
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerovskite oxides as a new family of tunable CO2 sorbents†
Seyedamin
Razavi
,
Vahid
Rahmanian
 ,
Runxia
Cai
,
Mahe
Rukh
,
Saad A.
Khan
and
Fanxing
Li
*
,
Runxia
Cai
,
Mahe
Rukh
,
Saad A.
Khan
and
Fanxing
Li
*
Chemical and Biomolecular Engineering Department, College of Engineering, North Carolina State University, 911 Partners Way, Raleigh, NC, USA. E-mail: fli5@ncsu.edu
First published on 26th February 2025
Abstract
CO2 adsorption using solid sorbents represents an attractive option for carbon capture due to its potential for low energy consumption, ease of operation, and higher stability. This study introduces perovskite oxides as a new family of highly tunable solid sorbents, with remarkable structural and compositional flexibility to tailor their sorption thermodynamics and kinetics. Using SrxLa1−xFeO3 (x = 0, 0.2, 0.5, and 0.7) as a model system, we demonstrated that varying the A-site composition of the perovskite oxide leads to substantial change in the CO2 adsorption and desorption behavior, allowing a tunable CO2 release within a temperature range of 75 to 500+ °C. A strong correlation between the oxide surface area and sorption capacity was also established. Despite the low surface area inherent to perovskite oxides, we managed to enhance their surface area from 3–5 m2 g−1 (prepared by a salt assisted reactive grinding method) to ∼30 m2 g−1 using electrospun nanofibers. Sr0.2La0.8FeO3 prepared via electrospinning exhibited a relatively moderate desorption temperature (onset: 120 °C and peak: 240 °C) and a CO2 sorption capacity of 0.68 wt%. Analysis of the adsorption isotherms indicates that CO2 is chemisorbed on the Sr0.2La0.8FeO3 sorbent at low CO2 partial pressures (0–1 kPa). Physisorption becomes dominant at higher pressures. Transmission electron microscopy (TEM) characterization revealed that perovskite synthesis via the electrospinning method results in the formation of perovskite nanorods. X-ray photoelectron spectroscopy indicates significantly higher surface La concentration when compared to oxides prepared with conventional methods. It was further determined that a higher surface La concentration is highly desirable for reversible CO2 sorption. This study demonstrates tunable perovskite oxides as a new family of CO2 sorbent materials and the potential to further enhance their surface area towards practical applications in CO2 capture and utilization.
1 Introduction
The primary driving force behind global climate change is the accumulation of carbon dioxide (CO2) in the Earth's atmosphere. As per the Intergovernmental Panel on Climate Change (IPCC), if no significant intervention occurs, CO2 emissions are anticipated to surge from the current rate of 36 Gt per year to a range between 48 and 55 Gt per year by 2050.1 This increase in emissions is primarily attributed to a substantial increase in energy demand, which is projected to grow by 40% to 150% during the same period. Recovering from an unavoidable excess of atmospheric CO2 concentrations demands proactive removal of CO2.2 Achieving this necessitates the deployment of technologies capable of effectively capturing and permanently eliminating CO2 from the atmosphere.3The primary technologies investigated for CO2 capture include cryogenic distillation, membrane separation, liquid absorption, and solid adsorption.4 Cryogenic distillation imposes significant limitations on CO2 capture due to its high energy requirements and the need for an initial step to eliminate compounds such as H2O, SOx, and NOx.4 Membrane separation is efficient when dealing with high concentrations of CO2. However, its effectiveness and selectivity decline when CO2 is not a substantial component in the stream. Solvent absorption is a well-established technology for CO2 capture.4 However, it comes with a significant energy cost for solvent regeneration and is susceptible to corrosion issues and volatility concerns.5–7 In this context, adsorption using solid sorbents emerges as an appealing alternative which offers potential to reduce the energy expenditure and to mitigate corrosion concerns. Its efficiency, coupled with the simplicity of operation, makes it a promising option for carbon capture from diluted CO2 streams.4
Capturing CO2 from ambient air, also known as Direct Air Capture (DAC), can complement point-source capture by addressing emissions from dispersed sources that are not reachable through point-source capture methods.8–10 Additionally, DAC can compensate for any residual emissions that escape during point-source capture.8–10 The majority of air capture methods employ solid sorbents. Each of the sorbents under investigation utilizes one of two potential adsorption mechanisms: physical or chemical adsorption. Physisorption involves the attraction of target molecules onto the surface of pore walls within a sorbent with high surface area, facilitated by van der Waals forces.11 The heat of adsorption in physisorption is relatively low, only slightly exceeding the heat of sublimation of the adsorbate. In chemisorption, the target gas undergoes a covalent chemical reaction to bind to specific sites on the sorbent, resulting in a significantly higher heat of adsorption that is approximately equivalent to the heat of the reaction.11 Notable adsorbents in physical adsorption include zeolites, activated carbon, alumina, and metal–organic frameworks.12 Their performance can be notably reduced by high humidity and elevated temperatures, especially in the cases of activated carbon and zeolites.4,13 MOFs offer the advantage of being highly flexible when it comes to their shape, pore structure, pore size, and surface properties.11 Like most sorbents, their sorption capacity and selectivity are influenced by temperature and humidity. In general, MOFs are better suited for storage purposes rather than for the capture or separation of CO2.11,14 The heat of adsorption of carbon dioxide on these types of sorbents ranges from −25 to −40 kJ per mole, close to the heat of sublimation (∼25 kJ per mole).11,12 This low heat of adsorption reduces the amount of energy needed to desorb a given quantity of carbon dioxide. Conversely, solid sorbents that capture carbon dioxide via a chemical process, such as bonding with an amine grafted or coated onto the sorbent's surface, exhibit significantly higher heats of adsorption. Depending on the specific amine employed, the heat of adsorption typically ranges between −60 and −100 kJ mol−1.11
This article reports perovskite oxides as a new family of solid sorbents for CO2 capture. Perovskites are appealing due to their remarkable stability and high compositional and structural flexibility, allowing for tunability in terms of their surface and bulk properties.15–19 Despite their flexibility and applications in thermal and electro catalysis, ceramic membranes, sensing, etc.20–22 perovskite oxides have not been explored as CO2 sorbents at low temperatures. Meanwhile, a number of previous studies have shown that alkali earth cations in perovskite oxides tend to enrich on the perovskite surface.23–28 This can lead to strong interaction with CO2.23–28 Although formation of bulk carbonate phases is not desirable, surface interactions between CO2 and surface alkali earth cations, whose coordination environment may be tunable by varying the perovskite composition, can offer interesting opportunities. In our current investigation, we utilized SrxLa1−xFeO3 (where x = 0, 0.2, 0.5, and 0.7) as a model system to illustrate how altering the A-site composition of perovskite oxide can significantly affect the behavior of CO2 adsorption and desorption. This variation allowed us to achieve adjustable CO2 release within a temperature range spanning from 180 to over 500 °C. We established a strong correlation between the surface area of the oxide and its sorption capacity. By employing an electrospinning method to create nanofibers known for their high surface areas,29,30 we successfully increased the surface area from 3 to 5 m2 g−1 (achieved through a salt-assisted reactive grinding method) to approximately 30 m2 g−1, resulting in a significant increase in the CO2 capture capacity (up to 0.68 wt%). This investigation highlights the potential of perovskite oxides as a customizable category of CO2 sorbent materials, with the possibility of further enhancing their surface area for practical applications in CO2 capture and combined CO2 capture and utilization.
2 Experimental section
2.1 Synthesis of SrxLa1−xFeO3 sorbents
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. To prevent the powders from adhering to the stainless-steel sample jar, we added 9 mL of ethanol (>99 vol% purity) to the mixture. The mixture was then ball-milled at 1200 rpm for 3 hours. Afterward, we dried the resulting wet mixture in the stainless-steel sample jar at 130 °C for 30 minutes to remove the ethanol. Following this step, the powder mixture was separated from the ZrO2 beads and calcined at 1000 °C in a muffle furnace for 10 hours to obtain the perovskite structure. Both the heating and cooling rates were set at 3 °C min−1. Finally, the perovskite samples were sieved to particle sizes below 250 μm for TGA testing. To enhance the surface area of perovskite materials, we employed the salt-grinding method, aiming to create a porous structure. The perovskite was blended with K2CO3 and 3 mm ZrO2 beads inside a stainless-steel sample jar, maintaining a specific mass ratio. Subsequently, the jar underwent ball milling at 1200 rpm for 24 hours, utilizing a high-energy ball mill to augment porosity. The resultant powder mixtures were subjected to a water rinse to eliminate K2CO3. Subsequently, the remaining moist particles were dried in an oven at 130 °C to yield the high-porosity perovskite materials.
:5:5 mass ratio. Additionally, 15.5 wt% PVP (polyvinylpyrrolidone) was added to the solution. The electrospinning process was conducted with a distance of 15 cm between the needle tip of the syringe and a grounded aluminum foil collector. An applied voltage of 20 kV was used, which was generated by a custom-made D.C. power supply. To control the injection rate, a syringe pump (New Era Pump System Inc., Model no. NE-300) was employed, maintaining a rate of 0.5 mL min−1. Following the electrospinning step, the resulting fibers were dried at 80 °C in an oven overnight and then collected. To obtain the Sr0.2La0.8FeO3 perovskite oxide, the collected fibers were subjected to calcination at 650 °C for 3 hours, employing a heating rate of 3 °C min−1 in air. It should be noted that while our approach here entailed a lab scale setup, industrial production lines for electrospinning have been designed and implemented industrially, enabling the large-scale manufacturing of electrospun nanofibers for applications such as air and water filtration, biomedical devices, and energy storage systems. Many of these approaches use multi-needle or nozzle-less systems, incorporating real-time monitoring for quality assurance and developing solvent-free or environmentally friendly methods to reduce costs and minimize environmental impact.32,33
:1. To prevent the powders from adhering to the stainless-steel sample jar, we added 9 mL of ethanol (>99 vol% purity) to the mixture. The mixture was then ball-milled at 1200 rpm for 3 hours. Afterward, we dried the resulting wet mixture in the stainless-steel sample jar at 130 °C for 30 minutes to remove the ethanol. Following this step, the powder mixture was separated from the ZrO2 beads and calcined at 1000 °C in a muffle furnace for 10 hours to obtain the perovskite structure. Both the heating and cooling rates were set at 3 °C min−1. Finally, the perovskite samples were sieved to particle sizes below 250 μm for TGA testing. To enhance the surface area of perovskite materials, we employed the salt-grinding method, aiming to create a porous structure. The perovskite was blended with K2CO3 and 3 mm ZrO2 beads inside a stainless-steel sample jar, maintaining a specific mass ratio. Subsequently, the jar underwent ball milling at 1200 rpm for 24 hours, utilizing a high-energy ball mill to augment porosity. The resultant powder mixtures were subjected to a water rinse to eliminate K2CO3. Subsequently, the remaining moist particles were dried in an oven at 130 °C to yield the high-porosity perovskite materials.
:5:5 mass ratio. Additionally, 15.5 wt% PVP (polyvinylpyrrolidone) was added to the solution. The electrospinning process was conducted with a distance of 15 cm between the needle tip of the syringe and a grounded aluminum foil collector. An applied voltage of 20 kV was used, which was generated by a custom-made D.C. power supply. To control the injection rate, a syringe pump (New Era Pump System Inc., Model no. NE-300) was employed, maintaining a rate of 0.5 mL min−1. Following the electrospinning step, the resulting fibers were dried at 80 °C in an oven overnight and then collected. To obtain the Sr0.2La0.8FeO3 perovskite oxide, the collected fibers were subjected to calcination at 650 °C for 3 hours, employing a heating rate of 3 °C min−1 in air. It should be noted that while our approach here entailed a lab scale setup, industrial production lines for electrospinning have been designed and implemented industrially, enabling the large-scale manufacturing of electrospun nanofibers for applications such as air and water filtration, biomedical devices, and energy storage systems. Many of these approaches use multi-needle or nozzle-less systems, incorporating real-time monitoring for quality assurance and developing solvent-free or environmentally friendly methods to reduce costs and minimize environmental impact.32,33
2.2 Sorbent evaluation
The CO2 adsorption capacity of SrxLa1−xFeO3 was investigated using a TA Instruments SDT 650 thermogravimetric analyzer (TGA) at a temperature of 35 °C. Samples weighing between 30 and 40 mg were loaded into an Al2O3 crucible with a 6.5 mm inner diameter and subsequently placed inside the TGA apparatus. The samples were then subjected to heating at a rate of 20 °C per minute until reaching 550 °C under an Ar atmosphere with a purity of 99.999% and a flow rate of 200 mL min−1. This heating step was maintained for 60 minutes to eliminate any adsorbed species. Following the cooling of the samples to the desired analysis temperature, a 60-minute equilibration period was allowed before switching the gas flow to a mixture comprising 10% CO2 and 90% Ar at a flow rate of 200 mL min−1. This CO2 exposure was conducted for a duration of 30 minutes. Subsequently, to initiate the desorption step and release CO2 species from the surface, the temperature was increased, and pure argon was employed. Cycle adsorption and desorption stability experiments were conducted under similar conditions.2.3 Sorbent characterization
The surface morphology of the catalyst was analyzed using a Thermo Fisher Scientific Verios 460L Field Emission Scanning Electron Microscope (SEM) operating at 2 kV. Samples were mounted onto conductive carbon tape, and secondary electron images were captured without the application of any coating.
In order to collect in situ IR data, FTIR spectroscopy was employed using a Thermo Fisher Nicolet iS50 FTIR, which was equipped with a DiffusIR sample chamber from Pike Technologies. The process began by loading the sample into an in situ cell and purging it with Ar gas at 600 °C for a duration of 30 minutes. After this step, the sample was cooled to a temperature of 25 °C. To obtain the background spectrum, the instrument was used to collect the spectrum in an Ar stream at 25 °C, which was then automatically subtracted from the sample spectrum. CO2 reaction gas was introduced into the reaction cell at a flow rate of 40 mL min−1, and spectra were collected repeatedly until the spectrum curve stabilized.
3 Results and discussion
3.1 Sorbent phase purity and the effect of preparation methods on sorbent surface areas
To determine the effect of A-site compositions on perovskites' CO2 sorption properties, a simple solid state synthesis method was adopted first to prepare the perovskite samples for screening purpose. This is followed with X-ray diffraction analysis (XRD) to confirm their phase purity. As shown in Fig. 1a, the XRD patterns for all the synthesized samples exhibited well-defined diffraction peaks, signifying their phase purity.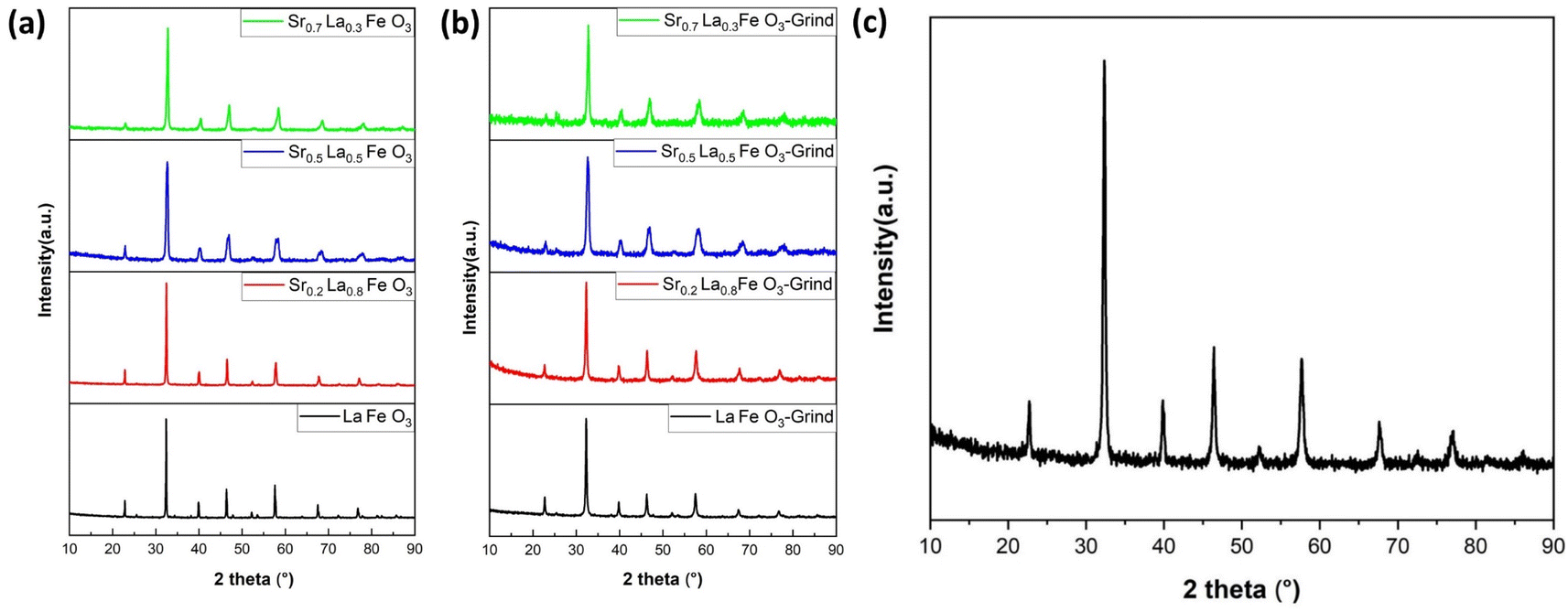 | ||
| Fig. 1 XRD results of SrxLa1−xFeO3 (x = 0, 0.2, 0.5, 0.7) perovskite oxide. (a) Before grinding with salt and (b) after grinding with salt. (c) XRD result of Sr0.2La0.8FeO3 (electrospun). | ||
One of the key challenges associated with the use of perovskites as adsorbents for CO2 capture is their inherently low surface area, which consequently results in a limited adsorption capacity. To address this limitation, we adopted two strategies, i.e. reactive grinding and creating nanofibrous structure via electrospinning, to increase their surface area. Using potassium as an alkali additive in reactive grinding increases surface area and adsorption capacity. Electrospinning with surfactants and templates, followed by calcination, creates perovskite oxides with higher surface areas and potentially controllable nanostructures.
Table 1 summarizes the BET surface areas of SrxLa1−xFeO3 (x = 0, 0.2, 0.5, 0.7) perovskite oxides before and after grinding with salt. As can be seen, before grinding, the perovskite's surface area averaged 3 to 5 m2 g−1. In comparison, the surface area increased nearly five-fold on average after reactive grinding. To further enhance the surface area of the perovskite, we employed the electrospinning technique. The BET results demonstrated the efficacy of this approach, with the surface area of the as-prepared Sr0.2La0.8FeO3 increasing to nearly 30 m2 g−1. This finding underscores the efficacy of electrospinning as a synthesis approach for enhancing the surface area. As illustrated in Fig. 1b and c, following the reactive grinding and electrospinning process, the perovskite materials have maintained their phase purity.
| Sample | S BET (m2 g−1) | V p (cm3 g−1) | S p (Å) |
|---|---|---|---|
| LaFeO3 | 3.43 | 0.0091 | 128.3 |
| Sr0.2La0.8FeO3 | 2.38 | 0.0076 | 143 |
| Sr0.5La0.5FeO3 | 3.55 | 0.0079 | 101.3 |
| Sr0.7La0.3FeO3 | 2.93 | 0.0063 | 108.8 |
| LaFe-grind | 15.76 | 0.075 | 164.5 |
| Sr0.2La0.8FeO3-grind | 15.56 | 0.098 | 213 |
| Sr0.5La0.5FeO3-grind | 23.36 | 0.132 | 206 |
| Sr0.7La0.3FeO3-grind | 14.18 | 0.079 | 201 |
| Sr0.2La0.8FeO3-electrospun | 29.55 | 0.119 | 143.5 |
3.2 Tunability of sorbents' surface properties for CO2 adsorption
The Sr percentage in the perovskite A-site is fully adjustable in SrxLa1−xFeO3 perovskite oxides. Given the high basicity of Sr2+, adjusting this ratio significantly influences the surface characteristics and sorption properties of the sorbents. Our research demonstrates that increasing the amount of strontium in the A-site makes the perovskite surface progressively more basic, facilitating the formation of more strontium carbonate and raising the desorption temperature, as shown in Fig. 2a. The TGA results indicate that the onset temperature for CO2 desorption, determined based on the second order derivative of the weight as a function of temperature, increased with increasing strontium content from 75 to 140 °C. Meanwhile, the peak desorption temperature also varied significantly with the strontium content: the Sr0.2La0.8FeO3 sample desorbs at around 120 °C (180 °C after grinding, likely due to residual potassium), while the Sr0.7La0.3FeO3 sample desorbs at over 500 °C (Fig. 2b). Thus, increasing strontium in the A-site primarily elevates the desorption temperature, with only a modest impact on adsorption capacity. Since a lower desorption temperature leads to lower energy consumption, we selected the sample with the lowest strontium content and then focused on enhancing the perovskite's surface area to improve carbon dioxide adsorption.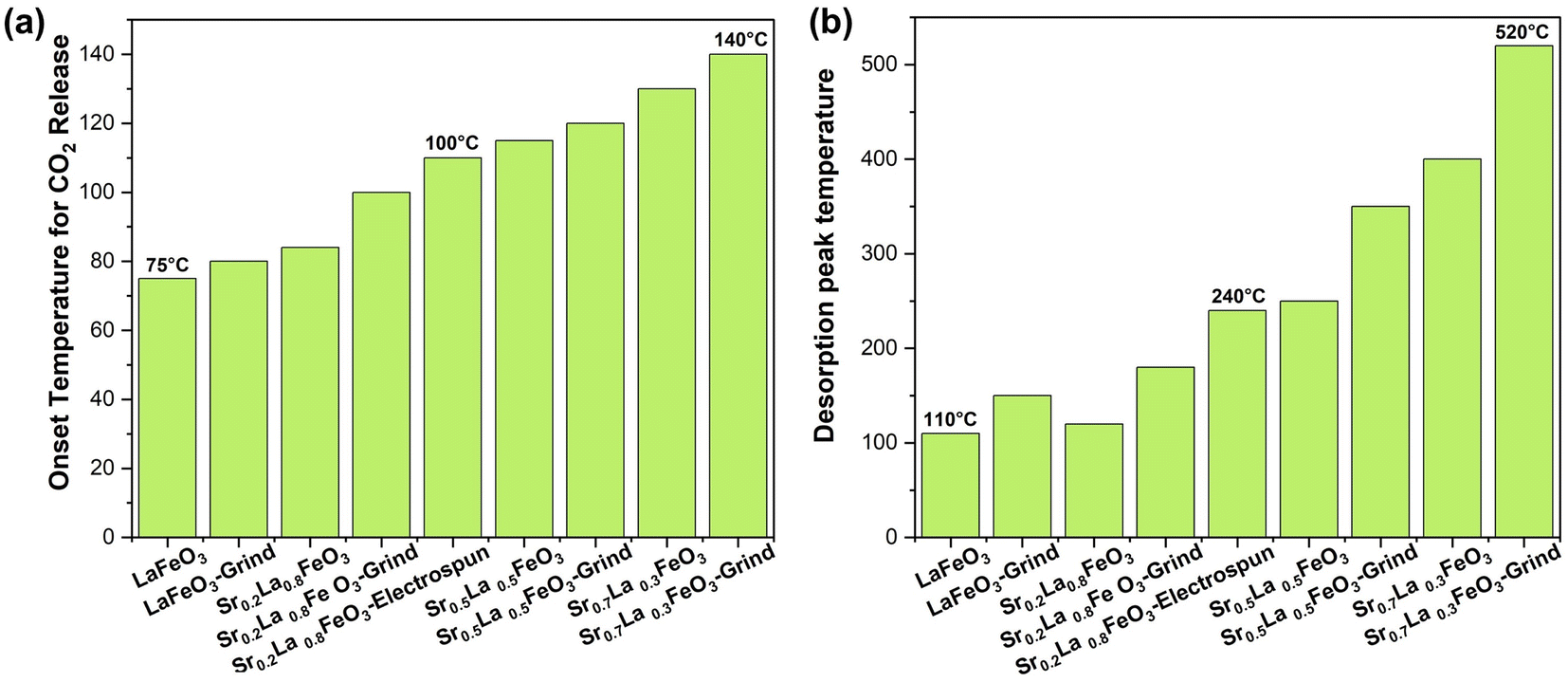 | ||
| Fig. 2 TGA results of SrxLa1−xFeO3 perovskite oxide. (a) Onset temperature for CO2 release of SrxLa1−xFeO3 perovskite oxide (x = 0, 0.2, 0.5, 0.7). (b) Desorption peak temperature of SrxLa1−xFeO3 perovskite oxide (x = 0, 0.2, 0.5, 0.7). | ||
3.3 Effect of the synthesis method on sorption capacity
Although sorbents prepared via SSR showed excellent phase purity and tunable sorption properties, their sorption capacities are rather limited (∼0.05 wt%). This is largely due to their low surface areas which are intrinsic to perovskite oxides. As shown in Table 1. We chose the Sr0.2La0.8FeO3 sample and increased its surface area by grinding it with potassium carbonate. Before grinding, the perovskite demonstrates an adsorption capacity of ∼0.05 wt%, as revealed by TGA analysis. Subsequent to the grinding process, the adsorption capacity more than tripled to ∼0.18 wt%. Fig. 3a shows the cycling stability of the sorbent. As can be seen, the adsorption capacity remained constant throughout the test, confirming its durability. | ||
| Fig. 3 (a) Cyclic CO2 uptake of Sr0.2La0.8FeO3 (reactive grinding) at 35 °C using CO2/Ar (10/90) via a gravimetric method. (b) In situ XRD analysis of Sr0.2La0.8FeO3 (electrospun) at 650 °C over varying exposure times. (c) Cyclic CO2 uptake of Sr0.2La0.8FeO3 (electrospun) at 35 °C using 0.4% CO2via a gravimetric method. (d) Correlation between surface area and adsorption capacity for Sr0.2La0.8FeO3 (grinding with salt) and Sr0.2La0.8FeO3 (electrospun). | ||
To further enhance the surface area of the perovskite, we employed the electrospinning technique. In this method, our objective is to determine the optimal calcination temperature that maximizes both phase purity and surface area. We conducted in situ XRD across a temperature range spanning from 400 to 700 °C. At 700 °C, we observed a surface area of approximately 17 m2 g−1, accompanied by the presence of a pure phase. However, at 550 °C, while we achieved a surface area of around 35 m2 g−1, we did not attain phase purity. It was therefore determined that the most suitable calcination temperature is 650 °C, as it allows us to achieve both phase purity and a high surface area (Fig. 3b). We conducted a cyclic experiment using TGA on Sr0.2La0.8FeO3 synthesized through the electrospinning method. This material exhibited excellent stability throughout the repeated cycles of adsorption and desorption (Fig. 3c and S1a†) at a low concentration of carbon dioxide (0.4 vol% CO2 balance Ar). As shown in Fig. 3c, the sample demonstrates notable stability, with its adsorption capacity exhibiting consistency across 55 cycles. This observation underscores the effectiveness of the Sr0.2La0.8FeO3 sorbent (synthesized via electrospinning).
As can be seen from Fig. 3d, sorption capacity is directly related to the sorbent surface area. In fact, sorbent capacities are proportional to their surface areas for the Sr0.2La0.8FeO3 samples prepared by SSR and reactive grinding. However, the electrospun sample does not follow this linear correlation: its sorption capacity (∼0.68 wt%) is more than three times greater than that achieved through the reactive grinding method, although its surface area only doubled. This variance in adsorption capacity is therefore likely attributable to differences in both surface area and the nature of the surface adsorption sites. The difference in surface properties is further discussed in the next section.
3.4 Effect of the synthesis method on sorbents' surface and sorption properties
The SEM image in Fig. 4a depicts the electrospun nanofibrous mat before the calcination process of Sr0.2La0.8FeO3, confirming that the electrospinning conditions indeed produce fibers that are devoid of aggregates, demonstrating a homogeneous structure.34 However, post-calcination, the fibrous mat is broken up, revealing a rod-like or short nanofiber shape, as evidenced in Fig. 4b. This is akin to shearing nanofiber dispersions to break them into smaller fibers that still provide a high surface area.35,36 This transformation occurs during the calcination process, wherein the decomposition of polyvinylpyrrolidone (PVP) takes place and the metal precursors stabilize to form a distinctive nanorod structure (Fig. 4c).37 To further characterize the material, an elemental analysis was performed using energy-dispersive X-ray spectroscopy (EDS), resulting in the distribution of atoms represented in the color map depicted in Fig. 4d. The EDS element images confirm the presence of all elements within the Sr0.2La0.8FeO3 composition, illustrating their well-dispersed distribution throughout the nanorod structure.37 The high aspect ratios of the nanorod structure result in a greater surface area compared to cluster materials obtained through reactive grinding, as shown in the TEM images of ground materials in Fig. S2.† In addition to offering more active sites for CO2 adsorption, the change in morphology and hence surface termination/elemental compositions can potentially alter the surface adsorption chemistry, as elaborated next.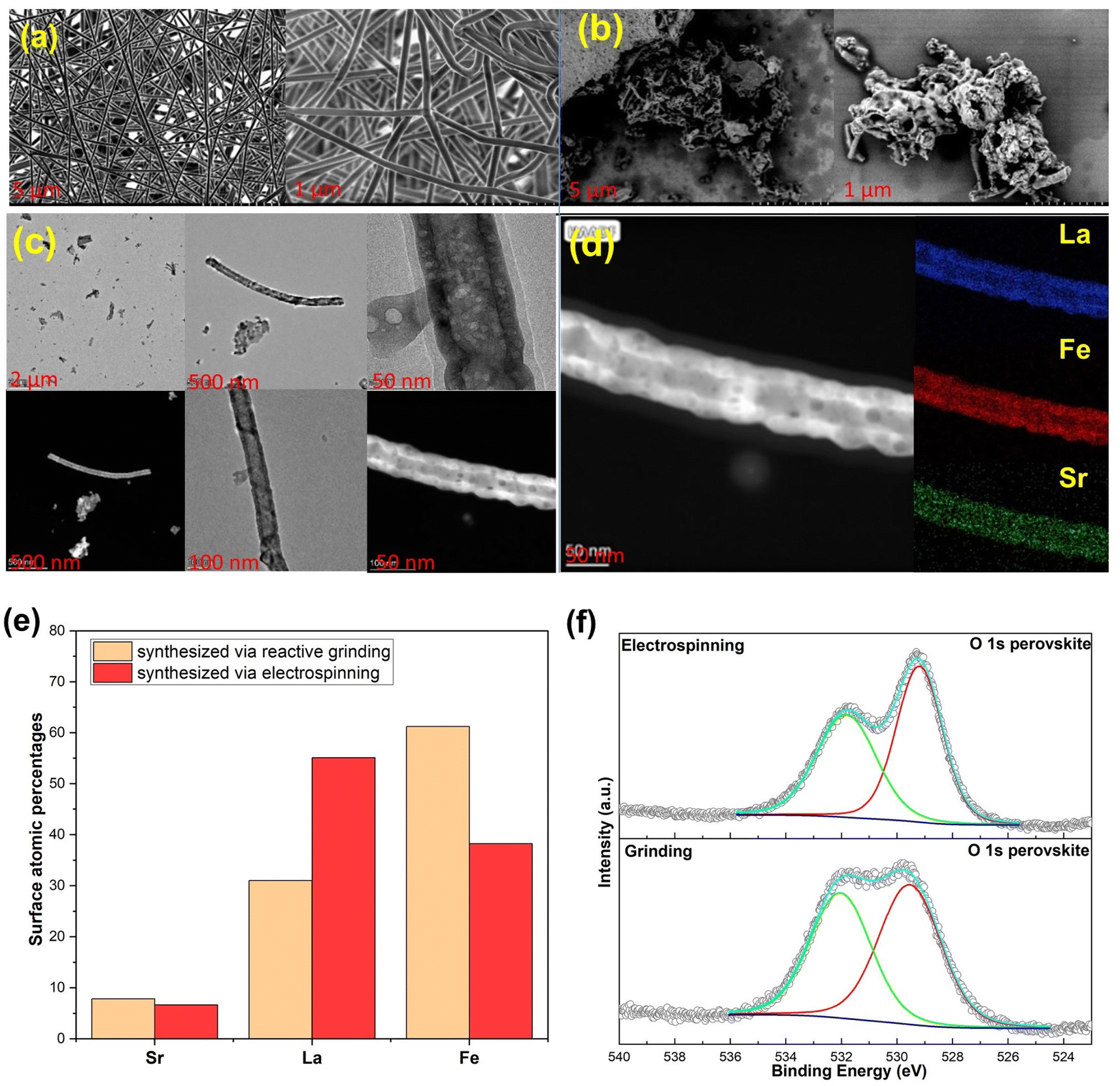 | ||
| Fig. 4 (a) SEM image of Sr0.2La0.8FeO3 prepared by electrospinning before calcination; (b) post-calcination; (c) TEM images of the electrospun sample; (d) corresponding elemental mapping by EDS; (e) comparison of the XPS derived near surface atomic surface percentages between samples prepared via electrospinning and grinding methods; (f) comparisons of the O 1s spectra of the electrospun and reactive ground samples. | ||
XPS analysis was conducted to assess the near surface elemental distribution of the samples prepared via electrospinning and reactive grinding methods. According to elemental composition analysis via XPS (Fig. 4e), the atomic percentages (carbon and oxygen free basis) are as follows: for Sr0.2La0.8FeO3 (synthesized via reactive grinding): the composition is 7.84% for Sr, 31.03% for La, and 61.24% for Fe. For Sr0.2La0.8FeO3 (synthesized via electrospinning), the composition is 6.65% for Sr, 55.09% for La, and 38.25% for Fe. These results suggest distinct elemental distributions between the two synthesis methods. Notably, electrospinning results in higher surface concentrations of A site cations, i.e. Sr and La, whereas the grinding method shows a higher percentage of Fe. Another interesting aspect is that the electrospun sample exhibited a significantly higher near surface La/Sr ratio (∼8.3) than the reactive ground sample (∼4). In fact, the near surface Sr concentration is higher in the reactive ground sample (7.84%) than that in the electrospun sample (6.65%). This points to (a) the CO2 sorption capacity may directly correlate with the surface concentration of A-site cations; (b) La is likely to be responsible for a notable fraction of the observed CO2 sorption. The latter is somewhat surprising given that Sr was originally envisioned as the active site for CO2 sorption.
The O 1s spectra for both electrospinning and grinding exhibit two distinct peaks (Fig. 4f). The peak at the lower binding energy can be attributed to lattice oxygen species (O2−),38–41 whereas the high binding energy peak is attributable to oxygen associated with carbonates42 or hydroxyls.43 As can be seen, the electrospun sample shows a weaker carbonate peak (high B.E. to low B.E. peak area ratio = 2/3) compared to the ground sample (peak area ratio = 1/1). Further analysis of the Sr 3d region confirms the presence of strontium carbonate (SrCO3) on both samples (Fig. S3 and S4†). Regarding the Sr 3d5/2 region, the broad peak suggests the presence of Sr(II) in various chemical environments. Typically, the dominant peak at 134.46 eV for the electrospinning method and 134.23 eV for the reactive grinding method are attributed to SrCO3. Additionally, the peak at around 132.37 eV for the reactive grinding method and 132.71 eV for the electrospinning method confirm the presence of strontium oxide (SrO) within the perovskite lattice.44 On the other hand, in the La 3d region, the binding energies observed at 834.19 eV (La 3d5/2) and 838.1 eV (La 3d3/2) for the reactive grinding method and 833.92 eV (La 3d5/2) and 837.78 eV (La 3d3/2) for the electrospinning method align with the known values for La(III) in similar perovskite structures. These peaks likely correspond to La(III) oxide or hydroxide.44 These results indicate that the high basicity of the Sr cation may lead to the formation of stable carbonates that are difficult to regeneration/decompose. In comparison, La cations would be largely available for reversible CO2 sorption/desorption.
To further clarify the role of surface A-site cations, the adsorption enthalpies of the two sorbents were also measured. This involved calculating the isosteric heat of CO2 adsorption through the utilization of the Clausius–Clapeyron equivalent adsorption heat equation.45
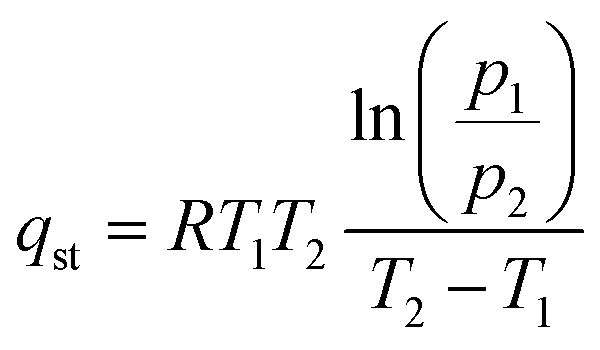 | (1) |
The isosteric heat of adsorption (qst) is calculated using the formula, where R represents the universal gas constant (8.314 J mol−1 K−1), T (T1 = 298 K; T2 = 273 K) denotes the adsorption temperature, and P is the adsorption pressure. The qst values for the prepared adsorbents are determined based on the CO2 adsorption data obtained at 0 and 25 °C. CO2 adsorption was carried out from 0 to 120 kPa, as shown in Fig. 5a and b. We note that in the case of adsorption isotherm measurement, the surface area normalized adsorption capacities for the electrospun and ground samples are similar. In contrast, the TGA data indicated that the electrospun sample exhibited significantly higher surface normalized CO2 sorption capacity (nearly 100% higher than that of the ground sample). This is due to differences in experimental conditions: the TGA measurement was performed to determine the reversible sorption capacity under temperature swings and at a relatively low CO2 partial pressure (0.1 atm). In contrast, the adsorption isotherm was obtained at low temperatures for up to 1.2 atm of CO2. Since the samples were pretreated at a high temperature to decompose the stable carbonates prior to the isothermal measurement, it is quite possible that some of the stable carbonates that formed during the isothermal measurement would not contribute to the reversible sorption capacity. Additional TGA tests further confirmed this hypothesis as the reactive ground sample showed a significantly higher amount of stable carbonates that act as a spectator phase and do not contribute to the usable CO2 capacity under the standard adsorption/desorption conditions (Fig. S6†).
 | ||
| Fig. 5 (a and b) CO2 adsorption isotherms of Sr0.2La0.8FeO3 at 0 and 25 °C. (c) CO2 isosteric heats of adsorption. (d) Comparison of isosteric heat of adsorption vs. adsorption capacity per surface area for electrospun and ground samples. (e) In situ IR spectra of CO2 adsorbed on Sr0.2La0.8FeO3 (electrospinning) monitored as a function of temperature. (f) In situ IR spectra of CO2 adsorbed on electrospun and ground Sr0.2La0.8FeO3 at 25 °C. | ||
For electrospun Sr0.2La0.8FeO3 at adsorption capacities below 0.0375 mmol g−1, it is difficult to accurately determine the heat of adsorption due to instrumentation limitations in measuring CO2 uptake at low pressures (Fig. S7a and b†). We note that this low-pressure limitation is not an issue for the ground sample since it requires significantly higher CO2 partial pressure to achieve the sample adsorption capacity. That said, based on two sets of measurements for the electrospun sample in the low-pressure range (Fig. S7c†), the heat of adsorption for the electrospun sample was approximately 85 kJ mol−1 at the lowest measurable pressure (corresponding to ∼0.028 mmol g−1 CO2 uptake). Between 0.028 and 0.035 mmol g−1, the heat capacity steadily dropped along with an increase in CO2 uptake, and the average heat of adsorption within this range is ∼70 kJ mol−1, corresponding to chemisorption. This indicates a strong adsorption affinity, with both data sets clearly supporting a chemisorption process characterized by adsorption enthalpies exceeding 60 kJ mol−1. Conversely, for adsorption capacities beyond this threshold, the adsorption enthalpies quickly decrease to below 40 kJ mol−1, indicating physisorption. Overall, the qst value shows a noticeable decline as adsorption progresses, eventually stabilizing nearly 10 kJ mol−1 for the electrospun Sr0.2La0.8FeO3. This phenomenon occurs likely because CO2 would preferentially adsorb on stronger adsorption sites, e.g. undercoordinated Sr–O sites. For Sr0.2La0.8FeO3 synthesized via reactive grinding, the majority of its adsorption capacity corresponds to chemisorption, with physisorption accounting for only a small fraction of its overall adsorption. The significantly different enthalpy of adsorption profiles between the two samples can potentially be explained by the difference in near surface cation compositions. The XPS results indicate that the electrospun sample has a higher concentration of La near the surface, which acts as the adsorption site and exhibits lower adsorption energy than Sr. On the other hand, in the ground sample, the high Sr concentration leads to strong adsorption energy. However, a notable fraction of the undercoordinated Sr–O sites would contribute to the formation of stable carbonates that do not contribute to usable CO2 sorption in a practical thermal swing adsorption process, as indicated from the TGA measurements.
To better understand the difference in the adsorption process between the two samples, we calculated the potential site densities on the 111 and 001 surfaces of the electrospun and ground samples and compared these with the experimentally measured adsorption capacity. In our calculations, we assumed that the entire surface could adsorb CO2 molecules, consistent with monolayer adsorption, using a cubic structure with an approximate distance of 6.7 Å between A-site cations. The calculated adsorption capacities per surface area, based on these assumptions, are approximately 0.0041 mmol CO2 per m2 for the 001 surface and 0.00429 mmol CO2 per m2 for the 111 surface. The experimental adsorption capacities per surface area of Sr0.2La0.8FeO3 synthesized via electrospinning and grinding are around 0.0053 mmol CO2 per m2 and 0.0051 mmol CO2 per m2, respectively. Based on Fig. 5d, comparing the experimental and calculated adsorption capacities per surface area indicates that both samples exhibit monolayer adsorption.
To unveil the disparity in the interaction between CO2 molecules and adsorption sites, FTIR was conducted for both samples. Fig. 5e shows the in situ FTIR spectra of Sr0.2La0.8FeO3 (electrospinning) at increasing temperature, measured immediately after 10% CO2 pulses. At each temperature level, the FTIR results in Fig. 5e confirm the presence of both monodentate and bidentate carbonate species. As the temperature increases, the absorbance intensity decreases, indicating a decrease in carbonate content. Notably, beyond 175 °C, one of the monodentate carbonate peaks diminishes, reflecting its weaker bonding compared to bidentate carbonate. This suggests that bidentate adsorption dominates at low CO2 coverage. With increasing CO2 coverage, some bidentate species are transformed into monodentate configuration. The disappearance of the carbonate peaks at 240 °C is consistent with the TGA measurements, which showed an onset temperature for CO2 desorption at 100 °C and a peak desorption temperature of 240 °C. This further confirms that the high near surface La concentration in the electrospun sample inhibits the formation of stable carbonate species and enhances the reversibility of the CO2 sorption/desorption. These findings also correspond well with the XPS results and enthalpy of adsorption measurements for the electrospun sample.
To compare the binding of CO2 on the surface of the electrospun and ground samples, we performed the in situ analysis with 10% CO2 balance Ar flowing at a rate of 40 mL min−1 operated at 25 °C (Fig. 5f). The carbonate peaks in these two samples exhibit distinct peak locations, stemming from differing surface properties and structures resulting from the electrospinning and grinding methods employed. The greater distance between the bidentate peaks in the electrospun sample compared to the ground sample indicates weaker carbonate species in the electrospun sample.46 This corroborates well with the isotherm analysis (Fig. 5c and d). Overall, it is evident that the higher surface La concentration in the electrospun sample provides favorable adsorption sites for reversible sorption and desorption of CO2. On the other hand, a high surface Sr concentration, e.g. on the reactive ground sample, would lead to highly stable carbonate species that are not desirable for low temperature CO2 sorption applications. These findings provide valuable insights into tailoring materials for improved adsorption performance.
4 Conclusions
CO2 adsorption using solid sorbents offers a promising approach to carbon capture due to low energy requirements, operational simplicity, and high stability. This study introduces perovskite oxides as a tunable family of solid sorbents with exceptional structural and compositional flexibility to optimize sorption thermodynamics and kinetics. Using SrxLa1−xFeO3 (x = 0, 0.2, 0.5, and 0.7) as a model system, we demonstrated that varying the A-site composition enables a wide range of tunable CO2 desorption temperatures (75–500+ °C). A strong correlation between surface area and sorption capacity was also established, along with effective methods to enhance the oxide surface area: a salt-assisted grinding method resulted in samples with 16 m2 g−1 whereas an electrospinning method enhanced surface areas to ∼30 m2 g−1 from 3–5 m2 g−1.Among the synthesized materials, Sr0.2La0.8FeO3 prepared via electrospinning exhibited the highest performance, with a moderate desorption temperature (onset: 120 °C and peak: 240 °C) and a sorption capacity of 0.68 wt%. Adsorption isotherm analysis revealed that CO2 chemisorption dominates at low pressures (0–1 kPa), while physisorption becomes significant at higher pressures. TEM analysis indicated the formation of perovskite nanorods through electrospinning. XPS and (in situ) IR characterization studies, coupled with experimental measurements, revealed that near surface La concentrations play a crucial role in reversible CO2 sorption. The significant over-representation of La on the surface of the electrospun sample compared to other preparation methods led to a superior surface normalized reversible CO2 sorption capacity.
This study highlights tunable perovskite oxides as a promising new class of CO2 sorbents. While the demonstrated capacity remains lower than that of typical solid sorbents reported to date (∼1.3–14%),47–52 their high stability and tunability, combined with opportunities for further optimization of surface area and composition, position these oxides as potential candidates for practical applications in CO2 capture and utilization.
Data availability
Data supporting this article have been included as part of the ESI.†Author contributions
Seyedamin Razavi: investigation, and writing – original draft. Vahid Rahmanian: conceptualization – review & editing. Runxia Cai: conceptualization – review & editing. Mahe Rukh: conceptualization. Saad Khan: writing – conceptualization, reviewing and editing, supervision. Fanxing Li: conceptualization, writing – review & editing, supervision, project administration, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Novo Nordisk Foundation (Grant No. NNF22SA0078767) under the Biocatalyst Interactions with Gases program. We would like to acknowledge Dr Sonja Salmon's helpful comments and suggestions.References
- B. N. Firtescu, F. Brinza, M. Grosu, E. M. Doaca and A. A. Siriteanu, Front. Environ. Sci., 2023, 10, 965841 Search PubMed.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS PubMed.
- D. Welsby, J. Price, S. Pye and P. Ekins, Nature, 2021, 597, 230–234 Search PubMed.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 Search PubMed.
- M. Yates, J. Blanco, P. Avila and M. Martin, Microporous Mesoporous Mater., 2000, 37, 201–208 CrossRef CAS.
- B. B. Saha, S. Koyama, I. I. El-Sharkawy, K. Habib, K. Srinivasan and P. Dutta, J. Chem. Eng. Data, 2007, 52, 2419–2424 CrossRef CAS.
- H. Yang, Z. Xu, M. Fan, R. Gupta, R. B. Slimane, A. E. Bland and I. Wright, J. Environ. Sci., 2008, 20, 14–27 Search PubMed.
- T. Ashirov and A. Coskun, Chimia, 2024, 78, 415–422 Search PubMed.
- C. W. Jones, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 31–52 Search PubMed.
- A. Goeppert, M. Czaun, G. S. Prakash and G. A. Olah, Energy Environ. Sci., 2012, 5, 7833–7853 RSC.
- D. M. Ruthven, S. Farooq and K. S. Knaebel, Pressure Swing Adsorption, John Wiley & Sons, 1996 Search PubMed.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CrossRef CAS PubMed.
- F. Brandani and D. M. Ruthven, Ind. Eng. Chem. Res., 2004, 43, 8339–8344 CrossRef CAS.
- R. Wu, E. Carrejo, M. S. Reza, E. Woods, S. Razavi, S. Park, F. Li and W. J. Sagues, Fuel, 2024, 373, 132372 Search PubMed.
- L. Brody, M. Rukh, R. Cai, A. Saberi Bosari, R. Schomäcker and F. Li, J. Phys.: Energy, 2023, 5, 035004 CAS.
- R. Cai, K. Yang, X. Wang, M. Rukh, A. S. Bosari, E. Giavedoni, A. Pierce, L. Brody, W. Tang, P. R. Westmoreland and F. Li, Energy Environ. Sci., 2024, 17, 6279–6290 RSC.
- A. Sayari, Y. Belmabkhout and R. Serna-Guerrero, Chem. Eng. J., 2011, 171, 760–774 CrossRef CAS.
- C.-H. Yu, C.-H. Huang and C.-S. Tan, Aerosol Air Qual. Res., 2012, 12, 745–769 CrossRef CAS.
- M. Li, M. Zhao, F. Li, W. Zhou, V. K. Peterson, X. Xu, Z. Shao, I. Gentle and Z. Zhu, Nat. Commun., 2017, 8, 13990 CrossRef CAS PubMed.
- Y. Cao, J. Liang, X. Li, L. Yue, Q. Liu, S. Lu, A. M. Asiri, J. Hu, Y. Luo and X. Sun, Chem. Commun., 2021, 57, 2343–2355 RSC.
- B. Koo, K. Kim, J. K. Kim, H. Kwon, J. W. Han and W. Jung, Joule, 2018, 2, 1476–1499 CrossRef CAS.
- M. Rukh, R. Cai, L. Brody and F. Li, Chem. Eng. J., 2024, 501, 157545 CrossRef CAS.
- Y. Zhu, W. Zhou, Z. G. Chen, Y. Chen, C. Su, M. O. Tadé and Z. Shao, Angew. Chem., 2015, 127, 3969–3973 CrossRef.
- C. Borca, B. Xu, T. Komesu, H.-K. Jeong, M. Liu, S.-H. Liou and P. Dowben, Surf. Sci., 2002, 512, L346–L352 CrossRef CAS.
- W. Lee and B. Yildiz, ECS Trans., 2013, 57, 2115 CrossRef.
- E. J. Crumlin, E. Mutoro, Z. Liu, M. E. Grass, M. D. Biegalski, Y.-L. Lee, D. Morgan, H. M. Christen, H. Bluhm and Y. Shao-Horn, Energy Environ. Sci., 2012, 5, 6081–6088 RSC.
- A. Nenning, A. K. Opitz, C. Rameshan, R. Rameshan, R. Blume, M. Hävecker, A. Knop-Gericke, G. n. Rupprechter, B. Klötzer and J. r. Fleig, J. Phys. Chem. C, 2016, 120, 1461–1471 CrossRef CAS PubMed.
- Y. Chen, W. Jung, Z. Cai, J. J. Kim, H. L. Tuller and B. Yildiz, Energy Environ. Sci., 2012, 5, 7979–7988 RSC.
- C. D. Saquing, J. L. Manasco and S. A. Khan, Small, 2009, 5, 944–951 CrossRef CAS PubMed.
- C. A. Bonino, M. D. Krebs, C. D. Saquing, S. I. Jeong, K. L. Shearer, E. Alsberg and S. A. Khan, Carbohydr. Polym., 2011, 85, 111–119 CrossRef CAS.
- B. V. Farias, T. Pirzada, R. Mathew, T. L. Sit, C. Opperman and S. A. Khan, ACS Sustain. Chem. Eng., 2019, 7, 19848–19856 CrossRef CAS.
- J. Xue, T. Wu, Y. Dai and Y. Xia, Chem. Rev., 2019, 119, 5298–5415 CrossRef CAS PubMed.
- G. Zheng, J. Jiang, X. Wang, W. Li, J. Liu, G. Fu and L. Lin, Mater. Des., 2020, 189 CAS.
- D. Chen and Y. Zhu, Nanoscale Res. Lett., 2017, 12, 1–5 CrossRef CAS PubMed.
- T. Pirzada, Z. Ashrafi, W. Xie and S. A. Khan, Adv. Funct. Mater., 2019, 30 Search PubMed.
- V. Rahmanian, M. Z. Ahmad Ebrahim, S. Razavi, M. Abdelmigeed, E. Barbieri, S. Menegatti, G. N. Parsons, F. Li, T. Pirzada and S. A. Khan, J. Mater. Chem. A, 2024, 12, 214–226 RSC.
- Y. Cao, B. Lin, Y. Sun, H. Yang and X. Zhang, J. Alloys Compd., 2015, 624, 31–39 CrossRef CAS.
- Y. Teraoka, H.-M. Zhang and N. Yamazoe, Chem. Lett., 2000, 19, 5 Search PubMed.
- G. Kulkarni, C. Rao and M. Roberts, J. Phys. Chem., 1995, 99, 3310–3316 CrossRef CAS.
- A. Carley, M. Roberts and A. Santra, J. Phys. Chem. B, 1997, 101, 9978–9983 CrossRef CAS.
- J. Fierro and L. G. Tejuca, Appl. Surf. Sci., 1987, 27, 453–457 CrossRef CAS.
- T. H. Fleisch, R. F. Hicks and A. T. Bell, J. Catal., 1984, 87, 398–413 CrossRef.
- J. Fierro, Catal. Today, 1990, 8, 153–174 CrossRef CAS.
- X. Xu, W. Wang, W. Zhou and Z. Shao, Small Methods, 2018, 2, 1800071 CrossRef.
- S. Richard, A. Mitchell, C. Evans, J. Whitaker, A. Thomson and A. Keith, Greenhouse gas removal methods and their potential UK deployment, Department for Business, Energy & Industrial Strategy, London, 2021, 120pp Search PubMed.
- G. Busca and V. Lorenzelli, Mater. Chem., 1982, 7, 89–126 CrossRef CAS.
- P. Priyadarshini, G. Rim, C. Rosu, M. Song and C. W. Jones, ACS Environ. Au, 2023, 3, 295–307 CrossRef CAS PubMed.
- G. Rim, P. Priyadarshini, M. Song, Y. Wang, A. Bai, M. J. Realff, R. P. Lively and C. W. Jones, J. Am. Chem. Soc., 2023, 145, 7190–7204 CrossRef CAS PubMed.
- A. Holewinski, M. A. Sakwa-Novak and C. W. Jones, J. Am. Chem. Soc., 2015, 137, 11749–11759 CrossRef CAS PubMed.
- M. Zhao, J. Xiao, W. Gao and Q. Wang, J. Energy Chem., 2022, 68, 401–410 CrossRef CAS.
- J. Wang, H. Huang, M. Wang, L. Yao, W. Qiao, D. Long and L. Ling, Ind. Eng. Chem. Res., 2015, 54, 5319–5327 CrossRef CAS.
- X. Zhu, W. Xie, J. Wu, Y. Miao, C. Xiang, C. Chen, B. Ge, Z. Gan, F. Yang, M. Zhang, D. O'Hare, J. Li, T. Ge and R. Wang, Chem. Soc. Rev., 2022, 51, 6574–6651 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08481k |
| This journal is © The Royal Society of Chemistry 2025 |