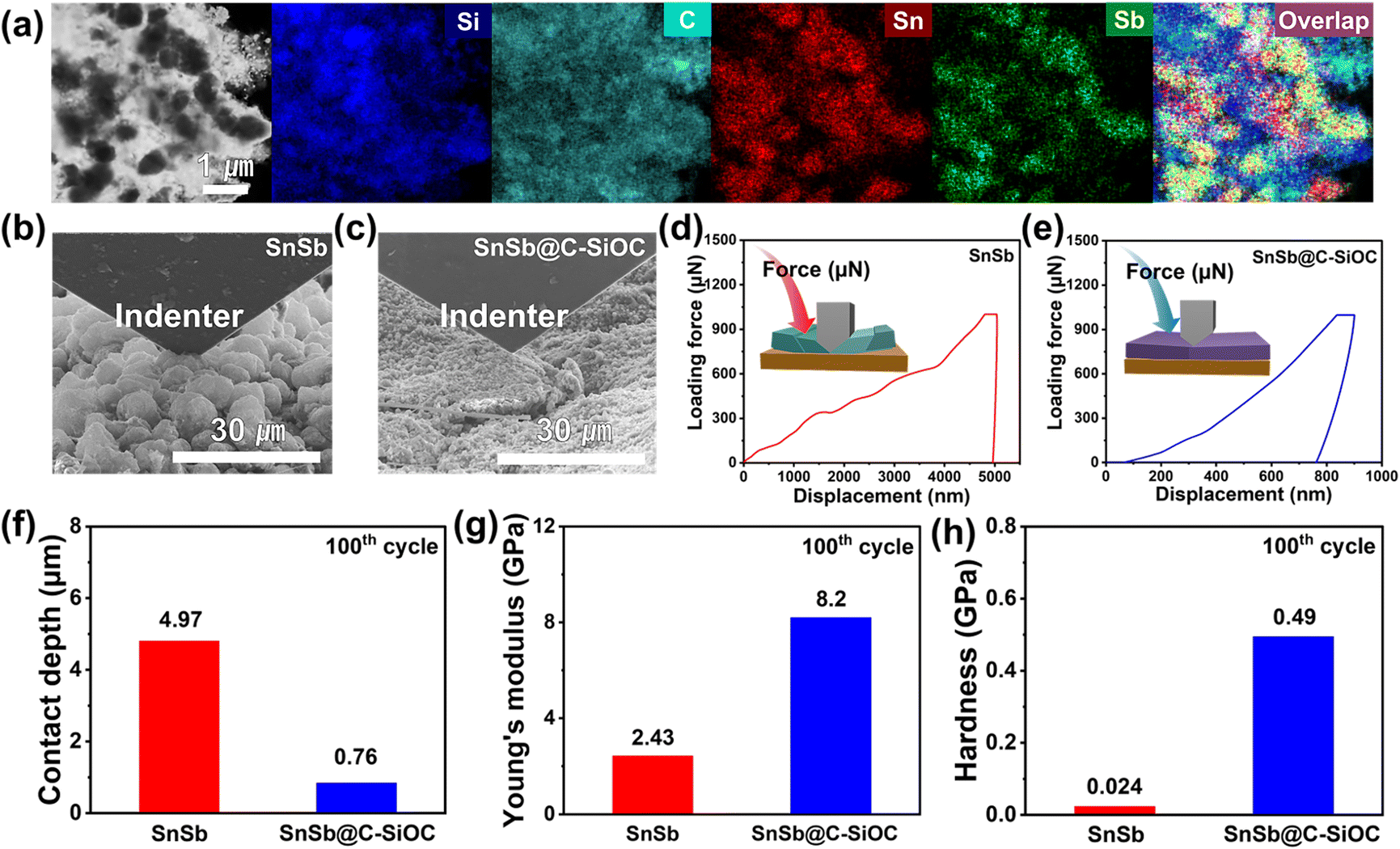 | ||
| Fig. 6 (a) TEM-EDS elemental mapping images of SnSb@C-SiOC after the 100th cycle. (b and c) SEM images from the nanoindentation analysis of SnSb-based electrodes. (d and e) Loading force–displacement curves, (f) contact depth, (g) Young's modulus (h) hardness of the pure SnSb and SnSb@C-SiOC electrodes after the 100th cycle. | ||
To determine the mechanical characteristics, the SnSb-based electrodes were pressed by the indenter tip (Fig. 6d, e and S13c, d, ESI†). The loading force (maximum value of 1000 μN) and displacement were recorded. After the 2nd cycle, the pure SnSb and SnSb@C-SiOC electrodes exhibited nearly the same contact depths (Fig. S13e, ESI†). However, after the 100th cycle, the contact depth into the electrodes was significantly higher in SnSb (4.97 μm) than in SnSb@C-SiOC (0.76 μm), indicating that the mechanical strength of the SnSb@C-SiOC electrode was well-maintained compared to that of the pure SnSb electrode (Fig. 6f). To confirm the correlation between the loading force and displacement, Young's modulus and hardness of the electrodes were calculated using eqn (3) and (4).46,47
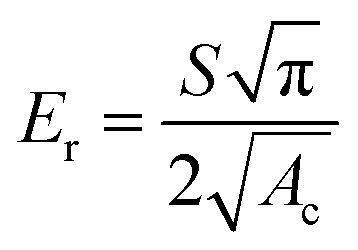 | (3) |
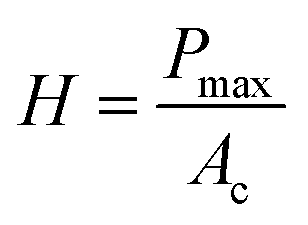 | (4) |
Furthermore, to investigate the effect of the C–SiOC shell on surface morphology after multiple cycles, the surfaces of SnSb and SnSb@C-SiOC electrodes after the 100th cycle were analyzed using SEM and atomic force microscopy (AFM) (Fig. S14, ESI†). The SEM and AFM analysis results revealed distinct differences in surface roughness, uniformity, and the presence of cracks and protrusions between the two SnSb-based electrodes. The pure SnSb electrode showed a significantly rough and non-uniform surface with significant height variations, indicating poor surface uniformity. Additionally, numerous protrusions and potential microcracks were observed, suggesting that the SEI layer may have suffered damage or undergone non-uniform formation upon several cycles, which can compromise long-term stability. In contrast, the SnSb@C-SiOC electrode exhibited a smoother and more homogeneous surface with fewer height variations and minimal protrusions, indicating a well-preserved surface morphology. This uniformity and the absence of extensive cracking or large protrusions suggest that the SnSb@C-SiOC electrode retains a more stable and intact SEI layer due to the presence of the C–SiOC shell, contributing to enhanced cycling stability.
To evaluate the practical application of the SnSb@C-SiOC anode, a full cell was assembled using Na3V2(PO4)3 (NVP) as the cathode (Fig. 7a) at an N/P ratio of 1.1:1. NVP was selected because of its high-voltage operation and superior cycling performance.48 The initial voltage profiles are shown in Fig. 7b. Prior to the full cell test, the SnSb@C-SiOC anode was pre-sodiated in the voltage range of 0.001–2.0 V at 0.1 A g−1. Furthermore, the NVP cathode was pre-cycled in the voltage range of 1.9–4.0 V at 0.1C (1C = 100 mA g−1), respectively. Considering the operating voltages of SnSb@C-SiOC and NVP, the full cell was operated within a voltage range of 1.4–3.7 V (vs. Na/Na+). In the GCD profiles, the SnSb@C-SiOC‖NVP full cell exhibited two distinct voltage-sloping regions, consistent with the approximately two-step electrochemical reaction of the SnSb@C-SiOC anode (Fig. 7c). The reversible sodiation/desodiation behavior was further confirmed by the dQ/dV plot of the full cell, which revealed two pairs of clear redox peaks at 3.3/3.4 and 3.1/3.4 V, respectively (Fig. 7d). The full cells were tested at current densities ranging from 0.1 to 5.0C. The full cell delivered relatively high reversible capacities of 110, 105.4, 102.7, 99.7, 93.7, and 70 mA h g−1 at 0.1, 0.2, 0.5, 1.0, 2.0, and 5.0C, respectively (Fig. 7e), with nearly 100% capacity recovery when the current density returned to 1.0C. Due to its good high-rate capability, the full cell retained a high reversible capacity of 75 mA h g−1 with a high CE of over 99%, even after long-term operation for 200 cycles at 1.0C (Fig. 7f). The reasonably good electrochemical performance of the full cell demonstrated that the SnSb@C-SiOC nanohybrid served as a promising anode material for advanced SIBs.
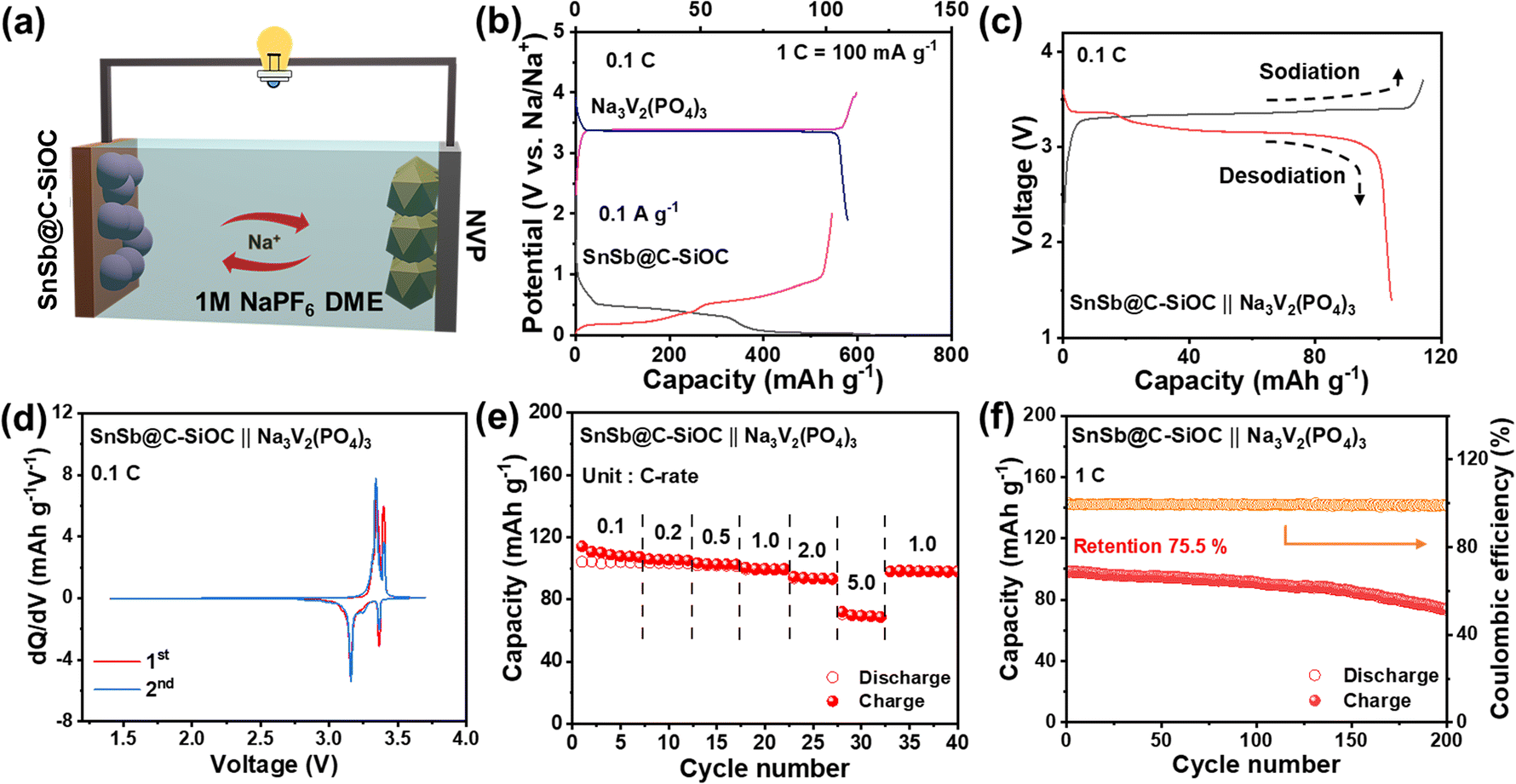 | ||
| Fig. 7 (a) Schematic of the full cell configuration comprising the SnSb@C-SiOC anode and Na3V2(PO4)3/C (NVP) cathode. (b) GCD profiles of the NVP and SnSb@C-SiOC electrodes. (c) GCD profiles of the full cell at 0.1C. (d) dQ/dV plots for the 1st and 2nd cycles of the full cell at 0.1C. (e) Rate performance of the full cell at various C-rates from 0.1 to 5.0C. (f) Cycling performance of the full cell at 1.0C for 200 cycles. | ||
3 Conclusions
In this study, an SnSb-based heterostructured anode was synthesized through two-step heat treatment using a SnSbOx@PDA precursor dispersed in silicone oil. The SnSb@C-SiOC nanohybrid exhibited a yolk–shell type structure with uniform encapsulation of SnSb nanoparticles within a mesoporous C–SiOC coating layer. With a high content of bimetallic SnSb (∼84%), the resulting nanohybrids exhibited a high reversible capacity of 445.6 mA h g−1 at 2 A g−1, significantly improved cycling stability, and excellent rate capability up to 3 A g−1 in the SIB. The internal void spaces in the yolk–shell structure effectively accommodated the massive volume variations of SnSb, leading to the enhanced structural stability of the SnSb@C-SiOC nanohybrid. Additionally, the mesoporous and highly conductive nature of the C–SiOC bi-layered shell not only provided abundant redox-active sites but also enabled efficient Na-ion transfer, thereby improving the reversible capacity and rate capability of the nanohybrid anode. Simultaneously, the chemically and electrochemically stable bi-layered shell prevented direct contact between the SnSb electrode and the electrolyte, creating a robust and durable SEI layer that restrained irreversible decomposition of the electrolyte and unfavorable metallic dissolution during cycling. Thus, the SnSb@C-SiOC anode demonstrated promising full-cell performance when combined with a Na3V2(PO4)3 cathode.This study highlights how surface modification using a heterostructure engineering approach can significantly enhance the performance of alloy-type anode materials, suggesting their potential for large-scale applications in SIBs. Further optimization for both reduced environmental impact and improved battery performance of such advanced anode materials could ensure their viability in green and sustainable energy applications.
4 Experimental
4.1 Materials synthesis
:divinylbenzene = 10:1, v/v) by ultrasonication and vigorous stirring. Subsequently, two-step pyrolysis was conducted at 500 °C for 4 h. Thereafter, the mixture was placed in a tube furnace under an inert Ar atmosphere at 900 °C for 1 h. Finally, the SnSb@C-SiOC nanohybrid was collected and ground into fine powder.
:5) atmosphere. After pyrolysis, the SnSb alloy was ground into fine powder.
4.2 Materials characterization
XRD was conducted using a MiniFlex (Rigaku, Japan) with a Cu Kα radiation source (λ = 1.5417 Å), within 2θ range of 10–80° at a scan rate of 4° min−1. Raman spectra were obtained using an inVia Raman microscope (Renishaw Inc., UK) with a 532 nm laser beam. TGA was performed using an SDT-Q600 (TA Corp., USA). FE-SEM was performed using Inspect-F (FEI, USA). DLS was performed using a NanoSAQLA (Otsuka, Japan). TEM equipped with EDS was conducted on Talos F200X (FEI, USA) operated at an accelerating voltage of 200 kV. XPS (Nexsa, Thermo Fisher Scientific, USA) was utilized to detect chemical energy states using monochromated Al Kα radiation (1486.6 eV) under pressure of 2.0 × 10−8 mbar. XPS depth profile was generated using Ar+ sputtering (0.5 nm s−1, 2 kV). Binding energies were calibrated based on C 1s peak at 284.6 eV. ICP-OES (iCAP 6000 Series, Thermo, USA) was performed to determine the elemental composition of the samples. Nanoindentation was conducted using an Hysitron PI-85 (Bruker, USA) equipped with FE-SEM (Nova Nano SEM, FEI, USA) under an Ar-filled atmosphere. AFM analysis was conducted using a Park NX-10 (Park Systems, South Korea) located in an Ar-filled glove box.4.3 Electrochemical measurements
A coin-type (CR2032) half-cell was fabricated to examine the electrochemical behavior of SnSb-based active materials. Active materials (80 wt%), Super P (10 wt%), and poly (acrylic acid) (10 wt%) binder were mixed with ethanol to prepare a homogeneous coating slurry. Then, the slurry was cast onto a Cu foil current collector and subsequently dried at 80 °C for 12 h. Typical areal mass loadings of the active materials were adjusted to ∼2.0–2.5 mg cm−2. The half-cell was assembled in an Ar-filled glovebox. Metallic Na and glass fiber membranes (GF/D) were used as the reference/counter electrode and separator, respectively. Organic electrolyte comprised 1 M NaPF6 dissolved in 1,2-dimethoxyethane (DME, 99.5%, Sigma-Aldrich). CV and GCD tests were conducted at 30 °C, with a potential window ranging from 0.001 to 2.0 V (vs. Na/Na+). EIS was performed using a VSP-300 Potentiostat (BioLogic, France) in the frequency range of 10–1 MHz at an AC amplitude of 5 mV. For full-cell tests, Na3V2(PO4)3 and SnSb@C-SiOC were used as the cathode and anode, respectively, with a negative-to-positive (N/P) electrode ratio of 1.1:1.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
J. M. Im and H. Lim contributed equally to this work. J. M. Im: methodology, formal analysis, data curation, writing – review & editing, writing – original draft. H. Lim: formal analysis, investigation, writing – original draft. H. Kim: data curation, investigation. Y. C. Kang: supervision. Y. Hwa: data curation, writing – original draft. S.-O. Kim: conceptualization, validation, supervision, writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Korea Institute of Science and Technology (KIST) Institutional Program (No. 2E33941 and 2V10180). This study was also supported by National Research Foundation of Korea (NRF) grants (No. RS-2024-00404414 and RS-2024-00427700) and the National Research Council of Science & Technology (NST) grant (No. 2710024139) funded by the Korean Government (MSIT).References
- K. Song, C. Liu, L. Mi, S. Chou, W. Chen and C. Shen, Small, 2021, 17, e1903194 CrossRef.
- S. Qiao, Q. Zhou, M. Ma, H. K. Liu, S. X. Dou and S. Chong, ACS Nano, 2023, 17, 11220–11252 CrossRef PubMed.
- D. Kim, H. Kim, H. Lim, K. J. Kim, H. G. Jung, D. Byun, C. Kim and W. Choi, Int. J. Energy Res., 2020, 44, 11473–11486 CrossRef.
- J. Liu, L. Yu, C. Wu, Y. Wen, K. Yin, F.-K. Chiang, R. Hu, J. Liu, L. Sun, L. Gu, J. Maier, Y. Yu and M. Zhu, Nano Lett., 2017, 17, 2034–2042 CrossRef PubMed.
- J. Yang, X. Guo, H. Gao, T. Wang, Z. Liu, Q. Yang, H. Yao, J. Li, C. Wang and G. Wang, Adv. Energy Mater., 2023, 13, 2300351 CrossRef.
- Y. N. Ko and Y. C. Kang, Chem. Commun., 2014, 50, 12322–12324 RSC.
- M. Zhu, Y. Jiang, X. Yang, X. Li, L. Wang and W. Lü, ACS Appl. Nano Mater., 2023, 6, 13503–13512 CrossRef.
- S. Sarkar, A. Chaupatnaik, S. D. Ramarao, U. Subbarao, P. Barpanda and S. C. Peter, J. Phys. Chem. C, 2020, 124, 15757–15768 CrossRef.
- A. Verdianto, H. Lim, J. Park and S.-O. Kim, J. Alloys Compd., 2023, 942, 168950 CrossRef.
- J. Song, P. Yan, L. Luo, X. Qi, X. Rong, J. Zheng, B. Xiao, S. Feng, C. Wang, Y.-S. Hu, Y. Lin, V. L. Sprenkle and X. Li, Nano Energy, 2017, 40, 504–511 CrossRef.
- H. Gu, L. Yang, Y. Zhang, C. Wang, X. Zhang, Z. Xie, J. Wei and Z. Zhou, Energy Storage Mater., 2019, 21, 203–209 CrossRef.
- X. Yang, Y. Zhu, D. Wu, M. Li, Y. He, L. Huang and M. Gu, Adv. Funct. Mater., 2022, 32, 2111391 CrossRef CAS.
- J. Qin, T. Wang, D. Liu, E. Liu, N. Zhao, C. Shi, F. He, L. Ma and C. He, Adv. Mater., 2018, 30, 1704670 CrossRef.
- Y. Zheng, T. Zhou, C. Zhang, J. Mao, H. Liu and Z. Guo, Angew Chem. Int. Ed. Engl., 2016, 55, 3408–3413 CrossRef CAS PubMed.
- H. Lim, S. Yu, W. Choi and S.-O. Kim, ACS Nano, 2021, 15, 7409–7420 CrossRef CAS PubMed.
- G. Zhang, S. Zeng, L. Duan, X. Zhang, L. Wang, X. Yang, X. Li and W. Lü, ChemElectroChem, 2020, 7, 4663–4671 CrossRef CAS.
- H. Lv, S. Qiu, G. Lu, Y. Fu, X. Li, C. Hu and J. Liu, Electrochim. Acta, 2015, 151, 214–221 CrossRef CAS.
- Y. Dong, M. Hu, Z. Zhang, J. A. Zapien, X. Wang, J.-M. Lee and W. Zhang, ACS Appl. Nano Mater., 2019, 2, 1457–1465 CrossRef CAS.
- Z. Wu, W. Lv, X. Cheng, J. Gao, Z. Qian, D. Tian, J. Li, W. He and C. Yang, Chemistry, 2019, 25, 2604–2609 CrossRef CAS.
- H. Lim, H. Kim, S.-O. Kim, K. J. Kim and W. Choi, Chem. Eng. J., 2021, 404, 126581 CrossRef CAS.
- H. Lim, S. Yu, W. Chang, K. Y. Chung, W. Choi and S.-O. Kim, Adv. Sci., 2024, 11, 2408450 CrossRef CAS.
- M. S. Tahir, M. Weinberger, P. Balasubramanian, T. Diemant, R. J. Behm, M. Lindén and M. Wohlfahrt-Mehrens, J. Mater. Chem. A, 2017, 5, 10190–10199 RSC.
- J. Dang, R. Zhu, S. Zhang, L. Yang, X. Chen, H. Wang and X. Liu, Small, 2022, 18, e2107869 CrossRef.
- W. Ma, K. Yin, H. Gao, J. Niu, Z. Peng and Z. Zhang, Nano Energy, 2018, 54, 349–359 CrossRef CAS.
- D. Cheng, L. Yang, J. Liu, R. Hu, J. Liu, K. Pei, M. Zhu and R. Che, J. Mater. Chem. A, 2019, 7, 15320–15332 RSC.
- D. Cheng, A. Wei, L. Ye, G. Xu, L. Tan, B. Lu and Y. Chen, ACS Sustainable Chem. Eng., 2022, 10, 12177–12187 CrossRef CAS.
- M. Kim and J. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 9036–9045 CrossRef CAS.
- Z. Yi, N. Lin, W. Zhang, W. Wang, Y. Zhu and Y. Qian, Nanoscale, 2018, 10, 13236–13241 RSC.
- A. Y. Mohamed, S. J. Lee, Y. Jang, J. S. Kim, C. S. Hwang and D.-Y. Cho, J. Phys.:Condens. Matter, 2020, 32, 065502 CrossRef CAS.
- X. Zhang, X. Hou and Y. Liu, J. Electrochem. Soc., 2020, 167, 155501 CrossRef CAS.
- L. Li, K. H. Seng, D. Li, Y. Xia, H. K. Liu and Z. Guo, Nano Res., 2014, 7, 1466–1476 CrossRef CAS.
- B. Feng, T. Long, C. Yang, K. Wang, Z. Wang and Y.-L. Ding, ACS Appl. Energy Mater., 2022, 5, 14107–14118 CrossRef CAS.
- T. Li, U. Gulzar, X. Bai, M. Lenocini, M. Prato, K. E. Aifantis, C. Capiglia and R. Proietti Zaccaria, ACS Appl. Energy Mater., 2019, 2, 860–866 CrossRef CAS.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597 RSC.
- Y. Li, Y. Meng, M. Xiao, X. Liu, F. Zhu and Y. Zhang, J. Mater. Sci.:Mater. Electron., 2019, 30, 12659–12668 CrossRef CAS.
- S. Qiu, L. Xiao, X. Ai, H. Yang and Y. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 345–353 CrossRef CAS.
- S. Qiu, X. Wu, L. Xiao, X. Ai, H. Yang and Y. Cao, ACS Appl. Mater. Interfaces, 2016, 8, 1337–1343 CrossRef CAS.
- J.-H. Choi, C.-W. Ha, H.-Y. Choi and S.-M. Lee, J. Ind. Eng. Chem., 2018, 60, 451–457 CrossRef CAS.
- L. Xiao, Y. Cao, J. Xiao, W. Wang, L. Kovarik, Z. Nie and J. Liu, Chem. Commun., 2012, 48, 3321–3323 RSC.
- H. Jia, M. Dirican, C. Chen, J. Zhu, P. Zhu, C. Yan, Y. Li, X. Dong, J. Guo and X. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 9696–9703 CrossRef CAS.
- Q. Yao, Y. Zhu, C. Zheng, N. Wang, D. Wang, F. Tian, Z. Bai, J. Yang, Y. Qian and S. Dou, Adv. Energy Mater., 2023, 13, 2202939 CrossRef CAS.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, ACS Cent. Sci., 2015, 1, 449–455 CrossRef CAS.
- X. Du, Y. Gao, Z. Hou, X. Guo, Y. Zhu and B. Zhang, ACS Appl. Energy Mater., 2022, 5, 2252–2259 CrossRef CAS.
- Y. Wang, F. Liu, G. Fan, X. Qiu, J. Liu, Z. Yan, K. Zhang, F. Cheng and J. Chen, J. Am. Chem. Soc., 2021, 143, 2829–2837 CrossRef CAS.
- Y. Tesfamhret, H. Liu, Z. Chai, E. Berg and R. Younesi, ChemElectroChem, 2021, 8, 1516–1523 CrossRef CAS.
- J. Menčík, D. Munz, E. Quandt, E. R. Weppelmann and M. V. Swain, J. Mater. Res., 1997, 12, 2475–2484 CrossRef.
- Y. Jeoun, K. Kim, S.-Y. Kim, S.-H. Lee, S.-H. Huh, S. H. Kim, X. Huang, Y.-E. Sung, H. D. Abruña and S.-H. Yu, ACS Energy Lett., 2022, 7, 2219–2227 CrossRef CAS.
- M. K. Sadan, H. Kim, C. Kim, S. H. Cha, K.-K. Cho, K.-W. Kim, J.-H. Ahn and H.-J. Ahn, J. Mater. Chem. A, 2020, 8, 9843–9849 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08119f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |