
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review on photocatalytic methane conversion systems: from fundamental mechanisms to the emerging role of ferroelectric materials
Yiming Lei
 ,
Xavier Sala,
Jordi García-Antón and
Jose Muñoz
*
,
Xavier Sala,
Jordi García-Antón and
Jose Muñoz
*
Departament de Química (Unitat de Química Inorgànica), Facultat de Ciències, Universitat Autònoma de Barcelona (UAB), Cerdanyola del Valles, 08193 Barcelona, Spain. E-mail: JoseMaria.Munoz@uab.cat
First published on 18th March 2025
Abstract
Methane (CH4) conversion is a promising strategy for reducing greenhouse gases, synthesizing high-value-added chemicals, and thus achieving carbon neutralization. However, due to its inertness, CH4 requires high energy input to initiate the conversion process, which always leads to excessive energy consumption and catalyst deactivation. Along these lines, the use of sunlight as energy input has demonstrated enormous potential to help overcome the uphill thermodynamics of methane conversion under mild reaction conditions. Nevertheless, the high recombination rate of photoinduced charge carriers still hampers photocatalytic efficiency. In the past years, ferroelectric photocatalysts have attracted much attention for dealing with sluggish charge separation/transfer dynamics, presenting themselves as excellent materials for enhancing photocatalytic CH4 conversion rates under mild conditions. Consequently, this work reviews and discusses four pivotal photocatalytic CH4 conversion routes, including (i) dry reforming of methane (DRM), (ii) partial oxidation of methane (POM), (iii) non-oxidative coupling of methane (NOCM), and (iv) oxidative coupling of methane (OCM). In particular, special attention has been paid to the recent advances in ferroelectric-assisted photocatalytic CH4 conversion to elucidate the influence of polarized ferroelectrics in the charge transfer mechanisms. Finally, the main challenges in the field are highlighted, also presenting possible strategies to overcome them in order to encourage more in-depth research on ferroelectric-assisted methane conversion in the future.
 Yiming Lei | Yiming Lei is a PhD student in the Department of Chemistry at Universitat Autònoma de Barcelona, Spain (2023–current). He is awarded by the China Scholarship Council (CSC). His current research focuses on the functionalization of 2D Xene materials represented by 2D germanene derivatives for real implementations including (bio)sensor and catalysis. |
 Jose Muñoz | Dr Jose Muñoz received his PhD in chemistry from Universitat Autònoma de Barcelona (UAB). Afterward, he carried out two postdoctoral stays: the first one at the Materials Science Institute of Barcelona (ICMAB, 2016–2019) with a “Juan de la Cierva” fellow, and the second one at the Central European Institute of Technology (CEITEC, 2020–2022) with an MSCA-IP grant. In 2023 he returned to the UAB as an independent researcher with a “Ramón y Cajal” senior grant. His research interests combine 3D printing technology, 2D materials, and molecular engineering for the development of biosensors and multiresponsive devices. |
1. Introduction
The rapid global population growth and rising living standards cause the high consumption of fossil fuels, leading to a non-negligible energy crisis and environmental issues.1,2 It is predicted that compared to 2018, the total energy demand will increase by about 40% in 2024.3 Fossil fuels as the dominant energy source will be depleted after 70 years, meaning that the current energy-industry system cannot be maintained without developing an alternative energy source.4 Besides, a series of environmental issues like climate change and air pollution owing to the over-exploitation of natural resources are the most complex challenges faced today.5 Therefore, the development of alternative energy resources is extremely urgent due to the increasing growth in energy demand and the necessity to reduce environmental problems.6,7 With a sufficient reserve of methane (CH4) hydrate and shale gas, the utilization of methane for industrial production becomes a reliable strategy for replacing petroleum.8,9 Besides the abundant natural resources, the diversity of synthesis pathways to obtaining CH4 is also a great advantage to utilizing this high-quality fuel gas. For instance, natural gas is expected to increase from 35.57 Tcf (trillion cubic feet) in 2024 to 42.07 Tcf by 2050, meaning that the chemical synthesis of high-value-added products will be possible on a large scale if suitable methane conversion technologies are realized.10,11 In addition, biogas—composed of ∼60% CH4—can be generated from animal excrement, wastewater, and food debris via anaerobic digestion, which is a clean and green chemistry route.12 In this regard, CH4 from biogas can be purified to obtain biomethane as a promising renewable energy for further chemical transformation. Another environmentally friendly production route for preparing CH4 is CO2 reduction (or hydrogenation) reactions. CH4 can be made from CO2 captured from industrial sources, combined with hydrogen obtained from water splitting using surplus renewable energy. This offers additional opportunities for large-scale CH4 conversion while reducing the use of fossil fuels and the global greenhouse effect. Thus, the diverse sustainable sources of CH4 are beneficial for its utilization towards the production of high value-added chemicals, oxygenated products including alcohols and aldehydes chemicals, and even longer chain compounds like long chain alkane.Methane conversion to added-value chemical products can be classified into two different strategies: indirect and direct routes. While indirect routes include (i) dry reforming of methane (DRM) and (ii) steam reforming of methane (SRM), direct routes are based on (iii) partial oxidation of methane (POM), (iv) oxidative coupling of methane (OCM), and (v) nonoxidative coupling of methane (NOCM). The main difference between these two strategies is the fact that indirect routes need to firstly produce syngas (a mix of CO and H2) for the production of high-value-added products from methanol (CH3OH) conversion, as summarized in Table 1.13 Importantly, these strategies have been shown to be useful in addressing excess greenhouse gas emissions without disrupting current infrastructure.14–19
| Conversion process | Product | Reaction pathway | ΔG0 (298 K) [kJ mol−1] |
|---|---|---|---|
| DRM | CO + H2 (syngas) | CH4 + CO2 → 2CO + 2H2 | 170.7 |
| CH3COCH3 | 2CH4 + CO2 → CH3COCH3 + H2O | 114.1 | |
| SRM | CO + H2 (syngas) | CH4 + H2O → CO + 3H2 | 142.1 |
| CH3OH | CH4 + H2O → CH3OH + H2 | 117.3 | |
| C2H5OH | 2CH4 + H2O → C2H5OH + 2H2 | 161.7 | |
| POM | CO + H2 (syngas) | 2CH4 + O2 → 2CO + 4H2 | −173 |
| CH3OH | 2CH4 + O2 → 2CH3OH | −222.6 | |
| HCHO | 2CH4 + O2 → 2HCHO + 2H2 | −103.6 | |
| OCM | C2H4 | 2CH4 + O2 → C2H4 + 2H2O | −287.6 |
| C2H6 | 4CH4 + O2 → 2C2H6 + 2H2O | −320 | |
| NOCM | C2H6 | 2CH4 → C2H6 + H2 | 68.6 |
| C6H6 | 6CH4 → C6H6 + 9H2 | 433.9 |
However, because of the thermodynamic stability of methane (440 kJ mol−1 for C–H bond cracking) due to its symmetrical tetrahedral geometry, it is difficult to convert methane into high-density energy fuels or chemicals taking into account other small hydrocarbon molecules with low chemical bond energy like 413 and 412 kJ mol−1 in propane and ethane, respectively.14,20,21 To date, the thermocatalytic reaction, carried out at high temperature or pressure, is still the main industrial route of methane conversion.22 It is undeniable that thermocatalytic processes often cause high energy consumption and catalyst poisoning (generally due to the rapid carbon deposition and catalyst sintering).23 To overcome these hurdles, increasing attention is paid to photocatalytic methane conversion technologies.
Compared to traditional thermocatalysis and its reaction conditions, photocatalysis is a promising technology that can be carried out under ambient conditions, using photons to drive chemical processes.24–27 Light illumination causes energetic charge carriers to move across the photocatalyst, significantly reducing the activation energy and overcoming the thermodynamic barrier.28–31 Thus, photocatalytic methane conversion with solar energy as the only energy input during the reaction process can reduce energy consumption to the greatest extent possible. Typically, over the conventional semiconductor surfaces, the light-driven catalytic reactions can be viewed as three fundamental steps as follows (Fig. 1a):32,33 (i) when the light irradiates the semiconductor-based photocatalysts, if the energy of the photons is equal to or greater than the semiconductor band gap (Eg), photons can be captured to excite photoelectrons. (ii) Then, the photoelectrons transfer and occupy the conduction band (CB). And the holes stay at the valence band (VB). Afterward, electrons migrate to the reduction centres, while holes transfer to oxidation sites. (iii) Finally, the adsorbed reactants will interact with electrons and holes to compete with the photocatalytic redox reactions. And several possible products should be desorbed from the photocatalyst surface to ensure the subsequent reaction cycles. Notably, several factors can affect the whole photocatalytic efficiency. For example, many semiconductor-based photocatalysts have a narrow light response range, which means that they only absorb ultraviolet light and little visible light.34–37 Therefore, visible-infrared light energy (∼90% of the solar energy) is wasted, resulting in a low quantum efficiency.38–40 In addition, poor adsorption/activation of reactants at the few active sites or high charge transfer resistance in photocatalysts can lead to the recombination of photoexcited carriers before they can participate in the redox process, which is also a major challenge.41–44 In addition, difficulties in the absorption–desorption also limit the photocatalytic efficiency.45,46 Among these issues, the sluggish charge separation inhibits the solar conversion and hinders the catalytic process, so improving the charge transfer dynamics is a must to improve the photocatalytic activity. Although the design of co-catalysts47–50 and heterojunction structures51–54 got a lot of attention to accelerate the charge transfer process, the efficiency is still not satisfactory for commercial use. Consequently, rationally designed photocatalysts with intrinsic controllable photoinduced charge transfer behaviour are crucial. Under these circumstances, ferroelectrics have been identified as potential materials that might offer new opportunities to further promote charge separation and thus improve photocatalytic activity.
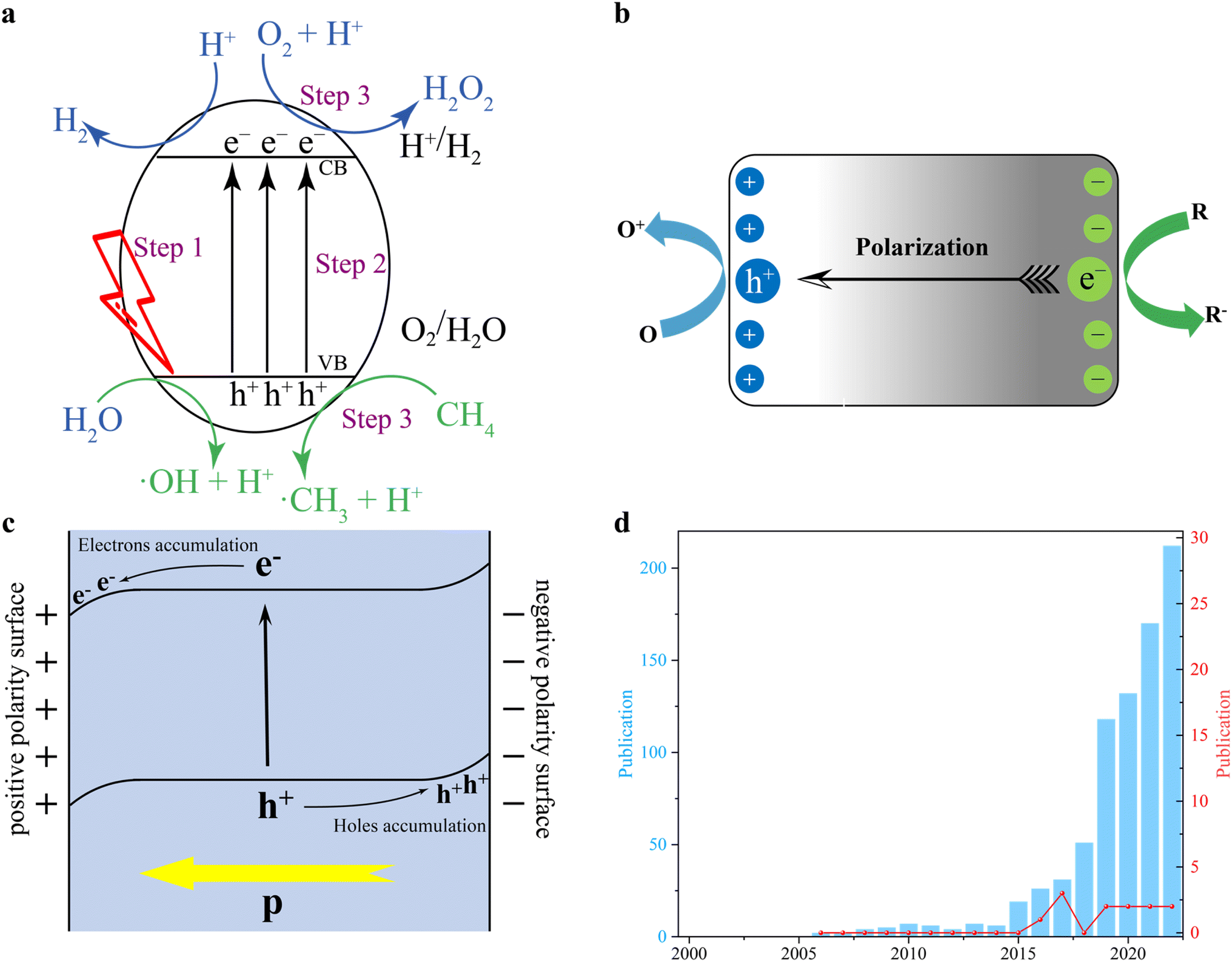 | ||
| Fig. 1 (a) Three basic steps of photo-driven catalytic reactions. Step 1: inducing photoexcited electrons and holes; Step 2: charges migration; Step 3: photo-driven redox reactions. (b) An illustration showing the impact of the polarization effect in the separation of charge carriers and subsequent redox reactions. (c) The band bending phenomenon in ferroelectric photocatalysts owing to polarization effect. (d) The blue bar chart is publications of ferroelectric, photocatalysts according to the search result in “Web of Science”. Key words: “photocatalysis” and “ferroelectric”. The red curve is “publications of photocatalytic methane conversion via ferroelectric photocatalysts”. | ||
In general, ferroelectric materials are polar non-centrosymmetric (NCS) materials with domain walls.55 These domain walls can generate an electric field induced by non-coincide positive–negative charge centres. And the NCS crystal structure endows ferroelectrics with spontaneous polarization ability even without external stimulation.56–61 Thus, an external electric field can simply adjust the polarization directions. However, high-temperature conditions might lead to the symmetric structure of the ferroelectric crystal, causing the materials to lose their ferroelectricity. The built-in electric field associated with the asymmetric domain wall (DW) polarity in ferroelectric materials facilitates the vectorial migration of photoexcited charge carriers (Fig. 1b).56–61 In addition, by controlling the polar facets, ferroelectric materials are able to further improve the ability of reactant adsorption and product desorption.41,62,63 Also, the selectivity of products as one of the most important factors for highly efficient photocatalytic methane conversion is adjusted by polarity switching at DWs.63,64 More importantly, the adsorbed reactants at specific active sites can generate several intermediates and charges with different polarity properties. In this case, their accumulation will lead to the band bending in different directions (Fig. 1c).65 Specifically, the positive polarity always caused an electron accumulation and thus a downward band bending. This can promote the flow of electrons and restrict hole movement.66,67 Conversely, the surface with negative polarity should lead to an upward band bending, since there is an electron depletion layer. Obviously, in this case, the electron transfer will be difficult and hole transfer is accelerated owing to the different concentrations of charges on both sides of the interface.68,69 Accordingly, the controllable properties of charge transfer in ferroelectric materials are beneficial for photocatalytic redox reactions with methane conversion no exception. Among the extensive library of ferroelectric materials—which can be mainly divided into three families, viz. inorganic, organic, and organic–inorganic hybrid ferroelectrics—inorganic and organic–inorganic hybrid ferroelectrics made of perovskite and its halide derivatives show, simultaneously, strong spontaneous polarization and optoelectrical properties.70–72 Thus, special interest has been focused on the implementation of perovskite-based ferroelectrics for photocatalytic approaches.
Although many publications on ferroelectric-assisted photocatalysis have been recently reported, there are fewer works on their implementation for photocatalytic methane conversion applications to advance toward the efficient production of value-added chemicals and energy sources (Fig. 1d). Therefore, this review aims to provide a comprehensive tutorial about the recent progress in ferroelectric-assisted photocatalytic methane conversion, pointing out the remarkable materials and merits of the most common CH4 conversion technologies. Finally, the main challenges in this emerging field and the corresponding solutions are listed. All in all, this review is hoped to provide in-depth insights to encourage the scientific community to further explore ferroelectric materials as potential photocatalysts for their implementation in the field of energy conversion in general, and methane conversion in particular.
2. Routes for photocatalytic methane conversion
2.1. Prominent semiconductors for photocatalytic methane conversion
Before reviewing the fundamental reaction mechanisms and key studies on different photocatalytic methane conversion strategies, it is essential to first introduce prominent examples of semiconductor photocatalysts capable of facilitating this conversion. Among the broad spectrum of semiconductor materials, two main families stand out due to their stability under redox catalytic reactions:73–75 metal oxides and nitrides. Indeed, the intrinsic properties of these materials are crucial for understanding why the C–H bond in CH4 can be cleaved photocatalytically (see Fig. 2).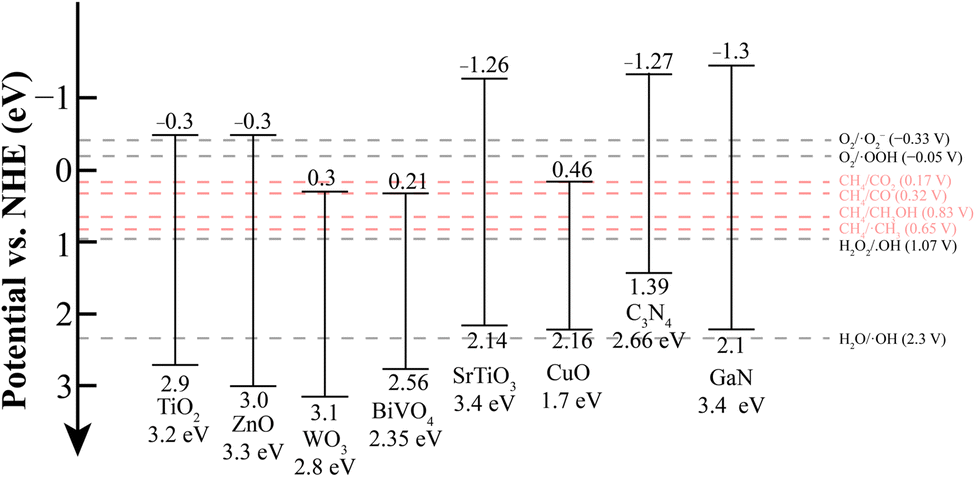 | ||
| Fig. 2 Energy band structures of semiconductors and the redox potentials of reactants and active species. | ||
2.2. Indirect methane conversion
Indirect methane conversion pathways encompass the oxidation of CH4 to syngas through either SRM or DRM technologies, in which the resulting mixture of CO and H2 serves as the precursor for their subsequent conversion into upgraded products. Generally, SRM utilizes steam (H2O) to produce syngas according to the formula CH4 + H2O → CO + 3H2,97 DRM involves the reaction of CH4 with CO2 by following the formula CH4 + CO2 → 2CO + 2H2.55Since the high H/C ratio of CH4 among hydrocarbons, SRM is a reliable technology to provide hydrogen energy at high operating temperatures.104 In brief, in the presence of CH4 and water, hydrogen production can be produced as shown in eqn (2-1). And some oxygenated products like CH3OH can also be directly formed by SRM through another reaction pathway shown in eqn (2-2). SRM is an endothermic reaction, which is always operated at temperatures of >700 °C, indicating that the catalysts for SRM should be stable under high-temperature conditions.105
| CH4 + H2O → CO + 3H2 | (2-1) |
| CH4 + H2O → CH3OH + H2 | (2-2) |
Several excellent works have tried to construct kinetic models for specific reaction conditions. Because of the endothermic nature, the fast transfer of both heat and mass is important to a highly efficient SRM process. In traditional industrial SRM reactors, the heat energy always is transferred from the furnace to the tube reactor, resulting in rapid reaction rates and thus very low effectiveness factor (η), compared to the diffusion rate of reactants and products over pellet-type catalysts.105 Abbas's group simulated and studied the SRM process based on a one-dimensional (1D) theoretical model.106 Theoretically, the activation energy for SRM was 257.01 kJ mol−1. High temperature, low pressure, and high H2O/CO2 ratio contributed to rapid CH4 conversion and high purity of H2. This work is consistent with the previous report from Xu et al., also showing a low η (<0.03) when using a small portion of catalyst in industrial-scale conditions.107 An early theoretical work studying the influence of SRM reactors on kinetics has also been investigated.108 Two computational fluid dynamics (CFD) models were developed for simulating the steam reforming and combustion of methane in microchannel reactors. The models included 20 mm in length and 0.7 mm side with 4 or 20 square microchannels. The results illustrated that the gaseous reactants were heated isothermally so that the complete CH4 combustion occurred at the channel entry within a short distance. The cross-flow disposition led to 80 °C of maximum temperature differences and an easier feed stream distribution, which was beneficial for the rapid reaction kinetics. In addition to the unique reaction vessels, the reaction temperature was also investigated from a kinetic perspective. Kechagiopoulos et al. clarified the reaction kinetics of SRM at low temperatures in conjunction with isotopic investigations.109 Temperature-programmed experiments of CH4 reforming and decomposition modes with isotopic investigations showed the cleavage of the C–H bond was the rate-determining step, but the steam-derived intermediates had a low influence on the whole reaction rate. The authors also proposed a thermodynamically consistent microkinetic model with the consideration of several surface pathways, indicating that –CH3 dehydrogenation on the catalyst surface could also limit the reaction rate.
Although SRM has become a mature industrial technology for obtaining hydrogen fuel, it still has several disadvantages such as mass/heat transfer and carbon deposition (coking). Coking often blocks reactors or breaks catalyst particles so as to cause deactivation. Experimentally, when the ratio of steam to carbon (S/C) is lower than 1, rapid and severe coke will inevitably occur.110 Meanwhile, the high temperature and pressure are also related to the carbon deposition problem.111 In the process of SRM industrialization, researchers try to use a high S/C ratio (2.5–3.0) to avoid the coke deposition and catalyst deactivation problem. However, the excessive partial pressure of steam reduces energy efficiency. In order to develop catalysts with high stability, the Boudouard reaction (including CH4 and CO cracking) should be considered to suppress coke formation. Additionally, the ratio and selectivity of products from SRM also need to be further controlled. In this case, the catalytic transformation of CH4 and CO2 into syngas via DRM might be an ideal alternative technology to SRM. DRM has the potential ability to achieve real carbon neutrality via the conversion of solar energy to chemical energy and mitigate the greenhouse effect. Considering the fact that the study about ferroelectric-assisted SRM systems is still a blank field, the SRM will not be further discussed in this review article. Some excellent works have comprehensively summarized and discussed the traditional thermal and advanced photo(thermal) catalytic SRM implementations. Interested readers are recommended to read ref. 97 and 112.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) than SRM (1:3). Thus, further downstream processes are contributed by the CO-rich syngas from DRM.114
:1) than SRM (1:3). Thus, further downstream processes are contributed by the CO-rich syngas from DRM.114| CH4 + CO2 → 2CO + 2H2 | (2-3) |
Generally, both traditional thermal catalysts and photocatalysts tend to activate reactants (CH4 and CO2) via a bifunctional route: (i) after CH4 is absorbed on reduction sites, dehydrogenation will occur (Fig. 3a). However, during the CH4 activation step, the cracking of the strong CH3–H(g) bond is very difficult because of the high binding energy (439.3 kJ mol−1), meaning the first step of CH4 dissociation is one of the rate-determining step (DRS).115 Regarding the CO2 molecules, as shown in Fig. 3b, they can be specifically adsorbed on three different reactive sites involving the metal (cocatalyst) surface, metal–support interface, and support. Although the conversion of CO2 to CO or carbonate precursors is simple and direct,116–118 the intrinsic properties of CO2 like non-polarity and thermodynamic stability lead to robust C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond with ∼750 kJ mol−1 of dissociation energy barrier. From the thermodynamic view, the activation of CO2 with high energy input is another RDS factor.103,119 Further, the strong chemical bonds in CH4 and CO2 make a high-temperature condition (>800 °C) necessary during the conventional thermal catalytic DRM process, resulting in the unavoidable sintering and carbon deposition on transition metal-based catalysts.120–122 It has been confirmed that the photocatalytic DRM can be carried out at low temperatures using semiconductor photocatalysts, which may be an effective strategy to avoid sintering and coking of catalysts.123
O bond with ∼750 kJ mol−1 of dissociation energy barrier. From the thermodynamic view, the activation of CO2 with high energy input is another RDS factor.103,119 Further, the strong chemical bonds in CH4 and CO2 make a high-temperature condition (>800 °C) necessary during the conventional thermal catalytic DRM process, resulting in the unavoidable sintering and carbon deposition on transition metal-based catalysts.120–122 It has been confirmed that the photocatalytic DRM can be carried out at low temperatures using semiconductor photocatalysts, which may be an effective strategy to avoid sintering and coking of catalysts.123
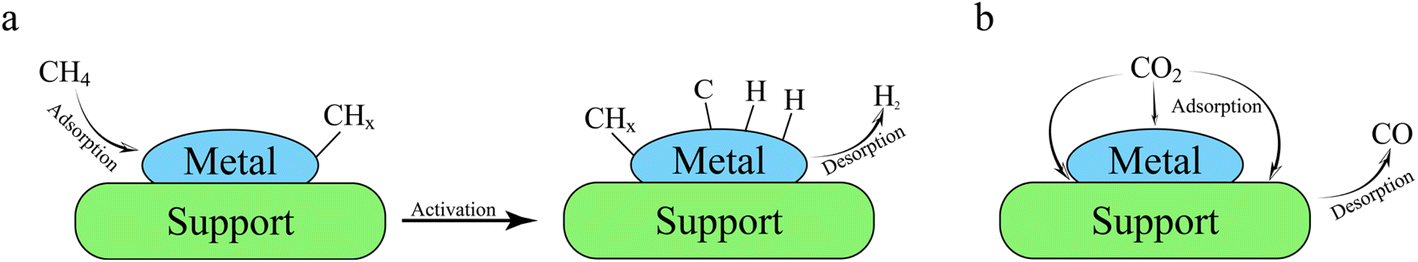 | ||
| Fig. 3 Illustration of reactants activation during DRM process: (a) CH4 adsorption and dissociation (cracking), generating H2 product. (b) CO2 adsorption on three different active sites including metal surface, metal–support interface, and support, and then direct generation and desorption of CO within one step. | ||
For the kinetic studies, several kinetic models have been proposed based on different adsorption states of the reactants, so that the rate-determining step (RDS) could be studied in different DRM systems. Here, three mainstream kinetic models for predicting the reaction rates are presented.
(1) Power-law model: the power-law model was constructed to provide reaction parameters without including reaction mechanisms. This kinetic model only assumed one situation, in which catalyst surfaces tend to adsorb more CH4 instead of CO2 so that the CH4 adsorption dominated the whole reaction. Thus, the parameters from this simple model are rough and sometimes are not consistent with the actual experimental results.
(2) Eley–Rideal (ER) model: the ER model investigated the DRM kinetics in the temperature range of 700–850 °C with the pressure ratio of CH4 to CO2 was 1. Besides, other competitive or reverse reactions were also taken into consideration.124 In this model, only one reactant (CH4 or CO2) is adsorbed on the catalyst surface to interact with another unadsorbed gaseous reactant. The ER model showed that both conversion processes, CH4 to H2 and active carbon (C) to CO (C + CO2 → CO), determine the reaction rate.
(3) Langmuir–Hinshelwood–Hougen–Watson (LHHW) model: although the ER model has considered a lot of factors including crucial parameters and common side reactions, the LHHW model is more accurate because the mechanistic steps in the LHHW model are consistent with practical experimental conditions and results.125 The greatest difference between LHHW and other models is that in the LHHW model, two reactants should be adsorbed on actives at the same time. In this case, the LHHW model regards both activation and cracking processes as the RDS. And another step is thermodynamic equilibrium. The LHHW model is proposed based on the activation process of two reactants and the interaction between CHx and O, which finally generates the desired syngas product. It has been confirmed that CHxO as an intermediate could be rapidly dissociated to CO and H2.126 However, water was always detected even though no water or steam was present in the initial reactants owing to the unavoidable RWGS (reverse water–gas shift) side reaction.98 Fortunately, although a small amount of water had a slight effect on the ratio of syngas (H2:CO), the RWGS with water generation leads to more CHxO active species and thus the yield rate of syngas.
2.2.2.1. Thermal catalytic DRM. Considering the fact that many photo(thermal)catalysts have been developed based on previous investigations of mature thermal DRM technology, some common thermal catalysts for conventional DRM reactions should be introduced before showing the advantages of ferroelectric-assisted photocatalytic DRM technology. During the thermal catalytic process, the selection of catalysts is important to achieve long-term and high conversion efficiency of methane. Although the cost of catalysts cannot be neglected, noble metals (e.g. Rh, Ru, Pt, Ir, and Pd) could be considered as suitable candidates due to their high activity and stability under harsh conditions for the DRM process.120–122 An early work reported a Ru/Al2O3 catalyst. Ru/Al2O3 exhibited 46% CH4 conversion and 77% H2 selectivity. Kinetic and temporal studies indicated that the Ru metals induced both CH4 and CO2 conversion processes. In addition, H and O species as intermediates were continuously provided by the Al2O3 substrate, so that catalyst aging (or active Ru oxidation) could be inhibited. Another example is the Ru-loaded lanthanum oxide (Ru/La2O3) for DRM application published by Cornaglia and co-workers.127 Ru/La2O3, which showed ∼15% conversion efficiency of CH4, had excellent stability for more than 80 h at high temperatures (823–903 K).
Furthermore, when considering industrial production, other requirements have been proposed, such as maintaining high activity while reducing the cost of catalysts, or achieving high stability and efficiency at the same time. In this case, transition metals have 3d orbitals with unpaired d-electrons. Especially, Ni-based catalysts have become the best choice owing to their low cost, high adsorption energy towards O and C atoms, and excellent activity for DMR.128–130 However, the strong cracking process of CH4 over Ni surface always leads to carbon deposition (CH4 → C* + 4H*), which can cover the active sites on the catalyst surface and finally result in inactivation. Because long-term stability is also pivotal for catalyst industrialization, several advantages have been proposed to improve the stability of Ni-based catalysts. For instance, the fabrication of bimetallic thermal catalysts has been used in Ni-based catalysts.131–133 The Ni-based alloy can resist carbon formation than single Ni.134 Zhang et al. reported NixAl1O2−δ mesoporous catalyst.135 The oxygen vacancy formed by the partially inverted structure of NiAl2O4 spinel can be used to activate CO2. Then CO2 reacted with deposited carbon atoms to avoid carbon deposition so that the stability was improved up to 50 h at 800 °C. Besides, in recent years, it has been reported that the position of carbon atoms could be tailored in interstitial or surface sites in/over Ni crystalline.136 And the interstitial atoms affect the electronic properties of surface atoms, further adjusting the catalytic behaviour. This provides a new idea to eliminate carbon deposition. In addition to reactants activation and long-term stability, other factors also determine the activity and selectivity of DRM implementation. Several works have discussed the impact of important factors in the DRM efficiency and product selectivity.137–139 However, the photocatalytic DRM systems did not get enough attention from the past works.
2.2.2.2. Photocatalytic DRM. Photocatalysis can reduce the harsh conditions of the traditional thermal DRM process by taking advantage of light illumination, as the energetic photoexcited charges effectively dissociate the C–H and C
O bonds and prevent carbon deposition under mild conditions instead of high temperature and pressure conditions to maintain the activity of the catalysts after several cycles.140–143 However, low-energy photons can hardly be used to directly excite photocatalysts to trigger reactions, so the infrared light energy has been overlooked for a long time.39,40,144 The photothermal DRM approach has led to an increase in DRM activity by integrating solar energy and thermal energy in the same reactor,145–147 and more studies have attempted to utilize both light and thermal energy from light irradiation to further improve the conversion rate of CH4. A representative work is the one reported by Shoji and co-workers employing SrTiO3-supported rhodium catalyst (Rh/STO).86 During DRM experiments, the limit of thermal catalytic DRM over Rh/STO was determined, suggesting the maximum H2 generation under dark and high-temperature conditions (Fig. 4a). However, under light irradiation without external thermal energy input, Rh/STO could overcome the limitation in the thermal system even at a mild temperature. On the other hand, no H2 could be detected from bare STO, meaning Rh was an important active centre for photothermal catalytic DRM. Rh remained at 1–2 nm of particle size before and after the DRM, indicating that there was no aggregation of Rh particles (Fig. 4b and c). And no carbon deposition was detected by STEM images. Thus, catalytic DRM with light energy as the only input energy source effectively enhanced the longevity of catalyst via inhibiting aggregation and coking. The charge transfer pathway during the DRM reaction was discussed by in situ EPR measurements. Trapped hole peaks in the VB were clearly observed under light irradiation (Fig. 4d). However, the trapped electrons in the CB were not obtained in Rh/STO, suggesting that the photoexcites electrons were injected into Rh. In N2, CO2/CH4 or CH4 atmosphere, the signal intensity of the hole decreased, indicating that the holes reacted with CH4 instead of CO2 (Fig. 4e). The subtract potential profile from Kelvin probe force microscope (KPFM) analysis implied that the electron could transfer from STO to Rh due to the gap in their Fermi levels (Fig. 4f–h). According to isotopic trace analysis, in the Rh/STO catalyst, the lattice oxygens (O2−) acted as mediators to drive DRM instead of protons. Thus, CH4 was selectively oxidized by surface lattice oxygen ions of STO, while CO2 was reduced by photoelectrons from STO over Rh particles, avoiding the recombination of charge carriers in the interior of STO (Fig. 4i).
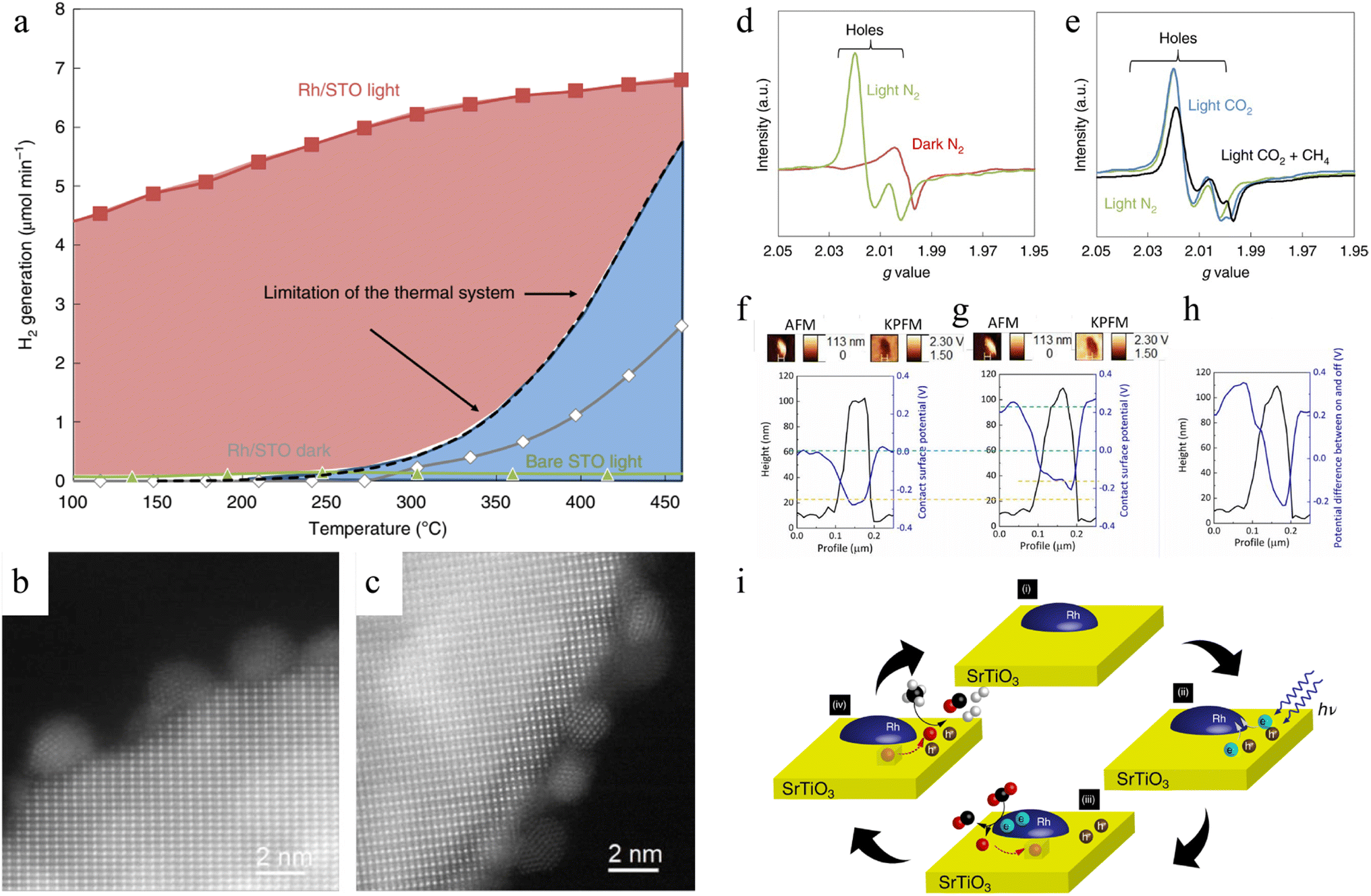 | ||
| Fig. 4 (a) H2 evolution efficiency and the limitation of the thermal DRM system. Reaction conditions: 150 W Hg–Xe lamp, 1% CH4 and 1% CO2 mixture in argon balance gas (4.5 μmol min−1). HAADF-STEM images of Rh/STO (b) before and (c) after DRM. ESR spectra of Rh/STO (d) in an N2 atmosphere; (e) in N2, CO2, or CH4/CO2 atmosphere. KPFM analysis of Rh/STO (f) before and (g) during UV light irradiation, and (h) potential difference between light-on and light-off conditions. (i) Charge carriers and oxygen ions transfer mechanisms.86 Copyright © 2020, Springer Nature. | ||
Photo(thermal) catalytic DRM technology realizes a highly efficient CH4 conversion under mild conditions because it can overcome the thermodynamic energy barrier with external light energy input and assistance.148,149 From this perspective, the charge-carrier recombination instead of CH4/CO2 activation will become the main challenge for the whole DRM process, since the random collision of electrons and holes will be aggravated by high-temperature conditions from external heating or light illumination.38 The collision of electrons and holes leads to their recombination and the waste of the captured photon energy before the charges take part in the photo(thermal) catalytic redox process.150,151 In this case, ferroelectric materials are able to inhibit the photoinduced charge recombination and promote charge transfer by constructing a built-in electric field during the photo(thermal)catalytic DRM process.
2.3. Direct methane conversion
Direct methane conversion technologies, including POM, OCM, and NOCM, rely on converting methane into added-value products without the need of producing syngas as intermediate fuel.152 Thus, these technologies are especially appealing when compared to traditional indirect methane conversion approaches since reduce energy consumption, improve selectivity, and minimize greenhouse gas emissions.153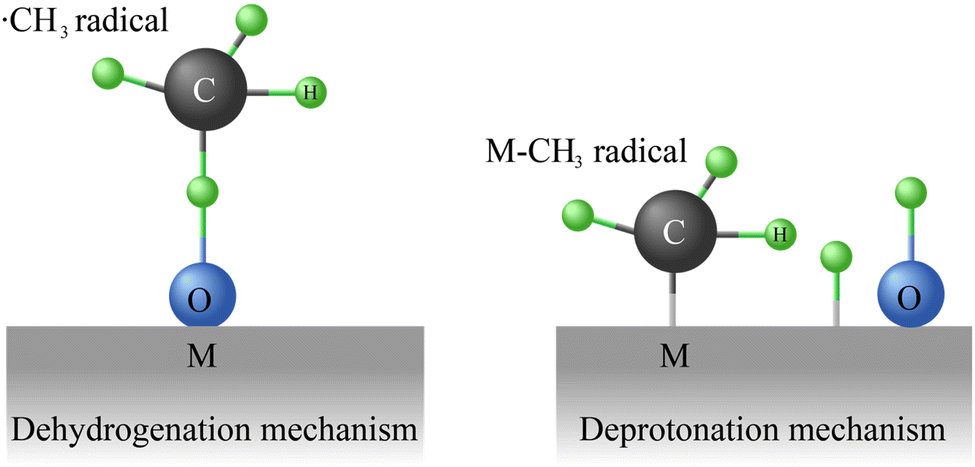 | ||
| Fig. 5 Dehydrogenation and deprotonation mechanism for CH4 activation during POM reactions. | ||
Several studies have discussed the kinetic model of POM over different catalysts. An early study elucidated the kinetics of the catalytic reactions over Pt catalysts,166 where the apparent activation energy of CH4 and O2 conversion is only 10 kJ mol−1, suggesting that the POM reaction is not temperature dependent, but completely determined by the transport behavior. Due to the excess of CH4 in this system, the efficiency of oxygen consumption, which is strongly influenced by mass transfer to the surface, appears to have become the RDS. However, the selectivity of the products (e.g. CO and H2) is strongly influenced by the temperature at the surface of the Pt catalysts.167 In addition to the noble metal catalysts, a microkinetic model of the CH4 oxidation reaction over PdO catalysts was proposed based on DFT results under atmospheric to moderate pressures, rare fuel, and low-temperature conditions.168 The adsorption of activated CH4 via dehydrogenation step over unsaturated surface Pd–O site pairs (denoted as Pdcus–Ocus) is a RDS during the initial stage of the POM reaction. Another focus of this work was the strong water-inhibiting effect, which persists up to high temperatures.169 The water inhibition could be considered as hydroxide formation leading to reduced availability of the Pdcus–Ocus site pairs. Thus, the presence of water vapor interacting with the catalyst particles was one of the main factors causing sluggish kinetics and even catalyst deactivation. Another kinetic model of Ni-based catalysts was also developed by comparing the numerical simulations with the experimental data in a flow reactor.170 The results showed that the initial temperature for CH4 conversion to CO2 and H2O was 723 K. However, there was no significant syngas evolution up to 880 K. At temperatures above 880 K, the formation of H2 and CO was increased, resulting in an equilibrium composition with CO2 and H2O. This behaviour indicated the indirect reaction pathway of syngas formation via the initial generation of H2O and CO2 during the CH4 oxidation process.171–173 Thus, the competition between the first and second steps determined the efficiency of the overall POM reaction, meaning that the CO2 dissociation and CO desorption were the RDSs for CO evolution at high temperatures. This review only shows some representative studies, including noble metal-/metal oxide-/transition metal-based catalysts and their kinetic models, which determine the reaction rate. More kinetic models of POM and discussion of reaction pathways over different catalysts have been well demonstrated by other comprehensive works and are therefore not shown in this review.174–176
2.3.1.1. Thermal catalytic POM. Various thermal catalysts using different oxidants for POM have been developed and reported over the last few decades. Early works used VOx and MoOx to convert CH4 to formaldehyde (desired product) and methanol (by-product) at 450 °C.177 The C–H bond activation over Pt and Pd sites has also been investigated via Periana's system using sulfuric acid (H2SO4) as the oxidant.157 The H2SO4 oxidant stabilized the methyl bisulfate product so that Pt2+ complex-based catalysts achieved ∼90% conversion and 81% selectivity. In addition to the noble metal-based catalysts, under mild conditions, zeolites with transition metal centres (e.g. Cu and Fe) could easily dissociate C–H bonds and were therefore developed for the oxidization of CH4 with N2O or O2 as oxidant (Fig. 6a).178,179 Metal zeolite was activated in O2 at 450 °C but reacted with CH4 without O2 at 200 °C. This stepwise process avoided the over-oxidation of CH4 at high temperatures (the first step) and thus achieved over 90% CH3OH selectivity in the second step (Fig. 6b). In addition, metal oxide-based supports have also been extensively studied for POM reactions. Some reports showed the real implementations of oxide-based thermal catalysts such as SiO2, Al2O3, MgO, ZrO2, and CeO2.180–184 For example, the Ni-loaded CeO2 catalysts were suitable for long-term POM at high temperatures, excellent redox capacity, and coke resistance behaviour. Bal et al. reported Ni/CeO2 catalyst, which could activate and totally convert methane at 400 and 800 °C, respectively.185 Due to the cooperation between Ni and CeO2, Ni/CeO2 could resist coke and produce syngas (H2
:CO = 2, Fig. 6c). Besides CeO2, iron oxides were also investigated as oxygen carrier candidates,186 but the oxygen species in iron oxide-based supports were too active to avoid the overoxidation of CH4 to CO2 and H2O.187 In addition, tungsten oxides (WO3) showed their superior stability and selectivity for specific products (e.g. syngas).188 However, one of the main challenges for WO3 thermal catalysts has been poor reducibility, suggesting that a high temperature is required to utilize the lattice oxygen in WO3 and initiate reactions.189 Despite the above advances in POM applications, the high stability and thermodynamic inertness of CH4 also make the oxidation reaction an extremely difficult task. Conventional industrial catalytic POM processes require high temperature (700–1000 °C) and pressure (2.5–3.5 MPa), which can cause oxidation of catalysts and non-negligible explosion risks due to the presence of O2.155
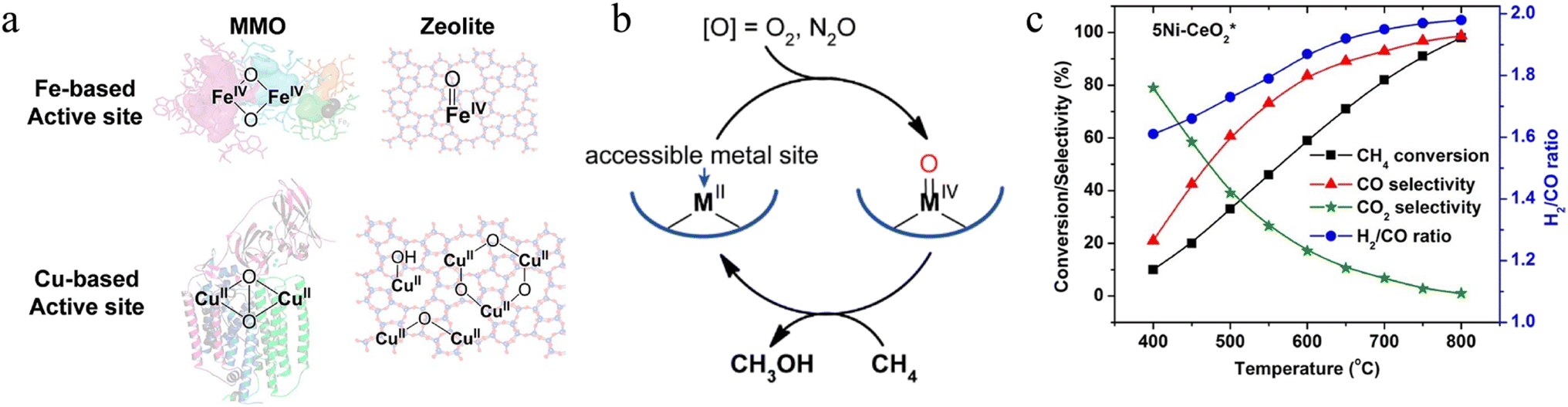 | ||
| Fig. 6 (a) Active site in enzymes and zeolites with Fe or Cu atoms. (b) Conceptual activation of CH4 at a high-spin high-valent metal-oxo species.178 Copyright © 2018, American Chemical Society. (c) Selectivity of Ni–CeO2 during POM reaction.185 Copyright © 2016, Elsevier B.V. | ||
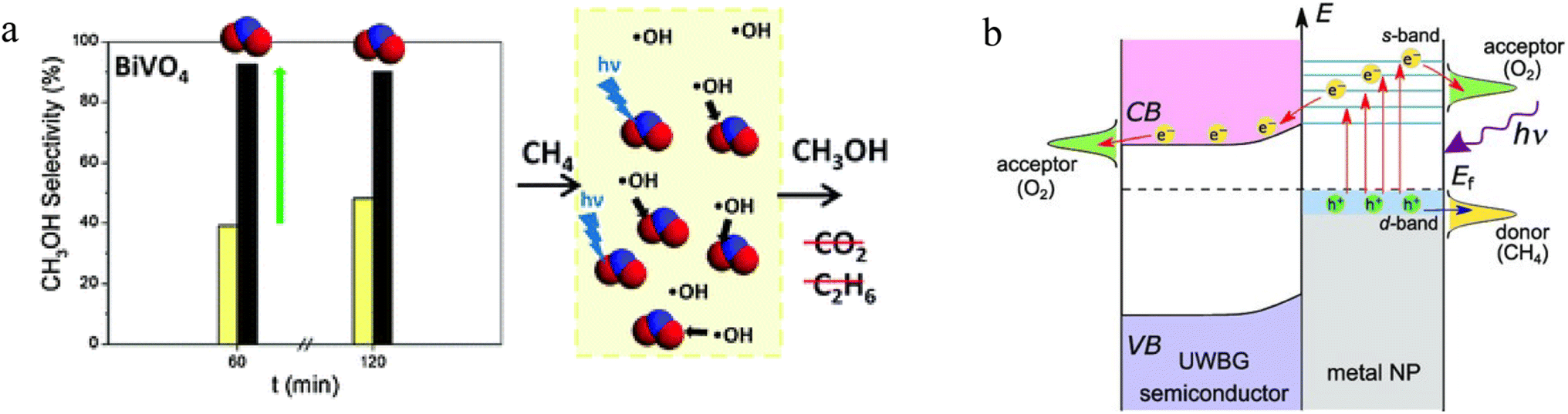
2.3.1.2. Photocatalytic POM. Many conventional semiconductor photocatalysts, such as TiO2, WO3, BiVO4, etc., have shown remarkable activity in CH4 oxidation.80,190,191 For example, Morante's group investigated the selectivity of BiVO4 photocatalyst in the presence of nitrite ions.192 The low concentration of nitrite was beneficial for the selective oxidation of CH4 to CH3OH. By reducing the free ˙CH3 radicals, nitrite as a ˙OH scavenger limited the subsequent undesirable oxidation process of CH3OH, thus greatly reducing the formation of CO2. Meanwhile, the nitrite ions could become a UV-filtering species, inhibiting water photolysis, which ultimately forms ˙OH. In this way, more than 90% selectivity towards CH3OH could be achieved over BiVO4 (Fig. 7a). As an ultrawide-bandgap semiconductor, strontium tantalate (Sr2Ta2O7) with Pd nanoparticles has also been used to perform POM reactions under low-temperature conditions.193 In this composite, O2 could accept the electrons from Sr2Ta2O7 to form radical species. In addition, the layered perovskite structure made Sr2Ta2O7 has high stability for immobilizing Pd nanoparticles (Fig. 7b). The hot electrons from Pd nanoparticles facilitated the activation of O2 and CH4. The photothermal effect was triggered by hot carrier relaxation. Thus, light illumination lowered the initial temperature of POM reactions to below 423 K and improved the catalytic performance to below 873 K to produce syngas. Other previous research has also demonstrated the excellent ability of photocatalysis to reduce the large heat energy during the POM process over various semiconductor-based photocatalysts such as g-C3N4, TiO2, and CuMoO4/SiO2.94,194 In addition, the recombination between electron–hole pairs is an unavoidable defect in the photocatalytic process. Compared to some photocatalytic reactions with simple products (e.g. water splitting, organic degradation), the selectivity of POM products is also a major challenge due to the diversity of products. In this case, ferroelectric materials show their ability to overcome these problems.
 radicals on the catalyst surface, which is a heterogeneous step. This is followed by a homogeneous step, during which
radicals on the catalyst surface, which is a heterogeneous step. This is followed by a homogeneous step, during which 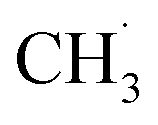 is coupled to produce ethane (C2H6). Finally, the production of ethylene (C2H4) is achieved by subsequent dehydrogenation of C2H6. During these steps, one of the difficulties is the energy required to dissociate the strong C–H bond (∼438 kJ mol−1), which is also present in other CH4 conversion technologies, suggesting that a sufficiently low activation energy barrier is required for the generation of
is coupled to produce ethane (C2H6). Finally, the production of ethylene (C2H4) is achieved by subsequent dehydrogenation of C2H6. During these steps, one of the difficulties is the energy required to dissociate the strong C–H bond (∼438 kJ mol−1), which is also present in other CH4 conversion technologies, suggesting that a sufficiently low activation energy barrier is required for the generation of 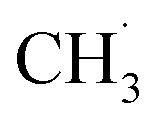 radicals and the subsequent coupling or dehydrogenation process.202 Overcoming the thermodynamic barriers often requires the introduction of O2 and high temperatures, which leads to the production of unwanted products, such as CO2 and graphitic carbon.85 In addition, C2H4 is more unstable than CH4, so consecutive oxidation of C2H4 is also a major challenge.203,204 Moreover, it is difficult to achieve efficient CH4 conversion while maintaining high selectivity for C2 products, because the complete and partial oxidation of methane to COx and the combustion of C2H6 and C2H4 can occur simultaneously. Thus, the low selectivity and evolution rates of C2 products are major challenges for large-scale utilization.
radicals and the subsequent coupling or dehydrogenation process.202 Overcoming the thermodynamic barriers often requires the introduction of O2 and high temperatures, which leads to the production of unwanted products, such as CO2 and graphitic carbon.85 In addition, C2H4 is more unstable than CH4, so consecutive oxidation of C2H4 is also a major challenge.203,204 Moreover, it is difficult to achieve efficient CH4 conversion while maintaining high selectivity for C2 products, because the complete and partial oxidation of methane to COx and the combustion of C2H6 and C2H4 can occur simultaneously. Thus, the low selectivity and evolution rates of C2 products are major challenges for large-scale utilization.
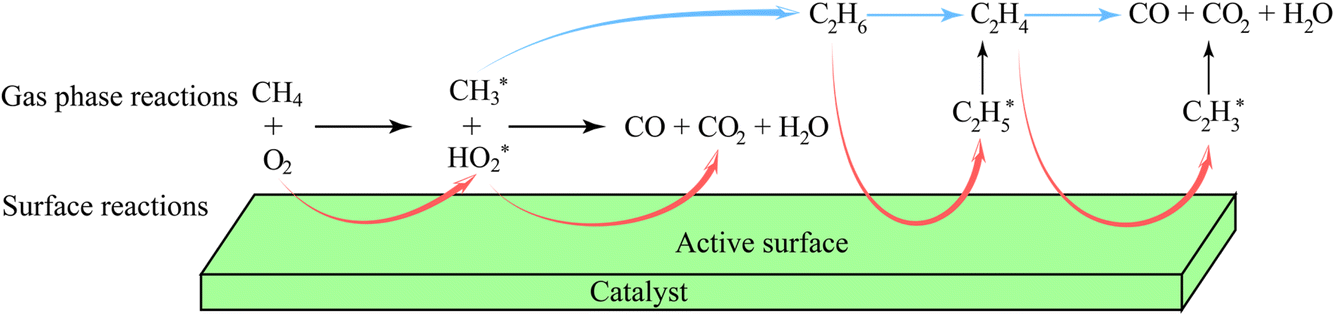 | ||
| Fig. 8 OCM reaction steps in the gas phase and over the catalyst surface. | ||
The actual reaction steps can be more complex than the theoretical three basic steps because both gas phase and catalyst surface reactions should be considered simultaneously to discuss the kinetic models and the RDSs.205–207 However, the currently proposed kinetic models are still incomplete and not able to fully explain the obtained experimental data.208,209 However, two typical assumptions based on the absence and presence of catalysts have been studied, leading to the development of many kinetic models in different catalytic systems. A representative kinetic scheme reflecting the OCM reaction under gas-phase conditions without heterogeneous catalysts has been demonstrated.210 According to this kinetic model, the time dependence of the compound concentrations could be calculated, thus determining the product selectivity as a function of the degree of conversion. Specifically, when there was an excess of CH4 in the gas phase, the concentration of active oxygen species interacting with 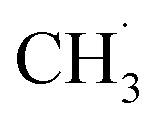 to form carbon oxides was low, resulting in a high selectivity for C2 hydrocarbon products. In contrast, under high O2 concentration conditions (e.g. CH4:O2 = 2), the formation of oxygen-containing radicals such as
to form carbon oxides was low, resulting in a high selectivity for C2 hydrocarbon products. In contrast, under high O2 concentration conditions (e.g. CH4:O2 = 2), the formation of oxygen-containing radicals such as  and ˙OH was promoted, leading to the low evolution rate of C2 hydrocarbon products. The authors summarized the optimum reaction conditions for achieving the maximum selectivity of C2 products. The reaction temperature should be 1000–1100 K, and the pressure ca. 10 bar while the ratio of CH4 to O2 was not less than 5–7. A total pressure higher than 10 bar was advantageous to produce the primary product C2H6 instead of the desired C2H4. A lower ratio between CH4 and O2 would make the non-selective reaction dominant. Based on the above study, further work has been carried out through dedicated experimental measurements, regression, sensitivity, and parameter contribution analyses were carried out to explore the optimal OCM reaction conditions.198,211–213 An interesting paper by Simoní Da Ros and co-workers summarizes some details of these works.208 In short, the updated kinetic model of OCM gas phase reaction systems has contributed to the optimization of the reaction parameters, and thus the operation of the product distribution.
and ˙OH was promoted, leading to the low evolution rate of C2 hydrocarbon products. The authors summarized the optimum reaction conditions for achieving the maximum selectivity of C2 products. The reaction temperature should be 1000–1100 K, and the pressure ca. 10 bar while the ratio of CH4 to O2 was not less than 5–7. A total pressure higher than 10 bar was advantageous to produce the primary product C2H6 instead of the desired C2H4. A lower ratio between CH4 and O2 would make the non-selective reaction dominant. Based on the above study, further work has been carried out through dedicated experimental measurements, regression, sensitivity, and parameter contribution analyses were carried out to explore the optimal OCM reaction conditions.198,211–213 An interesting paper by Simoní Da Ros and co-workers summarizes some details of these works.208 In short, the updated kinetic model of OCM gas phase reaction systems has contributed to the optimization of the reaction parameters, and thus the operation of the product distribution.
For the kinetic model in the presence of catalysts, Stansch et al. described a 1D homogeneous plug flow model comprising 10 reaction steps according to the experimental data over a La2O3/CaO catalyst with high activity and selectivity towards C2 hydrocarbons in a packed bed reactor.207 An important assumption in this kinetic model was the small effect in gas phase reactions, meaning that only a few cases where the gas phase reaction can be neglected, such as short contact time and low gas volume, were suitable for this model. Despite this drawback, the dynamics of the oxidative reaction were successfully represented by Hougen–Watson-type rate equations (eqn (3-1)). The power law rate equations determined the rates of dehydrogenation, C2H4/H2O reforming, and the WGSR (water–gas shift reaction, eqn (3-2) to (3-5)). The effects of O2 and CO2 on enhancing C2H4 selectivity via inhibiting C2H6 were also described (eqn (3-6)).
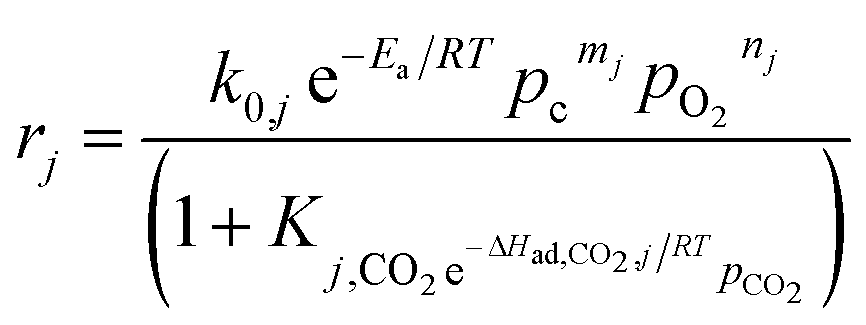 | (3-1) |
|
r1 = k0,1e−Ea,1/RTpC2H6
| (3-2) |
|
r2 = k0,2e−Ea,2/RTpC2H4m2pH2On2
| (3-3) |
|
r3 = k0,3e−Ea,3/RTpCOm3pH2On3
| (3-4) |
|
r4 = k0,4e−Ea,4/RTpC2H4m4pH2On4
| (3-5) |
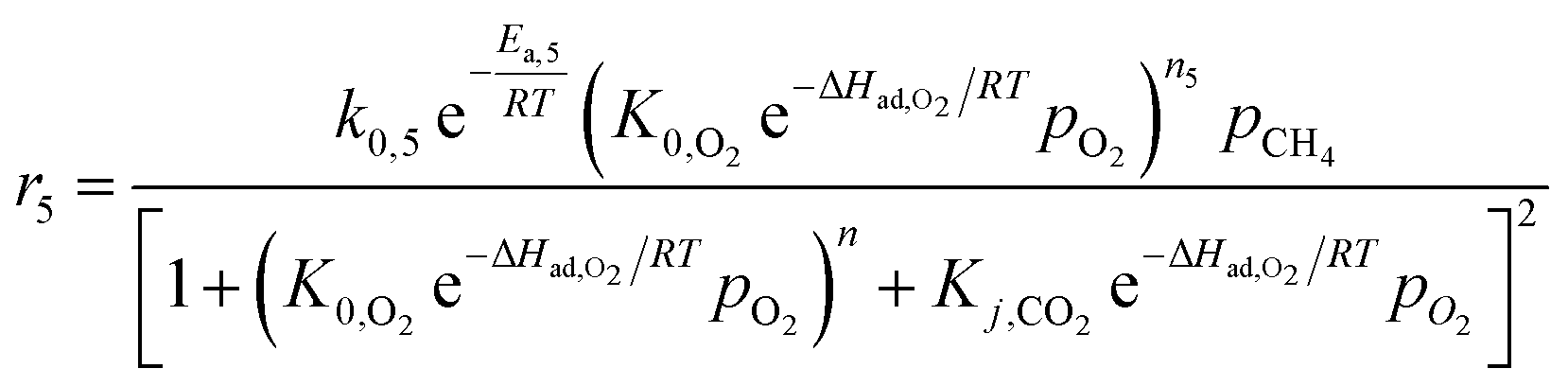 | (3-6) |
2.3.2.1. Thermal catalytic OCM. Considerable efforts have been made to develop catalysts for OCM to C2 hydrocarbons. For thermal catalysts, an early result was reported by Ito et al., who developed a lithium-doped magnesium oxide (Li/MgO) catalyst that had a high activity for the formation of ˙CH3 radicals in the presence of O2, resulting in high selectivity for C2 compounds (C2H4 and C2H6) and 28% conversion of CH4.218 Cu- and Fe-contained zeolite thermal catalysts have been widely studied with their dehydrogenation mechanism. For example, the Cu-/Fe-based catalysts have been used for OCM using N2O or H2O2 as strong oxidants.219,220 The Cu–O–Cu sites have been confirmed as the main active platform to activate CH4 via hydrogen abstraction, convert ˙CH3 and ˙OH radicals to CH3OH, and avoid catalyst inactivation by rapid desorption of products (Fig. 9a).221 Recently, IrO2 has been shown to be an effective catalyst for OCM application. Nørskov's group claimed that Ir coordinatively saturates with surface oxygen,159 which could be considered as an oxygen-promoted surface contributing to the high activity via the dehydrogenation process.222 However, another research with the opposite idea suggested that the (110) facet over IrO2 is able to dissociate C–H at low-temperature via the deprotonation mechanism. In addition to the controversial reaction mechanism, single IrO2 was not suitable for long-term OCM because overoxidation was unavoidable, resulting in undesired CO and CO2. Besides IrO2, other common thermal catalytic systems with the deprotonation process are noble metal-contained catalysts, which always exist in a low oxidation state so that proton acceptors can be accessible.156,223,224 Taking Pt as an example, an organic ligand-stabilized PtII complex was designed for the conversion of CH4 into methyl bisulfate (CH3OSO3H).157 Using H2SO4 as a strong oxidant, this complex achieved 90% conversion of CH4 and 81% selectivity of CH3OSO3H at 220 °C. Mechanistic studies showed that platinum-methyl, the intermediate of methyl ester, was produced via C–H activation of CH4 over PtII. And PtII was the most active oxidation of Pt for OCM (Fig. 9b). Other works on homogeneous Pt-based catalysts reported a similar mechanism, in which Pt oxidized to PtII active sites and PtIV–CH3 bond in an orderly manner. Finally, the reductive elimination produced CH3OH and regenerated PtII active sites.225–227 Although recent advances have been made in traditional thermocatalytic OCM, high temperatures (typically >600 °C) are always required for reactant activation, resulting in undesired products such as COx due to the high energy output property (exothermicity) of hydrocarbon combustion.228 Meanwhile, the stability of OCM catalysts is also an inevitable issue due to the excessive heat energy released during the reaction.
 | ||
| Fig. 9 (a) Transformation mechanism of CH4 into CH3OH on the Cu/Na-ZSM-5 zeolite.221 Copyright © 2019, American Chemical Society (b) OCM mechanism.157 Copyright © 1998, The American Association for the Advancement of Science. | ||
2.3.2.2. Photocatalytic OCM. Photocatalysis is a promising approach to achieving OCM reactions in mild conditions.21,28,229 However, studies of photocatalysis technology for realizing OCM are rare. Thus, in this section, a few noticeable and representative works will be summarized to display the great potential of photocatalytic OCM application. For instance, Tang's group designed a Pt/CuOx-loaded TiO2 photocatalyst with a 60% selectivity of C2H6 product in a gas flow reactor (Fig. 10a).85 In this nanohybrid, Pt as an electron acceptor facilitated the charge separation, while CuOx was an active site for photoexcited holes. Thus, deep dehydrogenation and overoxidation could be effectively avoided because of the low oxidation potential of photoinduced holes over the VB of CuOx clusters (Fig. 10b). A similar work to Pt/CuOx-loaded TiO2 photocatalyst was an Au–ZnO/TiO2 photocatalyst with an ethane production rate of ∼5000 μmol g−1 h−1 and 90% selectivity (Fig. 10c).228 These values are the highest efficiency in the previously published works. The modification of Au and TiO2 not only accelerated the charge transfer but benefited the O2 activation owing to the low adsorption energy and activation barrier at oxygen vacancies (Fig. 10d and e).230 The comprehensive investigation demonstrated that Au as a cocatalyst promotes the formation of ˙CH3 radicals and inhibits the overoxidation of CH4 to CO2 through the OCH3 intermediate (Fig. 10f–g). Another representative work was reported in 2019 by Liu et al.231 A plasma-assisted photocatalyst was developed for the oxidation of CH4 to C2H4 and CO via a new reaction route with CH4 and CO2 as reactants. Experimental analyses indicated that CO2 and CH4 tend to adsorb on TiO2 and Ag surfaces, separately. Because of the localized surface plasma resonance (LSPR) effect on Ag and photoelectric effect in TiO2, under a mild condition (simulated solar irradiation, 84.2 mW cm−2), the evolution rates of CO and C2H4 were 1149 and 686 μmol g−1 h−1, respectively. Apparently, the above studies successfully elucidate the great capacity of photocatalysis for OCM to obtain C2 products with high selectivity.
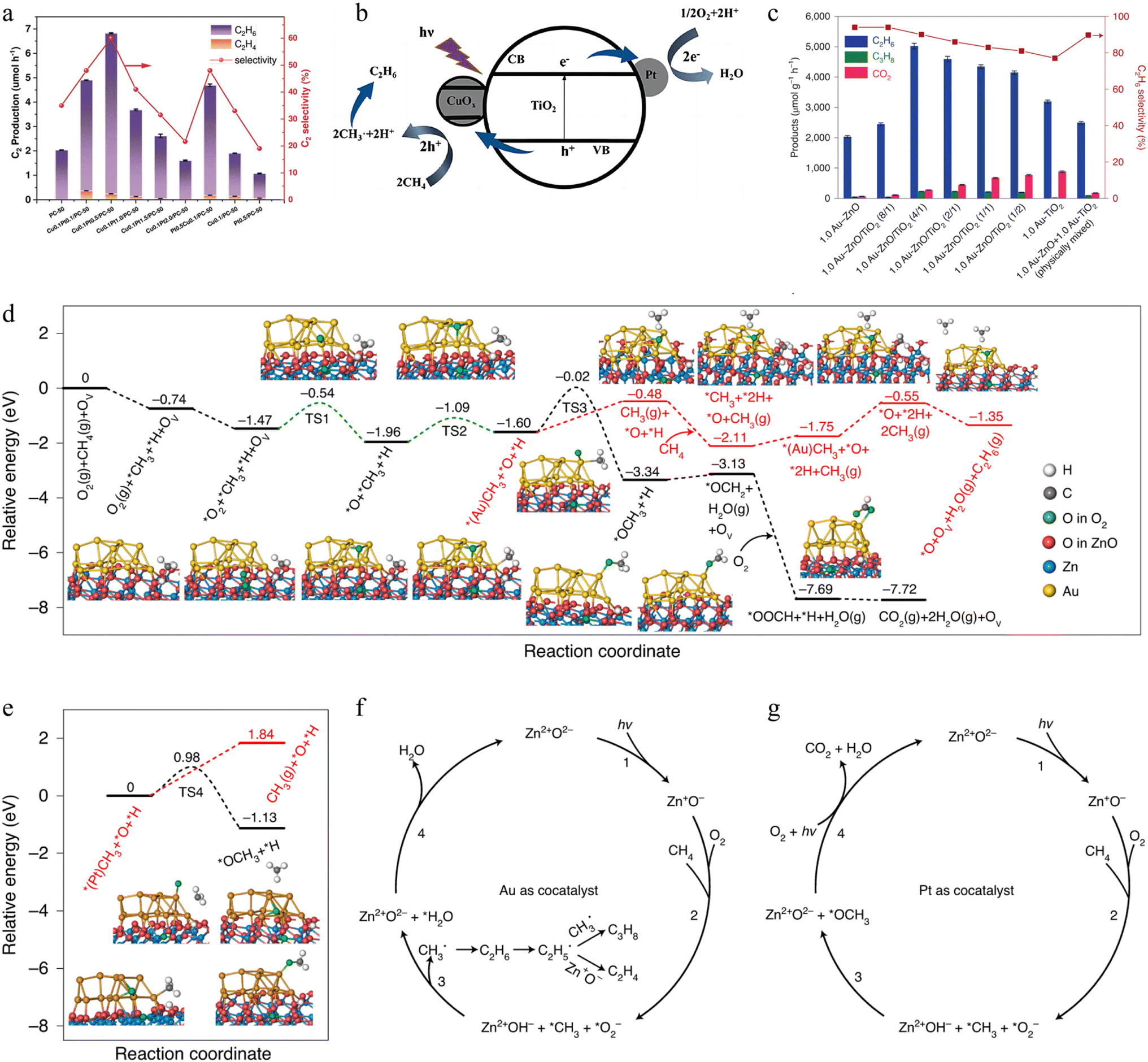 | ||
| Fig. 10 Copyright © 2020 Wiley. (a) C2 production and selectivity of photocatalytic OCM. Reaction conditions: O2/CH4 = 1:400, GHSV = 2400 h−1, 40 W 365 nm LED 40 W, 40 °C.85 (b) Photocatalytic OCM process.85 (c) Photocatalytic OCM activity. Reaction conditions: 69 mL min−1 of CH4 and 1 mL min−1 of 20% O2/N2, 300 W Xe lamp. Theoretical energy diagrams for (d) oxidizing CH4 to C2H6 or CO2 and (e) two key competing pathways. Proposed reaction process on (f) Au–ZnO and (g) Pt–ZnO.228 Copyright © 2021 Springer Nature. | ||
Several microkinetic models during NOCM have been proposed based on different kinds of catalysts. Dumesic's group studied the kinetic model of NOCM on PtSn surfaces with SiO2 or H-ZSM-5 supports via DFT calculation.232 Their model predicted that the full Pt surfaces adsorbed CH and C fragments and C–C coupling had the highest rate during the whole NOCM process. The RDS was the CH4 activation and product (H2) desorption over Pt species. Besides, the desorption of C2H6 as another main product also influenced the whole reaction rate to a large extent.
Kopyscinski et al. built another kinetic model based on a metal-free GaN/SBA-15 catalyst.233 Their kinetic model was proposed via a simple 1D model with the same observed rate as the forward rate, reflecting the reaction process away from the chemical equilibrium and without mass transfer limitation. The kinetic studies and also the isotope labelling experiments confirmed that the possible reaction pathway should be through a fast transformation from CH4 to 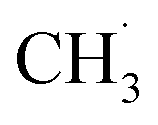 . Subsequently, the 2nd step was slow bond cleavage of C–H, forming
. Subsequently, the 2nd step was slow bond cleavage of C–H, forming 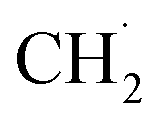 and finally coupling to C2H6. However, the author believed that the RDS should be the conversion step from
and finally coupling to C2H6. However, the author believed that the RDS should be the conversion step from 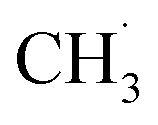 to
to 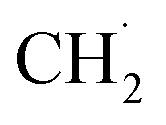 instead of the H–CH3 bond cracking since the apparent order of NOCM was ∼0 in CH4, indicating a fast adsorption process. More importantly, the authors claimed that active sites always fully adsorbed species. Their idea might be a basis for understanding and developing other metal-free catalysts.
instead of the H–CH3 bond cracking since the apparent order of NOCM was ∼0 in CH4, indicating a fast adsorption process. More importantly, the authors claimed that active sites always fully adsorbed species. Their idea might be a basis for understanding and developing other metal-free catalysts.
With the appearance of single-atom catalysts (SACs), lots of SACs have been used for NOCM so some related kinetic studies have been conducted. For instance, Pohar's group explored the micro-kinetics of NOCM to C2H6 over Pt/CeO2 catalyst.234 In the SAC system, the RDS step was similar to the above metallic catalysts, in which the abstraction of the first H˙ from a CH4 molecule and the sorption/desorption of the H2 product. However, one of the deficiencies in this model was the authors assumed the reactions only performed on Pt sites. Some works have proved that the active sites could also be the interface between Pt with CeO2.235 In the future, the synergistic effects between CeO2 support and Pt sites should be accounted for. Kim et al. also studied the kinetic model on SAC containing Fe single-atom.236 Similarly, they also claimed the formation of 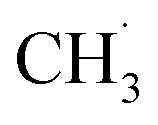 limited the whole NOCM efficiency. More importantly, they emphasized in the case of
limited the whole NOCM efficiency. More importantly, they emphasized in the case of 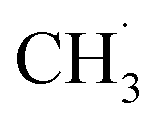 production, the activation barriers of CH4 needed abundant
production, the activation barriers of CH4 needed abundant 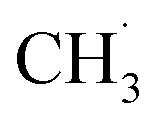 conditions. However, the C–C coupling required higher hydrogen transfer rates on carbon-attached structures under
conditions. However, the C–C coupling required higher hydrogen transfer rates on carbon-attached structures under 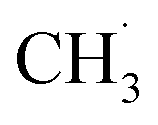 deficient conditions. This conflict suggested the optimal conditions for these two steps (CH4 conversion and C–C coupling) were difficult over a single catalyst. Therefore, the above works showed that there is still no perfect kinetic theory explaining all the catalytic NOCM systems. It is urgent to develop more theories and models to supply this area.
deficient conditions. This conflict suggested the optimal conditions for these two steps (CH4 conversion and C–C coupling) were difficult over a single catalyst. Therefore, the above works showed that there is still no perfect kinetic theory explaining all the catalytic NOCM systems. It is urgent to develop more theories and models to supply this area.
2.3.3.1. Thermal NOCM. The high thermodynamic barrier for NOCM means the high energy is essential to C–C coupling. Therefore, thermal catalytic technology is one of the common strategies to realize the NOCM reaction.237,238 Since the NOCM was first reported by Wang's group,239 different kinds of thermal catalysts have been designed and prepared for highly efficient and selective NOCM implementation. Metal- and alloy-based catalysts are one of the mainstream materials for NOCM. For instance, Soulivong's group reported a SiO2-supported HTa catalyst with an ethane selectivity of 98%. However, in this work, the CH4 conversion was less than 0.5%, which was not enough for industrialization.240 Similarly, Guo et al. realized CH4 conversion of 48% with C2H4, C6H6, and C10H8 with carbon selectivity of 53%, 22%, and 25% on Fe/SiO2 catalyst at 950 °C.241 The high CH4 conversion rate and good selectivity are attractive, but how to separate such diverse products seemly will be another problem. Varma prepared Pt–Bi bimetallic catalyst supported on ZSM-5 zeolite for NOCM, achieving more than 90% selectivity at 873 K, suggesting the potential of polymetallic or alloy systems for NOCM.242 Recently, due to the rapid development of molybdenum titanium carbide (MXene) family materials, noble metals presented by Pt have been anchored on the MXene substrate.243 It has been confirmed that the metal/MXene system could activate the C–H bond in CH4 molecular, then forming methyl radicals, which favoured desorption over subsequent dehydrogenation, so that coke deposition could be suppressed. In this case, the stability and CH4 conversion rate could be enhanced.
Although so many works have been reported in the thermal catalytic NOCM field. As mentioned before, the inevitable disadvantage of NOCM, the high-temperature conditions, still cannot be avoided, which causes large energy consumption, low selectivity, and short longevity of thermal catalysts. Hence, the photocatalytic NOCM at mild conditions attracts more and more attention for exploring a new NOCM route and thus achieving carbon neutrality.
2.3.3.2. Photocatalytic NOCM. Photocatalytic NOCM can convert CH4 to C2H6 and H2 using light energy instead of thermal energy, thereby reducing the reaction temperature and thus avoiding catalyst coking or deactivation. In addition, the energy barrier for CH4 activation can be lowered by photocatalysts. In 1998, Yoshida's group first reported the photocatalytic NOCM reaction over SiO2/Al2O3 composite.244 The SiO2/Al2O3 photocatalyst exhibited the highest CH4 conversion rate of 0.328 μmol h−1 under UV light irradiation. This study proved that the energy transfer from active sites (highly surface-dispersed Al species) to absorbed CH4 might be the crucial step for CH4 activation. Subsequently, a series of silica-based, metal oxide-based, and zeolite-based catalysts have been widely investigated in the photocatalytic NOCM field.89,245–247 Xiong et al. reported that the incorporation of Pd single atoms (SAs) into the lattice could overcome the limitations since the effect of O sites in VB was reduced.248 In this work, TiO2 was used as a proof-of-concept model. DFT calculations suggested that the TiO2 (001) facet was the best surface for the SAs modification because of the loose arrangement of O atoms that can form the M–O bond (Fig. 11a). In order to demonstrate the activity and selectivity toward CH4 to C2H6 conversion, a series of metal SA-modified TiO2 were prepared for photocatalytic NOCM (Fig. 11b). The highest activity and selectivity were observed for Pd SA-loaded TiO2. Therefore, the Pd1/TiO2 photocatalyst was further investigated. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) revealed the atomically dispersed Pd elements on the TiO2 support (Fig. 11c). The coordination of Pd was investigated by synchrotron radiation-based X-ray absorption near-edge structure (XANES) spectroscopy (Fig. 11d). Only a single peak at 1.53 Å could be detected on Pd/TiO2 corresponding to the coordination between Pd and O (Pd–O), indicating the presence of isolated Pd atoms in the photocatalyst. Thus, the coordination environment of Pd–O4 was formed through Pd SA binding with O atoms in TiO2 crystalline. Under ambient conditions with Xe lamp illumination, the photocatalytic NOCM results showed pristine TiO2 nanosheets exhibited a very low production rate of C2H6 (Fig. 11e). This could be attributed to the involvement of the lattice oxygen in CH4 conversion, resulting in a susceptible overoxidation to produce CO2 (Fig. 11f). In contrast, Pd/TiO2 exhibited a very high C2H6 production rate during the NOCM reactions. The improvement of the photocatalytic activity was due to Pd active sites instead of the lattice oxygen in TiO2, which suppressed the overoxidation of CH4 and accelerated the desorption of C2H6. To investigate the role of Pd SAs in CH4 dissociation and intermediate coupling via photogenerated holes, the energy barrier of the CH4–CH4 coupling on the active sites of Pd/TiO2 was studied by DFT calculation (Fig. 11g–h). Under dark conditions (Uh = 0.00), CH4 required higher energy for activation. Meanwhile, 1.74 eV of activation energy from CH4 to TS state was so high that hindered the whole NOCM. However, photogenerated holes would provide additional potential, so that the energy barrier of the step turns down (IM3 to IM4). In addition, the energy barriers of C–C coupling and C2H6 desorption via Pd SAs were also lowered. Thus, the photoexcited charge and the atomic-level reaction platform were advantageous for triggering the photocatalytic NOCM reaction with lower energy input and mild conditions. This work suggested the great advantages of photocatalysis in the activity and selectivity of NOCM.
 | ||
| Fig. 11 (a) Pristine TiO2, Pd1/TiO2, and Pdn/TiO2 models. (b) The metal contribution in VBM and photocatalytic NOCM performance. Reaction conditions: 0.1 MPa CH4, 300 W Xe lamp, room temperature. (c) STEM image and (d) Pd K-edge EXAFS spectra in R space of Pd1/TiO2. (e) The C2H6 production rates (f) and the corresponding selectivity of C2H6, C2H4, and CO2 in 0.1 MPa CH4. (g) Free-energy diagrams for C2H6 production on Pd1/TiO2. (h) Coordination environment during the DFT simulation.248 Copyright © 2022, Springer Nature. | ||
3. Key role of ferroelectric materials in photocatalytic reactions
3.1. Ferroelectric materials
Ferroelectricity was first discovered in rock salt in 1921.249 The ferroelectric material is one of the piezoelectric materials, which belongs to pyroelectrics and then dielectric materials in sequence. Unlike pyroelectric and piezoelectric materials, ferroelectrics have spontaneous dipole moments without an external heating source or applied stress activation. The dipole moments generate spontaneous polarization electric fields, which offer a sustainable driving force for the separation and transfer of charge carriers from bulk to the surface of catalysts. In general, ferroelectric materials can be classified into three main families, viz. inorganic, organic, and organic–inorganic hybrids. What follows summarizes the most representative ferroelectric-based photocatalysts utilized for CH4 conversion approaches.3.1.1.1. Ferroelectric oxides. According to different structures, ferroelectric oxides have four basic types: perovskite, pyrochlore, tungsten-bronzes, and layer-type bismuth compounds (also called Aurivillius).250 Among them, perovskite possesses higher polarization strength. Therefore, they are widely exploited in photocatalysis and photovoltaics. In ferroelectric oxides with perovskite structure, the ferroelectricity is from the electron clouds hybridization mechanism between adjacent ions, called displacive ferroelectricity.251 The photovoltaic effect in ferroelectric perovskite oxides was discovered in the 1950s and still gaining increasing attention.252 The homogeneous bulk region in ferroelectric semiconductors with inherent non-centrosymmetry could generate photocurrent and even photovoltage beyond the bandgap.253,254 Therefore, the photovoltage from ferroelectrics depended on the energy band structure instead of the built-in potential induced by p–n junctions or Schottky junctions at the interface.55 This property could be not only used in photovoltaics but also extended to photocatalysis. In fact, several studies have applied photoinduced charges and polarization from ferroelectric perovskite oxides to improve photocatalytic efficiency.255–257 These works confirmed that perovskite oxides were effective in both photocatalytic reduction and oxidation reactions, providing experimental support for fully utilizing ferroelectric oxides in various photocatalytic fields. More details about published work in photocatalytic methane conversion fields will be further introduced in the following sections. Currently, the main deficiency in conventional ferroelectric oxide perovskites is the narrow light responding range and unsatisfactory conductivity, limiting the photovoltaic and photocatalytic efficiency. In this case, the bandgap engineering promotes the development of ferroelectric perovskite into organic–inorganic halide perovskites as shown below.
3.1.2.1. Liquid crystals (LCs). LCs are a crucial part of the family called soft condensed matter materials. Thermodynamically, the state of LCs is located between an isotropic liquid and a crystalline phase.258 They are partially ordered but have anisotropic physical properties, such as electric conductivity or magnetic susceptibility while maintaining flowability.259 The mainstream methods for producing LCs are based on thermotropic phases and lyotropic phases, which are achieved by changing the intensive variable of state and the dopant concentration in the isotropic solvent, respectively. The ferroelectric properties in the LC family should be attributed to the chiral smectic C (SmC) phase, which is one of the molecular arrangements in LCs (the other two structures are nematic and smectic A phase).260 The chiral SmC is similar to SmC structure, but owing to the chiral properties, Meyer et al. presented the idea that a chiral smectic C mesophase should be ferroelectric, which has been experimentally confirmed in 1975 via reporting practical ferroelectric LCs.261 Owing to the particular chiral SmC phase with helical structure, ferroelectric LC shows spontaneous polarization and molecular switching in microsecond order. These attractive properties lead to wide applications in modern photonic fields including grating,262 lensing,263 beam steering,264 etc. Unfortunately, the aforementioned promising advantages of ferroelectric LCs are still not utilized in the whole catalytic field. Considering the fact that more and more studies tried to regulate the ferroelectricity/ferromagnetism in LCs,265,266 the ferroelectric LC family could be a potential candidate for achieving efficient photocatalytic implementations.
3.1.2.2. Ferroelectric polymers. In addition to ferroelectric LCs, the organic ferroelectric solid is another great family belonging to the class of organic ferroelectric materials. In this family, ferroelectric polymer is one of the most widely developed organic ferroelectrics and has been used in organic solar cells,267 ferroelectric memory,268 piezoelectric devices,269 and electrocaloric refrigeration devices.270 Among them, poly(vinylidene fluoride) (PVDF) and corresponding copolymers/terpolymers are the most representative ferroelectric polymers. PVDF and the co-/ter-polymers had high dielectric constant so that they are regarded as promising thin film-type materials for high-energy-density capacitors.271 In the photocatalytic field, PVDF is also developed as the source of ferroelectric fields, which are used to improve charge-carrier transfer behaviours. In brief, through the ferroelectric effect, PVDF-based photocatalysts have promoted different photocatalytic redox reactions, such as overall water splitting,38,272 CO2 photoreduction,273 and advanced oxidation.274 Although there is no work on PVDF-based photocatalytic systems for methane conversion implementation, in consideration of the great ability of PVDF and other ferroelectric polymers, organic ferroelectric polymers should be applied as substrates or cocatalysts for controlling charge migration pathways in future studies.
Although the low-molecular-mass compound is also a kind of organic ferroelectrics, since it is difficult for the steric to reorient dipoles, causing an extremely larger coercive field than that of other ferroelectrics, the number of the low-molecular-mass compound with ferroelectricity is rare so that the real-implementation of the low-molecular-mass compound is limited.275 Therefore, the low-molecular-mass compound represented by thiourea,276 the 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) free radical,277 and 1,6-bis(2,4-dinitrophenoxy)-2,4-hexadiyne278 are not listed as an independent sub-section.
3.1.3.1. Ferroelectric metal–organic complex (MOC). Since Valasek reported the ferroelectrics in Rochelle salt,279 tremendous efforts have been made to design and fabricate novel molecular-based ferroelectric materials. Although the ferroelectric oxides family is well-known due to their strong intrinsic ferroelectric effect, they are heavy, weak mechanically flexible, and not environmentally friendly.280 Owing to the ferroelectricity of organic molecules, organic ferroelectric materials gradually enter researchers' version as a new generation of ferroelectric material. If a metal–organic complex (MOC) can show ferroelectricity via modulating dipole moments of its components, this MOC belongs to the family of organic ferroelectric materials. Further, according to their crystal structures, ferroelectric MOC can be divided into (i) discrete (0D), (ii) 1D chain, (iii) 2D layer structure, and (iv) 3D framework. It is true that several ferroelectric MOCs have been reported by molecular design based on the addition of chiral/polar molecules/ions or structurally flexible ligands or ions, but the ferroelectric MOC is still at the early step since the number of developed MOCs is not abundant. Some works have reviewed the advances of ferroelectric MOC and also predicted their potential real-applicat ion. Detailed descriptions can be seen in these excellent review articles.280,281 However, in the catalytic field, studies about ferroelectric MOC are scarce, meaning that it is urgent to perform some theoretical studies based on thermodynamic theories and crystallographic requirements to offer fundamental support for exploring and developing ferroelectric MOC in photocatalytic areas.
3.1.3.2. Organic–inorganic perovskites. To a large extent, the prosperity of basic theory and practical applications in the field of ferroelectric materials should be attributed to the conventional inorganic ferroelectric perovskites with high stability and tunable ferroelectric properties.282 Subsequently, molecular ferroelectrics are flexible, cheap, and environmental-friendly, and also have biocompatibility and homochirality so the appearance of molecular ferroelectrics attracted our version, causing a novel family of hybrid organic–inorganic perovskite ferroelectric (HOIPF) materials.283,284 Compared to traditional inorganic ferroelectrics, HOIPFs have larger piezoelectricity coefficients, lower toxicity, and controllable chemical structures although their ferroelectricity is weaker. Besides, the ferroelectric quantum theory and Berry phase calculation also clarify their potential in semiconductor areas.285 Semiconductor properties transform HOIPFs into a new generation of photonic device platforms.286 For instance, some Pb-based or lead-free HOIPFs had tunable bandgap and high mobility similar to common inorganic ferroelectric semiconductors.287,288 A series of functional HOIPFs including (cyclohexylaminium)2PbBr4,289 (R)- and (S)-3-(fluoropyrrolidinium)MnBr3 (ref. 290) possess outstanding luminescent properties, such as high-purity luminescence, wider luminescence range, and high photoluminescence efficiency.291 Besides, the HOIPFs-based optoelectronic applications (e.g. bulk photovoltaic and self-powered photodetection) are also gradually explored with tremendous efforts on the photoresponse effects from HOIPFs and multifunctional hybrid ferroelectric systems.292,293 Although HOIPFs still face some challenges like mechanical weakness, low stability, and short resistivity, the disadvantages do not stop the expansion of the applications of HOIPFs towards photocatalytic fields. Actually, some lead-free halide perovskites (e.g. Cs2AgInCl6 and Cs3BiBr6) have been used for photocatalytic advanced oxidation reactions,294,295 photoreduction reactions,296 and even photocatalytic organic synthesis.297
Considering the number of practical examples of ferroelectric photocatalysts developed for methane conversion is very limited, it is necessary to emphasize the great effect of ferroelectric-assisted photocatalytic systems in improving methane conversion efficiency. In subsequent sections, the representative works about aforementioned ferroelectrics, especially ferroelectric perovskite and ferroelectric oxides, for methane conversion implementations will be introduced for further advancing the ferroelectrics-based catalytic community.
3.2. Key role of ferroelectric materials in photocatalytic reactions
Ferroelectric materials play a crucial role in photocatalytic reactions owing to their inherent domain walls (DWs), which arise from breaking-symmetry. These DWs influence charge transfer,310 local surface potential,311 and even surface adsorption/activation ability,312 all of which are essential for improving photocatalytic efficiency. Thus, this section summarizes the basic mechanism of DWs influencing photocatalytic reactions, supported by both experimental evidence and theoretical predictions.A prominent example for confirming the promotion of bulk charge separation is based on the SrBi4Ti4O15 ferroelectric photocatalyst.255 In this work, Huang and co-workers visualized the intrinsic ferroelectric polarization of SrBi4Ti4O15 nanosheet via KPFM, which was rationalized by DFT calculations. The KPFM images showed DWs with an amorphous shape (Fig. 12a). The bright and dark areas corresponded to different polarization directions. DFT calculations revealed that the effective masses of e− and h+ along the [100] orientation were smaller than along other orientations, suggesting the higher migration rates of e− and h+ along this axis, which was consistent with the polarization direction in SrBi4Ti4O15. Therefore, ferroelectric polarization promoted charge transfer. Similarly, Park et al.314 prepared K0.5Na0.5NbO3 with a permanent polarization via a corona electric field polarization method. With external polarization, the time-resolved photoluminescence (PL) spectra of K0.5Na0.5NbO3 presented a decrease in the decay time from 1.64 to 3.31 ns (Fig. 12b). This result suggests that the ferroelectric polarization field inhibited the recombination between electrons and holes.
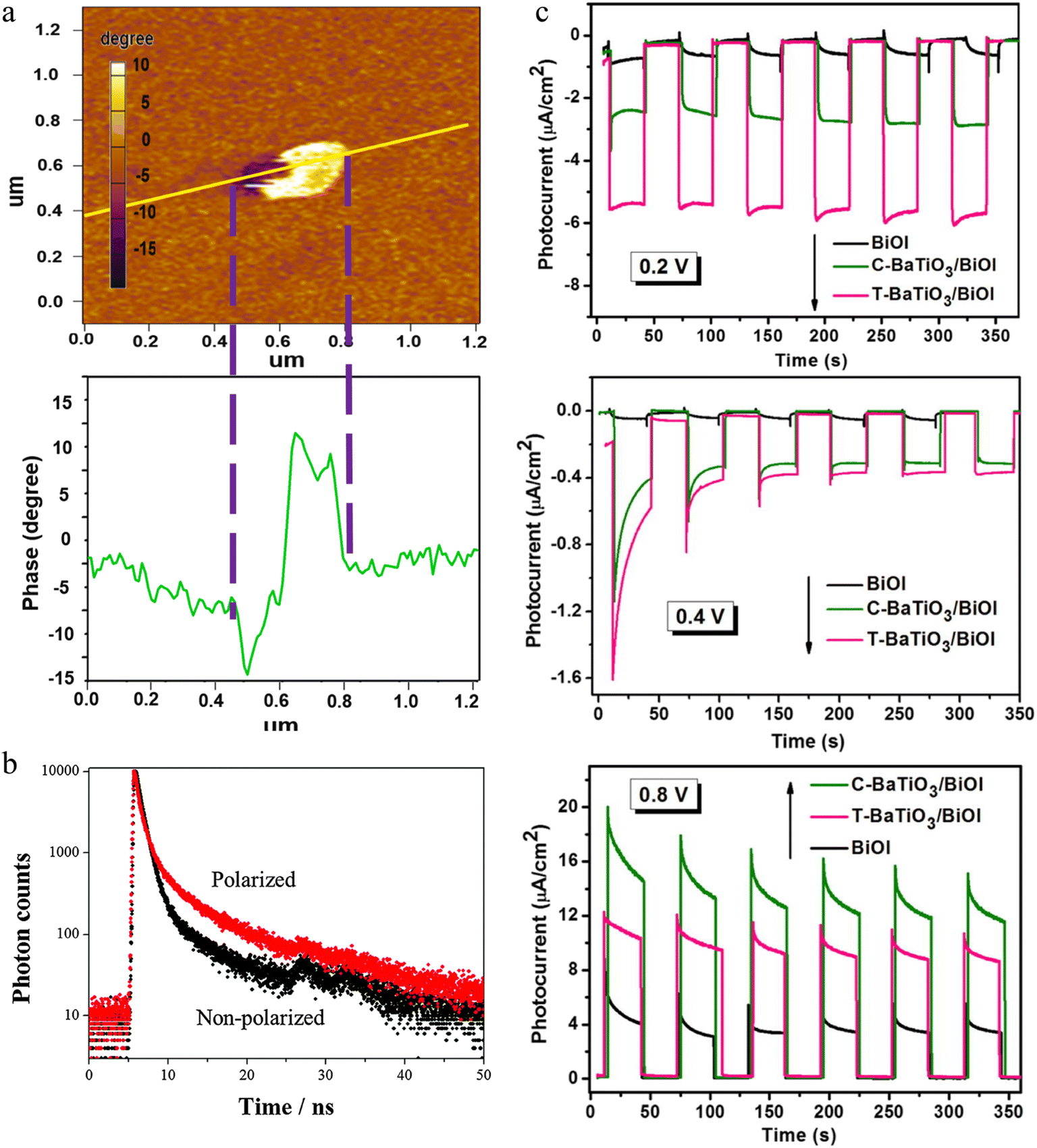 | ||
| Fig. 12 (a) Phase maps (top) and corresponding curve (bottom) of the piezoelectric response.255 Copyright © 2018 Elsevier. (b) Decay profile of PL from K0.5Na0.5NbO3.314 Copyright © 2014 Royal Society of Chemistry. (c) Transient photocurrent responses at different bias voltages.67 Copyright © 2017 Elsevier. | ||
Beyond the bulk charge separation, the ferroelectric effect can also promote charge transfer at the interface of heterojunctions. Considering the short time scale of interfacial charge migration, the electrochemical analysis becomes a reliable method to monitor the effect of ferroelectric materials. For example, Huang's group designed a heterojunction containing ferroelectric tetragonal-phase BaTiO3 (T-BaTiO3) and narrow-band-gap semiconductor BiOI (T-BaTiO3/BiOI).67 They applied photoelectrochemical (PEC) measurements to further investigate the polarization-induced field. Interestingly, with higher bias voltage, the photocurrent of T-BaTiO3/BiOI samples showed clear changes. In contrast, control experiments displayed stable current intensity, indicating that polarization could adjust charge transfer and be adjusted by external bias voltage (Fig. 12c). Other similar works also prepared ferroelectrics-based heterojunctions for accelerating the surface charge mobility during different photocatalytic reactions.58,315,316 In the field of methane conversion, although mechanistic studies are still at the early stage, state-of-the-art characterization methods like PL and PEC analysis have recently been used to clarify the role of ferroelectric materials in charge transfer kinetics. These results will be discussed in detail in Section 4.
 | ||
| Fig. 13 (a) Surface topography after Ag+ reduction (left) and Mn2+ oxidation (right). (b) PFM phase images of monoclinic and tetragonal elemental-doped BiVO4. Surface topographic images (left), inverse pole figure maps (mid), and topographic line profiles (right) of (c) monoclinic and (d) tetragonal elemental-doped BiVO4.317 Copyright © 2016 Royal Society of Chemistry. (e) AFM images of rutile TiO2 supported by BaTiO3 before (left) and after (right) a photochemical reaction, and (f) their heights.318 Copyright © 2010 American Chemical Society. (g) O2 dissociative binding and (h) NO binding on negatively (left) and positively (right) poled RuO2–PbTiO3. (i) Space electron redistribution, presenting covalent binding between NO and positively poled CrO2–PbTiO3.319 Copyright © 2015 American Chemical Society. | ||
Another potential role of ferroelectric materials is to provide high-density surface active sites as discussed in Section 1. The conversion of CO2 into CO2˙− bidentate radical is promoted by the electrons on positively polarized surfaces/sites.147 Further, based on the CO2˙− intermediate, the interaction between CO2˙− and H+ to form HCOO˙− and finally HCOOH is achieved via a proton-coupled electron mechanism.154 Accordingly, CH4 dissociation/cracking should tend to occur on negatively polarized surfaces/sites. In the case of ferroelectrics, the reduction and oxidation reactions will take place at different polarized surfaces/sites, inhibiting the recombination between photoexcited electrons and holes.
4. Ferroelectric-assisted photocatalytic methane conversion
4.1. Ferroelectric-assisted photocatalytic DRM
Recently, a Fe2O3-loaded BaTiO3 photocatalyst (denoted as BFxy, where xy is the BaTiO3/Fe2O3 molar ratio) is reported for the DRM reaction.320 The photoluminescence signals, a feature of the recombination rate of charge carriers, emitted by all the samples were measured (Fig. 14a). BaTiO3 showed the lowest charge recombination rate due to its intrinsic ferroelectric property. On the contrary, Fe2O3 showed the maximum charge recombination rate due to the narrow band gap of Fe2O3. The heterostructure and charge transfer across to interface of BF were confirmed by PL spectra. In this case, the ferroelectric effect from BaTiO3 endowed BF composites with a higher charge separation/transfer efficiency than Fe2O3. Thus, the DRM activity of BF31 showed the highest reactant conversion efficiency (Fig. 14b). Besides BaTiO3, TaN as another ferroelectric material was also developed for photocatalytic DRM reaction.146 In this work, Liu's group explored the effect of TaN as the photocatalyst substrate with a Pt NPs loading (Pt/TaN). Under visible light illumination, Pt/TaN showed improved photocatalytic DRM activity. The convergent beam electron diffraction (CBED) characterization exhibited the polarization phenomenon in the TaN substrate (Fig. 14a). Obviously, the polarity would lead to a built-in electric field, facilitating the photoexcited charge separation. On the contrary, TiN and ZrN as the reference samples did not show any polarized property and thus had lower activity than Pt/TaN (Fig. 14b and c). Accordingly, this study also proved the ineligible enhancement role of ferroelectrics with electrostatic field in photocatalytic efficiency. | ||
| Fig. 14 (a) PL spectra and (b) CO2 conversion efficiency of BaTiO3, Fe2O3, and BFxy. Reaction conditions: 40 psi containing 45% CO2, 45% CH4, and 10% He, 125 W Hg lamp, 35 °C.320 Copyright © 2019, Springer Nature. (a) CBED pattern of TaN. Catalytic DRM performance of (b) Pt/ZrN and (c) Pt/TiN. Reaction conditions: 300 W Xe lamp, CH4/CO2 = 1 (20.0 mL min−1), 500 °C.146 Copyright © 2018, Wiley. | ||
Until now, a large number of ferroelectric materials have been developed in photocatalytic fields.86,321 Unfortunately, in photocatalytic DRM implementations, the previous reports prefer to pay more attention to the impact of cocatalysts or the heterojunction with controllable energy band structure in the DRM process instead of investigating the ferroelectric effect and enhanced charge transfer dynamics. With the development of ferroelectric materials, the ferroelectric effect is expected to be maintained even under harsh reaction conditions (high temperatures or pressures),154 meaning that ferroelectrics can be widely used in different photo(thermal) catalytic applications. Therefore, it is hoped to report more in-depth research on the improvement of DRM reactions from the view of the ferroelectric effects soon. Our previous literature also discussed and emphasized the role of the ferroelectric field especially in DRM applications, the potential interested audience is recommended to browse ref. 55.
4.2. Ferroelectric-assisted photocatalytic POM
As mentioned above, the permanent polarization generated from ferroelectrics creates a built-in electric field. The strong and reliable driving force in an electric field adjusts the transfer path of electrons and holes to different active sites, separately. As a result, more charge carriers tend to complete the redox reaction instead of recombination. An early work was reported by Yi's group in 2013,322 where NaNbO3 polar semiconductors were used to enhance the POM activity. Scanning electron microscopy (SEM) images showed the preparation of NaNbO3 nanocuboids with ordered b-axis orientation (NNB-1, Fig. 15a and b). To investigate the role of ferroelectric property, the disordered random distribution of NaNbO3 (NNB-8) was also obtained (Fig. 15c). In general, the NaNbO3 crystal showed antiferroelectric and ferroelectric properties in the direction of the c-axis and b-axis, respectively, implying an internal polarization in NaNbO3 nanocuboids along the b-axis.323 Instead, NaNbO3 nanocuboids with a disordered nanostructure would induce a randomly distributed local bias electric field. In photocatalytic CH4 oxidation experiments, with Pt loading as cocatalyst via impregnation and photodeposition methods (denoted as Pt-NNB-1, Pt-NNB-1-LD, Pt-NNB-8, and Pt-NNB-8-LD), the CO2 evolution rate of NNB-1 was much higher than that of NNB-8 (Fig. 15d). Interestingly, pure NNB-1 had higher activity than that Pt-NNB-1 and Pt-NNB-1-LD. The reason was investigated by SEM, which showed that the Pt loading process caused the destruction of the ordered structure of NNB-1 (Fig. 15e). Therefore, the structure orderliness was a crucial factor for CH4 oxidation because it was a prerequisite for maintaining the strong ferroelectric effect (Fig. 15f). And the above results also confirmed the great ability of polar semiconductors with built-in electric fields to enhance CH4 oxidation compared to conventional cocatalysts (e.g. Pt). To further investigate the ferroelectric effect, photoelectrochemical (PEC) measurements were used to detect the photocurrent responses of different NaNbO3 samples. As shown in Fig. 15g and h, the authors used photoelectrochemical measurements, which displayed higher photovoltage and photocurrent response on ordered NNB-1 compared to disordered NNB-8, to confirm the built-in electric field of each crystal unit with consistent direction was beneficial for the directional charge transfer. Thus, the advantage of ordered ferroelectrics in charge separation/transfer dynamics was proved in this work. | ||
| Fig. 15 SEM images of NaNbO3 samples with (a) and (b) ordered, and (c) disordered nano cuboid assemblage. (d) CO2 evolution from POM reaction. Reaction conditions: 1 mL mixture of 10% CH4 and 90% O2, 300 W Xe lamp. Along the direction of the arrow, the samples are without catalyst, Pt-NNB-1, Pt-NNB-1-LD, NNB-8, Pt-NNB-8, Pt-NNB-8-LD, and NNB-1, respectively. (e) SEM image of Pt–NNB-1 sample prepared by the impregnation method. (f) Illustration of charge distribution in single crystal with ordered and disordered arrangement structures. (g) Open circuit photovoltage curves. (h) Photocurrent responses of ordered NNB-1 and disordered NNB-8.322 Copyright © 2014, Royal Society of Chemistry. | ||
Furthermore, to achieve the high selectivity of POM over photocatalysts, precise control of the surface polarity on ferroelectric materials is a credible strategy after the separation of electron–hole pairs is completed. In the CH4 to CH3OH conversion process, CH4 can interact with ˙OH on the oxidizing surface with negative polarity to produce CH3OH.22 The state of the surface is then switched to positive polarization with reducibility, allowing the O2 molecules to complete the dissociation. Due to the addition of O2, the surface is ready to oxide CH4 again. The whole cycle is therefore advantageous for the partial oxidization of CH4 to CH3OH instead of other products. However, Arvin Kakekhani et al. suggested that this theory also has some potential challenges according to DFT results. Firstly, the surface oxygens could couple with the neighbouring oxygen to form O2. In fact, the barrier to O2 leaving the surface is much lower than that for direct oxidation of CH4, suggesting that the dissociated oxygen may not participate in the oxidation reaction. Secondly, before CH3OH leaves the ferroelectric materials, the products undergo a reverse reaction to dissociate back into CH4 and surface oxygens when the surface state changes to positive polarization. Some groups have attempted to overcome these shortcomings in various photocatalytic applications by using monolayers of transition metal oxides.324 As an example, ZnO monolayer on PbTiO3 ferroelectric photocatalysts. Through the dynamically tuned catalysis mechanism, the energy barrier for CO2 conversion was obviously lowered.325 In addition, the CrO2-based monolayer also promotes NOx dissociation and CO oxidation over the PbTiO3 substrate. And the switchable polarity between strong and weak binding achieved rapid dissociation and easy desorption simultaneously, avoiding oxygen and sulfur poisoning.319 Recently, Yi et al. investigated the influence of polar and non-polar facets in methane oxidation based on ZnO ferroelectric photocatalysts.326 The ZnO nanosheets and nanorods were prepared to compare the influence of facets with different polar states (Fig. 16a and b). Compared to nanosheets with polar facets, ZnO nanorods mostly contained non-polar facets (Fig. 16c). The photocatalytic oxidation of CH4 showed distinct morphology dependence characteristics (Fig. 16d). Due to the polarization in ZnO nanosheets, the methane could be rapidly oxidated. However, ZnO nanorods showed a lower rate of photocatalytic CH4 oxidation under the same conditions. The CH4 oxidation followed the quasi-first-order reaction kinetics (Fig. 16e). The rate constants were 0.01 and 8.01 × 10−4 min−1 for ZnO nanosheets and nanorods, respectively. These results indicated the significant enhancement effect of polar facets over ZnO nanosheets in CH4 oxidation. High-energy polar facets are thermodynamically more reactive than non-polar facets, inducing polarity in stable and symmetric CH4 molecules. During the PEC measurements, ZnO nanosheets had higher photocurrent (Fig. 16f), which could be attributed to more polar facets in ZnO nanosheets. Furthermore, the authors proposed the relationship between CH4 activation and polar facets in ZnO nanostructures. As a ferroelectric material, the polar semiconductor property of ZnO could be viewed as alternately stacking multilayers of Zn2+ and O2− ions with tetrahedral coordination structure along the c-axis. This unique structure endowed ZnO with a built-in electric field. When CH4 was adsorbed on the m-plane of ZnO, the H3Cδ−–Hδ+ was polarized due to the interaction between ˙CH3 and ˙H and active sites over the m-facets. Meanwhile, the light illumination activated the C–H bond in CH4, the formation of free ˙CH3 and ˙H. On the other hand, the generation of oxygen species (activation of O2 molecules) would take place on the polar (0001) plane, forming ˙O2− radicals. Finally, ˙O2− would oxidize the intermediate to produce the products (CO2 and H2O). Although the products in this work were not high value-added chemicals, the above work indeed demonstrates the potential of an epitaxial monolayer of a transition metal oxide in the activation of reactants over the specific facet with polarization. Thus, the development of specific surface layers is beneficial to fully exploit the ferroelectric photocatalysts in POM application.
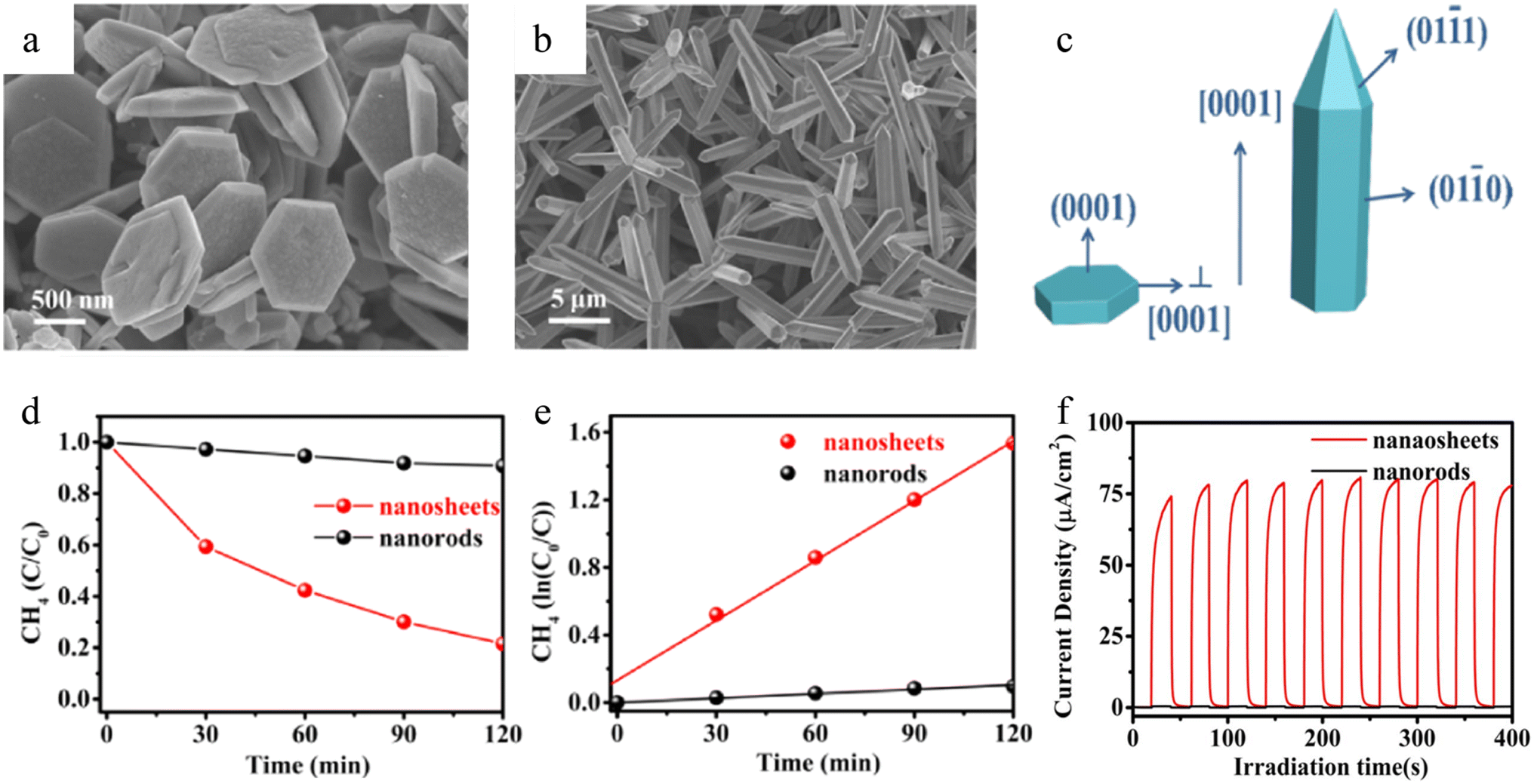 | ||
| Fig. 16 FESEM of the ZnO (a) nanosheets and (b) nanorods. (c) Growth models of ZnO. (d) Time course and (e) reaction kinetic plots of POM under simulated solar light over the ZnO. Reaction conditions: 200 ppm CH4, Xe lamp (200 mW cm−2). (f) Photocurrent responses of ZnO.326 Copyright © 2019, American Chemical Society. | ||
4.3. Ferroelectric-assisted photocatalytic OCM
Compared to the conventional thermal catalytic OCM process, photocatalytic OCM focuses on the inhibition of competitive reaction and overoxidation under mild conditions. From this perspective, ferroelectric materials are a suitable candidate for enhancing photocatalytic OCM efficiency. However, few studies reported that ferroelectric materials are used in the thermal catalytic or photocatalytic OCM field. Thus, in this section, the feasibility and possible advantages of ferroelectric-assisted OCM systems will be discussed to encourage more ferroelectrics to be developed for OCM applications. Firstly, both ferroelectric-based photocatalysts and ferroelectric cocatalysts could form an electric field due to intrinsic spontaneous polarization. The electric field would function as the above noble metal cocatalysts that become the charge acceptor via adjusting the electrons and holes to transfer onto the surface with different polarized states. Secondly, ferroelectric polarization could avoid some unfavorable effects from O2 by actively toggling the surface–absorbate interaction. Using PbTiO3 as an example, after the dissociation of O2 in the reactants, each oxygen adsorbate could bind to the surface oxygen atom, forming a surface O2 molecule. In negative polarization, the surface O2 molecules have very low dissociative binding energy. On the contrary, the interaction between positively polarized surface and surface O2 molecules is strong. Table 2 exhibits the binding energies for the surface O2 molecule.22 A positive number means the binding is favourable. Conversely, a negative number indicates a metastable state that is more unfavourable than O2. Indeed, this is meaningful for photocatalytic OCM applications because the overoxidation and inactivation from adsorbed O2 could be eliminated by controlling the polar state on ferroelectric catalysts (or cocatalysts). Finally, similar to the above-mentioned plasma-assisted photocatalysts, some novel reaction pathways of the OCM process might be found in the ferroelectric-assisted photocatalytic systems.231 Because the local surface state was changed by polarization, the localized electric field in ferroelectric materials probably led to some different intermediates or initiated OCM reactions by other oxidants except O2 and H2O2. Thereby, it is feasible to efficiently convert CH4 into high-value-added C2 products over photocatalysts containing ferroelectric materials.| Polarization status | Intact binding energy [eV] | Dissociative binding energy [eV] |
|---|---|---|
| Positive | 2.4 | 2.6 |
| Paraelectric | 0.2 | −0.8 |
| Negative | 0 | 0.4 |
4.4. Ferroelectric-assisted photocatalytic NOCM
Although photocatalytic NOCM has many advantages, unfortunately, the evolution rate of C2H6 from the photocatalytic NOCM reaction is almost an unexplored field. It is very urgent to design more efficient photocatalysts for NOCM. Several aspects should be considered for the reasonable development of NOCM photocatalysts. First, a strong local electric field may be needed to easily polarize and nucleophilically or electrophilically attack CH4. From this perspective, ferroelectric material is a suitable candidate to improve the efficiency of photocatalytic NOCM. However, few studies used ferroelectric materials in the NOCM field. Second, metallic co-catalysts are dynamically necessary for the occurrence of the half-reaction of H2 generation because the large overpotential inhibits the release of H2 across the bare oxide surface. In order to simultaneously address the above challenges, an Au/ZnO porous nanosheet with a local electric field and polar facet was designed for photocatalytic NOCM by Long's group.327 A ZnO crystal was tailored to expose [001] facet with polarized property, beneficial for the CH4 activation (Fig. 17a). Au/m-ZnO showed a bandgap absorption at ca. 395 nm and a broad absorption peak from plasmonic Au NPs (Fig. 17b). The as-prepared Au/m-ZnO samples achieved the highest evolution rates of C2H6 and H2. Further, the reaction mechanism of NOCM was investigated under different reactive atmospheres. Compared to the pure CH4 atmosphere, the C2H6 production was increased by O2 and H2O and decreased by CO2. However, the H2 evolution rate was decreased in the order of pure CH4 > CH4 + H2O > CH4 + CO2 > CH4 + O2. Thus, it could be assumed that CO2 did not favor the NOCM because CO2 is competitive for photoelectrons and inhibits the dehydrogenation process, which is an important process for CH4 activation. The decrease in H2 production could be attributed to the side reaction CH4 + CO2 → CH3COO− + H+, which would cause the stoichiometric mismatch between H2 and C2H6. Under O2-free conditions, C2H6 and H2 yields could remain stoichiometrically matched (C2H6:H2 = 1:1), indicating that the photocatalytic NOCM proceeds by a dehydrogenative process. In situ electron spin resonance (ESR) results showed the coupling between electron-occupied oxygen vacancy (VO+) and diamagnetic oxygen ion (O−) according to the characteristic peaks at 1.960 and 1.957 (Fig. 17c and d). However, light illumination did not change the VO+ signal, indicating the lattice O atoms have no effect on the NOCM process. Thus, the dehydrogenative coupling occurred at the Zn2+ atoms. CH4 was polarized by the Zn plane, resulting in Hδ+–CH3δ− polarization to generate ˙CH3 radical and H+ via the photoelectron transfer between ZnO and CH4. The plasmonic hot electrons on Au NPs promoted the H2 formation. DFT studies calculated the energy barrier of NOCM on Au/ZnO photocatalyst (Fig. 17e). The results showed the advantage of the polar Zn(001) surface in the generation of C2H6. The charge analysis showed the interaction in charge transfer between ZnO and Au, which means that Au activated the ZnO surface and enhanced the CH4 chemisorption. Meanwhile, the CH4-TPD results proved the enhancement of the CH4 dissociation on the (001) facet by the electric field coupling (Fig. 17f). Therefore, this work provided convincing evidence for the important role of the synergistic effect between ZnO ferroelectric material and Au plasma in accelerating the CH4 activation and charge-carrier interaction.
 | ||
| Fig. 17 (a) SEM image of Au/m-ZnO-4.8. (b) UV-vis spectra. In situ ESR spectra of (c) Au/m-ZnO-4.8 under CH4 atmosphere and (d) O deficiency under CH4/H2O atmospheres. (e) Energy barrier of NOCM on Zn3O3H3(001)/Au(111). (f) CH4-TPD patterns.327 Copyright © 2018, Royal Society of Chemistry. | ||
5. Potential role of ferroelectric effect during photocatalytic methane conversion
Based on the previous studies with convincing evidence of the influence of ferroelectric materials on photocatalytic processes, the potential effects of ferroelectrics on the photocatalytic methane conversion field can be summarized as follows:5.1. Promoting charge transfer
As a general effect, the polarization and corresponding built-in electric field are always mentioned and widely utilized when researchers purposely introduce ferroelectric materials into different photocatalytic systems. Considering that photocatalytic methane conversion systems must follow the general steps of photocatalysis, the sustainable internal electric field derived from ferroelectric properties should enhance charge-carrier transfer kinetics in this specific process.328 Meanwhile, as mentioned before (Section 3.3.2), different polar surfaces will lead to the separation of reduction and oxidation processes. This also adjusts the transfer routes of electrons and holes towards different surface sites, decreasing the recombination possibility. In addition, the spatial selectivity of charges over the ferroelectrics surface leads to energy band bending at the interface when the polarized surface contacts with solution.329 This characteristic has been widely used in both photocatalytic and electrocatalytic systems, because the negative surface/electrode causes upward band bending, promoting the charge migration from electrolyte/reactants to catalysts. On the contrary, the positive surface/electrode leads to downward band bending, enhancing charge transfer from ferroelectric catalysts to electrolyte/reactants.3305.2. Promoting reactant adsorption/activation
Although light illumination can directly promote chemical bond cracking, the high C–H bond energy of this small hydrocarbon molecule (CH4: ∼440 kJ mol−1) might arouse a suspicion: is the light energy enough to complete the CH4 association? In this case, the photoexcited radical species (˙OH and h+) should be more reliable in order to activate CH4 molecules. From this view, ferroelectric materials are able to create a negatively polarized surface, which is beneficial to oxygen-containing active species, and further oxidizes CH4 to intermediates or CH3OH.147 Depending on the atmosphere in the reaction systems, the intermediates (i.e. ˙CH3) will take part in the NOCM or OCM to obtain CO, CH3OH, or C2 products like C2H6 as reviewed in the previous sections. Another potential effect for accelerating reactant activation over ferroelectric surfaces might be the adsorption–desorption enhancement. It has been reported that the adsorption–desorption properties of ferroelectric materials can be tuned according to the molecules polarity.331,332 Even if CH4 is a non-polar molecule, products like CO and CH3OH are polar molecules. This suggests the surface states can be automatically switched during CH4 conversion processes, promoting reactant adsorption and subsequent activation, ensuring long-term reaction cycles. Further, DFT calculations have confirmed that 2D ferroelectric materials are able to control the adsorption energies, electron transfer, and magnetic moment of adsorbed reactant molecules so that there was a polarization-dependent gas adsorption behaviour over the monolayer surface.3335.3. Improving selectivity
Ferroelectric materials are able to switch reaction pathways to obtain desired products without stopping the reaction process and changing catalysts, demonstrating the great potential for achieving high selectivity during methane conversion systems.334 One approach to control the selectivity is the surface polarization. When the absorbance is saturated, the negative surface state will be switched to a positive state reducing oxygen species so that the overoxidation can be inhibited. Therefore, complex methane conversions can achieve high selectivity over a polarized surface.335 Besides, the selective adsorption of reactants on the polarized surface can effectively lift the limitations imposed by Sabatier's principle.336 For instance, the ferroelectric BiFeO3 could reduce the charge recombination rate from 17 to 0.6 s−1 so that increased 4.4 times the photoelectric efficiency.337 The improvement should be attributed to the compensation effect through electronic reconstruction when a built-in electric field promotes electron–hole separation.319,338 The changes in electron distribution lead to different abilities of polarized surfaces towards oxidation or reduction reactions. Thus, the switchable ferroelectric polarization allows us to control the surface affinity to desired redox reactions. Because ferroelectric materials always have large band gaps and dielectric constants, using ferroelectrics as triggers to control the dissociation of specific small hydrocarbon molecules has been reported as a strategy to tune the chemical selectivity.325 The ferroelectric matrix could dynamically reduce the reaction barrier through polarization manipulation to affect the local reaction pathways.339 In addition to changing the surface properties and reaction routes, recently, a novel idea to control the selectivity based on ferroelectric materials is using a ferroelectric field to immobilize active sites (i.e. cocatalyst on photocatalyst).340,341 To be specific, during methane conversion reactions, depositing at cocatalyst with a strong ability towards CH4 cracking over a holes-accumulated surface under the assistance of a ferroelectric field promoted the directional oxidation of CH4 molecules and the reversible reactions could be inhibited. However, the real implementation of ferroelectric photocatalysts with ideal selectivity is still challenging due to the large gap between theoretical predictions and practical paradigms. Up to now, these predictions have been mainly realized in CO2RR and NRR systems or ferroelectric electrocatalysts. These works make us hold strong confidence that ferroelectric systems should be widely and practically utilized in photocatalytic methane conversion reactions in the near future.6. Conclusions and outlook
The utilization of CH4 is one of the crucial steps in the transition from a fossil fuel energy system to a sustainable energy system. The mainstream sources for renewably obtaining CH4 are currently based on anaerobic digesters, which convert cow manure into natural gas, and CO2-based green chemistry transfer routes, such as biochemical, (photo)electrochemical, and photo(thermal) catalytic methods.342 Globally, more than 50% of H2 is produced through the reforming of CH4.343 Additionally, by producing higher hydrocarbons, various conversion routes of methane are crucial to reducing the greenhouse gas and achieving carbon neutralization. Therefore, clean production and sustainable utilization of CH4 seem like a complete and ideal strategy to deal with energy shortages and environmental problems.However, the C–H bond strength in CH4 is the biggest obstacle to converting CH4 into added-value products. As mentioned above, many thermal catalysts have been developed to overcome this thermodynamic challenge. Kinetic models and reaction pathways in these thermal catalytic systems are continuously being elucidated. Nevertheless, the high temperature needed for CH4 conversion processes leads to the inactivation of thermal catalysts and even causes excessive CO2 emissions. Fortunately, with the extensive studies of environmentally friendly photocatalysts, the harsh reaction conditions needed in thermal catalytic methane conversion are being gradually reduced, indicating that carbon neutralization becomes possible. Currently, an important problem to be solved in photocatalytic methane conversion is the insufficient efficiency mainly due to the slow charge transfer. In this regard, the ferroelectric-based photocatalysts are envisioned to play a key role in manipulating the evolution efficiency and selectivity of products from CH4 conversion by regulating the transfer kinetics. Many efforts have been made in this interdisciplinary field so far, but some obstacles and deficiencies still exist as follows.
6.1. Development of novel photocatalysts using ferroelectric materials
An increasing number of ferroelectric-assisted photocatalytic systems have been designed and fabricated to participate in reactions that produce the desired chemicals and clean energy fuels (e.g., H2 from overall water splitting and CO from CO2 reduction). However, in the catalytic methane conversion field, research based on ferroelectric materials is still rare. Thus, the excellent ability of ferroics should be further developed for improving CH4 conversion efficiency. One of the possible reasons is that some ferroelectric-based semiconductors do not have suitable energy band structures to generate photoinduced charge carriers with high redox capability. In addition, the work function of certain ferroelectric materials is not sufficient to promote the reduction process when electrons are transferred to the ferroelectric co-catalyst surface. Thus, energy band engineering is important for the design of ferroelectrics-supported photocatalytic systems. However, it is not practical to synthesize and experimentally test all candidate ferroelectrics to obtain the details of the energy band and work function. DFT calculations and hybrid functional calculations are useful to determine the electronic structure and predict ideal ferroelectric materials with sufficient redox ability and polarized state. However, the high computational cost of these methods should be considered for high throughput calculations. In addition, machine learning (ML) has recently become a highly efficient technique in materials data engineering to estimate various parameters (e.g., Eg, CB, and VB) in materials. Thus, with the improvement and supplementation of organic/inorganic databases, the ML method could provide guidance for the discovery and design of novel energy materials.6.2. Emphasize the role of the ferroelectric effect
Ferroelectric materials have been widely used in various photo(thermal)catalytic applications, but some works paid too much attention to other properties (e.g., optical properties, absorption/desorption abilities) in as-designed ferroic-based photocatalysts instead of their intrinsic ferroelectric effect. In particular, some outstanding works shown in this review also used ferroelectric materials such as BiVO4 and SrTiO3,86,192 but the authors only systematically investigated the influence of loaded co-catalyst or elemental doping on the photocatalytic activity or selectivity. To some extent, the reason for this problem is that the ferroelectric effect was not considered in the key points during the initial design process. However, it is known that the photocatalytic activity could be affected by various factors, and even a small factor could have a non-negligible effect on the overall photocatalytic efficiency. Therefore, the function of the natural ferroelectric effect and the accompanying built-in electric field should gain more interest when using ferroelectric materials not only for methane conversion reactions but also for photocatalytic applications in general.6.3. Exploring new effects from ferroelectric materials
During the photocatalytic reactions, the electron spin phenomenon plays an important role in electron migration and thus photocatalytic efficiency. The electron spin could be regulated by external fields like magnetic and electric fields.344 In detail, the spin–orbit coupling is able to change the spin orientation of both electrons and holes from semiconductors. Then the recombination between photoinduced charges will be inhibited. The electrons scatter can gradually decrease the impact from spin polarization, meaning that the spin polarization with a longer lifetime can effectively promote charge separation and transfer. From this perspective, a series of ferroelectrics with a magnetic response called the magnetoelectric effect, could be a new path toward ferroelectrics-based control of electron spin. It has been confirmed that some single-phase ferroelectric materials like BiFeO3 could adjust the spin behaviour via the interaction between intrinsic coupling between electric and magnetic order parameters.345,346 Some works also discussed the influence of spin polarization in the photocatalytic process.344 However, in the photocatalytic field, the investigation on the control of photoexcited electrons spin via ferroelectric materials is still scarce, and this blank is no exception in methane conversion areas. With the development of theories about ferroelectric materials, the spin-related mechanisms in ferroelectrics should obtain more attention for full utilization in practical photocatalytic reactions.6.4. Design of advanced reaction vectors
To further exploit the advantage of ferroelectric materials for boosting photocatalytic methane conversion, the design or update of reactors must be as important as the development of ferroelectric materials. It has been confirmed that different polarization states lead to the variation of adsorption/desorption behaviour towards reactants on ferroelectric materials. Thus, if the updated reactors are able to periodically control the surface state of ferroelectric photocatalysts, the activity, and selectivity for specific hydrocarbon products, especially C2 products, will be significantly improved. Similarly, there are some successful examples of piezoelectric/pyroelectric-assisted photocatalytic systems, proving that this proposal is reasonable and feasible. For example, the introduction of an external ultrasonic device to excite the piezoelectric effect, which forms a strong electric field in photocatalysts and thus improves the photocatalytic efficiency. In addition to the external auxiliary device, advanced flow reactors are more effective than conventional batch reactors. Although the operation, cost, and safety are major challenges for the design of flow reactors, some mature flow systems in thermal catalysis are ideal references for photocatalytic gas–solid two-phase reactions. Additionally, some membrane reactors also have unique advantages in separating catalysts from products after reaction, lowering the operating temperature, breaking the restriction of thermodynamic balance, and improving the selectivity and activity. Membrane reactors are expected to achieve the coupling of different photocatalytic applications. Therefore, if more effort is devoted to the development of reactors and assisted devices, ferroelectric-assisted photocatalytic CH4 conversion will realize industrial application at an early date.6.5. Coupling of other photocatalytic reactions
Nowadays, (i) most products of methane conversion are raw materials for downstream reactions, and (ii) ferroelectric-based materials have been widely used for photocatalytic applications beyond methane conversion. Taking these two points into account, if some advanced reactors could separate specific products of methane conversion, at least two photocatalytic reactions including methane conversion could be simultaneously and continuously conducted on ferroelectric photocatalysts. In this case, the cost of industrial production could be significantly reduced by eliminating the transportation and storage of intermediate raw materials. Meanwhile, other labour costs and space occupied by chemical production equipment, which are difficult to calculate, could also be saved by reaction coupling. However, to realize this goal, it is necessary to fabricate excellent (ferroelectric-based)photocatalysts and the aforementioned novel reaction vectors to overcome the complex requirements of various catalytic reactions.6.6. Investigation of enhancement and reaction mechanism
Compared with much scientific research focused on the development of novel photocatalysts, few mechanisms are known about each methane conversion technology. The transient intermediates and local reaction pathways need to be studied in detail because the surface state of ferroelectric materials may be changed during CH4 conversion. Beyond the reaction mechanism, little evidence has been provided to directly show the effect of polarization on charge transfer dynamics at the nano or micro-scale. Although some works claimed that electrons and holes would separate to different locations under the influence of the ferroelectric effect, more characterization data should be provided to demonstrate the migration or bias of photoexcited charge carriers. For this problem, a series of advanced characterization methods, such as in situ Fourier transform infrared spectroscopy (FT-IR), in situ X-ray photoelectron spectroscopy, piezoresponse force microscopy (PMF), and molecular probe technology, were integrated with DFT-based calculations to clarify both the enhancement and the reaction mechanism in the photocatalytic field. It is expected these advanced characterization methods will provide a deeper insight into the ferroelectric-assisted methane conversion field.It is undeniable that some high-level methane conversion routes are still at a very early stage and that ferroelectric-assisted catalysis, especially in the photocatalytic field, is far from industrial applications. However, this multidisciplinary field could develop rapidly with the presence of a new generation of advanced characterization techniques and chemical synthesis strategies. It is expected that more studies on ferroelectric-assisted photocatalytic methane conversion will be carried out in the coming years.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. L. acknowledges the program of China Scholarships Council (No. 202206250016). X. S. thanks ICREA for the ICREA Academia Prize 2020. X. S. and J. G.-A. thank MINECO/FEDER for financial support (PID2019-104171RB-I00). J. M. is indebted to the Ramón y Cajal Program (RYC2021-033820-I fellowship) funded by MCIN/AEI/10.13039/501100011033 and by the European Union “NextGenerationEU/PRTR”.References
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984 CrossRef CAS PubMed.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372 CrossRef CAS PubMed.
- H. Weldekidan, V. Strezov and G. Town, Renewable Sustainable Energy Rev., 2018, 88, 184 CrossRef CAS.
- J. O. Metzger and A. Hüttermann, Naturwissenschaften, 2009, 96, 279 CrossRef CAS PubMed.
- S. Morales, R. Miranda, D. Bustos, T. Cazares and H. Tran, J. Anal. Appl. Pyrolysis, 2014, 109, 65 CrossRef CAS.
- J. Ran, H. Zhang, J. Qu, J. Shan, K. Davey, J. M. Cairney, L. Jing and S. Qiao, Small, 2021, 17, 2100296 CrossRef CAS PubMed.
- B. Xia, Y. Zhang, J. Ran, M. Jaroniec and S. Qiao, ACS Cent. Sci., 2021, 7, 39 CrossRef CAS PubMed.
- B. L. Farrell, V. O. Igenegbai and S. Linic, ACS Catal., 2016, 6, 4340 CAS.
- E. McFarland, Science, 2012, 338, 340 CAS.
- U.S., Energy Information Administration (EIA), Annual Energy Outlook 2019, 2019.
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296 CAS.
- D. E. Holmes and J. A. Smith, Adv. Appl. Microbiol., 2016, 1–61 CAS.
- X. Li, C. Wang and J. Tang, Nat. Rev. Mater., 2022, 7, 617 CAS.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497 CAS.
- H. Song, X. Meng, Z. J. Wang, Z. Wang, H. Chen, Y. Weng, F. Ichihara, M. Oshikiri, T. Kako and J. Ye, ACS Catal., 2018, 8, 7556 CAS.
- X. Yu, V. De Waele, A. Löfberg, V. Ordomsky and A. Y. Khodakov, Nat. Commun., 2019, 10, 700 CAS.
- M. A. Gondal, A. Hameed, Z. H. Yamani and A. Arfaj, Chem. Phys. Lett., 2004, 392, 372 CAS.
- H. Yoshida, K. Hirao, J. I. Nishimoto, K. Shimura, S. Kato, H. Itoh and T. Hattori, J. Phys. Chem. C, 2008, 112, 5542 CAS.
- W. Zhang, Y. Yu and Z. Yi, J. Mater. Sci., 2017, 52, 5106 CAS.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521 CAS.
- L. Yuliati and H. Yoshida, Chem. Soc. Rev., 2008, 37, 1592 CAS.
- A. Kakekhani and S. Ismail-Beigi, Phys. Chem. Chem. Phys., 2016, 18, 19676 CAS.
- Y. Song, E. Ozdemir, S. Ramesh, A. Adishev, S. Subramanian, A. Harale, M. Albuali, B. A. Fadhel, A. Jamal, D. Moon, S. H. Choi and C. T. Yavuz, Science, 2020, 367, 777 CrossRef CAS PubMed.
- D. Kong, Y. Zheng, M. Kobielusz, Y. Wang, Z. Bai, W. Macyk, X. Wang and J. Tang, Mater. Today, 2018, 21, 897 CrossRef CAS.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645 RSC.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731 RSC.
- K. Shimura and H. Yoshida, Catal. Surv. Asia, 2014, 18, 24 CrossRef CAS.
- J. Xie, R. Jin, A. Li, Y. Bi, Q. Ruan, Y. Deng, Y. Zhang, S. Yao, G. Sankar, D. Ma and J. Tang, Nat. Catal., 2018, 1, 889 CAS.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201 CAS.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2018, 140, 526 CAS.
- Y. Lei, Z. Jia, H. Hu, L. Liu, J. Ye and D. Wang, Catalysts, 2022, 12, 1323 CAS.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72 CAS.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229 CAS.
- T. M. Khedr, K. Wang, D. Kowalski, S. M. El-Sheikh, H. M. Abdeldayem, B. Ohtani and E. Kowalska, J. Environ. Chem. Eng., 2022, 10, 107838 CAS.
- P. Jiménez-Calvo, V. Caps and V. Keller, Plasmonic Au-based junctions onto TiO2, gC3N4, and TiO2-gC3N4 systems for photocatalytic hydrogen production: Fundamentals and challenges, Renewable Sustainable Energy Rev., 2021, 149, 111095 Search PubMed.
- Z. Zhao, H. Tan, H. Zhao, Y. Lv, L. J. Zhou, Y. Song and Z. Sun, Chem. Commun., 2014, 50, 2755 CAS.
- R. Leary and A. Westwood, Carbon, 2011, 49, 741 CAS.
- B. Dai, J. Fang, Y. Yu, M. Sun, H. Huang, C. Lu, J. Kou, Y. Zhao and Z. Xu, Adv. Mater., 2020, 32, 1906361 CAS.
- L. Liu, S. Ouyang and J. Ye, Angew. Chem., Int. Ed., 2013, 52, 6689 CAS.
- X. Xu, F. Luo, W. Tang, J. Hu, H. Zeng and Y. Zhou, Adv. Funct. Mater., 2018, 28, 1804055 Search PubMed.
- L. Yin, M. Zhao, H. Hu, J. Ye and D. Wang, Chin. J. Catal., 2017, 38, 1307 CAS.
- J. Tan, X. Wang, W. Hou, X. Zhang, L. Liu, J. Ye and D. Wang, J. Alloys Compd., 2019, 792, 918 CrossRef CAS.
- M. Ye, X. Wang, E. Liu, J. Ye and D. Wang, ChemSusChem, 2018, 11, 1606 CrossRef CAS PubMed.
- J. Li, Z. Wang, H. Chen, Q. Zhang, H. Hu, L. Liu, J. Ye and D. Wang, Catal. Sci. Technol., 2021, 11, 4953 RSC.
- P. Li, L. Liu, W. An, H. Wang, H. Guo, Y. Liang and W. Cui, Appl. Catal., B, 2020, 266, 118618 CrossRef CAS.
- A. Meng, L. Zhang, B. Cheng and J. Yu, Adv. Mater., 2019, 31, 1807660 CrossRef PubMed.
- C. C. Hu and H. Teng, J. Catal., 2010, 272, 1 CAS.
- C. Liu, J. Qian, Y. Ye, H. Zhou, C.-J. Sun, C. Sheehan, Z. Zhang, G. Wan, Y.-S. Liu, J. Guo, S. Li, H. Shin, S. Hwang, T. B. Gunnoe, W. A. Goddard and S. Zhang, Nat. Catal., 2020, 4, 36 Search PubMed.
- Z. Jia, S. Ning, Y. Tong, X. Chen, H. Hu, L. Liu, J. Ye and D. Wang, ACS Appl. Nano Mater., 2021, 4, 10485 CAS.
- H. Zhao, P. Jiang and W. Cai, Chem.–Asian J., 2017, 12, 361 CAS.
- C. Han, L. Ge, C. Chen, Y. Li, X. Xiao, Y. Zhang and L. Guo, Appl. Catal., B, 2014, 147, 546 CAS.
- Q. Yu, Y. Dai, Y. Ling, Q. Wu, Z. Zhang and B. Feng, J. Environ. Chem. Eng., 2022, 10, 108693 CAS.
- J. Yang, T. Liu, H. Zhou, W. Cao, C. Chen, X. He, C. Jiang, Y. Li and Y. Wang, J. Alloys Compd., 2022, 900, 163440 CAS.
- H. Gao, P. Zhang, J. Hu, J. Pan, J. Fan and G. Shao, Appl. Surf. Sci., 2017, 391, 211 CAS.
- Y. Lei, J. Ye, J. García-Antón and H. Liu, Chin. J. Catal., 2023, 53, 72 CAS.
- P. S. Bednyakov, T. Sluka, A. K. Tagantsev, D. Damjanovic and N. Setter, Sci. Rep., 2015, 5, 15819 CAS.
- X. Liu, F. Zhang, P. Long, T. Lu, H. Zeng, Y. Liu, R. L. Withers, Y. Li and Z. Yi, Adv. Mater., 2018, 30, 1801619 Search PubMed.
- H. Li, X. Quan, S. Chen and H. Yu, Appl. Catal., B, 2017, 209, 591 CAS.
- H. Simons, A. B. Haugen, A. C. Jakobsen, S. Schmidt, F. Stöhr, M. Majkut, C. Detlefs, J. E. Daniels, D. Damjanovic and H. F. Poulsen, Nat. Mater., 2018, 17, 814 CAS.
- B. Dai, Y. Yu, Y. Chen, H. Huang, C. Lu, J. Kou, Y. Zhao and Z. Xu, Adv. Funct. Mater., 2019, 29, 1970105 CrossRef.
- X. Chen, Y. Li, X. Pan, D. Cortie, X. Huang and Z. Yi, Nat. Commun., 2016, 7, 12273 CrossRef CAS PubMed.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082 CAS.
- J. C. Yang, Y. L. Huang, Q. He and Y. H. Chu, J. Appl. Phys., 2014, 116, 066801 CrossRef.
- A. S. Pisat, G. S. Rohrer and P. A. Salvador, Semicond. Sci. Technol., 2017, 32, 103001 CrossRef.
- Y. Cui, J. Briscoe and S. Dunn, Chem. Mater., 2013, 25, 4215 CrossRef CAS.
- H. Kanda, N. Shibayama, A. J. Huckaba, Y. Lee, S. Paek, N. Klipfel, C. Roldán-Carmona, V. I. E. Queloz, G. Grancini, Y. Zhang, M. Abuhelaiqa, K. T. Cho, M. Li, M. D. Mensi, S. Kinge and M. K. Nazeeruddin, Energy Environ. Sci., 2020, 13, 1222 RSC.
- H. Huang, S. Tu, X. Du and Y. Zhang, J. Colloid Interface Sci., 2018, 509, 113 CrossRef CAS PubMed.
- C. Paillard, X. Bai, I. C. Infante, M. Guennou, G. Geneste, M. Alexe, J. Kreisel and B. Dkhil, Adv. Mater., 2016, 28, 5153 CrossRef CAS PubMed.
- F. Chen, H. Huang, L. Guo, Y. Zhang and T. Ma, Angew. Chem., 2019, 131, 10164 CrossRef.
- W. Song, P. A. Salvador and G. S. Rohrer, ACS Appl. Mater. Interfaces, 2018, 10, 41450 CrossRef CAS PubMed.
- O. Kwon, D. Seol, D. Lee, H. Han, I. Lindfors-Vrejoiu, W. Lee, S. Jesse, H. N. Lee, S. V. Kalinin, M. Alexe and Y. Kim, Adv. Mater., 2018, 30, 1703675 CrossRef PubMed.
- J. He, Y. Zhao, S. Jiang and S. Song, Sol. RRL, 2021, 5, 2000446 CAS.
- P. Wang, R. Shi, J. Zhao and T. Zhang, Adv. Sci., 2024, 11, 2305471 CrossRef CAS PubMed.
- J. Wang and S. Wang, Coord. Chem. Rev., 2022, 453, 214338 CrossRef CAS.
- Q. Luo, C. Lu, L. Liu and M. Zhu, Green Energy Environ., 2023, 8, 406 CAS.
- X. Yu, V. L. Zholobenko, S. Moldovan, D. Hu, D. Wu, V. V. Ordomsky and A. Y. Khodakov, Nat. Energy, 2020, 5, 511 CrossRef CAS.
- H. Song, X. Meng, S. Wang, W. Zhou, X. Wang, T. Kako and J. Ye, J. Am. Chem. Soc., 2019, 141, 20507 CrossRef CAS PubMed.
- K. Wang, L. Luo, C. Wang and J. Tang, Chin. J. Catal., 2023, 46, 103 CrossRef CAS.
- G.-X. Dong, M.-R. Zhang, K. Su, Z.-L. Liu, M. Zhang and T.-B. Lu, J. Mater. Chem. A, 2023, 11, 9989 CAS.
- S. Murcia-López, K. Villa, T. Andreu and J. R. Morante, ACS Catal., 2014, 4, 3013 Search PubMed.
- J. Yang, J. Hao, J. Wei, J. Dai and Y. Li, Fuel, 2020, 266, 117104 CrossRef CAS.
- S. Murcia-López, K. Villa, T. Andreu and J. R. Morante, ACS Catal., 2014, 4, 3013 CrossRef.
- H. Wei, J. Cai, Y. Zhang, X. Zhang, E. A. Baranova, J. Cui, Y. Wang, X. Shu, Y. Qin, J. Liu and Y. Wu, RSC Adv., 2020, 10, 42619 RSC.
- F. P. Koffyberg and F. A. Benko, J. Appl. Phys., 1982, 53, 1173 CrossRef CAS.
- X. Li, J. Xie, H. Rao, C. Wang and J. Tang, Angew. Chem., Int. Ed., 2020, 59, 19702 CrossRef CAS PubMed.
- S. Shoji, X. Peng, A. Yamaguchi, R. Watanabe, C. Fukuhara, Y. Cho, T. Yamamoto, S. Matsumura, M.-W. Yu, S. Ishii, T. Fujita, H. Abe and M. Miyauchi, Nat. Catal., 2020, 3, 148 CAS.
- G. A. Ozin and F. Hugues, J. Phys. Chem., 1982, 86, 5174 CrossRef CAS.
- W. Hill, B. N. Shelimov and V. B. Kazansky, J. Chem. Soc., Faraday Trans. 1, 1987, 83, 2381 RSC.
- L. Yuliati, T. Hattori, H. Itoh and H. Yoshida, J. Catal., 2008, 257, 396 CrossRef CAS.
- J. Wen, J. Xie, X. Chen and X. Li, Appl. Surf. Sci., 2017, 391, 72 CrossRef CAS.
- D. Masih, Y. Ma and S. Rohani, Appl. Catal., B, 2017, 206, 556 CAS.
- N. Li, Y. Li, R. Jiang, J. Zhou and M. Liu, Appl. Surf. Sci., 2019, 498, 143861 CrossRef CAS.
- A. Muhammad, M. Tahir, S. S. Al-Shahrani, A. Mahmood Ali and S. U. Rather, Appl. Surf. Sci., 2020, 504, 144177 CAS.
- S. Shi, Z. Sun, C. Bao, T. Gao and Y. H. Hu, Int. J. Energy Res., 2020, 44, 2740 CAS.
- Y. Zhou, L. Zhang and W. Wang, Nat. Commun., 2019, 10, 506 CAS.
- L. Li, S. Fan, X. Mu, Z. Mi and C.-J. Li, J. Am. Chem. Soc., 2014, 136, 7793 CAS.
- H. Song, X. Meng, Z. Jun Wang, H. Liu and J. Ye, Joule, 2019, 3, 1606 CAS.
- K. Wittich, M. Krämer, N. Bottke and S. A. Schunk, ChemCatChem, 2020, 12, 2130 CAS.
- G. C. Behera, K. Parida, N. F. Dummer, G. Whiting, N. Sahu, A. F. Carley, M. Conte, G. J. Hutchings and J. K. Bartley, Catal. Sci. Technol., 2013, 3, 1558 CAS.
- E. Ramírez, R. Soto, R. Bringué, M. Iborra and J. Tejero, Ind. Eng. Chem. Res., 2020, 59, 20676 Search PubMed.
- J. Li, Y. He, L. Tan, P. Zhang, X. Peng, A. Oruganti, G. Yang, H. Abe, Y. Wang and N. Tsubaki, Nat. Catal., 2018, 1, 787 CAS.
- C. D. Thomas, A. Cameron, R. E. Green, M. Bakkenes, L. J. Beaumont, Y. C. Collingham, B. F. N. Erasmus, M. F. de Siqueira, A. Grainger, L. Hannah, L. Hughes, B. Huntley, A. S. van Jaarsveld, G. F. Midgley, L. Miles, M. A. Ortega-Huerta, A. Townsend Peterson, O. L. Phillips and S. E. Williams, Nature, 2004, 427, 145 CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87 CAS.
- S. D. Angeli, G. Monteleone, A. Giaconia and A. A. Lemonidou, Int. J. Hydrogen Energy, 2014, 39, 1979 CAS.
- Y.-L. Lee, K. Lee, C. Hyun Ko and H.-S. Roh, Chem. Eng. J., 2022, 431, 134299 CAS.
- S. Z. Abbas, V. Dupont and T. Mahmud, Int. J. Hydrogen Energy, 2017, 42, 2889 CAS.
- J. Xu and G. F. Froment, AIChE J., 1989, 35, 97 CAS.
- G. Arzamendi, P. M. Diéguez, M. Montes, J. A. Odriozola, E. F. Sousa-Aguiar and L. M. Gandía, Chem. Eng. J., 2009, 154, 168 CAS.
- P. N. Kechagiopoulos, S. D. Angeli and A. A. Lemonidou, Appl. Catal., B, 2017, 205, 238 CAS.
- D. Pashchenko and I. Makarov, Energy, 2021, 222, 119993 CAS.
- D. Pashchenko, Int. J. Energy Res., 2020, 44, 438 CrossRef CAS.
- Z. Zhu, W. Guo, Y. Zhang, C. Pan, J. Xu, Y. Zhu and Y. Lou, Carbon Energy, 2021, 3, 519 CrossRef CAS.
- W. Li, J. Zhang and W. Wang, Coord. Chem. Rev., 2024, 503, 215638 CrossRef CAS.
- P. Gronchi, P. Centola and R. D. Rosso, Appl. Catal., A, 1997, 152, 83 CrossRef CAS.
- B. Christian Enger, R. Lødeng and A. Holmen, Appl. Catal., A, 2008, 346, 1 CrossRef.
- C. Papadopoulou, H. Matralis and X. Verykios, in Catalysis for Alternative Energy Generation, 2012, pp. 57–127 Search PubMed.
- A. Álvarez, M. Borges, J. J. Corral-Pérez, J. G. Olcina, L. Hu, D. Cornu, R. Huang, D. Stoian and A. Urakawa, ChemPhysChem, 2017, 18, 3135 Search PubMed.
- L. F. Bobadilla, V. Garcilaso, M. A. Centeno and J. A. Odriozola, ChemSusChem, 2017, 10, 1193 CAS.
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35 RSC.
- R. O. da Fonseca, R. C. Rabelo-Neto, R. C. C. Simões, L. V. Mattos and F. B. Noronha, Int. J. Hydrogen Energy, 2020, 45, 5182 CAS.
- B. Faroldi, J. Múnera, J. M. Falivene, I. R. Ramos, Á. G. García, L. T. Fernández, S. G. Carrazán and L. Cornaglia, Int. J. Hydrogen Energy, 2017, 42, 16127 CrossRef CAS.
- J. Yang, J. Wang, J. Zhao, Y. Bai, H. Du, Q. Wang, B. Jiang and H. Li, J. CO2 Util., 2022, 57, 101893 CAS.
- C. Agrafiotis, H. von Storch, M. Roeb and C. Sattler, Renewable Sustainable Energy Rev., 2014, 29, 656 CAS.
- M. F. Mark, W. F. Maier and F. Mark, Chem. Eng. Technol., 1997, 20, 361 CAS.
- Y. Kathiraser, U. Oemar, E. T. Saw, Z. Li and S. Kawi, Chem. Eng. J., 2015, 278, 62 CAS.
- E. le Saché and T. R. Reina, Prog. Energy Combust. Sci., 2022, 89, 100970 Search PubMed.
- C. Carrara, J. Múnera, E. A. Lombardo and L. M. Cornaglia, Top. Catal., 2008, 51, 98 CAS.
- J.-H. Park and T.-S. Chang, Catal. Lett., 2019, 149, 3148 CAS.
- I. de Dios García, A. Stankiewicz and H. Nigar, Catal. Today, 2021, 362, 72 Search PubMed.
- K. Nagaoka, Appl. Catal., A, 2004, 268, 151 CAS.
- B. Pawelec, S. Damyanova, K. Arishtirova, J. L. G. Fierro and L. Petrov, Appl. Catal., A, 2007, 323, 188 CAS.
- M. García-Diéguez, I. S. Pieta, M. C. Herrera, M. A. Larrubia and L. J. Alemany, Catal. Today, 2011, 172, 136 CrossRef.
- B. Steinhauer, M. R. Kasireddy, J. Radnik and A. Martin, Appl. Catal., A, 2009, 366, 333 CrossRef CAS.
- F. Menegazzo, M. Signoretto, F. Pinna, P. Canton and N. Pernicone, Appl. Catal., A, 2012, 439–440, 80 CrossRef CAS.
- S. Zhang, M. Ying, J. Yu, W. Zhan, L. Wang, Y. Guo and Y. Guo, Appl. Catal., B, 2021, 291, 120074 CAS.
- K. Y. Kim, J. H. Lee, H. Lee, W. Y. Noh, E. H. Kim, E. C. Ra, S. K. Kim, K. An and J. S. Lee, ACS Catal., 2021, 11, 11091 CrossRef CAS.
- A. Abdulrasheed, A. A. Jalil, Y. Gambo, M. Ibrahim, H. U. Hambali and M. Y. Shahul Hamid, Renewable Sustainable Energy Rev., 2019, 108, 175 CAS.
- O. Muraza and A. Galadima, Int. J. Energy Res., 2015, 39, 1196 Search PubMed.
- Y. Gao, J. Jiang, Y. Meng, F. Yan and A. Aihemaiti, Energy Convers. Manage., 2018, 171, 133 CAS.
- A. Tavasoli and G. Ozin, Joule, 2018, 2, 571 Search PubMed.
- A. V. Tavasoli, M. Preston and G. Ozin, Energy Environ. Sci., 2021, 14, 3098 CAS.
- F. Pan, X. Xiang, W. Deng, H. Zhao, X. Feng and Y. Li, ChemCatChem, 2018, 10, 940 CAS.
- M. Mao, Q. Zhang, Y. Yang, Y. Li, H. Huang, Z. Jiang, Q. Hu and X. Zhao, Green Chem., 2018, 20, 2857 CAS.
- L. Zhu, T. Ding, M. Gao, C. K. N. Peh and G. W. Ho, Adv. Energy Mater., 2019, 9, 1900250 Search PubMed.
- B. Han, W. Wei, L. Chang, P. Cheng and Y. H. Hu, ACS Catal., 2016, 6, 494 CAS.
- H. Liu, H. Song, W. Zhou, X. Meng and J. Ye, Angew. Chem., Int. Ed., 2018, 57, 16781 CAS.
- M. A. Boda, R. L. Withers, Y. Liu, J. Ye and Z. Yi, J. Mater. Chem. A, 2022, 10, 22977 CAS.
- Y. Li, D. Li, H. Liu, Y. Lei, R. Zhao, D. He, Z. Zheng, H. Luo and A. Liu, Catal. Sci. Technol., 2024, 14, 2722 CAS.
- H. Liu, X. Meng, W. Yang, G. Zhao, D. He and J. Ye, Chin. J. Catal., 2021, 42, 1976 CAS.
- N. Ma, N. Tanen, A. Verma, Z. Guo, T. Luo, H. (Grace) Xing and D. Jena, Appl. Phys. Lett., 2016, 109, 212101 CrossRef.
- Y. Zhao, J. Qiao, Z. Yu, P. Yu, K. Xu, S. P. Lau, W. Zhou, Z. Liu, X. Wang, W. Ji and Y. Chai, Adv. Mater., 2017, 29, 1604230 CrossRef PubMed.
- H. Song and J. Ye, Trends Chem., 2022, 4, 1094 CrossRef CAS.
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296 CAS.
- C. Peng, G. Reid, H. Wang and P. Hu, J. Chem. Phys., 2017, 147, 030901 CrossRef PubMed.
- D. Hu, V. V. Ordomsky and A. Y. Khodakov, Appl. Catal., B, 2021, 286, 119913 CrossRef CAS.
- S. Yuan, Y. Li, J. Peng, Y. M. Questell-Santiago, K. Akkiraju, L. Giordano, D. J. Zheng, S. Bagi, Y. Román-Leshkov and Y. Shao-Horn, Adv. Energy Mater., 2020, 10, 2002154 CrossRef CAS.
- R. A. Periana, D. J. Taube, S. Gamble, H. Taube, T. Satoh and H. Fujii, Science, 1998, 280, 560 CrossRef CAS PubMed.
- R. Horn and R. Schlögl, Catal. Lett., 2015, 145, 23 CrossRef CAS.
- A. A. Latimer, A. R. Kulkarni, H. Aljama, J. H. Montoya, J. S. Yoo, C. Tsai, F. Abild-Pedersen, F. Studt and J. K. Nørskov, Nat. Mater., 2017, 16, 225 CrossRef CAS PubMed.
- A. A. Latimer, H. Aljama, A. Kakekhani, J. S. Yoo, A. Kulkarni, C. Tsai, M. Garcia-Melchor, F. Abild-Pedersen and J. K. Nørskov, Phys. Chem. Chem. Phys., 2017, 19, 3575 CAS.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521 CAS.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CAS.
- A. I. Olivos-Suarez, À. Szécsényi, E. J. M. Hensen, J. Ruiz-Martinez, E. A. Pidko and J. Gascon, ACS Catal., 2016, 6, 2965 CrossRef CAS.
- Á. Szécsényi, G. Li, J. Gascon and E. A. Pidko, ACS Catal., 2018, 8, 7961 CrossRef PubMed.
- J. Kim, M. S. Abbott, D. B. Go and J. C. Hicks, ACS Energy Lett., 2016, 1, 94 CAS.
- C. R. H. de Smet, M. H. J. M. de Croon, R. J. Berger, G. B. Marin and J. C. Schouten, Appl. Catal., A, 1999, 187, 33 CAS.
- D. Hickman, J. Catal., 1992, 138, 267 CAS.
- J. Haber and E. M. Serwicka, React. Kinet. Catal. Lett., 1987, 35, 369 CAS.
- G. Groppi, W. Ibashi, M. Valentini and P. Forzatti, Chem. Eng. Sci., 2001, 56, 831 CAS.
- K. Delgado, L. Maier, S. Tischer, A. Zellner, H. Stotz and O. Deutschmann, Catalysts, 2015, 5, 871 CAS.
- R. Schwiedernoch, S. Tischer, C. Correa and O. Deutschmann, Chem. Eng. Sci., 2003, 58, 633 CAS.
- V. R. Choudhary, A. M. Rajput and B. Prabhakar, J. Catal., 1993, 139, 326 CAS.
- D. Dissanayake, J. Catal., 1991, 132, 117 CAS.
- G. A. Karim and G. Zhou, Int. J. Hydrogen Energy, 1993, 18, 125 CAS.
- A. Mosayebi, J. Taiwan Inst. Chem. Eng., 2020, 114, 36 CAS.
- K. J. Warren and J. R. Scheffe, Mater. Today Energy, 2018, 9, 39 Search PubMed.
- A. V. de Vekki and S. T. Marakaev, Russ. J. Appl. Chem., 2009, 82, 521 CrossRef.
- K. T. Dinh, M. M. Sullivan, P. Serna, R. J. Meyer, M. Dincă and Y. Román-Leshkov, ACS Catal., 2018, 8, 8306 CrossRef CAS.
- M. H. Groothaert, P. J. Smeets, B. F. Sels, P. A. Jacobs and R. A. Schoonheydt, J. Am. Chem. Soc., 2005, 127, 1394 CrossRef CAS PubMed.
- C. Berger-Karin, J. Radnik and E. V. Kondratenko, J. Catal., 2011, 280, 116 CrossRef CAS.
- L. Li, S. He, Y. Song, J. Zhao, W. Ji and C.-T. Au, J. Catal., 2012, 288, 54 CrossRef CAS.
- Y. Hu and E. Ruckenstein, J. Catal., 1996, 158, 260 CrossRef CAS.
- T. V. Choudhary and V. R. Choudhary, Angew. Chem., Int. Ed., 2008, 47, 1828 CAS.
- C. T. Au, H. Y. Wang and H. L. Wan, J. Catal., 1996, 158, 343 CAS.
- R. K. Singha, A. Shukla, A. Yadav, L. N. Sivakumar Konathala and R. Bal, Appl. Catal., B, 2017, 202, 473 CAS.
- J. Saupsor, C. Pei, H. Li, S. Wongsakulphasatch, P. Kim-Lohsoontorn, S. Ratchahat, W. Kiatkittipong, S. Assabumrungrat and J. Gong, Energy Fuels, 2021, 35, 11580 CAS.
- Y. Feng, N. Wang and X. Guo, Fuel, 2021, 290, 120094 CrossRef CAS.
- L. Zeng, Z. Cheng, J. A. Fan, L.-S. Fan and J. Gong, Nat. Rev. Chem., 2018, 2, 349 CAS.
- S. Kim, J. Lauterbach and E. Sasmaz, ACS Catal., 2021, 11, 8247 CrossRef CAS.
- I. Grčić, J. Marčec, L. Radetić, A.-M. Radovan, I. Melnjak, I. Jajčinović and I. Brnardić, Environ. Sci. Pollut. Res., 2021, 28, 18354 Search PubMed.
- S. Sugiyama, Y. Hayashi, I. Okitsu, N. Shimoda, M. Katoh, A. Furube, Y. Kato and W. Ninomiya, Catalysts, 2020, 10, 559 CAS.
- S. Murcia-López, K. Villa, T. Andreu and J. R. Morante, Chem. Commun., 2015, 51, 7249 Search PubMed.
- H. Jiang, X. Peng, A. Yamaguchi, S. Ueda, T. Fujita, H. Abe and M. Miyauchi, Sol. RRL, 2019, 3, 1900076 Search PubMed.
- K. R. Thampi, J. Kiwi and M. Grätzel, Catal. Lett., 1988, 1, 109 CAS.
- F. Schüth, Science, 2019, 363, 1282 Search PubMed.
- C. Mesters, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 223 Search PubMed.
- H. Cruchade, I. C. Medeiros-Costa, N. Nesterenko, J.-P. Gilson, L. Pinard, A. Beuque and S. Mintova, ACS Catal., 2022, 12, 14533 CrossRef CAS.
- B. Beck, V. Fleischer, S. Arndt, M. G. Hevia, A. Urakawa, P. Hugo and R. Schomäcker, Catal. Today, 2014, 228, 212 CrossRef CAS.
- C. Karakaya, H. Zhu, C. Loebick, J. G. Weissman and R. J. Kee, Catal. Today, 2018, 312, 10 CrossRef CAS.
- J. Sun, J. Thybaut and G. Marin, Catal. Today, 2008, 137, 90 CrossRef CAS.
- J. H. Lunsford, Angew. Chem., Int. Ed. Engl., 1995, 34, 970 CrossRef CAS.
- E. E. Wolf, J. Phys. Chem. Lett., 2014, 5, 986 CrossRef CAS PubMed.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497 CrossRef CAS PubMed.
- Y. Gao, L. Neal, D. Ding, W. Wu, C. Baroi, A. M. Gaffney and F. Li, ACS Catal., 2019, 9, 8592 CrossRef CAS.
- Y. Simon, F. Baronnet, G. M. Côme and P. M. Marquaire, Stud. Surf. Sci. Catal., 2004, 147, 571 CrossRef CAS.
- P. M. Couwenberg, Q. Chen and G. B. Marin, Ind. Eng. Chem. Res., 1996, 35, 3999 CAS.
- Z. Stansch, L. Mleczko and M. Baerns, Ind. Eng. Chem. Res., 1997, 36, 2568 CrossRef CAS.
- S. Da Ros, T. Barbalho Fontoura, M. Schwaab, N. Castro de Jesus and J. Pinto, Processes, 2021, 9, 2196 CrossRef CAS.
- M. Y. Sinev, Z. T. Fattakhova, V. I. Lomonosov and Y. A. Gordienko, J. Nat. Gas Chem., 2009, 18, 273 CrossRef CAS.
- H. Zanthoff and M. Baerns, Ind. Eng. Chem. Res., 1990, 29, 2 CrossRef CAS.
- Q. Chen, P. M. Couwenberg and G. B. Marin, Catal. Today, 1994, 21, 309 CrossRef CAS.
- Q. Chen, J. H. B. J. Hoebink and G. B. Marin, Ind. Eng. Chem. Res., 1991, 30, 2088 CrossRef CAS.
- Q. Chen, P. M. Couwenberg and G. B. Marin, AIChE J., 1994, 40, 521 CrossRef CAS.
- O. V. Buyevskaya, M. Rothaemel, H. W. Zanthoff and M. Baerns, J. Catal., 1994, 146, 346 CrossRef CAS.
- P. N. Kechagiopoulos, J. W. Thybaut and G. B. Marin, Ind. Eng. Chem. Res., 2014, 53, 1825 CrossRef CAS.
- R. H. Nibbelke, J. Scheerova, M. H. J. M. Decroon and G. B. Marin, J. Catal., 1995, 156, 106 CrossRef CAS.
- J. Sun, J. Thybaut and G. Marin, Catal. Today, 2008, 137, 90 CAS.
- T. Ito and J. H. Lunsford, Nature, 1985, 314, 721–722 Search PubMed.
- B. Wood, J. Catal., 2004, 225, 300 CAS.
- C. Hammond, N. Dimitratos, J. A. Lopez-Sanchez, R. L. Jenkins, G. Whiting, S. A. Kondrat, M. H. ab Rahim, M. M. Forde, A. Thetford, H. Hagen, E. E. Stangland, J. M. Moulijn, S. H. Taylor, D. J. Willock and G. J. Hutchings, ACS Catal., 2013, 3, 1835 CAS.
- J.-F. Wu, X.-D. Gao, L.-M. Wu, W. D. Wang, S.-M. Yu and S. Bai, ACS Catal., 2019, 9, 8677 Search PubMed.
- Z. Liang, T. Li, M. Kim, A. Asthagiri and J. F. Weaver, Science, 2017, 356, 299 Search PubMed.
- N. Basickes, T. E. Hogan and A. Sen, J. Am. Chem. Soc., 1996, 118, 13111 CAS.
- T. Zimmermann, M. Soorholtz, M. Bilke and F. Schüth, J. Am. Chem. Soc., 2016, 138, 12395 CrossRef CAS PubMed.
- I. H. Hristov and T. Ziegler, Organometallics, 2003, 22, 1668 CrossRef CAS.
- T. M. Gilbert, I. Hristov and T. Ziegler, Organometallics, 2001, 20, 1183 CrossRef CAS.
- S. Chempath and A. T. Bell, J. Am. Chem. Soc., 2006, 128, 4650 CrossRef CAS PubMed.
- S. Song, H. Song, L. Li, S. Wang, W. Chu, K. Peng, X. Meng, Q. Wang, B. Deng, Q. Liu, Z. Wang, Y. Weng, H. Hu, H. Lin, T. Kako and J. Ye, Nat. Catal., 2021, 4, 1032 CrossRef CAS.
- H. Song, X. Meng, Z. Wang, H. Liu and J. Ye, Joule, 2019, 3, 1606 CrossRef CAS.
- H. Li, J. Li, Z. Ai, F. Jia and L. Zhang, Angew. Chem., Int. Ed., 2018, 57, 122 CrossRef CAS PubMed.
- N. Li, R. Jiang, Y. Li, J. Zhou, Q. Ma, S. Shen and M. Liu, ACS Sustain. Chem. Eng., 2019, 7, 11455 CAS.
- D. Gerceker, A. H. Motagamwala, K. R. Rivera-Dones, J. B. Miller, G. W. Huber, M. Mavrikakis and J. A. Dumesic, ACS Catal., 2017, 7, 2088 CAS.
- K. Dutta, M. Shahryari, C.-J. Li and J. Kopyscinski, Appl. Catal. A Gen., 2023, 663, 119319 CAS.
- D. Bajec, A. Kostyniuk, A. Pohar and B. Likozar, Chem. Eng. J., 2020, 396, 125182 CAS.
- M. Kottwitz, Y. Li, R. M. Palomino, Z. Liu, G. Wang, Q. Wu, J. Huang, J. Timoshenko, S. D. Senanayake, M. Balasubramanian, D. Lu, R. G. Nuzzo and A. I. Frenkel, ACS Catal., 2019, 9, 8738 CAS.
- S. K. Kim, H. W. Kim, S. J. Han, S. W. Lee, J. Shin and Y. T. Kim, Commun. Chem., 2020, 3, 58 CrossRef CAS PubMed.
- S. F. Moya, R. L. Martins, A. Ota, E. L. Kunkes, M. Behrens and M. Schmal, Appl. Catal., A, 2012, 411–412, 105 CAS.
- H. Sheng, E. P. Schreiner, W. Zheng and R. F. Lobo, ChemPhysChem, 2018, 19, 504 CrossRef CAS PubMed.
- L. Wang, L. Tao, M. Xie, G. Xu, J. Huang and Y. Xu, Catal. Lett., 1993, 21, 35 CrossRef CAS.
- D. Soulivong, S. Norsic, M. Taoufik, C. Coperet, J. Thivolle-Cazat, S. Chakka and J.-M. Basset, J. Am. Chem. Soc., 2008, 130, 5044 CrossRef CAS PubMed.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng, M. Wei, D. Tan, R. Si, S. Zhang, J. Li, L. Sun, Z. Tang, X. Pan and X. Bao, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- Y. Xiao and A. Varma, ACS Catal., 2018, 8, 2735 CAS.
- Z. Li, Y. Xiao, P. R. Chowdhury, Z. Wu, T. Ma, J. Z. Chen, G. Wan, T.-H. Kim, D. Jing, P. He, P. J. Potdar, L. Zhou, Z. Zeng, X. Ruan, J. T. Miller, J. P. Greeley, Y. Wu and A. Varma, Nat. Catal., 2021, 4, 882 CrossRef CAS.
- Y. Kato, H. Yoshida and T. Hattori, Chem. Commun., 1998, 2389–2390 RSC.
- S. Murcia-López, M. C. Bacariza, K. Villa, J. M. Lopes, C. Henriques, J. R. Morante and T. Andreu, ACS Catal., 2017, 7, 2878 CrossRef.
- Y. Kato, N. Matsushita, H. Yoshida and T. Hattori, Catal. Commun., 2002, 3, 99 CrossRef CAS.
- S. Wu, L. Wang and J. Zhang, J. Photochem. Photobiol., C, 2021, 46, 100400 CrossRef CAS.
- W. Zhang, C. Fu, J. Low, D. Duan, J. Ma, W. Jiang, Y. Chen, H. Liu, Z. Qi, R. Long, Y. Yao, X. Li, H. Zhang, Z. Liu, J. Yang, Z. Zou and Y. Xiong, Nat. Commun., 2022, 13, 2806 CrossRef CAS PubMed.
- T. Sakthivel, G. Venugopal, A. Durairaj, S. Vasanthkumar and X. Huang, J. Ind. Eng. Chem., 2019, 72, 18 CAS.
- M. M. Vijatovic Petrovic and J. D. Bobic, in Magnetic, Ferroelectric, and Multiferroic Metal Oxides, Elsevier, 2018, pp. 35–49 Search PubMed.
- X. Deng, Y. Wu, Z. Gao and G. Zhou, J. Mater. Chem. A, 2024, 12, 567 CAS.
- A. G. Chynoweth, Phys. Rev., 1956, 102, 705 CAS.
- A. M. Glass, D. von der Linde, D. H. Auston and T. J. Negran, J. Electron. Mater., 1975, 4, 915 CAS.
- A. M. Glass, D. von der Linde and T. J. Negran, Appl. Phys. Lett., 1974, 25, 233 CAS.
- S. Tu, Y. Zhang, A. H. Reshak, S. Auluck, L. Ye, X. Han, T. Ma and H. Huang, Nano Energy, 2019, 56, 840 CAS.
- Y. Wang, M. Zhang, J. Liu, H. Zhang, F. Li, C. Tseng, B. Yang, G. Smith, J. Zhai, Z. Zhang, S. Dunn and H. Yan, Adv. Energy Mater., 2020, 10, 2001802 CAS.
- Y. AlSalka, L. I. Granone, W. Ramadan, A. Hakki, R. Dillert and D. W. Bahnemann, Appl. Catal., B, 2019, 244, 1065 CAS.
- I. Dierking and S. Al-Zangana, Nanomaterials, 2017, 7, 305 Search PubMed.
- I. Dierking, Nanomaterials, 2018, 8, 453 Search PubMed.
- S. K. Gupta, D. Budaszewski and D. P. Singh, Eur. Phys. J.: Spec. Top., 2022, 231, 673 CAS.
- R. B. Meyer, L. Liebert, L. Strzelecki and P. Keller, J. Phys., Lett., 1975, 36, 69 CrossRef.
- S. K. Gupta, Z. Sun, H.-S. Kwok and A. K. Srivastava, Liq. Cryst., 2020, 47, 1162 CrossRef CAS.
- S. Mukherjee, Z. Yuan, Z. Sun, A. Li, C. Kang, H.-S. Kwok and A. K. Srivastava, Opt. Express, 2021, 29, 8258 CrossRef PubMed.
- P. Berthele, E. Gros, B. Fracasso and J.-L. de B. de la Tocnaye, Ferroelectrics, 1998, 214, 117 CrossRef.
- S. Al-Zangana, M. Turner and I. Dierking, J. Appl. Phys., 2017, 121, 085105 CrossRef.
- A. Mertelj, D. Lisjak, M. Drofenik and M. Čopič, Nature, 2013, 504, 237 CrossRef CAS PubMed.
- Y. Yuan, T. J. Reece, P. Sharma, S. Poddar, S. Ducharme, A. Gruverman, Y. Yang and J. Huang, Nat. Mater., 2011, 10, 296 CrossRef CAS PubMed.
- B. B. Tian, J. L. Wang, S. Fusil, Y. Liu, X. L. Zhao, S. Sun, H. Shen, T. Lin, J. L. Sun, C. G. Duan, M. Bibes, A. Barthélémy, B. Dkhil, V. Garcia, X. J. Meng and J. H. Chu, Nat. Commun., 2016, 7, 11502 CrossRef CAS PubMed.
- Y. Liu, H. Aziguli, B. Zhang, W. Xu, W. Lu, J. Bernholc and Q. Wang, Nature, 2018, 562, 96 CrossRef CAS PubMed.
- B. Neese, B. Chu, S.-G. Lu, Y. Wang, E. Furman and Q. M. Zhang, Science, 2008, 321, 821 CrossRef CAS PubMed.
- H. Li, F. Liu, B. Fan, D. Ai, Z. Peng and Q. Wang, Small Methods, 2018, 2, 1700399 CrossRef.
- Y. Cui, Z. Wang, B. Li, Y. Yan, R. Xu, M. Meng and Y. Yan, Nano Energy, 2022, 99, 107429 CrossRef CAS.
- Z. Wei, T. Ji, X. Zhou, J. Guo, X. Yu, H. Liu and J. Wang, Small, 2023, 19, 2304202 CrossRef CAS PubMed.
- C. Yang, P. Wang, J. Li, Q. Wang, P. Xu, S. You, Q. Zheng and G. Zhang, Chem. Eng. J., 2021, 417, 129340 CrossRef CAS.
- S. Horiuchi and Y. Tokura, Nat. Mater., 2008, 7, 357 CrossRef CAS PubMed.
- A. L. Solomon, Phys. Rev., 1956, 104, 1191 CrossRef CAS.
- D. Bordeaux, J. Bornarel, A. Capiomont, J. Lajzerowicz-Bonneteau, J. Lajzerowicz and J. F. Legrand, Phys. Rev. Lett., 1973, 31, 314 CrossRef.
- P. Gruner-Bauer and E. Dormann, J. Phys.: Condens. Matter, 1992, 4, 5599 CAS.
- J. Valasek, Phys. Rev., 1921, 17, 475 CAS.
- Y. Sekine, R. Akiyoshi and S. Hayami, Coord. Chem. Rev., 2022, 469, 214663 CAS.
- T. Hang, W. Zhang, H.-Y. Ye and R.-G. Xiong, Chem. Soc. Rev., 2011, 40, 3577 CAS.
- T. Zhang, K. Xu, J. Li, L. He, D. W. Fu, Q. Ye and R. G. Xiong, Natl. Sci. Rev., 2023, 10, nwac240 CAS.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250 CAS.
- X.-B. Han, C.-Y. Chai, B.-D. Liang, C.-C. Fan and W. Zhang, CrystEngComm, 2022, 24, 1507 CAS.
- N. A. Spaldin, J. Solid State Chem., 2012, 195, 2 CAS.
- H. Huyan, L. Li, C. Addiego, W. Gao and X. Pan, Natl. Sci. Rev., 2019, 6, 669 CrossRef CAS PubMed.
- W. Zheng, X. Wang, X. Zhang, B. Chen, H. Suo, Z. Xing, Y. Wang, H. Wei, J. Chen, Y. Guo and F. Wang, Adv. Mater., 2023, 35, 2205410 CrossRef CAS PubMed.
- D. Li, J. Shi, Y. Xu, Y. Luo, H. Wu and Q. Meng, Natl. Sci. Rev., 2018, 5, 559 CrossRef CAS.
- Z. Sun, X. Liu, T. Khan, C. Ji, M. A. Asghar, S. Zhao, L. Li, M. Hong and J. Luo, Angew. Chem., Int. Ed., 2016, 55, 6545 CrossRef CAS PubMed.
- J.-X. Gao, W.-Y. Zhang, Z.-G. Wu, Y.-X. Zheng and D.-W. Fu, J. Am. Chem. Soc., 2020, 142, 4756 CrossRef CAS PubMed.
- C. Ji, S. Wang, L. Li, Z. Sun, M. Hong and J. Luo, Adv. Funct. Mater., 2019, 29, 1805038 CrossRef.
- K. T. Butler, J. M. Frost and A. Walsh, Energy Environ. Sci., 2015, 8, 838 RSC.
- Y. Sun, G. Niu, W. Ren, X. Meng, J. Zhao, W. Luo, Z.-G. Ye and Y.-H. Xie, ACS Nano, 2021, 15, 10982 CAS.
- C. Wang, Y. Ding, B. Liu, B. Weng, J. Hofkens and M. B. J. Roeffaers, Chem. Commun., 2023, 59, 3122 RSC.
- K. Li, S. Li, W. Zhang, Z. Shi, D. Wu, X. Chen, P. Lin, Y. Tian and X. Li, J. Colloid Interface Sci., 2021, 596, 376 CAS.
- J. San Martin, N. Dang, E. Raulerson, M. C. Beard, J. Hartenberger and Y. Yan, Angew. Chem., Int. Ed., 2022, 61, e202205572 CAS.
- X. Zhu, Y. Lin, J. San Martin, Y. Sun, D. Zhu and Y. Yan, Nat. Commun., 2019, 10, 2843 Search PubMed.
- K. Chang, J. Liu, H. Lin, N. Wang, K. Zhao, A. Zhang, F. Jin, Y. Zhong, X. Hu, W. Duan, Q. Zhang, L. Fu, Q.-K. Xue, X. Chen and S.-H. Ji, Science, 2016, 353, 274 CAS.
- W. Ding, J. Zhu, Z. Wang, Y. Gao, D. Xiao, Y. Gu, Z. Zhang and W. Zhu, Nat. Commun., 2017, 8, 14956 CAS.
- A. Chandrasekaran, A. Mishra and A. K. Singh, Nano Lett., 2017, 17, 3290 CAS.
- J. Muñoz, Adv. Mater., 2024, 36, 2305546 Search PubMed.
- Q. Yang, W. Xiong, L. Zhu, G. Gao and M. Wu, J. Am. Chem. Soc., 2017, 139, 11506 CAS.
- K. L. Seyler, D. Zhong, D. R. Klein, S. Gao, X. Zhang, B. Huang, E. Navarro-Moratalla, L. Yang, D. H. Cobden, M. A. McGuire, W. Yao, D. Xiao, P. Jarillo-Herrero and X. Xu, Nat. Phys., 2018, 14, 277 Search PubMed.
- B. Huang, G. Clark, E. Navarro-Moratalla, D. R. Klein, R. Cheng, K. L. Seyler, D. Zhong, E. Schmidgall, M. A. McGuire, D. H. Cobden, W. Yao, D. Xiao, P. Jarillo-Herrero and X. Xu, Nature, 2017, 546, 270 CAS.
- C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang, Y. Wang, Z. Q. Qiu, R. J. Cava, S. G. Louie, J. Xia and X. Zhang, Nature, 2017, 546, 265 CAS.
- L. Kou, Y. Ma, T. Liao, A. Du and C. Chen, Phys. Rev. Appl., 2018, 10, 024043 CAS.
- Y. Lei, X. Sala, J. García-Antón and J. Muñoz, Small Methods, 2024 Search PubMed.
- Y. Lei, Á. Campos-Lendínez, X. Sala, J. García-Antón and J. Muñoz, Small Struct., 2025, 6, 2400240 CAS.
- J. Muñoz, M. Palacios-Corella, I. J. Gómez, L. Zajíčková and M. Pumera, Adv. Mater., 2022, 34, 2206382 Search PubMed.
- J. L. Giocondi and G. S. Rohrer, Top. Catal., 2008, 49, 18 CAS.
- G. F. Nataf, M. Guennou, J. Kreisel, P. Hicher, R. Haumont, O. Aktas, E. K. H. Salje, L. Tortech, C. Mathieu, D. Martinotti and N. Barrett, Phys. Rev. Mater., 2017, 1, 074410 Search PubMed.
- K. Garrity, A. M. Kolpak, S. Ismail-Beigi and E. I. Altman, Adv. Mater., 2010, 22, 2969 CrossRef CAS PubMed.
- M. Du, W. Liu, N. Liu, Y. Ling and S. Kang, Nano Energy, 2024, 124, 109495 CrossRef CAS.
- S. Park, C. W. Lee, M.-G. Kang, S. Kim, H. J. Kim, J. E. Kwon, S. Y. Park, C.-Y. Kang, K. S. Hong and K. T. Nam, Phys. Chem. Chem. Phys., 2014, 16, 10408 RSC.
- F. Wu, Y. Yu, H. Yang, L. N. German, Z. Li, J. Chen, W. Yang, L. Huang, W. Shi, L. Wang and X. Wang, Adv. Mater., 2017, 29, 1701432 CrossRef PubMed.
- W. Li, F. Wang, M. Li, X. Chen, Z. Ren, H. Tian, X. Li, Y. Lu and G. Han, Nano Energy, 2018, 45, 304 CAS.
- R. Munprom, P. A. Salvador and G. S. Rohrer, J. Mater. Chem. A, 2016, 4, 2951 CAS.
- N. V. Burbure, P. A. Salvador and G. S. Rohrer, Chem. Mater., 2010, 22, 5831 CAS.
- A. Kakekhani and S. Ismail-Beigi, ACS Catal., 2015, 5, 4537 CAS.
- N. P. Zangeneh, S. Sharifnia and E. Karamian, Environ. Sci. Pollut. Res., 2020, 27, 5912 CAS.
- Z. Du, C. Petru, X. Yang, F. Chen, S. Fang, F. Pan, Y. Gang, H.-C. Zhou, Y. H. Hu and Y. Li, J. CO2 Util., 2023, 67, 102317 CAS.
- L. Jiang, Y. Zhang, Y. Qiu and Z. Yi, RSC Adv., 2014, 4, 3165 CAS.
- R. Von der Mühll, A. Sadel and P. Hagenmuller, J. Solid State Chem., 1984, 51, 176 Search PubMed.
- K. Garrity, A. Kakekhani, A. Kolpak and S. Ismail-Beigi, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 045401 Search PubMed.
- B. O. Alawode and A. M. Kolpak, J. Phys. Chem. Lett., 2016, 7, 1310 CAS.
- Z. Li, M. A. Boda, X. Pan and Z. Yi, ACS Sustain. Chem. Eng., 2019, 7, 19042 CAS.
- L. Meng, Z. Chen, Z. Ma, S. He, Y. Hou, H.-H. Li, R. Yuan, X.-H. Huang, X. Wang, X. Wang and J. Long, Energy Environ. Sci., 2018, 11, 294 RSC.
- B. Dai, Y. Chen, S. Hao, H. Huang, J. Kou, C. Lu, Z. Lin and Z. Xu, J. Phys. Chem. Lett., 2020, 11, 7407 CrossRef CAS PubMed.
- A. M. Schultz, Y. Zhang, P. A. Salvador and G. S. Rohrer, ACS Appl. Mater. Interfaces, 2011, 3, 1562 CrossRef CAS PubMed.
- H. S. Kushwaha, A. Halder and R. Vaish, J. Mater. Sci., 2018, 53, 1414 CrossRef CAS.
- Y. Yun and E. I. Altman, J. Am. Chem. Soc., 2007, 129, 15684 CAS.
- M. H. Zhao, D. A. Bonnell and J. M. Vohs, Surf. Sci., 2008, 602, 2849 CAS.
- X. Tang, J. Shang, Y. Ma, Y. Gu, C. Chen and L. Kou, ACS Appl. Mater. Interfaces, 2020, 12, 39561 CAS.
- T. L. Wan, L. Ge, Y. Pan, Q. Yuan, L. Liu, S. Sarina and L. Kou, Nanoscale, 2021, 13, 7096 CAS.
- J. Y. Y. Loh, N. P. Kherani and G. A. Ozin, Nat. Sustainability, 2021, 4, 466 Search PubMed.
- W. Ding, J. Lu, X. Tang, L. Kou and L. Liu, ACS Omega, 2023, 8, 6164 CAS.
- J. Xie, C. Guo, P. Yang, X. Wang, D. Liu and C. M. Li, Nano Energy, 2017, 31, 28 CAS.
- C. Noguera, J. Phys.: Condens. Matter, 2000, 12, R367 CAS.
- A. M. Kolpak, I. Grinberg and A. M. Rappe, Phys. Rev. Lett., 2007, 98, 166101 Search PubMed.
- C. Zhen, J. C. Yu, G. Liu and H.-M. Cheng, Chem. Commun., 2014, 50, 10416 CAS.
- F. Chen, Z. Ren, S. Gong, X. Li, G. Shen and G. Han, Chem.– Eur. J., 2016, 22, 12160 CAS.
- A. J. Welch, I. A. Digdaya, R. Kent, P. Ghougassian, H. A. Atwater and C. Xiang, ACS Energy Lett., 2021, 1540 CAS.
- J. Baltrusaitis, I. Jansen and J. D. Schuttlefield Christus, Catal. Sci. Technol., 2014, 4, 2397 CAS.
- R. Li, L.-P. Qiu, S.-Z. Cao, Z. Li, S.-L. Gao, J. Zhang, S. Ramakrishna, Y.-Z. Long, R. Li, L.-P. Qiu, S.-Z. Cao, Z. Li, S.-L. Gao, J. Zhang, Y.-Z. Long and S. Ramakrishna, Adv. Funct. Mater., 2024, 2316725 CAS.
- P. Rovillain, R. de Sousa, Y. Gallais, A. Sacuto, M. A. Méasson, D. Colson, A. Forget, M. Bibes, A. Barthélémy and M. Cazayous, Nat. Mater., 2010, 9, 975 CAS.
- A. Kumar, J. F. Scott and R. S. Katiyar, Appl. Phys. Lett., 2011, 99, 062504 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |