
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh performance La2NiO4+δ oxygen and Ni–Ce0.9Gd0.1O2−δ fuel electrodes for thin film reversible solid oxide cells†
Adeel Riaz a,
Juande Sirventb,
Juan Zueco-Vincellec,
Fjorelo Buzib,
Silvère Panisseta,
Alexander Stangla,
Laetitia Rapennea,
Federico Baiuttib,
Michel Mermouxd,
Miguel Angel Laguna-Berceroc,
Albert Tarancónbe and
Mónica Burriel*a
a,
Juande Sirventb,
Juan Zueco-Vincellec,
Fjorelo Buzib,
Silvère Panisseta,
Alexander Stangla,
Laetitia Rapennea,
Federico Baiuttib,
Michel Mermouxd,
Miguel Angel Laguna-Berceroc,
Albert Tarancónbe and
Mónica Burriel*a
aUniv. Grenoble Alpes, CNRS, Grenoble INP, LMGP, 38000 Grenoble, France. E-mail: monica.burriel@grenoble-inp.fr
bCatalonia Institute for Energy Research (IREC), Jardins de les Dones de Negre 1, 08930 Sant Adrià del Besòs, Barcelona, Spain
cInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, Zaragoza 50009, Spain
dUniv. Grenoble Alpes, Univ. Savoie Mont Blanc, CNRS, Grenoble INP, LEPMI, Grenoble, 38000, France
eICREA, 23 Passeig Lluis Companys, 08010 Barcelona, Spain
First published on 12th May 2025
Abstract
Thin film reversible solid oxide cells (TF-rSOCs) are attracting a great deal of interest as they promise to operate at much lower temperatures (400–600 °C) than state-of-the-art commercial fuel electrode-supported cells (600–800 °C). However, in all-ceramics TF-rSOCs the high polarization resistance of the electrodes limits the cell performance. To overcome this limitation, high performing oxygen and fuel electrodes were selected and their nanostructure was optimized. Thin nanoporous films of La2NiO4+δ were deposited as oxygen electrode by Pulsed Injection Metal Organic Chemical Vapor Deposition (PI-MOCVD) with different thickness. As for the fuel electrode, thin films of Ni–Ce0.9Gd0.1O2−δ (NiCGO) were deposited by Pulsed Laser Deposition (PLD) at various temperatures and pO2. The electrochemical activity of the oxygen and fuel electrodes was measured by Electrical Conductivity Relaxation (ECR) and Electrochemical Impedance Spectroscopy (EIS), respectively. The optimized electrodes were then deposited on a YSZ single crystal electrolyte and the cell was measured in fuel cell and electrolysis modes showing high performance with a power density of 70 mW cm−2 at 0.7 V and a current density of −44 mA cm−2 at 1.3 V at a low operating temperature of 600 °C. These results demonstrate the potential of using these materials as electrodes in TF-rSOCs.
Introduction
Reversible solid oxide cells (rSOC) are gaining a lot of interest as they offer an innovative solution, operating alternately under SOFC and SOEC modes to produce oxygen and hydrogen fuels by consuming excess electrical power (in electrolysis mode) and providing electrical power when needed (in fuel cell mode).1–3 During periods of high renewable energy output from solar or wind sources, excess electricity can be transformed into synthetic fuels, which are conducive to efficient storage, easy transport, and can be converted back to electricity and heat during peak demand or when renewable energy is scarce. In industrial context, such as steel or glass manufacturing, using H2 generated from SOECs can significantly reduce carbon emissions. In transportation, rSOC based range extenders (secondary source of power generation used in electric vehicles) offer benefits including extended driving range, rapid refuelling, and fuel adaptability for electric vehicles.4 Additionally, rSOC systems, with fast dynamic response time, enable the alternating operation of the SOEC and SOFC processes, contributing to highly efficient, green, and environmentally friendly conversion processes with ideally zero greenhouse gas emissions. This makes rSOCs important energy conversion devices for building future low-carbon energy supply systems. This makes it a potential candidate for building a future low-carbon energy supply system.rSOCs, developed based on SOFC and SOEC research, integrate the features of power generation and fuel production into a single unit. Their performance is intricately connected to the electrochemical activity and stability of the electrode materials, thereby necessitating an in-depth understanding of the reaction mechanisms of the rSOC electrodes. As the electrode and electrolyte materials for SOFC and SOEC are mostly similar,1,5–10 the biggest challenge in rSOC operation is the electrochemical performance and stability of the electrodes under alternating operating voltages and their long-term electrochemical and mechanical stability under cyclic stress. High temperature operation leads to interdiffusion and reaction of the electrodes, especially the oxygen electrode, with the electrolyte. This leads to the formation of secondary phases at the electrode/electrolyte interface which impede the reaction and diffusion pathways and significantly decrease the performance of the cell. In SOECs, another factor leading to performance degradation is the delamination of the oxygen electrode due to the high oxygen partial pressures (pO2) at the electrode/electrolyte interface. Moreover, in hypo-stoichiometric electrodes, where ionic transport is dominated by oxygen vacancies, the high pO2 at the interface can lead to a depletion of the vacancies and a drop in the oxygen evolution reaction kinetics.11 There is also a relatively higher overpotential at the oxygen electrode compared to the fuel electrode. Therefore, the choice of materials, the microstructure and long duration cyclic stability in both SOFC and SOEC modes need to be optimized.
Thin films are being developed to decrease the overpotential losses and hence allow to lower the operational temperatures of SOCs to avoid electrochemical degradation and high temperature operation costs.12–19 All thin film cells (TF-rSOCs), also known as, micro-solid oxide cells (μ-SOCs) are being investigated for use in high performance portable devices and small mobile applications.20–25 μ-SOCs show higher volumetric power densities and faster startup times. However, the current collection is one of the major issues.21 Initial μ-SOFC devices were developed using thin layers of porous metals (mostly Pt) as electrodes (anode and cathode) on suspended yttria stabilized zirconia (YSZ) membranes which were integrated into a Si substrate.26–28 These devices work at lower temperatures (<550 °C), however, there is a significant variability in the reported maximum power values as summarized by Garbayo et al.29 Dewetting of Pt is well known at temperatures higher than 400 °C, which causes the variability in the reported values.29 Tsuchiya et al. used LSCF (La1−xSrxCo1−yFeyO3−δ) as cathode in their μ-SOFCs, however, the anode used was still Pt and the power dropped to 86% of the maximum value in less than an hour.30 Garbayo et al.29 reported the first all-ceramics based μ-SOFC using LSC (La1−xSrxCoO3−δ) and CGO (CexGd1−xO2−δ) as cathode and anode, respectively, achieving a power density of 100 mW cm−2 at 750 °C under pure H2 as fuel and air as oxidant. Develos-Bagarinao et al.31 deposited by Pulsed Laser Deposition (PLD) a 300 nm thick composite oxygen electrode composed of a LSFC-CGO vertically aligned nanostructure (VAN) and covered by a 1 μm nanoporous LSC layer. A 2 μm thick CGO barrier layer (BL) had been previously deposited by PLD on top of a dense 2 μm YSZ electrolyte. Platinum was used as current collector and the cell showed a high performance of 3.3 W cm−2 at 700 °C. However, the stability test at 1 A cm−2 exhibited a rapid degradation in the first 10 hours and after 250 hours the power density at 0.7 V dropped to 1.4 W cm−2. In a different study Lee et al.24 deposited a complete thin film SOFC on anodized aluminum oxide by consecutive sputtering of 650 nm Ni-YSZ, 1400 nm of YSZ, 400 nm CGO BL and a 600 nm porous composite VAN of LSCF-YSZ. The cell exhibited a good performance with 1.47 W cm−2 at 650 °C. A short degradation study of 7 hours did not show any degradation at 0.2 A cm−2 and 600 °C.
Typically, perovskite-based materials such as LSC, LSCF and their composites with doped ceria have been preferred in SOFCs due to their significantly higher electrical conductivity and good overall performance.8,12,32 However, these materials suffer from strong Sr segregation at grain boundaries and surfaces leading to reduced performance over time.33 These structures also have an oxygen sub-stoichiometry which can lead to a buildup of pO2 at the electrolyte/electrode interface in SOEC mode resulting in delamination of the electrode layer.34 In this sense La2NiO4+δ (L2NO4), a Ruddlesden–Popper phase material with alternating layers or perovskite and rock-salt structure, is a promising alternative to perovskite-based oxygen electrode materials for intermediate-low temperature operation due to its mixed ionic and electronic conductivity, high oxygen exchange activity and low activation energy.17,35–37 Interestingly, L2NO4 is able to accommodate over-stoichiometric oxygen as interstitials, which can help prevent an increase in the pO2 at the electrode/electrolyte interface and avoid delamination of the oxygen electrode layer in SOEC mode34,38 making it an ideal candidate for rSOCs. The performance of L2NO4 has been tested previously on fuel electrode-supported commercial cells. Lee et al.39 achieved the highest power density, reporting a value of 485 mW cm−2 at 700 °C for a 16 μm L2NO4 layer prepared via screen-printing. Vibhu et al.40,41 tested L2NO4 separately in SOFC and SOEC modes achieving a power density of 760 mW cm−2 (at 0.6 V) in fuel cell mode and a current density of 1.51 A cm−2 (1.5 V) in electrolysis mode at 800 °C. They observed a large degradation at 800 °C in both SOFC and SOEC modes of 227 mV kh−1 (+1 A cm−2) and 113 mV kh−1 (−1 A cm−2), respectively. The significant performance degradation was mainly attributed to the formation of secondary phases during the synthesis process due to the high sintering temperature (∼1200 °C). One of the advantages of fabricating thin film electrodes is that the synthesis temperature can be drastically decreased (∼600 °C) thus avoiding the formation of insulating secondary phases. By using metal organic chemical vapor deposition, we previously fabricated thin films with a highly porous microstructure, significantly enhancing the electrode's electrochemical activity.17,42 This increase was linked to a boost of the intrinsic activity, via the exposure of a large number of different extended defects on the lateral surface of the columns as well as a largely enhanced active surface area. Going from a nano-columnar to a dendrite-like nanostructure, the strong ramification leads to an even larger active surface area, which further decreases the polarization resistance. However, the performance of L2NO4 thin films in rSOCs has not yet been evaluated.
In the case of the fuel electrode, state-of-the-art materials are mainly based on porous metallic phases43 and metal–electrolyte composites (cermets).44–46 Nevertheless, such structures present several limitations in terms of structural stability at high temperatures and sulphur poisoning.47 Such instabilities may include nickel coarsening, as well as crack formation due to volumetric changes result of reduction/oxidation cycles during cell operation. Moreover, their integration in planar configurations or thin-film based devices is hindered by the poor in-plane percolation of porous layers and by fabrication incompatibilities of the metallic phases with typical thin film deposition techniques. There is an increasing effort in developing novel thin-film oxide materials that may reach the high performance of metallic-based fuel electrodes. State-of-the-art full ceramic fuel electrodes are based on ceria doped with Gd (as in CGO) or Sm (SDC).48–50 While doped-ceria presents an excellent catalytic activity upon the Hydrogen Oxidation Reaction (HOR), it shows limited electronic conductivity,51 which can lead to current percolation losses. To solve this limitation, integrating ceria in nanoengineered structures has been proposed as a feasible route for maximizing electrode performance.52 Such structures include mesoporous nanostructures,53 nanocomposites54,55 and vertically aligned heterostructures,56 even in combination with exsolved metallic particles.57 Among these strategies, the use of novel formulations of NiO–Ce0.9Gd0.1O2−δ (NiCGO) nanocomposites has been proposed in a recent publication, reporting remarkable electrode activity for surprisingly low contents of Ni.58 It should be noted that conventional NiCGO cermets present nickel contents over 30 vol% of NiO.59–61 However, a systematic study on the microstructural effects of the low Ni-containing layer on the HOR activity has not been carried out to date.
Herein, we report on the fabrication, optimization and electrochemical testing of highly active L2NO4 and NiCGO electrodes as potential oxygen and fuel electrodes, respectively, for TF-rSOCs. The effect of the different microstructural parameters of the layers on the final electrochemical performance is studied. Best performing layers are employed in a YSZ single crystal-supported cell, which is operated in SOFC and SOEC modes for validating rSOC operation.
Experimental methods
Thin film fabrication
Symmetrical thin film L2NO4 films were grown on commercial double side polished YSZ single crystal substrates (Crystec) with (100) orientation and a thickness of 0.5 mm by pulsed-injection metal organic chemical vapor deposition (PI-MOCVD). The precursor solution was prepared by dissolving commercial (Strem chemicals) powders La(tmhd)3 and Ni(tmhd)2 (tmhd = 2,2,6,6-tetramethylheptane-3,5-dionate) in m-xylene solution (Alfa Aesar) with a La/Ni ratio of 4.0 and a solution concentration of 0.02 mol l−1. The solution was injected into the reactor with a frequency of 4 Hz, opening time of 2 ms and a deposition (substrate) temperature of 600 °C. The carrier gas concentration was fixed at 34% Ar/66% O2 with a total pressure of 5 Torr. The thickness of the films was controlled by the number of pulses injected. To evaluate the performance in fuel cell and electrolysis modes, similar double side polished YSZ single crystals were used.NiCGO thin films were deposited in a pulsed laser deposition chamber (PVD Systems, PLD 5000) equipped with a 248 nmKrF excimer laser (Lambda Physics, COMPex PRO 205) on YSZ (100) substrates (Crystec GmbH). The deposition conditions are defined as follows: oxygen partial pressure of 5 mTorr for dense thin films, and 150/200 mTorr for nanoporous materials; 500/700 °C; target-substrate distance of 90 mm; laser fluency ≈0.8 J cm−2; laser frequency of 10 Hz. The total number of ablation pulses was adjusted for compensating the increase in the partial pressure, with a total of 1500, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 30000 for the depositions made at 5, 150 and 200 mT, respectively. A commercial target of NiO:Ce0.9Gd0.1O2 (3.5 vol% of NiO) was employed for the depositions. A thermal treatment was carried out on the sample deposited at 500 °C and 200 mT for 1 h in air ambient atmosphere at 700 °C.
000 and 30000 for the depositions made at 5, 150 and 200 mT, respectively. A commercial target of NiO:Ce0.9Gd0.1O2 (3.5 vol% of NiO) was employed for the depositions. A thermal treatment was carried out on the sample deposited at 500 °C and 200 mT for 1 h in air ambient atmosphere at 700 °C.
For the full cell measurements, the electrode active area was 0.64 cm2 on all cells. For current collection a gold layer was brush painted on both sides and heated to 250 °C to evaporate the organic components and produce porous films.
Structural characterization
Full cell Transmission Electron Microscopy (TEM) specimens were prepared in cross-sections using the semi-automated polishing tripod technique with a MultiPrep™ system (Allied High-Tech Products, Inc.). A PIPS II from GATAN was used for the final polishing. TEM and high resolution TEM (HRTEM) images were recorded with a JEOL JEM 2010 LaB6 microscope operating at 200 kV with a 0.19 nm point-to-point resolution. For STEM EDX mapping, a JEOL STEM 2100F 200 kV was used.
Functional characterization
For the ECR measurements, the samples were placed onto a 1/2′′ ceramic heating stage in a high temperature cell (Nextron). Ag electrodes were painted on the corners of the sample for better electrical contact. Electrical conductivity was measured in van-der-Pauw configuration using an excitation current of 1 μA. The sample was heated in a dry gas flow with mixed O2/N2 atmosphere at 1 atm and constant flow rate of 1000 mL min−1, whereas the pO2 was controlled via the flow ratio of highly pure oxygen to nitrogen. This high gas flow is used to minimise the chamber flush time (∼4.5 s). The pO2 was measured using a Rapidox 2100 gas analyser at the exit of the chamber.17 The change in conductivity, σ = 1/ρ, is measured with respect to an external stimulus such as a change in temperature or a change in the atmosphere (vacuum or change in oxygen partial pressure etc.). When the oxygen transport is dominated by the surface exchange reaction, the concentration of the oxygen ions in the film is uniform. Assuming linear surface exchange kinetics and using Fick's second law of diffusion for a thin film, we get:
 | (1) |
Integrating eqn (1) gives the oxygen concentration as a function of time and the normalized conductivity, with respect to initial and final oxygen concentration, which can be expressed as:
 | (2) |
In some cases, the oxidation or reduction process cannot be described by a single time constant (τ). By using a two τ model the fit is more accurate especially in the initial state after the abrupt change in pO2. A second exponential is introduced with an assumption that there are two separate processes taking place simultaneously:
 | (3) |
The value of τ2 is typically an order of magnitude higher than τ1. The shorter time constant can be related to the fast surface exchange process while the larger time constant can be due to a slower process which limits the oxygen diffusion at the film–substrate interface, chemical material degradation35 or due to different microstructures on the surface of the films.62 A supplementary analysis can be done by using an effective saturation time. This is a weighted average of the two saturation times:
| τeff = A1(τ1) + A2(τ2) | (4) |
The electrochemical performance of the NiCGO films was studied with a Novocontrol impedance spectrometer, in the frequency range of 1 MHz to 0.1 Hz, open circuit potential, an AC amplitude of 50 mV for a total of 150 points. The measurement was performed in symmetric configuration with the films deposited on both sides of the YSZ substrate. Porous gold paste (Fuel Cell Materials) was applied on top of the films to minimize undesired current percolation losses. The atmosphere set for the characterization was 100 mL min−1 of pure H2 passing through a bubbler. The measurements were carried out in a ProboStat station (NorECs) placed in a vertical furnace in the 550–700 °C temperature range. The samples were initially measured at 700 °C for ∼2.5 h for initial conditioning and stabilizing the Ni phases under the reducing atmosphere. After this period, the impedance measurements were carried out ramping down in temperature with a 50 °C step.
The measurements of full cells were performed in a commercial Fiaxell setup. The setup had to be modified as it was not adapted to small cells with small planar dimensions of 10 × 10 mm2. A dummy cell was placed on top of one of the H2 gas outlets to avoid flowing of the oxygen gas into this compartment. This helped in obtaining a good open circuit voltage (OCV) in the range of 1.05–1.1 V. All the measurements were done under a composition of 3% H2O/H2 on the fuel electrode side and dry air on the oxygen electrode side. EIS and j−V measurements were performed using a SP-300 Potentiostat from BioLogic.
Results and discussion
Nano-architectured L2NO4 thin film oxygen electrodes
L2NO4 nanocolumnar films with thicknesses ranging from 200 to 1800 nm were deposited by Pulsed Injection Metal Organic Chemical Vapor Deposition (PI-MOCVD) in a symmetric configuration on YSZ single crystal substrates. In order to create a nanocolumnar dendritic morphology, i.e., more porous films with a larger active surface, a first optimization consisted in lowering the deposition temperature from 650 °C (value used in our previous studies)17,42 to 600 °C. Fig. 1 presents the SEM images illustrating the morphology of nano-structured L2NO4 thin films of varying thicknesses, deposited at 600 °C. The top surface morphology reveals a highly porous microstructure for all film thicknesses (Fig. 1(a, c, e and g)). The cross-section SEM (Fig. 1(b, d, f and h)) images show a nano-columnar microstructure with open porosity. Films with thickness up to 1200 nm exhibit a distinct microstructure characterized by a thin, dense bottom layer overlaid with V-shaped nano-columns featuring intra-grain open porosity which substantially enhances the exposed surface area. Beyond this thickness the microstructure evolves into a dendrite-like morphology with smaller columns coalescing. Fig. 2 shows the XRD pattern of the L2NO4 thin films deposited on YSZ single crystals. The films grow polycrystalline, due to the high lattice mismatch between the cell parameters of L2NO4 and the unit cell of the YSZ single crystal. The observed L2NO4 peaks match to the ICDD reference 01-076-6116 (tetragonal phase). The presence of an impurity phase should be noted, indicated by 2θ peaks at 26.6°, 30.4°, and 63.2°. These peaks correspond to the La2O3 impurities (ICDD: 04-027-4353), likely resulting from excess La in the films. | ||
| Fig. 1 SEM images of L2NO4 thin films depicting the surface morphology and cross-sections of films with thicknesses of (a) and (e) 200 nm, (b) and (f) 400 nm, (c) and (g) 1200 nm, and (d) and (h) 1800 nm, respectively. | ||
 | ||
| Fig. 2 XRD spectra of L2NO4 thin films of various thicknesses (200, 400 and 1800 nm) grown on a YSZ single-crystal substrate. | ||
Electrical (i.e., in-plane conductance) and kinetic properties (i.e., oxygen exchange activity) were studied by Electrical Conductivity Relaxation (ECR) in L2NO4 films with different thickness. The experimental details can be found in Section 4.3. A representative example of a normalized conductivity transient is given in Fig. S1† for a pO2 step from 10–250 mbar, while the resulting effective time constants, τeff, are shown in Fig. 3(a) for various temperatures and film thicknesses. As highlighted in Fig. 3(b), at 300 °C, τeff decreases with increasing thickness up to 1200 nm. In agreement with previous results,17 we ascribe this improvement to the larger exposed surface area due to the open porosity in the nanostructured films. The difference in relaxation times between 400 nm and 1200 nm films is relatively small (approximately 15% at 300 °C). As the film thickness increases, grain coalescence may occur, leading to a reduction in the specific surface area (SSA), which in turn could lower the electrode's activity. Additionally, at this thickness, the film may transition from being surface-limited to operating in a mixed regime of surface and diffusion limitations.42 The electrical conductance of the films measured in the Van der Pauw configuration at a pO2 of 250 mbar in the temperature range of 250 to 400 °C is shown in Fig. 3(c). The conductance increases with film thickness (see Fig. 3(d)), possibly due to an increase in the thickness of the bottom dense layer on which the nanocolumns are rooted.17 The 1200 nm film exhibits the highest oxygen exchange activity, slightly higher compared to the 400 nm film, as indicated by its lower τeff. Additionally, the conductance of the 1200 nm film is twice that of the 400 nm film. Consequently, the 1200 nm thick L2NO4 film was identified as the best-performing oxygen electrode and was selected to construct the full cell and perform the SOFC and SOEC measurements.
 | ||
| Fig. 3 (a) Arrhenius plot of the effective time constant (τeff) obtained from ECR measurements during a pO2 change from 10 to 250 mbar, and (b) comparison of the τeff at different film thickness at 300 °C; (c) Arrhenius plot of the electrical conductance measured using the van der Pauw configuration at a pO2 of 250 mbar between 250–400 °C, and (d) comparison of the conductance of films with different thickness at 300 °C. | ||
Optimization of fuel electrode with Ni and doped ceria
NiCGO nanocomposite thin films were fabricated by pulsed laser deposition (PLD) on YSZ (100) single-crystal substrates. The target composition of NiCGO 2 vol% (after Ni reduction) was chosen following the results obtained by Park et al. for the development of an efficient low metal-containing cermet as fuel electrode in SOFCs.58 The films were grown under different operating conditions, as described in Table 1, resulting in variable microstructure (cf. later in the text).| Sample | O2 pressure (mTorr) | Temperature (°C) | Thermal treatment |
|---|---|---|---|
| NiCGO5mT–700C | 5 | 700 | — |
| NiCGO150mT–700C | 150 | 700 | — |
| NiCGO200mT–500C-TT | 200 | 500 | 700 °C 1 h |
Fig. 4(a) shows a top-view micrograph measured by Atomic Force Microscopy (AFM) of a NiCGO thin film deposited at 700 °C and 5 mTorr of oxygen partial pressure (NiCGO5mT–700C). The outcome was the fabrication of a dense electrode, as it can be clearly seen in the smooth topography represented in the micrograph. Similarly, Fig. 4(b) depicts the SEM micrograph obtained for the NiCGO thin film deposited at 700 °C and 150 mTorr (NiCGO150mT–700C). The change in the oxygen partial pressure aimed at increasing the porosity of the film,63 leading to higher surface area and thus improving the electrochemical activity of the electrode. The micrograph clearly shows the growth of a rougher surface with crystallite sizes in the range of 10–20 nm. The micrograph in Fig. 4(c) corresponds to the NiCGO200mT–500C-TT sample, fabricated with an oxygen partial pressure of 200 mTorr and at a lower temperature of 500 °C. Additionally, the sample was annealed at 700 °C for 1 h in order to promote the opening of the porous structure. This can be observed in Fig. S2,† which shows a comparison between the as deposited and thermally annealed microstructure, with the latter. The film presents similar microstructure than for NiCGO150mT–700C, with larger crystallites in the range of 25–40 nm in length and seemingly more exposed area due to increased roughness, resulting in a nanoporous surface.
 | ||
| Fig. 4 Microstructural characterization of as deposited NiCGO-based thin films showing (a) AFM micrograph of dense NiCGO5mT–700C; (b) SEM micrographs for the rest of the NiCGO thin films fabricated: NiCGO150mT–700C, and (c) NiCGO200mT–500C-TT. | ||
Information on the crystallographic nature of the NiCGO films was retrieved by XRD, as shown in Fig. 5. NiCGO5mT–700C shows epitaxial growth of the CGO cubic phase over the YSZ substrate, due to the unique presence of the (h00) reflections in the XRD pattern, as expected given the crystallographic match with the substrate fluorite structure. Minor presence of NiO (111) orientation might be discussed, as suggested by the peak shoulder located at 37.3°. The same comment applies to the NiCGO150mT–700C, film. Regarding the annealed NiCGO200mT–500CTT film, only the (200) diffraction peak is observed for the CGO phase, with apparent higher crystallinity. This is expected due to the similarities observed in the microstructure with NiCGO150mT–700C with the additional contribution of the thermal treatment performed. A low peak intensity likely reveals the presence of NiO with an apparent (220) orientation. The minor unidentified peak present at 36° could be originated by cross-contamination from the annealing treatment, while minor peaks in the 25–30° range (labelled with a star) are ascribed to artefact signals occasionally coming from the sample holder setup.
 | ||
| Fig. 5 XRD patterns of the NiCGO thin films studied. The star (*) indicates artefact diffraction signals from the setup. | ||
Fig. 6 shows the results of the electrochemical performance obtained for the NiCGO thin films studied under reducing conditions. The area specific resistance (ASR) evolution during the first hours of the NiCGO electrodes is presented in Fig. 6(a). NiCGO5mT–700C improved the initial electrochemical performance (≈4.6 Ω cm2) during the first stages of the characterization at 700 °C, most likely due to progressive reduction of the NiO present in the sample into metallic Ni, reaching a final ASR of ≈2.7 Ω cm2. An increase in the surface roughness and opening of the microstructure could also be responsible of the initial improvement observed. In the case of the NiCGO150mT–700C film, the polarization resistance increased over the first 2 hours of characterization despite being initially lower than for the fully dense NiCGO5mT–700C electrode (from ≈2.6 Ω cm2 to ≈6.8 Ω cm2). This could be attributed to changes in the electrode microstructure, e.g., due to grain growth and densification, inducing a reduction in the surface area and instability of the active phase. Further analysis on the evolution of the microstructure among the samples-beyond the scope of this work-would be desirable for deeper understanding on the material. Additionally, NiCGO200mT–500C-TT showed the lowest ASR among the films studied (≈1.1 Ω cm2 at 700 °C) with high stability over the first hours of the experiment, reaching a value of ≈0.9 Ω cm2 after conditioning. This suggests full reduction of the Ni species within the optimized nanoporous structure and the absence of microstructure evolution. Fig. 6(b) depicts representative Nyquist plots measured at 700 °C for all the samples after the initial conditioning finished, in line with the ASR values described. All the impedance spectra obtained present a main contribution of the polarization resistance dominated by a low-frequency arc, characteristic of the hydrogen oxidation reaction dominant for these materials. Slight asymmetries in the arcs are attributed to local and minor differences in the films of the symmetric cell measured.
 | ||
| Fig. 6 (a) ASR evolution with time measured in the beginning of the experiment, at 700 °C; (b) Nyquist plots obtained of the NiCGO samples at 700 °C under humidified hydrogen atmosphere; (c) evolution of the ASR with temperature for NiCGO based thin films fabricated under different conditions. | ||
Fig. 6(c) shows the Arrhenius analysis performed for all the NiCGO studied samples. A reference of a dense Ce0.8Sm0.2O2−δ (SDC) thin film grown under comparable conditions is added for comparison purposes.54 It can be clearly seen that the performance of all the NiCGO electrodes outperforms the one of dense SDC. In the case of fully dense NiCGO5mT–700C the improvement in the ASR is of one order of magnitude (from ≈20 Ω cm2 for SDC to ≈2.6 Ω cm2 at 700 °C) with comparable activation energy. This confirms the improvement in performance of the NiCGO composition irrespective of the microstructure. In the case of NiCGO150mT–700C, the ASR observed is the highest among the NiCGO films for the temperature range explored, which is in agreement with the ASR increase observed during the initial conditioning with respect to the other two films. Notably, NiCGO200mT–500C-TT shows the best performance of the films studied in this section. The ASR at 700 °C is 0.95 Ω cm2, a 20-fold factor below the ASR of that of dense SDC. The low values in ASR of NiCGO200mT–500C-TT could be attributed both to the increase in the activity resulting of the Ni content in the layer, and the retaining of the nanoporous structure observed in Fig. 4(c). The resulting NiCGO material was selected as the fuel electrode material for the thin film-based rSOCs electrochemical experiments.
Reversible SOCs using optimized thin film electrodes
To investigate the performance of thin film electrodes for TF-rSOCs, a 1000 nm L2NO4 oxygen electrode and a 120 nm NiCGO fuel electrode were deposited on a YSZ single crystal electrolyte. The surface morphology of the optimized L2NO4 oxygen electrode and NiCGO fuel electrode is shown in the SEM images in Fig. S3(a) and (b),† respectively. The L2NO4 surface shows a typical porous nano-columnar structure while the NiCGO surface shows a nano-porous surface. The cross-section of both electrodes is shown in the TEM images of Fig. 7. A thin dense layer of L2NO4 (∼7 nm) can be observed at the interface with YSZ (marked by green dotted lines), on top of which a porous nano-columnar microstructure is observed as shown in the TEM and high resolution TEM images in Fig. 7(a) and (b), respectively. The Selected Area Electron Diffraction (SAED) pattern shows rings corresponding to the L2NO4 polycrystalline phase (Fig. 7(c)), while a ring corresponding to the (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) plane of La2O3 impurity phase can also be observed (Fig. 7(d)). The NiCGO fuel electrode also has a thin dense layer at the interface with YSZ followed by porous film on top (Fig. 7(e)). The HRTEM image in Fig. 7(f) and SAED pattern in Fig. 7(g) confirms an epitaxial growth of the CGO layer on top of the YSZ electrolyte. STEM EDX mapping of the layer shows a good dispersion of the Ni particles throughout the CGO layer (Fig. 7(h)).
0) plane of La2O3 impurity phase can also be observed (Fig. 7(d)). The NiCGO fuel electrode also has a thin dense layer at the interface with YSZ followed by porous film on top (Fig. 7(e)). The HRTEM image in Fig. 7(f) and SAED pattern in Fig. 7(g) confirms an epitaxial growth of the CGO layer on top of the YSZ electrolyte. STEM EDX mapping of the layer shows a good dispersion of the Ni particles throughout the CGO layer (Fig. 7(h)).
 | ||
| Fig. 7 (a) TEM cross-section images of the full L2NO4 layer (dense layer marked by green dotted lines), (b) HRTEM image of L2NO4 at the interface with the YSZ electrolyte, and SAED patterns of L2NO4 film indexed with (c) L2NO4 theorical phase and (d) La2O3 phase. (e) TEM cross-section images of the full NiCGO layer, (f) HRTEM image of NiCGO at the interface with YSZ, (g) SAED pattern of NiCGO at the interface with the YSZ electrolyte, and (h) STEM EDX mapping of the NiCGO layer showing the Ni particles (green) incorporated in the CGO layer (blue for Ce). | ||
The electrochemical performance of the full cell in both, fuel cell and electrolysis cell modes, was tested in the temperature range of 500–600 °C. The j–V (current density–voltage) and j–P (current density–power density) curves in SOFC mode and j–V curves in SOEC mode are shown in Fig. 8. All measurements were carried out under a dry synthetic air flow on the oxygen electrode side and humidified hydrogen flow (3% H2O) on the fuel electrode side. An open circuit voltage (OCV) in the range of 1.0–1.1 V was obtained which indicates an adequate sealing and gas separation between the fuel electrode and oxygen electrode chambers. The OCV decreases when increasing the temperature, also according to the Nernst equation. Hydrogen acts as a carrier for the steam in SOEC mode, also avoiding Ni oxidation. Combined j–V curves can be obtained by using the same conditions for fuel cell and electrolysis cell measurements. The cell shows good performance in SOFC mode reaching current densities about 100 mA cm−2 and the power density reached a value of about 70 mW cm−2 at 600 °C and at an operating voltage of 0.7 V. At the same temperature, a current density of −44 mA cm−2 was obtained in SOEC mode at an operating voltage of 1.3 V.
 | ||
| Fig. 8 j–V and j–P curves of electrolyte-supported cell at different temperatures measured under dry synthetic air and humidified (3% H2O) hydrogen flows in SOFC and SOEC modes. | ||
EIS measurements were carried out at each temperature under the same conditions, namely OCV, 0.7 V (SOFC) and 1.3 V (SOEC). It is noticeable that in all the studied temperatures there is a decrease of Rs when changing the applied bias. We believe that this is related with a local increase of the sample environment temperature, possibly due to the small thickness of the electrodes. This temperature increase would also explain the observed differences on the polarization resistance decrease when polarizing the cell in both SOFC and SOEC modes. The EIS spectra were fitted with an equivalent circuit consisting of an inductive element (L1), in series with a resistor for the serial contribution (Rseries), and three R-CPEs for the fuel and oxygen electrodes and charge transfer resistance at the interface. The overall equivalent circuit can be represented as: [L1-Rseries – R2CPE2-R3CPE3-R4CPE4] and is graphically shown in each EIS graph. The EIS data was further treated by the Distribution of Relaxation Times (DRT) analysis technique, as shown in Fig. S4,† which gives more insight into the electrochemical processes taking place in the cell. DRT analysis shows at least three processes taking place in the cell. Taking this into account the EIS data were fitted with 3 R-CPEs here, as both the oxygen and fuel electrode are surface-limited due to the low thickness. The series and polarization resistance contributions are presented in an Arrhenius plot in Fig. 9(a). The contributions were assigned based on the maximum frequency (fmax) and the capacitance range. The EIS at 600 °C, 550 °C and 500 °C at OCV and different operating voltages are shown in Fig. 9(b), (c) and (d), respectively. The total resistance of the cell at OCV is 4.2, 7.1 and 12.1 Ω cm2 at 600, 550 and 500 °C, respectively. The largest contribution to the total resistance of the cell is the series resistance from the thick YSZ electrolyte reaching 2.9 Ω cm2 at 600 °C. The contribution at the high frequencies, shown in Fig. 9(d), is the charge transfer resistance at the electrode/electrolyte interface (ASR4) with a capacitance in the range of 10−5 F cm−2. ASR3 can be associated to the NiCGO fuel electrode with a capacitance in the range of 10−3 F cm−2 and frequency (fmax) range of 30–150 Hz. These values correspond to the ones obtained from the EIS measurements of the symmetrical cells. The fuel electrode shows a low activation energy of 0.57 eV. The ASR2 contribution would come from the L2NO4 oxygen electrode with a capacitance in the range of 10−2 to 10−3 F cm−2 and frequency (fmax) range of 1–10 Hz, in agreement with previously reported ranges for polycrystalline L2NO4 thin films.42,64 The L2NO4 oxygen electrode contribution has an ASR2 of 0.42 Ω cm2 at 600 °C and an activation energy of 1.09 eV. In both fuel cell and electrolysis cell operation mode, the polarization resistance contribution of L2NO4 remains constant. Based on our EIS analysis, we conclude that the biggest contribution to the total ASR arises from the oxygen diffusion through the single crystal electrolyte. Considering the expected theoretical power losses at the electrolyte, a significant performance enhancement can be expected by going to thinner electrolyte thicknesses (<1 μm)24 for the materials developed within this work.
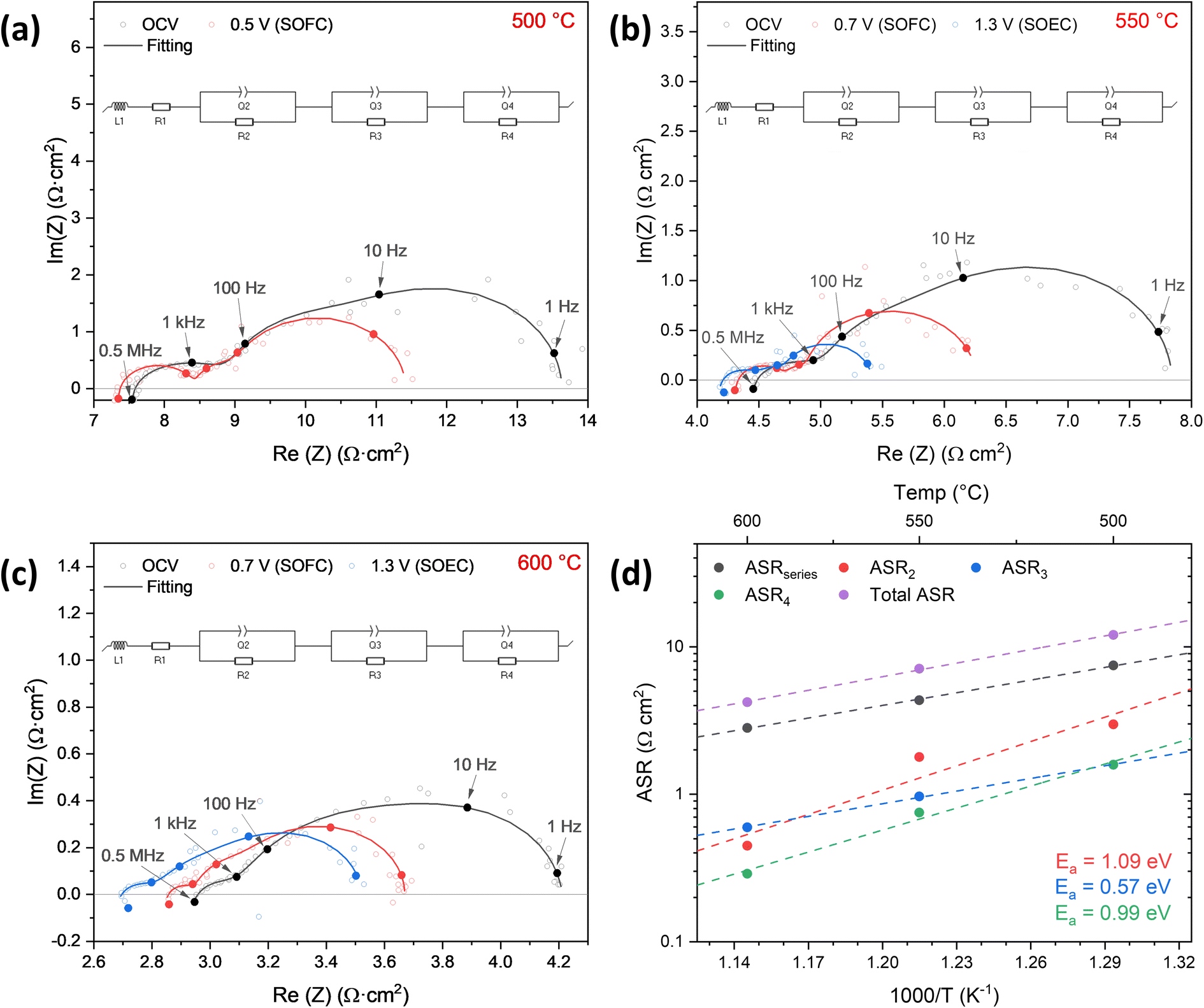 | ||
| Fig. 9 (a)–(c) Nyquist impedance diagrams at open circuit voltage (OCV), 0.7 V (SOFC) and 1.3 V (SOEC) of the electrolyte-supported cell at different temperatures measured under dry synthetic air and humidified (3% H2O) hydrogen flows. Insets show the equivalent circuit used for fitting the complex impedance diagrams; (d) Arrhenius plots of the series, polarization (ASRseries, ASR2, ASR3) and total area specific resistance. | ||
Conclusions
Thin films of L2NO4 and NiCGO were optimized and tested as potential oxygen and fuel electrodes, respectively, for thin film reversible solid oxide cells. L2NO4 thin films were deposited by PI-MOCVD with the thickness ranging from 200 nm to 1800 nm. The morphology of the thin film transitions from a nano-columnar structure to a dendritic-like nanostructure as their thickness increases. ECR and conductance measurements indicated that the optimal thickness is close to 1200 nm, with electrochemical activity improving due to the increased active surface area. NiCGO was deposited by PLD at different temperatures and pO2. Depositing at 700 °C resulted in a dense film while depositing at high pO2 and at a lower temperature of 500 °C with a post-deposition thermal anneal at 700 °C for 1 hour resulted in a relatively porous film. EIS measurements showed that the NiCGO porous film has the lowest polarization resistance of 0.9 Ω cm2 at 700 °C. The electrochemical performance of these optimized electrode thin films was subsequently evaluated for rSOC using electrolyte-supported YSZ cells. The cells exhibited good performance, achieving a power density of 70 mW cm−2 at 0.7 V and a current density of −44 mA cm−2 at 1.3 V, both at 600 °C in SOFC and SOEC modes, respectively. Overall, these electrodes demonstrate promising results for low-temperature TF-rSOCs.Data availability
All sample datasets and materials related to this work are made available under CC BY 4.0 license in the Zenodo repository: https://doi.org/10.5281/zenodo.15402717.Author contributions
Conceptualization: MB, FB, AT; investigation: AR, JS, JZV, LR, FB, SP, AS; formal analysis: AR, JS, JZV, SP, LR; methodology: AR, JS, JZV, FJ, SP, MB, FB; writing – original draft: AR, JS; writing – review & editing: AR, MB, AS, MALB, FB, SP, MM, LR, AT.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 Research and Innovation Program under grant agreements no. 824072 (Harvestore project) and no. 101017709 (EPISTORE project) and by the Centre of Excellence of Multifunctional Architectured Materials ‘CEMAM’ no. ANR-10-LABX-44-01 as part of the ‘Investments for the Future’ Program. This research has benefited from characterization equipment of the Grenoble INP – CMTC platform. The authors also acknowledge support from the Generalitat de Catalunya (2021-SGR-00750, NANOEN). This research has also received funding from PID2022-137626OB-C31 (MCIU/AEI/FEDER, UE), NextGenerationEU (PRTR-C17.I1) and T02_23R (DGA) grants.References
- V. Venkataraman, M. Pérez-Fortes, L. Wang, Y. S. Hajimolana, C. Boigues-Muñoz, A. Agostini, S. J. McPhail, F. Maréchal, J. Van Herle and P. V. Aravind, Reversible solid oxide systems for energy and chemical applications – review & perspectives, J. Energy Storage, 2019, 24, 100782 CrossRef.
- M. Moser, H.-C. Gils and G. Pivaro, A sensitivity analysis on large-scale electrical energy storage requirements in Europe under consideration of innovative storage technologies, J. Cleaner Prod., 2020, 269, 122261 CrossRef.
- S. He, Y. Zou, K. Chen and S. P. Jiang, A critical review of key materials and issues in solid oxide cells, Interdiscip. Mater., 2023, 2, 111–136 Search PubMed.
- L. Fang, F. Liu, H. Ding and C. Duan, High-Performance Reversible Solid Oxide Cells for Powering Electric Vehicles, Long-Term Energy Storage, and CO2 Conversion, ACS Appl. Mater. Interfaces, 2024, 16, 20419–20429 CAS.
- S. Y. Gómez and D. Hotza, Current developments in reversible solid oxide fuel cells, Renewable Sustainable Energy Rev., 2016, 61, 155–174 CrossRef.
- A. Hauch, R. Küngas, P. Blennow, A. B. Hansen, J. B. Hansen, B. V Mathiesen and M. B. Mogensen, Recent advances in solid oxide cell technology for electrolysis, Science, 2020, eaba6118 CrossRef CAS PubMed.
- P. Vinchhi, M. Khandla, K. Chaudhary and R. Pati, Recent advances on electrolyte materials for SOFC: a review, Inorg. Chem. Commun., 2023, 152, 110724 CrossRef CAS.
- M. Shen, F. Ai, H. Ma, H. Xu and Y. Zhang, Progress and prospects of reversible solid oxide fuel cell materials, iScience, 2021, 24, 103464 CrossRef CAS PubMed.
- J. A. Kilner and M. Burriel, Materials for Intermediate-Temperature Solid-Oxide Fuel Cells, Annu. Rev. Mater. Res., 2014, 44, 365–393 CrossRef CAS.
- M. B. Mogensen, M. Chen, H. L. Frandsen, C. Graves, J. B. Hansen, K. V. Hansen, A. Hauch, T. Jacobsen, S. H. Jensen, T. L. Skafte and X. Sun, Reversible solid-oxide cells for clean and sustainable energy, Clean Energy, 2019, 3, 175–201 CrossRef.
- P. Kim-Lohsoontorn, P. Prasopchokkul, A. Wongmaek, P. Temluxame, R. Visvanichkul, S. Bairak and N. Nuengjumnong, in High Temperature Electrolysis, ed. M. A. Laguna-Bercero, Springer International Publishing, Cham, 2023, pp. 277–312 Search PubMed.
- J. d. D. Sirvent, F. Buzi, F. Baiutti and A. Tarancón, in Nanoengineered Materials for Solid Oxide Cells, IOP Publishing, Bristol, UK, 2023, p. 1 Search PubMed.
- M. Acosta, F. Baiutti, A. Tarancón and J. L. MacManus-Driscoll, Nanostructured Materials and Interfaces for Advanced Ionic Electronic Conducting Oxides, Adv. Mater. Interfaces, 2019, 6, 1–15 Search PubMed.
- M. Machado, F. Baiutti, L. Bernadet, A. Morata, M. Nuñez, J. P. Ouweltjes, F. C. Fonseca, M. Torrell and A. Tarancón, Functional thin films as cathode/electrolyte interlayers: a strategy to enhance the performance and durability of solid oxide fuel cells, J. Mater. Chem. A, 2022, 10, 17317–17325 RSC.
- L. Bernadet, J. Segura-Ruiz, L. Yedra, S. Estrade, F. Peiró, D. Montinaro, M. Torrell, A. Morata and A. Tarancón, Enhanced diffusion barrier layers for avoiding degradation in SOFCs aged for 14000 h during 2 years, J. Power Sources, 2023, 555, 232400 CrossRef CAS.
- F. Buzi, K. Kreka, J. Santiso, L. Rapenne, Z. Sha, J. O. Douglas, F. Chiabrera, A. Morata, M. Burriel, S. Skinner, L. Bernadet, F. Baiutti and A. Tarancón, A Self-Assembled Multiphasic Thin Film as an Oxygen Electrode for Enhanced Durability in Reversible Solid Oxide Cells, ACS Appl. Mater. Interfaces, 2024, 16(33), 43462–43473 CrossRef CAS PubMed.
- A. Stangl, A. Riaz, L. Rapenne, J. M. Caicedo, J. de Dios Sirvent, F. Baiutti, C. Jiménez, A. Tarancón, M. Mermoux and M. Burriel, Tailored nano-columnar La2NiO4 cathodes for improved electrode performance, J. Mater. Chem. A, 2022, 10, 2528–2540 RSC.
- J. Zhang, S. Ricote, P. V. Hendriksen and Y. Chen, Advanced Materials for Thin-Film Solid Oxide Fuel Cells: Recent Progress and Challenges in Boosting the Device Performance at Low Temperatures, Adv. Funct. Mater., 2022, 32, 2111205 CrossRef CAS.
- F. Baiutti, F. Chiabrera, M. Acosta, D. Diercks, D. Parfitt, J. Santiso, X. Wang, A. Cavallaro, A. Morata, H. Wang, A. Chroneos, J. MacManus-Driscoll and A. Tarancon, A high-entropy manganite in an ordered nanocomposite for long-term application in solid oxide cells, Nat. Commun., 2021, 12, 2660 CrossRef CAS PubMed.
- A. Evans, A. Bieberle-Hütter, J. L. M. Rupp and L. J. Gauckler, Review on microfabricated micro-solid oxide fuel cell membranes, J. Power Sources, 2009, 194, 119–129 CrossRef CAS.
- F. Chiabrera, I. Garbayo, N. Alayo and A. Tarancón, Proc. SPIE, 2017, 10246, 102460S CrossRef.
- D. Beckel, A. Bieberle-Hütter, A. Harvey, A. Infortuna, U. P. Muecke, M. Prestat, J. L. M. Rupp and L. J. Gauckler, Thin films for micro solid oxide fuel cells, J. Power Sources, 2007, 173, 325–345 CrossRef CAS.
- A. Evans, A. Bieberle-Hütter, H. Galinski, J. L. M. Rupp, T. Ryll, B. Scherrer, R. Tölke and L. J. Gauckler, Micro-solid oxide fuel cells: status, challenges, and chances, Monatsh. Chem., 2009, 140, 975–983 CrossRef CAS.
- Y. H. Lee, H. Ren, E. A. Wu, E. E. Fullerton, Y. S. Meng and N. Q. Minh, All-Sputtered, Superior Power Density Thin-Film Solid Oxide Fuel Cells with a Novel Nanofibrous Ceramic Cathode, Nano Lett., 2020, 20, 2943–2949 CrossRef CAS PubMed.
- O. Hodjati-Pugh, A. Dhir and R. Steinberger-Wilckens, The development of current collection in micro-tubular solid oxide fuel cells—a review, Appl. Sci., 2021, 11, 1077 CrossRef CAS.
- U. P. Muecke, D. Beckel, A. Bernard, A. Bieberle-Hütter, S. Graf, A. Infortuna, P. Müller, J. L. M. Rupp, J. Schneider and L. J. Gauckler, Micro Solid Oxide Fuel Cells on Glass Ceramic Substrates, Adv. Funct. Mater., 2008, 18, 3158–3168 CrossRef CAS.
- Y. Yan, S. Rey-Mermet, Z. B. He, G. Deng and P. Muralt, Micro Solid Oxide Fuel Cell in Silicon Technology with Reinforced Electrolyte Membrane, Procedia Chem., 2009, 1, 1207–1210 CrossRef CAS.
- C.-W. Kwon, J.-W. Son, J.-H. Lee, H.-M. Kim, H.-W. Lee and K.-B. Kim, High-Performance Micro-Solid Oxide Fuel Cells Fabricated on Nanoporous Anodic Aluminum Oxide Templates, Adv. Funct. Mater., 2011, 21, 1154–1159 CrossRef CAS.
- I. Garbayo, D. Pla, A. Morata, L. Fonseca, N. Sabaté and A. Tarancón, Full ceramic micro solid oxide fuel cells: towards more reliable MEMS power generators operating at high temperatures, Energy Environ. Sci., 2014, 7, 3617–3629 RSC.
- M. Tsuchiya, B.-K. Lai and S. Ramanathan, Scalable nanostructured membranes for solid-oxide fuel cells, Nat. Nanotechnol., 2011, 6, 282–286 CrossRef CAS PubMed.
- K. Develos-Bagarinao, T. Ishiyama, H. Kishimoto, H. Shimada and K. Yamaji, Nanoengineering of cathode layers for solid oxide fuel cells to achieve superior power densities, Nat. Commun., 2021, 12, 3979 CrossRef CAS PubMed.
- C. Sun, J. A. Alonso and J. Bian, Recent Advances in Perovskite-Type Oxides for Energy Conversion and Storage Applications, Adv. Energy Mater., 2021, 11, 2000459 CrossRef CAS.
- B. Koo, K. Kim, J. K. Kim, H. Kwon, J. W. Han and W. Jung, Sr Segregation in Perovskite Oxides: Why It Happens and How It Exists, Joule, 2018, 2, 1476–1499 CrossRef CAS.
- M. S. Khan, X. Xu, R. Knibbe and Z. Zhu, Air electrodes and related degradation mechanisms in solid oxide electrolysis and reversible solid oxide cells, Renewable Sustainable Energy Rev., 2021, 143, 110918 CrossRef CAS.
- G. Garcia, M. Burriel, N. Bonanos and J. Santiso, Electrical Conductivity and Oxygen Exchange Kinetics of La2NiO4+δ Thin Films Grown by Chemical Vapor Deposition, J. Electrochem. Soc., 2008, 155, P28 CrossRef CAS.
- M. Burriel, G. Garcia, J. Santiso, J. A. Kilner, R. J. Chater and S. J. Skinner, Anisotropic oxygen diffusion properties in epitaxial thin films of La2NiO4+δ, J. Mater. Chem., 2008, 18, 416–422 RSC.
- R. K. Sharma, M. Burriel, L. Dessemond, V. Martin, J.-M. Bassat and E. Djurado, An innovative architectural design to enhance the electrochemical performance of La2NiO4+δ cathodes for solid oxide fuel cell applications, J. Power Sources, 2016, 316, 17–28 CrossRef CAS.
- M. A. Laguna-Bercero, H. Monzón, A. Larrea and V. M. Orera, Improved stability of reversible solid oxide cells with a nickelate-based oxygen electrode, J. Mater. Chem. A, 2016, 4, 1446–1453 RSC.
- Y. Lee and H. Kim, Electrochemical performance of La2NiO4+δ cathode for intermediate-temperature solid oxide fuel cells, Ceram. Int., 2015, 41, 5984–5991 CrossRef CAS.
- V. Vibhu, I. C. Vinke, R.-A. Eichel and L. G. J. (Bert) De Haart, Performance and Stability of Nickelates Based Oxygen Electrodes for Solid Oxide Cells, ECS Trans., 2021, 103, 1505 CrossRef CAS.
- V. Vibhu, I. C. Vinke, R.-A. Eichel, J.-M. Bassat and L. G. J. de Haart, La2Ni1−xCoxO4+δ (x = 0.0, 0.1 and 0.2) based efficient oxygen electrode materials for solid oxide electrolysis cells, J. Power Sources, 2019, 444, 227292 CrossRef CAS.
- S. Panisset, A. Riaz, A. Stangl, M. Burriel and D. Jauffres, Contribution of numerical modelling to design oxygen electrode for micro-solid oxide cells: a case study of high-performance nano-columnar La2NiO4 thin films, J. Power Sources, 2024, 593, 233951 CrossRef CAS.
- T. Setoguchi, K. Okamoto, K. Eguchi and H. Arai, Effects of Anode Material and Fuel on Anodic Reaction of Solid Oxide Fuel Cells, J. Electrochem. Soc., 1992, 139, 2875–2880 CrossRef CAS.
- W. H. Kan, A. J. Samson and V. Thangadurai, Trends in electrode development for next generation solid oxide fuel cells, J. Mater. Chem. A, 2016, 4, 17913–17932 RSC.
- M. C. Doppler, J. Fleig, M. Bram and A. K. Opitz, Hydrogen oxidation mechanisms on Ni/yttria stabilized zirconia anodes: separation of reaction pathways by geometry variation of pattern electrodes, J. Power Sources, 2018, 380, 46–54 CrossRef CAS.
- M. C. Doppler, J. Fleig, M. Bram and A. K. Opitz, Comparison of electrochemical hydrogen oxidation on different metal/ceramic model anodes and mechanistic implications, J. Phys.: Energy, 2019, 1, 035001 CAS.
- Y. Matsuzaki, The poisoning effect of sulfur-containing impurity gas on a SOFC anode: part I. Dependence on temperature, time, and impurity concentration, Solid State Ionics, 2000, 132, 261–269 CrossRef CAS.
- W. Jung, K. L. Gu, Y. Choi and S. M. Haile, Robust nanostructures with exceptionally high electrochemical reaction activity for high temperature fuel cell electrodes, Energy Environ. Sci., 2014, 7, 1685–1692 RSC.
- W. C. Chueh, Y. Hao, W. Jung and S. M. Haile, High electrochemical activity of the oxide phase in model ceria–Pt and ceria–Ni composite anodes, Nat. Mater., 2012, 11, 155–161 CrossRef CAS PubMed.
- W. Jung, J. O. Dereux, W. C. Chueh, Y. Hao and S. M. Haile, High electrode activity of nanostructured, columnar ceria films for solid oxide fuel cells, Energy Environ. Sci., 2012, 5, 8682–8689 RSC.
- J. H. Joo and G. M. Choi, Electrical conductivity of thin film ceria grown by pulsed laser deposition, J. Eur. Ceram. Soc., 2007, 27, 4273–4277 CrossRef CAS.
- C. Sun, Advances in nanoengineering of cathodes for next-generation solid oxide fuel cells, Inorg. Chem. Front., 2024, 11, 8164–8182 RSC.
- L. Almar, B. Colldeforns, L. Yedra, S. Estradé, F. Peiró, A. Morata, T. Andreu and A. Tarancón, High-temperature long-term stable ordered mesoporous Ni–CGO as an anode for solid oxide fuel cells, J. Mater. Chem. A, 2013, 1, 4531–4538 RSC.
- J. d. D. Sirvent, A. Carmona, L. Rapenne, F. Chiabrera, A. Morata, M. Burriel, F. Baiutti and A. Tarancón, Nanostructured La0.75Sr0.25Cr0.5Mn0.5O3–Ce0.8Sm0.2O2 Heterointerfaces as All-Ceramic Functional Layers for Solid Oxide Fuel Cell Applications, ACS Appl. Mater. Interfaces, 2022, 14, 42178–42187 CrossRef CAS PubMed.
- D. Burnat, G. Nurk, L. Holzer, M. Kopecki and A. Heel, Lanthanum doped strontium titanate – ceria anodes: deconvolution of impedance spectra and relationship with composition and microstructure, J. Power Sources, 2018, 385, 62–75 CrossRef CAS.
- J. Zamudio-García, F. Chiabrera, E. R. Losilla, D. Marrero-López and V. Esposito, Vertical heterostructures for symmetrical and reversible solid oxide fuel cells, Nano Energy, 2024, 131, 110293 CrossRef.
- J. Zamudio-García, F. Chiabrera, A. Morin-Martínez, I. E. Castelli, E. R. Losilla, D. Marrero-López and V. Esposito, Hierarchical exsolution in vertically aligned heterostructures, Nat. Commun., 2024, 15, 8961 CrossRef PubMed.
- J. H. Park, J.-H. Lee, K. J. Yoon, H. Kim, H.-I. Ji, S. Yang, S. Park, S. M. Han and J.-W. Son, A nanoarchitectured cermet composite with extremely low Ni content for stable high-performance solid oxide fuel cells, Acta Mater., 2021, 206, 116580 CrossRef CAS.
- U. P. Muecke, S. Graf, U. Rhyner and L. J. Gauckler, Microstructure and electrical conductivity of nanocrystalline nickel- and nickel oxide/gadolinia-doped ceria thin films, Acta Mater., 2008, 56, 677–687 CrossRef CAS.
- B. Iwanschitz, J. Sfeir, A. Mai and M. Schütze, Degradation of SOFC Anodes upon Redox Cycling: A Comparison Between Ni/YSZ and Ni/CGO, J. Electrochem. Soc., 2010, 157, B269 CrossRef CAS.
- I. W. Choi, W. Yu, S. Ryu, S. Lee, S. W. Cha and G. Y. Cho, Effect of nanostructured grains in co-sputtered Ni-GDC thin-film anode on methane conversion kinetics for low temperature solid oxide fuel cells operating on nearly dry methane, Ceram. Int., 2022, 48, 9083–9089 CrossRef CAS.
- G. Kim, S. Wang, A. J. Jacobson and C. L. Chen, Measurement of oxygen transport kinetics in epitaxial La2NiO4+δ thin films by electrical conductivity relaxation, Solid State Ionics, 2006, 177, 1461–1467 CrossRef CAS.
- A. Infortuna, A. S. Harvey and L. J. Gauckler, Microstructures of CGO and YSZ Thin Films by Pulsed Laser Deposition, Adv. Funct. Mater., 2008, 18, 127–135 CrossRef CAS.
- G. T. Kim, S. Wang, A. J. Jacobson, Z. Yuan and C. Chen, Impedance studies of dense polycrystalline thin films of La2NiO4+δ, J. Mater. Chem., 2007, 17, 1316–1320 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta08962f |
| This journal is © The Royal Society of Chemistry 2025 |