
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conversion of CO2 into porous metal–organic framework monoliths†
Kanchana
Sotho
 a,
Kentaro
Kadota
*b,
Takuya
Kurihara
c,
Thanakorn
Tiyawarakul
a,
Hiroki
Yamada
d,
Kanokwan
Kongpatpanich
a and
Satoshi
Horike
*abe
a,
Kentaro
Kadota
*b,
Takuya
Kurihara
c,
Thanakorn
Tiyawarakul
a,
Hiroki
Yamada
d,
Kanokwan
Kongpatpanich
a and
Satoshi
Horike
*abe
aDepartment of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology, Rayong 21210, Thailand
bDepartment of Chemistry, Graduate School of Science, Kyoto University, Kitashirakawa-Oiwakecho, Sakyo-ku, Kyoto 606-8502, Japan. E-mail: kadota.kentaro.7a@kyoto-u.ac.jp; horike.satoshi.3r@kyoto-u.ac.jp
cDivision of Material Chemistry, Graduate School of Natural Science and Technology, Kanazawa University, Kanazawa, Ishikawa 920-1192, Japan
dDiffraction and Scattering Division, Japan Synchrotron Radiation Research Institute (JASRI), Sayo, Hyogo 679-5198, Japan
eInstitute for Integrated Cell-Material Sciences, Institute for Advanced Study, Kyoto University, Yoshida-Honmachi, Sakyo-ku, Kyoto 606-8501, Japan
First published on 19th February 2025
Abstract
We demonstrate the one-pot conversion of CO2 into amorphous formate-based metal–organic frameworks (MOFs) that form grain-boundary-free monoliths with permanent porosity through hot-pressing. The local coordination geometries of metal ions are characterized using solid-state NMR and synchrotron total X-ray scattering analyses. Hot-pressing decreases the pore sizes of monoliths, enhancing the adsorption selectivity toward H2. The key for the formation of microporous monoliths is the coordination network in which formate, capable of adopting various coordination modes, is connected via stable metal–oxygen bonds.
 Kentaro Kadota | Dr Kentaro Kadota received his PhD in chemistry from Kyoto University in 2020, followed by a postdoctoral fellowship at the University of Oregon from 2020 to 2022. He returned to Kyoto in 2022 as an Assistant Professor at Kyoto University. He has received an Inoue Research Award for Young Scientists. His research interest focuses on the design and synthesis of molecular-based frameworks derived from CO2, as well as their applications for CO2 capture and utilization. |
1. Introduction
The conversion of CO2 into functional materials is essential for realizing a carbon-neutral society.1–3 The synthesis of organic polymers and carbon materials from CO2 as a feedstock has been extensively studied in the past few decades.4,5 In recent years, the synthesis of metal–organic frameworks (MOFs) from CO2 has gained attention.6 CO2 is converted into bridging linkers, such as formate (OCHO−), formylhydroborate,7 carbamate,8 and carboxylate.9 On the other hand, the inherent inertness of CO2 has restricted the structural diversity of CO2-derived linkers and functionality of the resultant MOFs. An approach to functionalize CO2-derived MOFs that does not rely on the structural diversity of CO2-derived linkers is highly demanded.Tailoring structural disorder offers a powerful route for functionalizing solid materials. Amorphous materials, such as glasses and gels, lack long-range structural order and exhibit various features, e.g., defects, isotropy, transparency, and high mechanical strength.10 Amorphous MOFs exhibit unique functionalities that are not achieved by the crystalline analogues. For example, grain-boundary-free monoliths with permanent porosity are formed via thermal and mechanical treatment, e.g., melt quenching and hot-pressing.11,12 The formability and processability of porous monoliths is attractive for enhanced volumetric gas storage capacity and recyclable heterogeneous catalysts.13,14 Meanwhile, few studies focus on amorphous CO2-derived MOFs and none of them show the formability of porous monoliths.7,15
In this work, we attempted to synthesize amorphous CO2-derived MOFs showing permanent porosity as the monolithic form. OCHO− was selected as a CO2-derived bridging linker. Borohydride (BH4−) readily converts CO2 into OCHO− by hydride transfer.7 The small steric hindrance of OCHO− allows for various coordination geometries, making it suitable for constructing an isotropic grain-boundary-free structure.16 Oxophilic Al3+ and Ga3+ ions were employed to form a strong coordination bond with OCHO− that is essential to preserve a stable porous structure through hot-pressing treatment.17
2. Experimental section
2.1. Synthesis of OCHO−-based MOFs from CO2
All chemicals were obtained from commercial suppliers and used without further purification. The powder samples of OCHO−-based MOFs, denoted as M-CO2 (M = Al3+ and Ga3+), were synthesized from CO2. Sodium borohydride (4.0 mmol) in anhydrous acetonitrile (MeCN, 20 mL) was reacted with CO2 (99.99%) at 2.0 MPa at 25 °C for 1.5 hours in a high-pressure reaction vessel. The resulting suspension was mixed with metal nitrate salts (1.0 mmol) in anhydrous methanol (MeOH, 20 mL) at 100 °C for 48 hours. The white precipitate was isolated by centrifugation, washed with MeOH, and dried under vacuum (45 and 26% yields for Al-CO2 and Ga-CO2).2.2. General characterization
Thermogravimetric analysis (TGA) was carried out using a Rigaku Thermo plus TG 8122 under N2 flow or air with a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) was carried out using a Hitachi High-Tech DSC 7200 instrument under N2 flow with a heating/cooling rate of 10 °C min−1. Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) was carried out using a JEOL JSM-7610F operated with an acceleration voltage of 200 kV. Transmission Electron Microscopy and Energy-Dispersive X-ray Spectroscopy (TEM-EDS) was carried out using a JEOL JEM-ARM 200F. Fourier transform infrared (FT-IR) spectra were collected using a Bruker ALPHA II FT-IR spectrometer with a Universal ATR accessory under a N2 atmosphere. Inductively coupled plasma-optical emission spectrometry (ICP-OES) was carried out using an Agilent 700 series. The supernatant (200 μL) was dried and digested in a solution of 2% nitric acid with a total volume of 5 mL before a measurement. The average static water contact angle was measured at 25 °C, using a contact angle goniometer by dropping 10 μL of deionized water on three different locations on a monolith. The contact angle of the droplet was analyzed using the Ossila contact angle software.2.3. X-ray analysis
Powder X-ray diffraction (PXRD) patterns were collected on a Rigaku SmartLab SE X-ray type with CuKα radiation (λ = 1.54059 Å). X-ray photoelectron spectroscopy (XPS) spectra were collected using a JEOL XPS instrument (JPS-9010 MC) with a Mg Kα and Al Kα source (1253.6 eV and 1486.6 eV) under high vacuum (10−7 Pa). All binding energy values were referenced to the C 1s peak (284.70 eV). Synchrotron variable-temperature (VT) PXRD patterns were collected using synchrotron radiation (λ = 0.99927 Å) employing a large Debye–Scherrer camera with semiconductor detectors on the BL02B2 beamline at the Super Photon Ring (SPring-8, Hyogo, Japan). Pair distribution function (PDF) analysis was performed using synchrotron scattering data. Total X-ray scattering was collected at 30 °C with two 2D CdTe detectors at the BL04B2 beamline in SPring-8. The incident energy was 112.9232 keV. G(r) was obtained from the Fourier transform of S(Q) with a Lorch modification function by using IgorPro software. X-ray absorption spectroscopy (XAS) including X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) regions was performed in the transmission mode at the BL14B2 beamline at SPring-8.2.4. NMR analysis
1H, 11B, and 13C solution NMR spectra were collected using a Bruker Avance III HD 600 MHz. The powder samples were digested in 1.0 M DCl/D2O/DMSO-d6 for NMR measurement. CHN elemental analysis was performed using a LECO TruSpec Micro Element Series with cystine standard. Solid-state 1H, 13C, and 27Al magic-angle spinning (MAS) NMR experiments were conducted on a JEOL JNM-ECZ600R spectrometer at a 14.1 T superconductor magnet at room temperature. 13C cross-polarization (CP)/MAS, 1H–13C 2D CP-heteronuclear correlation (HETCOR), 27Al single-pulse, Harn-echo, and 3QMAS measurements were performed using a JEOL 3.2 mm double resonance MAS probe at a MAS rate of 20 kHz. In the CP/MAS sequence, a 1H 90° pulse width of 2.3 μs and 1H and 13C spin-lock pulse strengths of 70 and 50 kHz with a contact time of 3 ms were used. A ramped-amplitude spin-lock pulse was used for 13C. 13C signals were acquired under 1H TPPM heteronuclear dipolar decoupling pulse irradiation with a pulse strength of 100 kHz. 27Al single-pulse and Hahn-echo MAS spectra were measured with 90° and 180° pulse lengths of 1.15 and 2.3 μs. The 27Al 3QMAS spectrum was obtained using the z-filter 3QMAS sequence. 0 quantum (Q)–3Q excitation, 3Q–0Q reconversion, and weak 90° pulse lengths of 3.3, 1.1, and 8.0 μs were used. 27Al quadrupolar line-shape analysis was performed using ssNake software.182.5. Catalytic activity of M-CO2 for CO2 cycloaddition
Epichlorohydrin (ECH, 12.5 mmol), M-CO2 (0.3–1.0 mol%), and tetrabutylammonium bromide (TBAB, 2.0 mol%) were added to the reaction vial (20 mL). CO2 (99.99%) was introduced into the reaction mixture under stirring at 0.1 MPa at 30 °C for 48 hours. The reaction using styrene oxide was carried out at 80 °C. The conversion of ECH into chloropropylene carbonate (CPC) was calculated based on 1H NMR analysis. The recovered catalyst was collected by centrifugation, followed by washing with fresh MeOH and dried in a vacuum.2.6. Preparation of M-CO2 monolith by hot-pressing
The powder sample of M-CO2 (20 mg) was introduced into a 7 mm diameter stainless steel die set. A monolith was prepared under air by hot-pressing at 145 °C and 50–55 kN for 2 hours. After pressure release, the die set was taken out to cool down to room temperature.2.7. Gas adsorption analysis
Gas adsorption isotherms were collected using a MicrotracBEL BELSORP-mini X for N2 at 77 K and CO2 at 195, 273, and 298 K. Brunauer–Emmett–Teller surface areas (SBET) were calculated from the N2 adsorption isotherms. H2 adsorption isotherms at 77 K were recorded on a MicrotracBEL BELSORP-mini II. The pore size distribution (PSD) was calculated using the non-local density functional theory (NLDFT) method based on the N2 adsorption isotherms. NH3 temperature-programmed desorption (TPD) was carried out using a MicrotracBEL BELCAT. NH3 was captured with 50 mg of powder sample at 40 °C for 30 minutes and the desorption profile was recorded using a TCD detector.3. Results and discussion
3.1. Synthesis and structural characterization of M-CO2
The synthesis is a one-pot, two-step procedure involving the insertion of CO2 into BH4− to afford formylhydroborate ([BH(OCHO)3]−) and subsequent solvothermal reaction that converts [BH(OCHO)3]− into OCHO− (Fig. 1).7 This method is classified as a type-II (one-pot) synthesis of CO2-derived MOFs according to the literature.6 The choice of metal ions and solvents was essential for obtaining amorphous porous structures. A synthetic attempt using Ce3+ ion instead of Al3+/Ga3+ resulted in a non-porous crystalline product (Fig. S1 and S2†). The larger ionic radius of Ce3+ led to a higher coordination number, forming a dense structure. Methoxide (MeO−), derived from MeOH, serves as a linker in both M-CO2 (details below). Control experiments using ethanol (EtOH) instead of MeOH as a synthetic solvent provided amorphous non-porous products consisting of OCHO− and Al3+ without EtO− (Fig. S3–S5†). This indicates that the smaller steric hindrance of MeO− is suitable for constructing the porous structure of M-CO2. | ||
| Fig. 1 Schematic illustration of the synthesis and proposed structures of M-CO2 (M = Al3+, Ga3+). Carbon and oxygen atoms derived from CO2 are highlighted. The symbol X in the structure of Ga-CO2 indicates either O2−, OH−, or OMe−. SEM images of M-CO2 are displayed, respectively. The scale bar indicates 1 μm. | ||
Acid-digested solution NMR was carried out to confirm the formation of OCHO−. The 1H NMR spectra of M-CO2 show peaks at 3.1 and 8.1 ppm corresponding to MeO− and OCHO−, respectively. The ratios of OCHO− and MeO− were calculated as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:1.8 for Al-CO2 and Ga-CO2 (Fig. S6†). The assignment was also confirmed by 13C NMR (Fig. S7†). The 11B NMR spectra showed that M-CO2 contained no boron species derived from NaBH4 (Fig. S8†). The FT-IR spectra exhibit the C
:1 and 1:1.8 for Al-CO2 and Ga-CO2 (Fig. S6†). The assignment was also confirmed by 13C NMR (Fig. S7†). The 11B NMR spectra showed that M-CO2 contained no boron species derived from NaBH4 (Fig. S8†). The FT-IR spectra exhibit the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of OCHO− at 1581 and 1652 cm−1 for Al-CO2 and Ga-CO2, respectively (Fig. S9†).19 The broad peak at 3420 cm−1 in Ga-CO2 was attributed to the O–H stretching vibration of OH−.20
O stretching vibration of OCHO− at 1581 and 1652 cm−1 for Al-CO2 and Ga-CO2, respectively (Fig. S9†).19 The broad peak at 3420 cm−1 in Ga-CO2 was attributed to the O–H stretching vibration of OH−.20
The metal content of M-CO2 was determined by pyrolysis under air. TGA heating up to 900 °C under a flow of air fully converted M-CO2 into corresponding metal oxides (Fig. S10 and S11†). The gravimetric metal contents were calculated as 19.3 and 59.1 wt% for Al-CO2 and Ga-CO2, respectively. Given the results of CHN elemental analysis, the chemical compositions were determined as [Al2(OCHO)3(OMe)3] for Al-CO2 and [Ga2O1.8(OHCO)0.5(OMe)0.9(OH)] for Ga-CO2. The gravimetric CO2 content was calculated as 46.8 and 9.3 wt% for Al-CO2 and Ga-CO2 (Table S1†).
The PXRD patterns exhibit broad features at 8.1, 10.8, and 12.3° for Al-CO2 and at 5.1° for Ga-CO2, which do not match the reported patterns of metal formate compounds (Fig. 2A and S12†).19,21,22 SEM-EDX exhibits spherical particles with a diameter of 5 and 2 μm for Al-CO2 and Ga-CO2 respectively and a homogeneous distribution of each metal element (Fig. 1 and S13, S14†). TEM-EDS also confirmed the homogeneous distribution of each element in M-CO2 (Fig. S15 and S16†). Solid-state 1H–13C CP-HETCOR NMR experiment was carried out to examine the intermolecular distance correlation between OCHO− and MeO−. The 1H–13C HETCOR NMR spectrum of Al-CO2 exhibits a clear correlation peak between 1H of OCHO− at 8.5 ppm and 13C of MeO− at 51.5 ppm (Fig. 2B). Ga-CO2 also exhibits the correlation between OCHO− and MeO− (Fig. S17†). The observed correlations indicate that both OCHO− and MeO− are homogeneously distributed to form the structure of M-CO2.
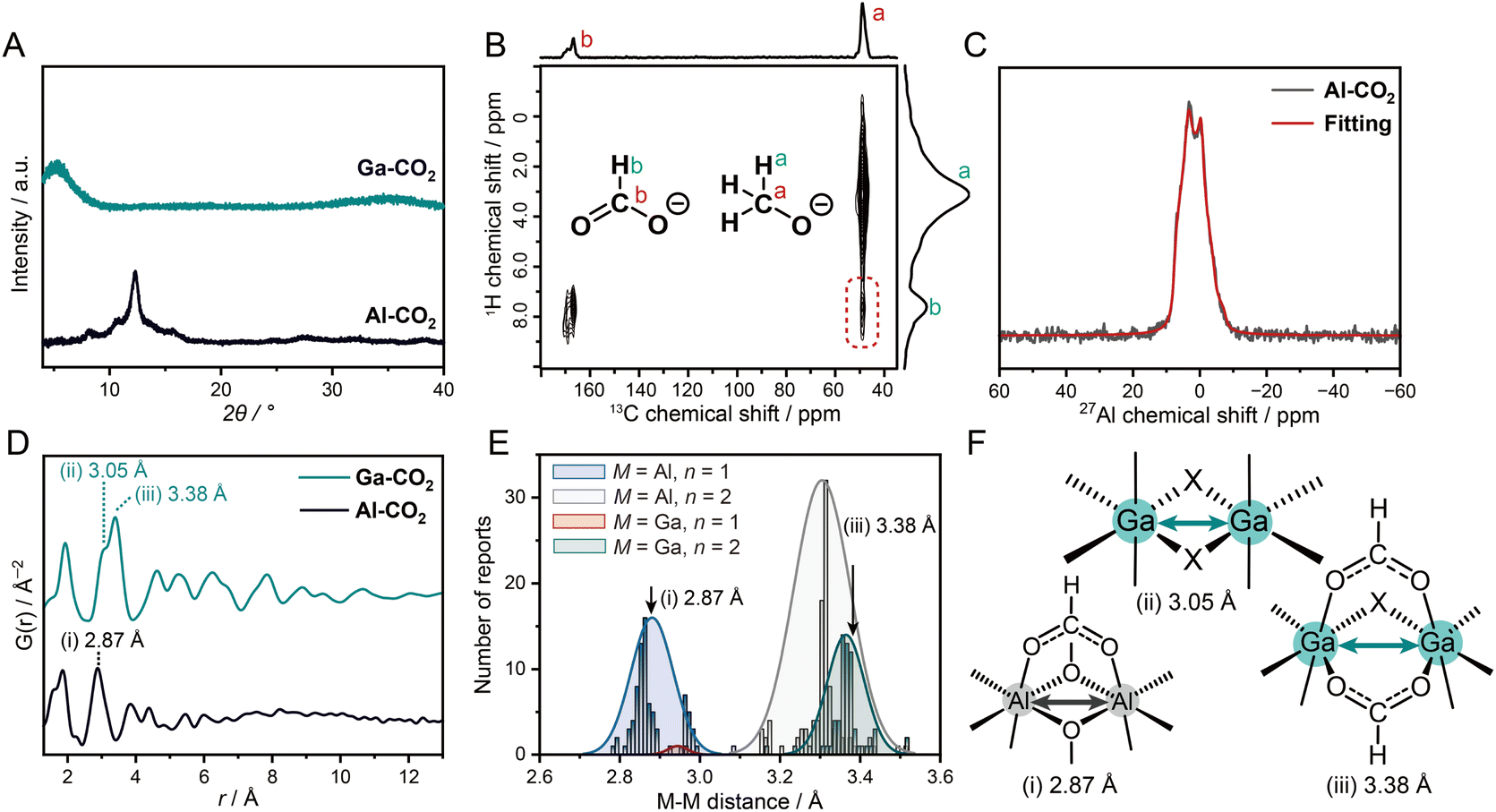 | ||
| Fig. 2 Structural characterization of M-CO2. (A) PXRD patterns of Al-CO2 and Ga-CO2. (B) 1H–13C CP-HETCOR NMR spectra of Al-CO2. The correlation between 1H of OCHO− and 13C of MeO− is highlighted as a green box. (C) 27Al 1D sliced 3QMAS NMR spectrum (black) and fitting (red) of Al-CO2. (D) PDF profiles of Al-CO2 and Ga-CO2. (E) Histograms of M–M distances for [M–(R–COO)n(X)3−n–M] with Gaussian distribution. (F) Proposed M–M building units of M-CO2. X = O2−, OH−, or OMe−. | ||
The local coordination geometry of Al3+ was characterized by 27Al MAS NMR. The 27Al NMR spectrum of Al-CO2 exhibits an asymmetric line shape due to the 27Al quadrupolar interaction (Fig. S18†). To analyze the coordination structure, we performed quadrupolar line-shape fitting to the 27Al 1D spectrum sliced at the peak center of the isotropic dimension (12.31 ppm) of the 3QMAS spectrum (Fig. 2C and S19†). The isotropic chemical shift δiso = 8.21 ppm, quadrupolar coupling constant CQ = 5.00 MHz, and asymmetry parameter η = 0.48 were obtained. The δiso value suggests that Al3+ forms an octahedral six-coordinated geometry.23,24 The 27Al MAS spectrum of [Al(OCHO)3], possessing the highly symmetric Al3+–6O coordination structure, shows a narrow peak with CQ ∼ 0 (Fig. S20†). The larger CQ value of Al-CO2 reflects an asymmetric coordination structure of Al3+ bound with both OCHO− and MeO−. The non-zero η suggests a low axial-symmetry structure around Al3+ due to the random distribution of the two linkers. The coordination geometry of Ga3+ in Ga-CO2 was characterized by synchrotron XAS. The EXAFS fitting on the first coordination shell confirmed that the coordination number of Ga3+ in Ga-CO2 was 5.6 ± 0.6, which is indicative of octahedral geometry (Fig. S21 and Table S2†). The formation of octahedral geometry in M-CO2 was also confirmed by XPS (Fig. S22 and S23†). The binding energies of O (1s), C (1s), and Al (2p3/2) were found to be 530.1, 288.0 and 71.6 eV in Al-CO2 which correspond to Al–O coordination and the presence of OHCO−.25Ga-CO2 shows binding energies of O (1s) and Ga (2p3/2) at 531.5 eV and 1118.9 eV which belong to Ga–O bond and Ga3+, respectively.
PDF analysis was performed to characterize the structural periodicity of M-CO2 (Fig. 2D). The peaks below 2 Å were assigned as the metal–oxygen bonds (1.85 and 1.95 Å for Al-CO2 and Ga-CO2).26,27 The peaks between 2.0 and 4.0 Å are assigned as metal–metal (M–M) correlations (2.87 Å for Al-CO2 and 3.05 and 3.38 Å for Ga-CO2). To figure out the bridging mode of M–M, we surveyed M–M distances of an Al3+/Ga3+-based coordination compound with octahedral geometry and R–COO− groups in the CCDC database. The histograms of M–M distances were plotted for the building units of [M–(R–COO)n(X)3−n–M] (M = Al3+, Ga3+; X = O2−/OH−/OMe−; n = 1, 2, Fig. 2E). We assign the peak at 2.87 Å in Al-CO2 as [Al–(OCHO)(OMe)2–Al] and 3.38 Å in Ga-CO2 as [Ga–(OCHO)2(X)–Ga] (Fig. 2E). The peak at 3.05 Å in Ga-CO2 is assigned as [Ga–(X)2–Ga], edge-sharing octahedral geometry (Fig. 2F and Tables S3, S4†). The mixed coordination of OCHO− and OMe− in Al-CO2 is consistent with the low axial-symmetry structure of Al3+ observed by 27Al MAS NMR. The longer-range periodicity was observed up to around 8 and 12 Å for Al-CO2 and Ga-CO2. The extended network is formed by connecting the M–M building units as proposed in Fig. 1.
3.2. Thermal and chemical stability
The TGA profiles under N2 indicate that there was no significant weight loss up to 190 and 180 °C for Al-CO2 and Ga-CO2 (Fig. S24†). The thermal stability is slightly lower than those of [M(OCHO)3] (decomposition temperatures: 200 and 230 °C for Al3+ and Ga3+).21 The thermal stability of Al-CO2 was studied by synchrotron variable-temperature PXRD (Fig. S25†). The crystallinity of Al-CO2 was maintained up to 200 °C and the intensity reduced when heated to 320 °C. The stability of common organic solvents was also studied. The PXRD pattern of Al-CO2 was intact after soaking in common organic solvents, e.g., tetrahydrofuran, chloroform, and toluene, for 24 h at 25 °C, respectively (Fig. S26†).3.3. Characterization of the defective open metal site (OMS) for CO2 cycloaddition
We evaluated the Lewis acidity of defective OMS in M-CO2 by NH3-TPD (Fig. 3). NH3 was completely released below 190 °C within 2 hours, indicating that the defective OMS in M-CO2 serves as a weak acid site.28 The total acidic sites were calculated as 0.59 and 2.25 mmol g−1 for Al-CO2 and Ga-CO2, respectively. The larger amount of acidic sites in Ga-CO2 was attributed to a defective OMS in the amorphous structure.15 Defective OMS serves as a catalytically active site for CO2 cycloaddition with epoxides. The catalytic activity of M-CO2 was evaluated with ECH as a model reaction. The solvent-free reaction of ECH and CO2 in the presence of 1.0 mol% of M-CO2 and TBAB as a co-catalyst at 30 °C yielded CPC with a high conversion of 94.4 and 91.6% for Al-CO2 and Ga-CO2 (Fig. 3 and S27†). The catalyst amount and reaction time were optimized in the range of 0.3–1.0 mol% and 12–72 h (Fig. S28 and Table S5†). The control experiment using only TBAB without M-CO2 catalysts resulted in a conversion of 67.5%. The catalytic activity of M-CO2 maintained the conversion exceeding 90% after three cycles, which is comparable to those of MOFs with catalytically active OMS (Table S7†).29–31 ICP-OES on the supernatant after the reactions confirms negligible leaching of metal ions from M-CO2 over three cycles (Al-CO2: 0.14–0.33 ppm and Ga-CO2: 0.07 ppm, Table S7†).32,33Al-CO2 also exhibited catalytic activity toward the CO2 cycloaddition of styrene oxide into styrene carbonate at 80 °C with conversion of 76.3% (Fig. S29 and Table S6†). Although the diffraction intensity of recycled Al-CO2 decreased in PXRD, N2 adsorption isotherms and SBET values were comparable after three cycles (Fig. S30 and S31†). In contrast, the recycled Ga-CO2 exhibited a large decrease in N2 adsorption, whereas the catalytic activity was preserved. This indicates that the catalytic reaction in Ga-CO2 mainly occurred at the particle surface rather than internal pores, which is consistent with the catalytic CO2 cycloaddition at the surface of dense MOFs.34 This is also supported by the smaller pore size of Ga-CO2 than Al-CO2 confirmed by N2 adsorption (Fig. S32†). The results indicate that CO2-derived MOFs serve as a catalyst for CO2 cycloaddition under ambient conditions.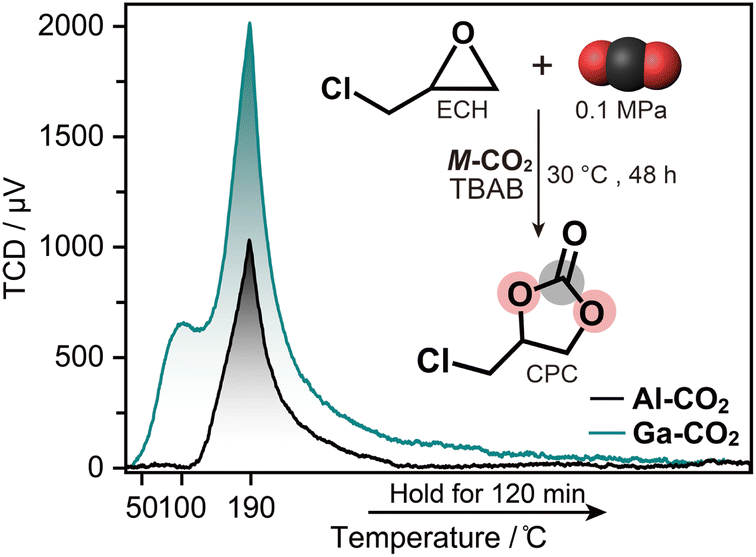 | ||
| Fig. 3 NH3-TPD profiles of M-CO2. Inset: conversion of ECH into CPC via CO2 cycloaddition reaction using M-CO2 catalysis with TBAB as a co-catalyst. | ||
3.4. Formability of transparent monoliths
Hot-pressing provided a transparent monolith of Al-CO2 (Fig. 4A). In contrast, the Ga-CO2 monolith prepared by hot-pressing under the same conditions was not transparent. The PXRD pattern of the Al-CO2 monolith exhibits broad features in the same peak positions before hot-pressing, indicative of preservation of the long-range order (Fig. S33†). The differential scanning calorimetry (DSC) profile of Al-CO2 ground powder does not display any features assignable to the glass transition in the temperature range from −50 to 150 °C (Fig. S34†). The SEM image of the Al-CO2 monolith displays a grain-boundary-free, smooth surface, resulting in the formation of a transparent monolith (Fig. 4B and S35†). The surface hydrophobicity of the M-CO2 monolith was tested by a contact angle analysis (Fig. 4C and S36†). The monolithic samples of Al-CO2 and Ga-CO2 exhibit water contact angles of 121 ± 0.4 and 116 ± 0.8°, which are categorized as hydrophobic surfaces as the angles are greater than 90°.35 The hydrophobic feature of M-CO2 is attributed to the presence of the methyl group of MeO−. The lower hydrophobicity of Ga-CO2 was due to the OH− group which can form hydrogen bonding with water molecules.35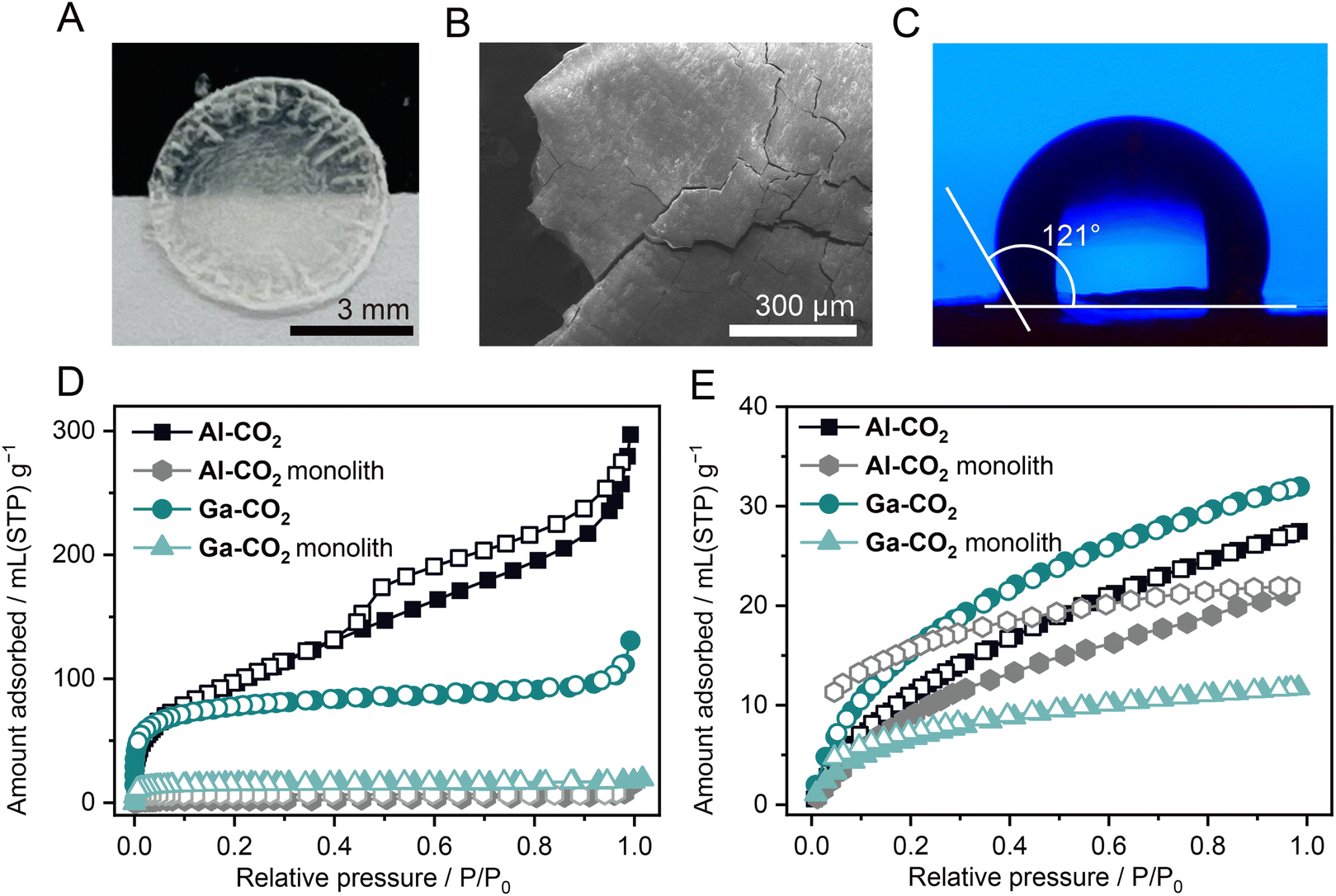 | ||
| Fig. 4 (A) Optical image of the transparent Al-CO2 monolith with a diameter of 7.0 mm. (B) SEM image of the Al-CO2 monolith. (C) Image of a water droplet on the surface of the Al-CO2 monolith. (D) N2 and (E) H2 adsorption isotherms at 77 K for M-CO2 powder and monoliths. Solid/open symbols indicate adsorption/desorption isotherms, respectively. | ||
3.5. Porous properties as powder and monoliths
The porous properties of MOFs as a monolithic form are also essential for wide applications such as catalytic processes and volumetric gas adsorption capacity.36–38 The gas adsorption measurements were carried out for both powder and monolith samples (N2 at 77 K, CO2 at 195 K, and H2 at 77 K, Fig. 4D, E and S37†). The gas uptake and SBET values calculated from the N2 adsorption isotherms are displayed in Table S8.† The N2 and CO2 uptake of M-CO2 monoliths largely decreased by 94 and 81% for Al-CO2 and 85 and 83% for Ga-CO2, respectively (Table S8†). This is consistent with the decrease of SBET values of M-CO2 monoliths, which are reduced by 91% for Al-CO2 and 80% for Ga-CO2. In contrast, M-CO2 monoliths exhibit a smaller decrease in H2 uptake compared to the reduction in N2 and CO2 uptake (22 and 63% for Al-CO2 and Ga-CO2, respectively). As the kinetic diameters increase (2.89, 3.30, and 3.65 Å for H2, CO2, and N2),39 larger decreases in gas uptake for both M-CO2 monoliths were observed. This indicates the pore size reduction of M-CO2 through hot-pressing, which is consistent with mechanical pressure-induced pore size reduction observed in flexible zeolitic imidazolate frameworks (ZIFs).40 The key for the preservation of the microporosity of M-CO2 as the monolithic form is attributed to the coordination network in which OCHO−, capable of adopting various coordination modes, is connected via stable Al/Ga–O bonds. Hybrid perovskites composed of OCHO− exhibit structural transformation in response to mechanical pressure, driven by alternation in the coordination modes of OCHO−.41 The flexibility of the coordination geometry of OCHO− originates from its low steric hindrance.16 Due to the coordination flexibility and stable Al/Ga–O bonds, hot-pressing does not collapse M-CO2 into a non-porous structure, and the microporous structure is maintained while the pore size decreases.4. Conclusions
We demonstrated the one-pot synthesis of amorphous MOFs with permanent porosity, [Al2(OCHO)3(OMe)3] (Al-CO2) and [Ga2O1.8(OHCO)0.5(OMe)0.9(OH)] (Ga-CO2), from CO2 by converting CO2 into OHCO−. The local coordination geometries of the metal center were revealed by solid-state NMR and synchrotron total X-ray scattering analysis. Al-CO2 formed a grain-boundary-free transparent microporous monolith via hot-pressing. Hot-pressing treatment mechanically reduced the pore size of M-CO2 monoliths, enhancing adsorption selectivity toward H2. The key for the formation of the porous monolith was attributed to the coordination flexibility of OHCO− and stable metal–oxygen bonds. These findings offer a new design guideline for CO2-derived porous materials by incorporating structural disorder.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
K. K., S. H. and K. S. designed the project. K. S., K. K., T. K., T. T., H. Y., and K. K. contributed to data collection and formal analyses. K. K., S. H. and K. S. wrote the manuscript and all the authors approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Japan Society of the Promotion of Science (JSPS) for a Grant-in-Aid for Early-Career Scientists (JP24K17696), the Japan Science and Technology Agency (JST) for a JST-ALCA-Next (JPMJAN24C4), Trans-formative Research Areas (A) “Supraceramics” (JP22H05147), fund for the Promotion of Joint International Research (International Collaborative Research, JP24K0112) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, and Thailand Science Research and Innovation (TSRI) under the fiscal year 2025 funding program (FRB680014/0457NM) and NSRF via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation, Thailand (B40G660034). The synchrotron radiation experiments were performed at BL02B2, BL04B2, and BL14B2 in SPring-8 with the approval of RIKEN (2023B1639, 2023B1721, and 2023B2001).Notes and references
- J. Artz, T. E. Muller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
- A. Otto, T. Grube, S. Schiebahn and D. Stolten, Energy Environ. Sci., 2015, 8, 3283–3297 RSC.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- X. Cheng, M. Wu, J. Li, W. Wang, N. Mitsuzaki and Z. Chen, Catal. Sci. Technol., 2023, 13, 3891–3900 RSC.
- B. Grignard, S. Gennen, C. Jerome, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- K. Kadota and S. Horike, Acc. Chem. Res., 2024, 57, 3206–3216 CrossRef CAS PubMed.
- K. Kadota, N. T. Duong, Y. Nishiyama, E. Sivaniah and S. Horike, Chem. Commun., 2019, 55, 9283–9286 RSC.
- K. Kadota, Y. L. Hong, Y. Nishiyama, E. Sivaniah, D. Packwood and S. Horike, J. Am. Chem. Soc., 2021, 143, 16750–16757 CrossRef CAS PubMed.
- B. Song, Y. Liang, Y. Zhou, L. Zhang, H. Li, N.-X. Zhu, B. Z. Tang, D. Zhao and B. Liu, J. Am. Chem. Soc., 2024, 146, 14835–14843 CrossRef CAS PubMed.
- Z. Guo, Z. Liu and R. Tang, Mater. Chem. Front., 2024, 8, 1703–1730 RSC.
- Y. Zhao, S. Y. Lee, N. Becknell, O. M. Yaghi and C. A. Angell, J. Am. Chem. Soc., 2016, 138, 10818–10821 CrossRef CAS PubMed.
- Y.-S. Wei, Z. Fan, C. Luo and S. Horike, Nat. Synth., 2023, 3, 214–223 CrossRef.
- J. Hou, A. F. Sapnik and T. D. Bennett, Chem. Sci., 2020, 11, 310–323 RSC.
- D. G. Madden, D. O'Nolan, N. Rampal, R. Babu, C. Camur, A. N. Al Shakhs, S. Y. Zhang, G. A. Rance, J. Perez, N. P. Maria Casati, C. Cuadrado-Collados, D. O'Sullivan, N. P. Rice, T. Gennett, P. Parilla, S. Shulda, K. E. Hurst, V. Stavila, M. D. Allendorf, J. Silvestre-Albero, A. C. Forse, N. R. Champness, K. W. Chapman and D. Fairen-Jimenez, J. Am. Chem. Soc., 2022, 144, 13729–13739 CrossRef CAS PubMed.
- Z. Fang, Y. Hu, B. Yao, Z. Z. Ye and X. S. Peng, J. Mater. Chem. A, 2024, 12, 6112–6122 RSC.
- A. D. Nicholas, A. Arteaga, L. C. Ducati, E. C. Buck, J. Autschbach and R. G. Surbella III, Chem.–Eur. J., 2023, 29, e202300077 CrossRef CAS PubMed.
- W. Xu, N. Hanikel, K. A. Lomachenko, C. Atzori, A. Lund, H. Lyu, Z. Zhou, C. A. Angell and O. M. Yaghi, Angew. Chem., Int. Ed., 2023, 62, e202300003 CrossRef CAS PubMed.
- S. G. J. van Meerten, W. M. J. Franssen and A. P. M. Kentgens, J. Magn. Reson., 2019, 301, 56–66 CrossRef CAS PubMed.
- V. N. Krasil'nikov, A. P. Tyutyunnik, I. V. Baklanova, A. N. Enyashin, I. F. Berger and V. G. Zubkov, CrystEngComm, 2018, 20, 2741–2748 RSC.
- M. Calatayud, S. E. Collins, M. A. Baltanás and A. L. Bonivardi, Phys. Chem. Chem. Phys., 2009, 11, 1397–1405 Search PubMed.
- Y.-Q. Tian, Y.-M. Zhao, H.-J. Xu and C.-Y. Chi, Inorg. Chem., 2007, 46, 1612–1616 CrossRef CAS PubMed.
- K. Kadota, N. T. Duong, Y. Nishiyama, E. Sivaniah and S. Horike, Chem. Commun., 2019, 55, 9283–9286 RSC.
- S.-Y. Yang, Y. Yan, B. Lothenbach and J. Skibsted, J. Phys. Chem. C, 2021, 125, 27975–27995 CrossRef CAS.
- M. Haouas, F. Taulelle and C. Martineau, Prog. Nucl. Magn. Reson. Spectrosc., 2016, 94–95, 11–36 CrossRef CAS PubMed.
- T. Song, Q. Liu, J. Liu, W. Yang, R. Chen, X. Jing, K. Takahashi and J. Wang, Appl. Surf. Sci., 2015, 355, 495–501 CrossRef CAS.
- J. A. van Bokhoven, A. M. J. van der Eerden and D. C. Koningsberger, J. Am. Chem. Soc., 2003, 125, 7435–7442 CrossRef CAS PubMed.
- P. Castro-Fernández, M. V. Blanco, R. Verel, E. Willinger, A. Fedorov, P. M. Abdala and C. R. Müller, J. Phys. Chem. C, 2020, 124, 20578–20588 CrossRef.
- A. Eskemech, H. Chand, A. Karmakar, V. Krishnan and R. R. Koner, Inorg. Chem., 2024, 63, 3757–3768 CrossRef CAS PubMed.
- P.-Z. Li, X.-J. Wang, J. Liu, J. S. Lim, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 2142–2145 CrossRef CAS PubMed.
- X.-Y. Li, L.-N. Ma, Y. Liu, L. Hou, Y.-Y. Wang and Z. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 10965–10973 CrossRef CAS PubMed.
- G. Zhang, G. Wei, Z. Liu, S. R. J. Oliver and H. Fei, Chem. Mater., 2016, 28, 6276–6281 CrossRef CAS.
- S. Kaewsai, S. Del Gobbo and V. D'Elia, ChemCatChem, 2024, 16, e202301713 CrossRef CAS.
- C. Chen, J. Zhang, G. Li, P. Shen, H. Jin and N. Zhang, Dalton Trans., 2014, 43, 13965–13971 RSC.
- J. L. Obeso, J. G. Flores, C. V. Flores, V. B. Lopez-Cervantes, V. Martinez-Jimenez, J. A. de Los Reyes, E. Lima, D. Solis-Ibarra, I. A. Ibarra, C. Leyva and R. A. Peralta, Dalton Trans., 2023, 52, 12490–12495 RSC.
- L.-H. Xie, M.-M. Xu, X.-M. Liu, M.-J. Zhao and J.-R. Li, Adv. Sci., 2020, 7, 1901758 CrossRef CAS PubMed.
- B. Yeskendir, J.-P. Dacquin, Y. Lorgouilloux, C. Courtois, S. Royer and J. Dhainaut, Mater. Adv., 2021, 2, 7139–7186 RSC.
- M. I. Nandasiri, S. R. Jambovane, B. P. McGrail, H. T. Schaef and S. K. Nune, Coord. Chem. Rev., 2016, 311, 38–52 CrossRef CAS.
- S. Vaidya, O. Veselska, A. Zhadan, M. Diaz-Lopez, Y. Joly, P. Bordet, N. Guillou, C. Dujardin, G. Ledoux, F. Toche, R. Chiriac, A. Fateeva, S. Horike and A. Demessence, Chem. Sci., 2020, 11, 6815–6823 RSC.
- G. Zheng, J. Wu, Y. Jia, G. Han and S. Zhang, J. Membr. Sci., 2024, 705, 122924 CrossRef CAS.
- S. Henke, M. T. Wharmby, G. Kieslich, I. Hante, A. Schneemann, Y. Wu, D. Daisenberger and A. K. Cheetham, Chem. Sci., 2018, 9, 1654–1660 RSC.
- S. Sobczak and A. Katrusiak, Inorg. Chem., 2019, 58, 11773–11781 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08744e |
| This journal is © The Royal Society of Chemistry 2025 |