
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New tetrabutylphosphonium organic ionic plastic crystals incorporating borate anions†
Haris Amir a,
Mega Karb,
Luke A. O'Dellb,
Maria Forsythb,
Małgorzata Swadźba-Kwaśnya and
John D. Holbrey*a
a,
Mega Karb,
Luke A. O'Dellb,
Maria Forsythb,
Małgorzata Swadźba-Kwaśnya and
John D. Holbrey*a
aThe QUILL Research Centre, School of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Stranmillis Road, Belfast BT9 5AG, UK. E-mail: j.holbrey@qub.ac.uk
bInstitute for Frontier Materials, Deakin University, Geelong, Victoria 3217, Australia
First published on 19th May 2025
Abstract
Organic plastic crystals have seen rapid growth in their development and potential application as solid-state electrolytes in electrochemical devices. Two tetrabutylphosphonium ([P4444]+) salts incorporating non-halogenated bis(butane-1,4-dioxy)borate ([B(bdo)2]−) and tetrakis(pyrazolyl)borate ([B(pyr)4]−) anions have been synthesised and their physicochemical properties characterised. Both [P4444][B(pyr)4] and [P4444][B(bdo)2] are solid under ambient conditions and display solid–solid thermal phase transitions suggesting the formation of plastic crystal phases, which was confirmed by observation of significant ionic conductivity and ion mobility in the solid state. [P4444][B(pyr)4] showed more extensive polymorphism and greater ionic conductivity, and from the 31P linewidth NMR the phosphonium cation exhibited diffusion at lower temperatures. Therefore, these are of interest as potential new halogen-free solid-state electrolyte materials.
Introduction
Organic ionic plastic crystals (OIPCs) consist of organic cations and usually symmetric anions.1,2 OIPCs ideally incorporate desirable electrolyte properties and high ionic mobility coupled with high thermal stability, non-volatility, and non-flammability.3 Lithium-metal cells containing OIPCs have been reported to have cycling stabilities and current density of around 0.05 and 0.5 mA cm−2 over more than 2000 cycles.4 However, restricted ion transport and electrode compatibility are challenges that still remain and drive current research.5The potential for structural modification in OIPCs mimics that of ionic liquids (ILs) in that both OIPCs and ILs exhibit local structural disorder, and this can, in principle, be modified or influenced by the nature of the ions incorporated into the materials. In the case of ILs, local structural disorder and poor solid-state packing of ions leads to suppression of melting points and, for OIPCs, to positional and rotational defects and disorder within a solid phase providing plasticity and enhanced diffusional vectors for ionic conductivity.2 Indeed, OIPCs can be considered as solid state analogues of ILs6 and salts incorporating N- and P-containing cations including ammonium,7 pyrolidinium,8 imidazolium,9 and phosphonium10–12 have been extensively studied.
The formation of an OIPC phase and its characteristics can, in principle, be modified through selective changes to the cation and anion to reduce specific cation–anion interactions, as with ILs. However, the link between ion size, shape, temperature-dependent ion dynamics, physical, and electrochemical properties is still unclear.11,13 The formation of an OIPC is reliant on achieving a balance between a coulombic attraction that is sufficient to allow retention of a long-range crystalline lattice and minimisation in site-specific associations to allow reorientation and disordering of ions within the lattice.11,14 It should also be noted that many IL materials can also crystallise with significant degrees of structural disorder within their unit cells (for example, see Dymon et al.15).
Incorporating boron atoms as components of electrolyte anions can provide a central site for functionalisation allowing new anion motifs to be developed,16 and can actively contribute to advantageous SEI (solid electrolyte interphase) chemistry through formation of thin, boron-rich stable layers that increase interface stability17,18 However, despite the desirability of borate anions as contributors towards high performance electrolytes and enhanced SEI chemistry, most work to-date on OIPCs and non-aqueous organic electrolytes in general19 has focused on materials containing fluorinated-borate anions, including the ubiquitous tetrafluoroborate19,20 and bis-fluoro and trifluoromethyl-sulfonylimide21 both of which may be used with dopants that facilitate ion conduction.22
With the global effort to transition away from fluorine-containing electrolytes and other materials in batteries, membranes and polymeric materials due to environmental impact, sustainability, and safety of fluorinated components, new halogen-free electrolytes are increasingly sought after for both liquid and solid-state applications.23
A goal of our on-going work exploring the utility of fluorine-free anions in ionic liquid applications,24 has been to explore whether new fluorine-free functional ionic materials incorporating functionalised borate anions can be developed, taking inspiration most significantly from the utility of bis(oxalato)borate ([BOB]−) and related anions.16 We describe here the synthesis and characterisation of two new tetrabutylphosphonium salts containing fluorine-free borate anions; tetrabutylphosphonium bis(butane-1,4-dioxy)borate ([P4444][B(bdo)2]), the 1,4-butanediol derived analogue of [P4444][B(edo)2], tetrabutylphosphonium bis(ethane-1,4-dioxy)borate ([P4444][B(edo)2]), recently described by Xu et al.12 that has an ortho-borate anion derived from ethylene glycol/boric acid and exhibits OIPC behaviour over an uncommonly wide temperature range (−59 to 158 °C), and tetrabutylphosphonium tetrakis(pyrazolyl)borate ([P4444][B(pyr)4]) that incorporates an N4–coordinated borate anion (Fig. 1). The thermal properties, solid-state conductivity, and ion dynamics determined via 1H, 31P and 11B solid-state wideline NMR have been investigated for these new organic ionic plastic crystals that have the potential to be used as ambient temperature solid-state electrolytes.
 | ||
| Fig. 1 Chemical structure of the two tetrabutylphosphonium salts under investigation, tetrabutylphosphonium tetrakis(pyrazolyl)borate ([P4444][B(pyr)4], left) and tetrabutylphosphonium bis(butane-1,4-dioxy)borate ([P4444][B(bdo)2], right). | ||
Experimental
Materials and methods
Acetonitrile (99.5%), acetone (99.5%), toluene (99.5%), absolute ethanol (99.5%), chloroform (99.5%), DMSO-d6 (99.5 atom% D), CDCl3 (99.8 atom% D), 1,4-butanediol (99%), sodium borohydride (98%), hexagonal boron nitride powder (98%) and boric acid (99%) were purchased from Sigma-Aldrich. Anhydrous sodium carbonate (99.5%) was purchased from Fisher Scientific, ammonium dihydrogen phosphate (NH4H2PO4) (98%) was purchased from Alfa Aesar, 1H-pyrazole (99%) was purchased from Doug Discovery (Fluorochem), and tetrabutylphosphonium chloride (95%) was purchased from TCI. All chemicals were used as received.Na[B(bdo)2], first reported as a crystalline salt by Gainsford and Kemmitt25 was prepared using a modification of the method described by Chiappe et al.26 for sodium bis(glycerol)borate using sodium carbonate rather than NaOH, and removing co-formed water by azeotropic distillation to inhibit competing hydrolysis of labile B–O bonds in the orthoborate anion. Na[B(bdo)2] is insoluble in toluene, allowing facile isolation. Na[B(pyr)4] was prepared from sodium borohydride and 1H-pyrazole as described by Chao and Moore27 using excess pyrazole as both solvent and to shift the substitution equilibrium to four pyrazole ligands on boron forming the tetrahedral [B(pyr)4]−. The two salts [P4444][B(pyr)4] and [P4444][B(bdo)2] were prepared from the corresponding sodium salts Na[B(pyr)4] and Na[B(bdo)2] by cation exchange with tetrabutylphosphonium chloride ([P4444]Cl).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio) were placed into a round bottom flask under nitrogen and heated to 125 °C. The evolution of H2 was monitored and, after 4 h., evolution of gas ceased and the reaction mixture was allowed to cool to ambient temperature. The excess pyrazole was extracted with acetone (100 ml) and the product was collected by filtration and dried overnight in vacuo at 80 °C, to give a white powder (yield 15.71 g, 65.6%).
:5 ratio) were placed into a round bottom flask under nitrogen and heated to 125 °C. The evolution of H2 was monitored and, after 4 h., evolution of gas ceased and the reaction mixture was allowed to cool to ambient temperature. The excess pyrazole was extracted with acetone (100 ml) and the product was collected by filtration and dried overnight in vacuo at 80 °C, to give a white powder (yield 15.71 g, 65.6%).1H (400 MHz; DMSO-d6; SiMe4) δ 7.10 (8H, s, N–CH), 6.68 (4H, s, C–CH); 13C{1H} (100 MHz; DMSO-d6; SiMe4) δ 143.59 (N–CH), 125.23 (CH); 11B (128 MHz; DMSO-d6; 15% BF3·OEt2) δ −6.68. Calc. for C12H12BN8Na: C, 51.64; H, 4.33; N, 40.15. Found: C, 51.62; H, 4.35; N, 40.17.
:1.3 ratio) were combined in CHCl3 (50 ml) and stirred for 24 h under ambient conditions. Deionised water (20 ml) was then added. mixed and the organic layer was removed and washed with deionised water (5 × 20 ml) to remove traces of Na+ and Cl−. The solvent was removed under reduced pressure by rotary evaporation followed by drying in vacuo at 80 °C to give [P4444][B(bdo)2] as pale yellow crystals (yield 2.87 g, 64.2%). 1H (400 MHz; CDCl3; SiMe4) δ 4.43 (8H, m, O–CH2), 2.93 (8H, m, –CH2–), 1.65 (8H, m, P–CH2–), 0.95 (16H, m, –CH2–), 0.34 (12H, t, –CH3); 13C{1H} (100 MHz; CDCl3; SiMe4) δ 60.44 (O–CH2), 27.86 (OCH2–CH2), 22.43 (d, JCP = 22 Hz, P−CH2), 17.39 (PCH2–CH2), 16.92 (–CH2–), 12.10 (–CH3); 11B (128 MHz; CDCl3; 15% BF3·OEt2) δ 2.15. 31P{1H} (162 MHz; CDCl3; 85% H3PO4) δ 32.99. MS (ESI) calcd for [C16H36P]+ m/z 259.3; found m/z 259.2; calcd for [C8H16BO4]− m/z 187.1; found m/z 187.1; Chloride content determined by XRF analysis was 219 ± 7 ppm.:1.3 ratio) in CHCl3 and produced the product as a cloudy soft solid (yield 4.15 g, 77.1%). 1H (400 MHz; CDCl3; Me4Si) δ 7.51 (8H, s, –N–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ), 7.46 (4H, s, CH–), 1.93 (8H, m, P–CH2–), 1.41 (16H, m, –CH2–), 0.92 (12H, t, –CH3); 13C{1H} (100 MHz; CDCl3; SiMe4) δ 138.46 (–NCH–), 134.50 (–N–CH = ), 103.28 (CHCH–), 24.15 (d, JCP = 12 Hz, P−CH2), 18.95 (PCH2–CH2), 18.48 (–CH2–), 13.90 (–CH3); 11B (128 MHz; CDCl3; 15% BF3·OEt2) δ −6.58. 31P{1H} (162 MHz; CDCl3; 85% H3PO4) δ 32.86. MS (ESI) calcd for [C16H36P]+ m/z 259.3; found m/z 259.2; calcd for [C12H12BN8]− m/z 279.1; found m/z 279.2; Chloride content determined by XRF analysis was 203 ± 9 ppm.
), 7.46 (4H, s, CH–), 1.93 (8H, m, P–CH2–), 1.41 (16H, m, –CH2–), 0.92 (12H, t, –CH3); 13C{1H} (100 MHz; CDCl3; SiMe4) δ 138.46 (–NCH–), 134.50 (–N–CH = ), 103.28 (CHCH–), 24.15 (d, JCP = 12 Hz, P−CH2), 18.95 (PCH2–CH2), 18.48 (–CH2–), 13.90 (–CH3); 11B (128 MHz; CDCl3; 15% BF3·OEt2) δ −6.58. 31P{1H} (162 MHz; CDCl3; 85% H3PO4) δ 32.86. MS (ESI) calcd for [C16H36P]+ m/z 259.3; found m/z 259.2; calcd for [C12H12BN8]− m/z 279.1; found m/z 279.2; Chloride content determined by XRF analysis was 203 ± 9 ppm.1H, 13C, 11B, and 31P NMR spectra and electro-spray mass spectrometry for Na[B(bdo)2], Na[B(pyr)4], [P4444][B(bdo)2] and [P4444][B(pyr)4] are shown in Fig. S1–16 in the ESI.†
Characterisation and analysis
 | (1) |
Elemental analysis (CHNS) was also performed on the sodium salts using a Perkin Elemer 2400 series 2 elemental analyser. Mass spectrometry measurements were performed using a Waters LCT Premier Instrument equipped with an Advion Nanomate Infusion system. All reported mass spec data are accurate within the range of 0–10 ppm. Chloride content in the [P4444]+ salts was measured directly by quantitative energy dispersive X-ray fluorescence analysis (Rigaku NEX QC + QuantEZ) with analysis carried out in triplicate with an error range of ± 9 ppm.
 | (2) |
Results and discussion
Two new tetrabutylphosphonium borate salts containing O–O chelated [B(bdo)2]− and N-coordinated [B(pyr)4]− anions (Fig. 1) have been prepared from [P4444]Cl by anion metathesis with the sodium salts of the respective anions. Na[B(bdo)2] was first reported as a crystalline polymeric salt by Gainsford and Kemmitt25 with the Na+ and K+ salts subsequently described as additives in polyether acid gas (CO2) scrubbing applications,29 while butyrolactone solutions of the methyltriethylammonium ([N1112]+) salt has been reported as a non-aqueous electrolyte formulations30 in the patent literature.We provide here a brief description of the thermal properties of the two [P4444]+ salts, highlighting the thermal characteristics to initially lead to the potential identification of OIPC behaviour. We then describe the ionic conductivities of the two salts, [P4444][B(pyr)4] and [P4444][B(bdo)2] and correlation of cation and anion dynamics determined by NMR spectroscopy, demonstrating a much more extensive region of conductivity in the solid (plastic) state than should be expected from the initial DSC thermal profile. This further suggests that, when searching for potential OIPC materials, using DSC as an initial screen may lead to many ‘misses’, particularly if the plastic crystal domain is large and extends to very low temperatures as was described for [P4444][B(edo)2],12 the ethylene glycol-chelated analogue of [P4444][B(bdo)2].
Thermal properties
TGA profiles, shown in Fig. 2, indicate that both salts are thermally stable to >150 °C. In comparison, [P4444][B(edo)2] reported by Xu et al.12 shows similar stability profile with weight loss starting around 150 °C and complete decomposition by 400 °C. | ||
| Fig. 2 TGA profiles from 50–600 °C of [P4444][B(pyr)4] (red) and [P4444][B(bdo)2] (blue). | ||
DSC reveals the presence of a number of transitions in the heating and cooling cycles for both [P4444]+ salts, and these were repeatable across multiple thermal cycles. Data is shown from the second cycle of each sample in Fig. 3. For both salts, multiple endothermic transitions are observable, with peak positions determined from onset of each peak on heating before the solids finally melt. [P4444][B(pyr)4] shows three transitions on heating as a broad, unresolved endothermic peak between ca. 50 °C (first onset) to 86 °C. In the cooling cycle, the reversible crystallisation and subsequent solid–solid transitions resolve into three sharp, and distinct exothermic events. [P4444][B(bdo)2] melts around 60 °C with a broad, transition that appears to contain two peaks at 58 and 60 °C, and on cooling crystallises after extensive super-cooling to <10 °C with at least two distinct events evident in the DSC profile.
 | ||
| Fig. 3 DSC scan of [P4444][B(pyr)4] (red) and [P4444][B(bdo)2] (blue) with a scan rate of 10 °C min−1 (endothermic up). | ||
The entropy of fusion (ΔSfus) for each phase transition was calculated using eqn (1) and is summarised in Table 1, where Tm are 69 °C and 86 °C for [P4444][B(bdo)2] and [P4444][B(pyr)4] respectively. The solid–solid (II–I) transition of [P4444][B(bdo)2] occurs at 58 °C (ΔS = 5 J K−1 mol−1) and is followed closely by melting (I–liquid) at 60 °C (ΔS = 7 J K−1 mol−1). [P4444][B(pyr)4] undergoes a first solid–solid transition (III–II) at the slightly higher temperature of 69 °C (ΔS = 2 J K−1 mol−1). The small ΔS suggests only small changes in molecular or ionic reorientation and motion occur. The second transition (II–I) at 79 °C has a more significant change in entropy associated with it (ΔS = 23 J K−1 mol−1) and is followed by melting (I–liquid) at 86 °C (ΔS = 5 J K−1 mol−1) where the change in entropy is of similar magnitude to the phase change from II–I.
| Sample | Transition | Peak (onset)/°C | ΔSfus/J K−1 mol−1 |
|---|---|---|---|
| [P4444][B(bdo)2] | II–I | 58 (48) | 5 |
| I–liquid | 61 (60) | 7 | |
| [P4444][B(pyr)4] | III–II | 69 (66) | 2 |
| II–I | 79 (74) | 23 | |
| I–liquid | 86 (83) | 5 |
The transition shown for [P4444][B(pyr)4] in Fig. 3 (and listed in Table 1) are all broad and poorly resolved merging over the range 58–86 °C, however, on cooling, these resolve into two distinct, well separated events at 68 °C and 57 °C, followed by broader and less exothermic transitions at 47 °C and finally at 40 °C.
For both salts, the DSC melting profiles show a long broad onset to melting which is characteristic of a gain in rotational or translational disorder prior to melting of the lattice and is consistent with plastic behaviour, as is the observation of multiple phase transitions. The melting entropy (ΔSmelt) for [P4444][B(bdo)2] (7 J K−1 mol−1) and [P4444][B(pyr)4] (5 J K−1 mol−1) are significantly lower than Timmermans' threshold (<20 J K−1 mol−1)31 thereby classifying these materials as plastic crystals.
Compared to [P4444][B(edo)2] (mp 158 °C), the mps of [P4444][B(pyr)4] (86 °C) and [P4444][B(bdo)2] (60 °C) are significantly reduced. In addition, on further cooling in the DSC, no further low temperature thermal events were observed. This contrasts to [P4444][B(edo)2] where an exceptionally low temperature first-order solid–solid transition at −59 °C was reported giving an OIPC phase range of almost 240 °C between ca. −59–158 °C.
However, it is important to note that multiple phase transitions alone are not definitive indicators of OIPC formation. While these transitions suggest the potential for orientational flexibility – a hallmark of OIPC behaviour – they are not the sole markers of plastic crystal characteristics. Additional factors, such as the material's ability to retain a pseudo-crystalline state across transitions and structural disorder at the molecular level are also critical in establishing a true OIPC.
Conductivity
The conductivities of [P4444][B(pyr)4] and [P4444][B(bdo)2] were determined by EIS between 20–70 °C in order to scan across the phase transitions observed by DSC. The samples were not heated above their respective melting points due to the barrel-cell not being designed for liquid samples. The solid state conductivity of [P4444][B(pyr)4] and [P4444][B(bdo)2] as a function of temperature is shown in Table 2 and in Fig. 4 as an Arrhenius plot (eqn (3)).
 | (3) |
| Temperature/K | σ/S cm−1 | |
|---|---|---|
| [P4444][B(bdo)2] | [P4444][B(pyr)4] | |
| a Between 303–323 K.b Between 313–343 K.c Between 293–313 K. | ||
| 293 | 1.55 × 10−7 | 8.85 × 10−7 |
| 303 | 5.20 × 10−7 | 1.57 × 10−6 |
| 313 | 9.13 × 10−7 | 3.07 × 10−6 |
| 323 | 1.87 × 10−6 | 4.57 × 10−6 |
| 333 | — | 6.87 × 10−6 |
| 343 | — | 8.89 × 10−6 |
| Ea | 51.9a | 32.2b, 47.4c |
| R2 | >0.9996 | >0.996 |
 | ||
| Fig. 4 Arrhenius plot of the conductivity of [P4444][B(pyr)4] (red) and [P4444][B(bdo)2] (blue). Dashed lines represent fits to the Arrhenius equation with activation energy, Ea, of 51.9 kJ mol−1 for [P4444][B(bdo)2] between 303–323 K, 32.2 kJ mol−1 for [P4444][B(pyr)4] between 312–343 K and 47.4 kJ mol−1 for [P4444][B(pyr)4] between 293–313 K (all with r2 > 0.996). | ||
The temperature-dependent conductivity measurements (Fig. 4) show that both salts are conductive at temperatures below the respective endothermic transitions observed by DSC (Table 1) at 50–86 °C for [P4444][B(pyr)4] and 50–60 °C for [P4444][B(bdo)2]. This suggests a high degree of disorder or defects enabling conductivity in the solid state at, and below, ambient temperature. Plastic crystal formation is supported by the ion mobilities determined from wide-line NMR spectroscopy described below and, with the absence of a large (ΔS ≫ 20 J K−1 mol−1) solid–solid transition below the II-I transition for [P4444][B(bdo)2] and below the III–II transition for [P4444][B(pyr)4] allows the tentative conclusion that these materials are also plastic crystals at lower temperatures with a possible plastic → ordered crystal transition below the lower limit of scanning on the DSC, e.g. below −75 °C.
It is important to note that while the wideline NMR data were collected down to 243 K (−30 °C), the conductivity measurements were limited to 293 K (20 °C). This temperature range was chosen to focus on the practical operating conditions relevant to solid-state electrolyte applications. However, extending conductivity measurements to lower temperatures in future studies would provide further insight into the ionic transport mechanisms across a broader temperature range.
[P4444][B(pyr)4] showed higher conductivity than [P4444][B(bdo)2] and, for both salts, the conductivities increase by an order of magnitude from 8.85 × 10−7 S cm−1 to 8.89 × 10−6 S cm−1 for [P4444][B(pyr)4] between 293–343 K, and from 1.55 × 10−7 S cm−1 to 1.87 × 10−6 S cm−1 between 293–323 K for [P4444][B(bdo)2].
It is notable that the conductivities of both [P4444][B(pyr)4] and [P4444][B(bdo)2] in the solid state are comparable to the reported values for other tetraalkylphosphonium-based OIPCs.10,12 For example, the conductivity of [P4444][B(bdo)2] at 293 K (1.55 × 10−7 S cm−1) is the same as that reported by Xu et al.12 for tetrabutylammonium bis(ethylene-dioxyl)borate. At 293 K, [P4444][bis(glycolato)borate] was reported to have a higher conductivity of 2.3 × 10−6 S cm−1, however for the corresponding tetrabutylammonium analogue, [N4444][bis(glycolato)borate], conductivity is reduced to 1.0 × 10−7 S cm−1. It is worth noting that [P4444][BOB], an IL at 293 K is 2.8 × 10−5 S cm−1. Compared to OIPCs containing smaller asymmetric tetraalkylphosphonium cations,10 [P4444][B(pyr)4] displayed greater conductivity than [P1444][BF4] and [P1224][BF4], while the less conductive [P4444][B(bdo)2] was intermediate between [P1224][BF4] and [P1444][BF4].
From the Arrhenius plot of log(conductivity) vs. reciprocal temperature shown in Fig. 4, both [P4444][B(bdo)2] and [P4444][B(pyr)4] show similar changes in conductivity with temperature. A small change in slope appears for [P4444][B(pyr)4] at 313 K which may be due to a change in the dominant ion transport mechanism or subtle phase behaviour changes within the plastic crystal domain. Fitting to the Arrhenius equation between 313–343 and 293–313 K, activation energies (Ea) of 32.2 and 47.4 kJ mol−1 respectively, with r2 > 0.996. For [P4444][B(bdo)2], a drop in conductivity can be seen at the lowest temperature measured (293 K, σ = 1.55 × 10−7 S cm−1) and Ea, determined between 303–323 K, was 51.9 kJ mol−1. The lower Ea of [P4444][B(pyrazole)4] compared to [P4444][B(bdo)2] is likely due to the larger, more bulky borate anion that will show a greater sensitivity to temperature change in comparison to the smaller [B(bdo)2]−.
Despite the absolute conductivity values of [P4444][B(bdo)2] and [P4444][B(pyr)4] (1.55 × 10−7–8.89 × 10−6 S cm−1 between 293–343 K) being greater than some analogues, they remain less conductive than fluorinated OIPCs (e.g., [P1444][FSA] > 1.70 × 10−3 S cm−1 at 313 K).10 This limitation reflects the trade-off between eliminating fluorine (for safety and sustainability) and achieving the highest ionic transport rates. Future work will focus on molecular and structural modifications to bridge this gap.
Wideline solid state nuclear magnetic resonance (NMR) spectroscopy
Cation and anion dynamics were investigated over the temperature range 243–343 K using static (wideline) 31P and 11B NMR spectroscopy, with spectral analysis modelled using Dmfit.28 For 31P spectra, with spin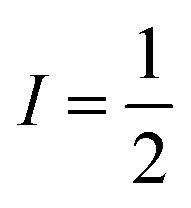 , the chemical shift anisotropy (CSA) model was applied. Whereas for 11B nuclei where quadrupolar interactions have a more significant influence on signals and the CSA model is less effective, dynamics were assessed by examining the full width at half maximum (FWHM) and linewidths of 11B signals.
, the chemical shift anisotropy (CSA) model was applied. Whereas for 11B nuclei where quadrupolar interactions have a more significant influence on signals and the CSA model is less effective, dynamics were assessed by examining the full width at half maximum (FWHM) and linewidths of 11B signals.
Wideline 31P and 11B NMR signals from [P4444][B(bdo)2] and [P4444][B(pyr)4] over the temperature range 243–343 K are shown in Fig. 5. At 243 K, the 31P NMR signal from [P4444][B(bdo)2] presents as a broad band (line width >5000 Hz) with a secondary sharp component, with both signals centred at the same chemical shift suggesting that this reflects dynamics rather than formation of different chemical species and is characteristic of a material containing a bulk of static components (the broad signal) and a small concentration of mobile species. On increasing temperature, from 243–343 K, the broad peak reduces in intensity and the sharp signal (line width <1000 Hz), representing mobile dynamic cations, becomes dominant from 303 K. For [P4444][B(pyr)4], the 31P signal is far less broad at 243 K with a line width of ca. 2000 Hz that sharpens with increasing temperature to <1000 Hz by 283–293 K mapping to the line width seen for [P4444][B(bdo)2].
 | ||
| Fig. 5 Stacked wide line static solid state 31P (top) and 11B (bottom) NMR signals from at various temperatures [P4444][B(bdo)2] (a, left) and [P4444][B(pyr)4] (b, right) at T = 243 K (green), 273 K (purple), 303 K (blue), and 343 K (black). | ||
The 31P signals for [P4444][B(bdo)2] and [P4444][B(pyr)4] were deconvoluted into the broad and sharp peak components using dmfit. The contributions of the two components from [P4444][B(bdo)2] at 253 K are shown in Fig. 6 and the line width of the dominant species (static or mobile) is plotted as a function of temperature in Fig. 7.
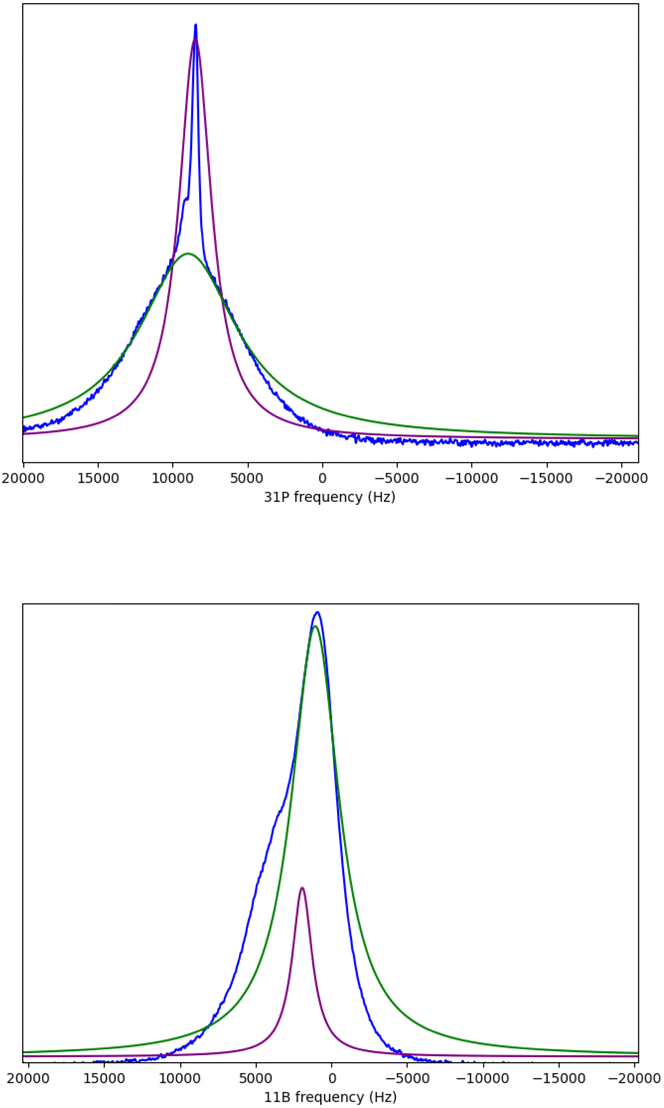 | ||
| Fig. 6 Solid state, wideline 31P (top) and 11B (bottom) NMR spectrum of [P4444][B(bdo)2] at 253 K (−20 °C) showing deconvolution into the broad (static) contribution in green and sharper (mobile) contributions in purple. | ||
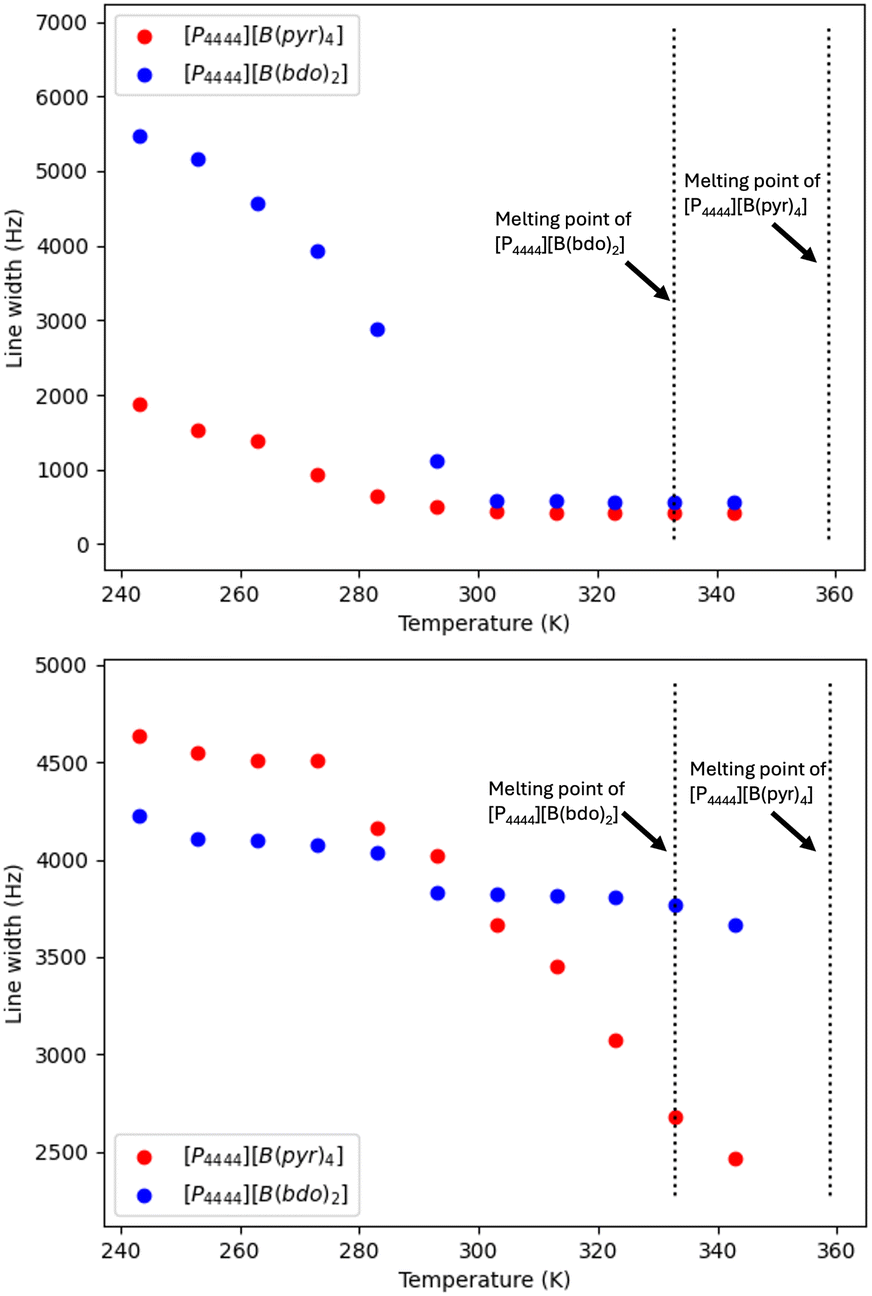 | ||
| Fig. 7 Line width (Hz) of 31P (top) and 11B (bottom) static NMR signals from [P4444][B(bdo)2] and [P4444][B(pyr)4] over the temperature range 243–343 K. | ||
For the [P4444]+ cation in both [P4444][B(bdo)2] and [P4444][B(pyr)4], a reduction in line width of the broad component as the temperature is raised from 243–293 K can be clearly seen, and from around 303 K, both solid salts contain dynamic [P4444]+ cations characterised by the sharp 31P NMR signal (line width <1000 Hz). The 31P NMR signal for [P4444][B(bdo)2] shows a large reduction in line width between 243–293 K, with a transformation of the NMR signal from static to almost wholly dynamic character. For [P4444][B(pyr)4], the change in line width is smaller, due to the presence of a larger dynamic component in the salt even at 243 K. It is notable that this increase in the cation dynamics and the switch to a single, sharp signal above 303 K for both salts does not appear to be associated with any of the thermal events observed from DSC (Fig. 3), but is consistent with the conductivity measured even below the transition temperatures.
Dynamics of the [B(bdo)2]− and [B(pyr)4]− anions were examined using the corresponding 11B NMR data shown in Fig. 5. Compared to the cation 31P NMR signals, there is less evidence for large changes in the 11B signals with temperature. For both [P4444][B(bdo)2] and [P4444][B(pyr)4], the 11B NMR presents a broad signal (ca. 500–1000 Hz) with a shoulder at higher frequency. For [P4444][B(pyr)4], the 11B NMR signal also contains some unresolved additional structure within the major signals. The asymmetric shape of the signal may be due to the sensitivity of the quadrupolar 11B to magnetic anisotropy from the solid state environment and from the quadrupolar N-atoms coordinating to the B-centre.
Deconvolution of the dominant symmetric signal within the composite 11B signal using dmfit allows separation of the broad and sharper peak components. These are shown independently in Fig. 6. The broader peak, corresponding to static solid boron nuclei, is the most dominant component at this temperature (253 K), this is repeated at each temperature between 243–343 K for 11B for [P4444][B(bdo)2] and [P4444][B(pyr)4] as shown in Fig. 5. The line width of the more dominant species (static or mobile) is plotted against temperature, as shown in Fig. 7.
The line width of the 11B signals in both OIPCs (Fig. 5) show very little change. The signal for [P4444][B(pyr)4] has a line width at half height of ca. 4500 Hz and is retained between 243–273 K before becoming sharper and narrower between 273–343 K with a monotonic reduction in line width of ca. 50% to 2500 Hz, which is of the same order as that reported from solid state studies of a range of boronate esters32 and it is unlikely that the borate anion is mobile so any mobile component from the interpretation of the conductivity results above could reflect rotation11 that would yield a more isotropic magnetic environment. For [P4444][B(bdo)2], the 11B NMR signal shows little variation with temperature, with the linewidth decreasing from ca. 4250–3750 Hz between 243–343 K. Again, this suggests that the borate anions are not dynamic and so are not the predominant charge carriers for the two OIPCs.
Conclusion
This work has introduced two new fluorine-free borate anion motifs through the synthesis of Na[B(pyr)4] and Na[B(dbo)2] and subsequent transformation to tetrabutylphosphonium salts [P4444][B(pyr)4] and [P4444][B(bdo)2]. The thermal behaviour of the [P4444]+ salts was studied using DSC, and an initial identification of potential organic ionic plastic crystal (OIPC) phases in both salts was made based on observation of multiple low-energy, reversible solid–solid transitions were observed between 50–90 °C. Conductivity measurements further supported this observation, revealing unexpectedly high ionic conductivities (1.57 × 10−6 S cm−1 for [P4444][B(pyr)4] and 5.20 × 10−7 S cm−1 at 303 K) comparable to other phosphonium-based OIPCs, even below the observed phase transition temperatures. These findings suggest a high degree of disorder in the room-temperature phase, confirmed by wideline solid-state NMR and linewidth analysis, which showed increased cation mobility between 240–293 K and indicate the potential to incorporate these and related12 new fluorine-free O–O chelated and N-coordinated borate anions into the development of advanced ionic electrolytes both as OIPCs and potentially as ILs.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Australian Research Council for support and funding through the IC180100049 (storEnergy).Notes and references
- (a) M. L. Thomas, K. Hatakeyama-Sato, S. Nanbu and M. Yoshizawa-Fujita, Energy Adv., 2023, 2, 748–764 RSC; (b) L. Jin, P. C. Howlett, J. M. Pringle, J. Janikowski, M. Armand, D. R. MacFarlane and M. Forsyth, Energy Environ. Sci., 2014, 7, 3352–3361 RSC; (c) Y. Abu-Lebdeh, P. Alarco, A. Abouimrane, L. Ionescu-Vasii, A. Hammami, M. Armand and J. New Mat, Electr. Sys., 2005, 8, 197 CAS; (d) Y. Abu-Lebdeh, A. Abouimrane, P.-J. Alarco and M. Armand, J. Power Sources, 2006, 154, 255–261 CrossRef CAS; (e) Y. Abu-Lebdeh, A. Abouimrane, P.-J. Alarco, I. Dividson and M. Armand, J. Power Sources, 2006, 159, 891–893 CrossRef CAS; (f) T. Rüther, J. Huang and A. F. Hollenkamp, Chem. Commun., 2007, 5226–5228 RSC; (g) Z.-B. Zhou and H. Matsumoto, Electrochem. Commun., 2007, 9, 1017–1022 CrossRef CAS; (h) Y. Abu-Lebdeh, E. Austin and I. J. Davidson, Chem. Lett., 2009, 38, 782–783 CrossRef CAS.
- F. Makhlooghiazad, R. Yunis, D. Mecerreyes, M. Armand, P. C. Howlett and M. Forsyth, Solid State Ionics, 2017, 312, 44–52 CrossRef CAS.
- P. C. Howlett, J. Sunarso, Y. Shekibi, E. Wasser, L. Jin, D. R. MacFarlane and M. Forsyth, Solid State Ionics, 2011, 204, 73–79 Search PubMed.
- (a) P. C. Howlett, Y. Shekibi, D. R. MacFarlane and M. Forsyth, Adv. Eng. Mater., 2009, 11, 1044–1048 Search PubMed; (b) A. Lennert, K. Wagner, R. Yunis, J. M. Pringle, D. M. Guldi and D. L. Officer, ACS Appl. Mater. Interfaces, 2018, 10, 32271–32280 Search PubMed.
- H. Zhu, D. R. MacFarlane, J. M. Pringle and M. Forsyth, Trends Chem., 2019, 1, 126–140 Search PubMed.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan and W. Zheng, et al., Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- (a) C. Shi, S. Li, W. Zhang, L. Qiu and F. Yan, J. Mater. Chem. A, 2013, 1, 13956–13962 RSC; (b) W. A. Henderson, D. M. Seo, Q. Zhou, P. D. Boyle, J.-H. Shin, H. C. De Long, P. C. Trulove and S. Passerini, Adv. Energy Mater., 2012, 2, 1343–1350 Search PubMed.
- D. R. MacFarlane, J. Huang and M. Forsyth, Nature, 1999, 402, 792–794 Search PubMed.
- M. Lee, Y.-H. Lee, J. H. Park and U. H. Choi, Org. Electron., 2017, 48, 241–247 Search PubMed.
- V. Armel, D. Velayutham, J. Sun, P. C. Howlett, M. Forsyth, D. R. MacFarlane and J. M. Pringle, J. Mater. Chem., 2011, 21, 7640–7650 RSC.
- L. Jin, K. M. Nairn, C. M. Forsyth, A. J. Seeber, D. R. MacFarlane, P. C. Howlett, M. Forsyth and J. M. Pringle, J. Am. Chem. Soc., 2012, 134, 9688–9697 Search PubMed.
- Y. Xu, A. Reinholdt, O. N. Antzutkin, M. Forsyth, P. Johansson and F. U. Shah, Cryst. Growth Des., 2024, 24, 8989–8998 Search PubMed.
- J. Adebahr, F. C. Grozema, S. W. Deleeuw, D. R. MacFarlane and M. Forsyth, Solid State Ionics, 2006, 177, 2845–2850 CrossRef CAS.
- (a) D. R. MacFarlane and M. Forsyth, Adv. Mater., 2001, 13, 957–966 Search PubMed; (b) H. Zhu, F. Chen, L. Jin, L. A. O'Dell and M. Forsyth, ChemPhysChem, 2014, 15, 3720–3724 CrossRef CAS PubMed.
- J. Dymon, R. Wibby, J. Kleingardner, J. M. Tanski, I. A. Guzei, J. D. Holbrey and A. S. Larsen, Dalton Trans., 2008, 2999–3006 RSC.
- (a) W. Xu and C. A. Angell, Electrochem. Solid-State Lett., 2001, 4, L3 Search PubMed; (b) K. Xu, S. Zhang, T. R. Jow, W. Xu and C. A. Angell, Electrochem. Solid-State Lett., 2001, 5, A26 CrossRef.
- (a) Z. Liu, A. Patra and N. Matsumi, ACS Appl. Energy Mater., 2025, 8, 3360–3368 CrossRef CAS; (b) J. Li, Y. Long, L. Li, F. Pu, W. Liao, X. Yu, H. Liao and X. Hu, New J. Chem., 2025, 49, 1755–1762 RSC; (c) N. R. Park, M. Zhang, B. Han, W. Li, K. Qian, H. Nguyen, S. Kumakura and Y. S. Meng, Adv. Energy Mater., 2024, 14, 2401968 CrossRef CAS.
- (a) D. Zhu, J. Xu, K. Ding, Q. Xu, P. Shi and Y. Min, Adv. Funct. Mater., 2023, 33, 2213822 Search PubMed; (b) L. Ma, J. Tan, Y. Wang, Z. Liu, Y. Yang, T. Gray, X. Zhang, M. Ye and J. Shen, Adv. Energy Mater., 2023, 13, 2300042 Search PubMed; (c) C. Bodin, J. Forero Saboya, P. Jankowski, K. Radan, D. Foix, C. Courrèges, I. Yousef, R. Dedryvère, C. Davoisne, M. Lozinšek and A. Ponrouch, Batteries Supercaps, 2022, 6, e202200433 Search PubMed; (d) J. Sheng, Q. Zhang, M. Liu, Z. Han, C. Li, C. Sun, B. Chen, X. Zhong, L. Qiu and G. Zhou, Nano Lett., 2021, 21, 8447–8454 Search PubMed; (e) Q. Xie, W. Li, A. Dolocan and A. Manthiram, Chem. Mater., 2019, 31, 8886–8897 CrossRef CAS.
- (a) K. Xu, Chem. Rev., 2004, 104, 4303–4418 Search PubMed; (b) K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- (a) C. S. M. Kang, O. E. Hutt and J. M. Pringle, ChemPhysChem, 2022, 23, e202200115 CrossRef CAS PubMed; (b) F. Nti, L. Porcarelli, G. W. Greene, H. Zhu, F. Makhlooghiazad, D. Mecerreyes, P. C. Howlett, M. Forsyth and X. Wang, J. Mater. Chem. A, 2020, 8, 5350–5362 Search PubMed; (c) R. Yunis, J. M. Pringle, X. Wang, G. M. A. Girard, R. Kerr, H. Zhu, P. C. Howlett, D. R. MacFarlane and M. Forsyth, J. Mater. Chem. A, 2020, 8, 14721–14735 Search PubMed; (d) N. Iranipour, D. J. Gunzelmann, A. J. Seeber, J. Vongsvivut, A. F. Hollenkamp, M. Forsyth and P. C. Howlett, J. Mater. Chem. A, 2017, 5, 24909–24919 Search PubMed; (e) T. J. Stockmann and Z. Ding, J. Phys. Chem. B, 2012, 116, 12826–12834 CrossRef CAS PubMed.
- (a) J. Sun, C. S. M. Kang, G. Huang, F. M. F. Vallana, A. Kumar, L. A. O'Dell, M. Galceran, O. Hutt, P. C. Howlett, M. Forsyth and J. M. Pringle, J. Mater. Chem. A, 2023, 11, 22329–22339 RSC; (b) Y. Garcia, L. Porcarelli, H. Zhu, D. Mecerreyes, M. Forsyth and L. A. O'Dell, J. Mater. Chem. A, 2024, 12, 4146–4158 Search PubMed.
- (a) J. Luo, O. Conrad and I. F. J. Vankelecom, J. Mater. Chem. A, 2013, 1, 2238–2247 RSC; (b) M. Forsyth, T. Chimdi, A. Seeber, D. Gunzelmann and P. C. Howlett, J. Mater. Chem. A, 2014, 2, 3993–4003 RSC; (c) X. Chen, H. Tang, T. Putzeys, J. Sniekers, M. Wubbenhorst, K. Binnemans, J. Fransaer, D. E. De Vos, Q. Li and J. Luo, J. Mater. Chem. A, 2016, 4, 12241–12252 RSC; (d) X. Li, Z. Zhang, S. Li, K. Yang and L. Yang, J. Mater. Chem. A, 2017, 5, 21362–21369 RSC.
- (a) Y. Xu, A. Filippov, S. Bhowmick, P. Johansson and F. U. Shah, Energy Adv., 2024, 3, 564–573 RSC; (b) G. Hernández, R. Mogensen, R. Younesi and J. Mindemark, Batteries Supercaps, 2022, 5, e202100373 CrossRef; (c) R. Mogensen, A. Buckel, S. Colbin and R. Younesi, Chem. Mater., 2021, 33, 1130–1139 CrossRef CAS; (d) L. O. S. Colbin, R. Mogensen, A. Buckel, Y. Wang, A. J. Naylor, J. Kullgren and R. Younesi, Adv. Mater. Interfaces, 2021, 8, 2101135 CrossRef CAS.
- (a) A. McGrogan, E. L. Byrne, R. Guiney, T. F. Headen, T. G. A. Youngs, A. Chrobok, J. D. Holbrey and M. Swadźba-Kwaśny, Phys. Chem. Chem. Phys., 2023, 25, 9785–9795 Search PubMed; (b) L. Wang, G. A. Nelson, J. Toland and J. D. Holbrey, ACS Sustainable Chem. Eng., 2020, 8, 13362–13368 CrossRef CAS; (c) L. Moura, L. C. Brown, M. Blesic and J. D. Holbrey, ChemPhysChem, 2017, 18, 3384–3389 CrossRef CAS PubMed.
- G. J. Gainsford and T. Kemmitt, Acta Crystallogr., Sect. C, 2005, 61, m136–m138 CrossRef PubMed.
- C. Chiappe, F. Signori, G. Valentini, L. Marchetti, C. S. Pomelli and F. Bellina, J. Phys. Chem. B, 2010, 114, 5082–5088 CrossRef CAS PubMed.
- S. Chao and C. E. Moore, Anal. Chim. Acta, 1978, 100, 457–467 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2001, 40, 70–76 CrossRef.
- C. R. Laroche, Composition comprising organoborates and physical solvents and use thereof for the removal of acid gases from hydrocarbon fluid streams, WO Pat. App. WO2017180285, 2017.
- Y. Washio, N. Takeishi, H. Shimamoto, K. Mori, N. Ushio, K. Shiono, T. Kishi and H. Samura, An electrolyte for use in electrolytic capacitors, Eur. Pat., App. EP381936, 1990 Search PubMed.
- J. Timmermans, J. Phys. Chem. Solids, 1961, 18, 1–8 CrossRef CAS.
- J. W. Weiss and D. L. Bryce, J. Phys. Chem. A, 2010, 114, 5119–5131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C, 11B, and 31P NMR spectra and electro-spray mass spectrometry for Na[B(bdo)2], Na[B(pyr)4], [P4444][B(bdo)2] and [P4444][B(pyr)4]. See DOI: https://doi.org/10.1039/d5ta00562k |
| This journal is © The Royal Society of Chemistry 2025 |