
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication of graphene oxide/silk protein core-sheath aerogel fibers for thermal management†
Wenjing
Xu
 a,
Chencheng
Ren
a,
Zhe
Wang
a,
Bimlesh
Lochab
b,
Jian
Liu
*acdef,
Yuanyuan
Zhang
*ac and
Limin
Wu
aefg
a,
Chencheng
Ren
a,
Zhe
Wang
a,
Bimlesh
Lochab
b,
Jian
Liu
*acdef,
Yuanyuan
Zhang
*ac and
Limin
Wu
aefg
aCollege of Chemistry and Chemical Engineering, Inner Mongolia University, Hohhot, Inner Mongolia 010021, P. R. China. E-mail: zyy@imu.edu.cn
bDepartment of Chemistry, Materials Chemistry Laboratory, School of Natural Sciences, Shiv Nadar Institution of Eminence, Gautam Buddha Nagar, Uttar Pradesh 201314, India
cInner Mongolia Key Laboratory of Rare Earth Catalysis, Inner Mongolia University, Hohhot, Inner Mongolia 010021, P. R. China
dDICP-Surrey Joint Centre for Future Materials, Department of Chemical and Process Engineering and Advanced Technology Institute, University of Surrey, Guildford, Surrey, GU2 7XH, UK. E-mail: jian.liu@surrey.ac.uk
eCollege of Energy Material and Chemistry, Inner Mongolia University, Hohhot, Inner Mongolia 010021, P. R. China
fInstitute for Green Chemistry and Environmental Science, Inner Mongolia University, Hohhot, Inner Mongolia 010021, P. R. China
gDepartment of Materials Science and State Key Laboratory of Molecular Engineering of Polymers, Fudan University, 200433 Shanghai, China
First published on 12th February 2025
Abstract
Effective thermal management in advancing electronics is hindered by phase change material (PCM) leakage. This study introduces coaxial wet spinning to encapsulate polyethylene glycol (PEG) within a graphene oxide/silk fibroin (GO/SF) core sheathed by thermoplastic polyurethane (TPU), forming a robust core–shell structure. The resulting GO/SF@TPU aerogel fibers exhibit exceptional tensile strength (17.21 MPa) and toughness (136.8 MJ m−3) while retaining shape stability under varying humidity. Subsequent PEG impregnation yields GO/SF@TPU-PEG composite phase change fibers (PCFs) with high latent heat (86.66 J g−1) and low thermal conductivity (0.0863 W m−1 K−1), enabling efficient thermal storage and insulation. These PCFs maintain a 26.4 °C temperature gradient at 100 °C, demonstrating superior thermal regulation. Combining mechanical durability, humidity resistance, and thermal efficiency, the fibers are ideal for smart textiles and wearable devices requiring lightweight, sustainable thermal management. This innovation addresses PCM leakage challenges and advances eco-friendly thermal insulation solutions, showcasing significant potential for next-generation electronics and energy-efficient applications.
Introduction
As electronic devices advance towards higher power densities, greater integration, and miniaturization, the challenge of excessive heat generation during operation has become increasingly pressing.1 This heat generation not only threatens the safety and reliability of these devices,2 but also necessitates the development of efficient thermal management materials to ensure their optimal performance and safety.3 Traditional thermal management methods are plagued by inefficiency, high energy consumption, and environmental unfriendliness, prompting an urgent need for materials that offer high efficiency, eco-friendliness, and energy savings. Phase change materials (PCMs) are emerging as key players in wearable thermal management. Yet, traditional PCMs, including paraffins4 hydrated salts,5 alcohols,6 and fatty acids7 often experience leakage issues during phase transitions, significantly limiting their practical applications.8 Therefore, selecting appropriate methods to construct shape-stabilized composite phase change materials (CPCMs) is essential for the practical use of PCMs. These composites, which integrate thermal energy storage (TES) with structural functionality, have attracted considerable attention in the field of thermal energy storage.9 Typically, they consist of low-melting-point PCMs, such as paraffins, encapsulated within polymer matrices.10 To enhance the stability and performance of CPCMs, researchers have explored various strategies, such as incorporating mesoporous materials like activated carbon and silica molecular sieves,11 carbon nanotube sponges,12 and expanded perlite.13 Phase change fibers (PCFs), which are intelligent materials containing PCMs, are currently recognized as promising platforms for latent heat storage due to their thermal energy storage and temperature regulation capabilities.14Aerogels, characterized by their high porosity, substantial pore volume, large specific surface area, and low thermal conductivity and density, have found extensive applications in various fields, including life sciences,15 aerospace,16 military,17 and thermal management.18,19 Currently, most macroscopic aerogels are monolithic, characterized by brittleness and poor structural integrity. There is a significant need for integrating diverse morphologies, improved machinability, and multiple functionalities. In this context, aerogel fibers, known for their excellent flexibility, weavability, and ease of integration, have emerged as a promising solution.20 Unlike traditional bulky aerogels, aerogel fibers can be engineered with tailored mechanical properties, enabling them to be bent, knotted, twisted, and woven.21 This makes them ideal ultra-insulating materials for wearable garments22,23 with inherent flame retardancy.24 These attributes have made aerogel fibers essential in thermal management. Aerogel fibers, are particularly effective for encapsulating PCMs, providing a viable approach for fabricating PCFs. The unique properties of aerogels allow them to fully exploit the nanoconfinement of PCMs, thus enhancing the performance and stability of phase change fibers.
Graphene, a two-dimensional material composed of sp2-bonded carbon atoms, has garnered widespread attention due to its exceptional mechanical, thermal, chemical, electrical, and optical properties.25 As a novel nanomaterial, graphene features a single layer of conjugated carbon atoms arranged in a honeycomb lattice, exhibiting outstanding electrical conductivity, high thermal conductivity, and flexibility.26 Graphene oxide (GO), a graphene derivative rich in oxygen-functional groups, finds extensive applications in electronics, energy storage, and biomedical fields.27 GO enhances processability while simultaneously improving interfacial adhesion with textile substrates.28 Additionally, incorporating GO sheets can stabilize molten polyethylene glycol (PEG) during solid–liquid phase transitions, making it an effective smart heat storage system.29 Silk fibroin (SF), a natural protein produced by silkworms and spiders, is a biopolymer renowned for its excellent biocompatibility and degradability.30 Its unique three-dimensional network structure provides superior insulating properties, making it suitable for applications in biomedicine,31 textiles,32 and insulation.33 SF-based aerogels, with their high specific surface area and porosity,34 contribute to enhanced energy storage density and exhibit low thermal conductivity, offering excellent insulation.35 Additionally, SF has demonstrated UV absorption capabilities,36 effectively functioning as a sunscreen to protect skin from UV damage. GO can be well-dispersed in SF solutions and uniformly distributed within SF scaffolds.37 The incorporation of SF into GO aerogels results in through-pore structures, providing an abundance of pore channels. This combination leads to composite aerogels with superior thermal stability and insulating properties.38 However, the poor mechanical strength of current aerogel fibers limits their practical application in wearable textiles. To enhance the mechanical integrity and wearability of GO-based aerogel fibers, an elastic protective layer is formed on the surface, aiming to bolster overall strength and flexibility.
Polyethylene glycol (PEG), also known as poly(oxyethylene), is a water-soluble polyether polymer39 widely utilized as an organic phase change material in various sectors, including textiles, architectural structures, and solar energy.40 Its high latent heat value, stable thermal properties, minimal thermal hysteresis, and non-toxic nature make it a prime candidate for the fabrication of composite phase change materials. PEG's biodegradable nature minimizes environmental impact, aligning with current ecological trends.41 Additionally, its excellent chemical stability and thermal properties, coupled with its ability to bond well with other materials such as graphene,42 have established it as a key component of high-performance composites.
In this study, we have successfully designed and fabricated composite phase change fibers (PCFs) with a core–shell structure for efficient environmental thermal management and storage applications. Employing coaxial wet spinning technology, we incorporated a GO/SF composite core, known for its exceptional tensile properties, and selected thermoplastic polyurethane (TPU) as the shell material due to its superior mechanical strength and ductility.43 The resulting GO/SF@TPU aerogel fibers demonstrated an impressive tensile strength of up to 17.21 MPa and a toughness of 136.8 MJ m−3. Subsequently, the aerogel fibers were impregnated with PEG, a cost-effective PCM known for its excellent thermal storage capacity, to form the composite structure of GO/SF@TPU-PEG PCFs. A comprehensive evaluation of their insulating properties, thermal stability, and thermal cycling was conducted to validate the practical potential of these materials in thermal management. The results revealed excellent thermal storage and temperature regulation capabilities, a low thermal conductivity of 0.0863 W m−1 K−1, and a melting latent heat of 86.66 J g−1. Furthermore, this fabrication process proved to be highly efficient and economically viable, with the produced composite PCFs exhibiting superior insulation effects and energy storage efficiency. These fibers can be further woven into fabrics, opening new possibilities for the development of smart thermal regulation textiles in wearable devices.
Results and discussion
We developed a novel method utilizing coaxial wet spinning and post-processing techniques to manufacture TPU-encapsulated GO/SF aerogel fibers. As illustrated in Fig. 1a, the initial materials consisted of GO solutions, GO/SF mixtures, and TPU solutions, each individually loaded into two 20 mL syringes. These solutions were extruded through fixed outer (17G) and inner (22G) nozzles into a coagulation bath, employing coaxial wet spinning to successfully fabricate core–shell structured fibers with GO/SF as the core and TPU as the shell. Following freeze-drying, TPU-encapsulated GO/SF aerogel fibers were obtained. The injection speed of the outer solution was set at 300 μL min−1, while the inner flow rate was 800 μL min−1, ensuring optimal spinnability and facilitating the continuous production of long fibers. This process can be scaled up by adjusting the process parameters, as depicted in the schematic of the collected fibers in Fig. 1b. The cross-sectional scanning electron microscope (SEM) images of the resulting aerogel fibers, as presented in Fig. 1c, illustrate the direct fabrication of core–shell structured fibers in a one-step process. This method effectively eliminates the need for elaborate steps traditionally involved in converting aerogel precursors into hollow polymer fibers. Significant differences are observed when compared to hollow TPU aerogel fibers, as shown in Fig. S1.† From the scanning electron microscopy (SEM) images, the cross-section of the coaxial fibers reveals uniformly fine pores (Fig. 1f), with the wall thickness of the core–shell fibers ranging from 20 to 65 μm. Fig. S2† illustrates the rough morphological characteristics of the fiber surface, which provides an additional protective layer for the internal graphene fibers, significantly enhancing the overall mechanical strength of the composite. In contrast, the core GO exhibits a relatively porous structure (Fig. 1d). The surface of the GO aerogel fibers features a relatively dense outer layer (Fig. S3†), whereas the introduction of SF results in a more porous exterior with prominent through-pore structures (Fig. S4†). The weaving process is depicted in Fig. 1e. Additionally, Fig. 1g shows the surface morphology of the fibers after knotting, confirming the durability and weave-ability of the aerogel fibers. Fig. 1h displays the energy-dispersive X-ray spectroscopy (EDS) elemental mapping of the GO/SF@TPU aerogel fiber cross-section, clearly illustrating the uniform distribution of carbon (C), oxygen (O), and nitrogen (N) across the fiber cross-section.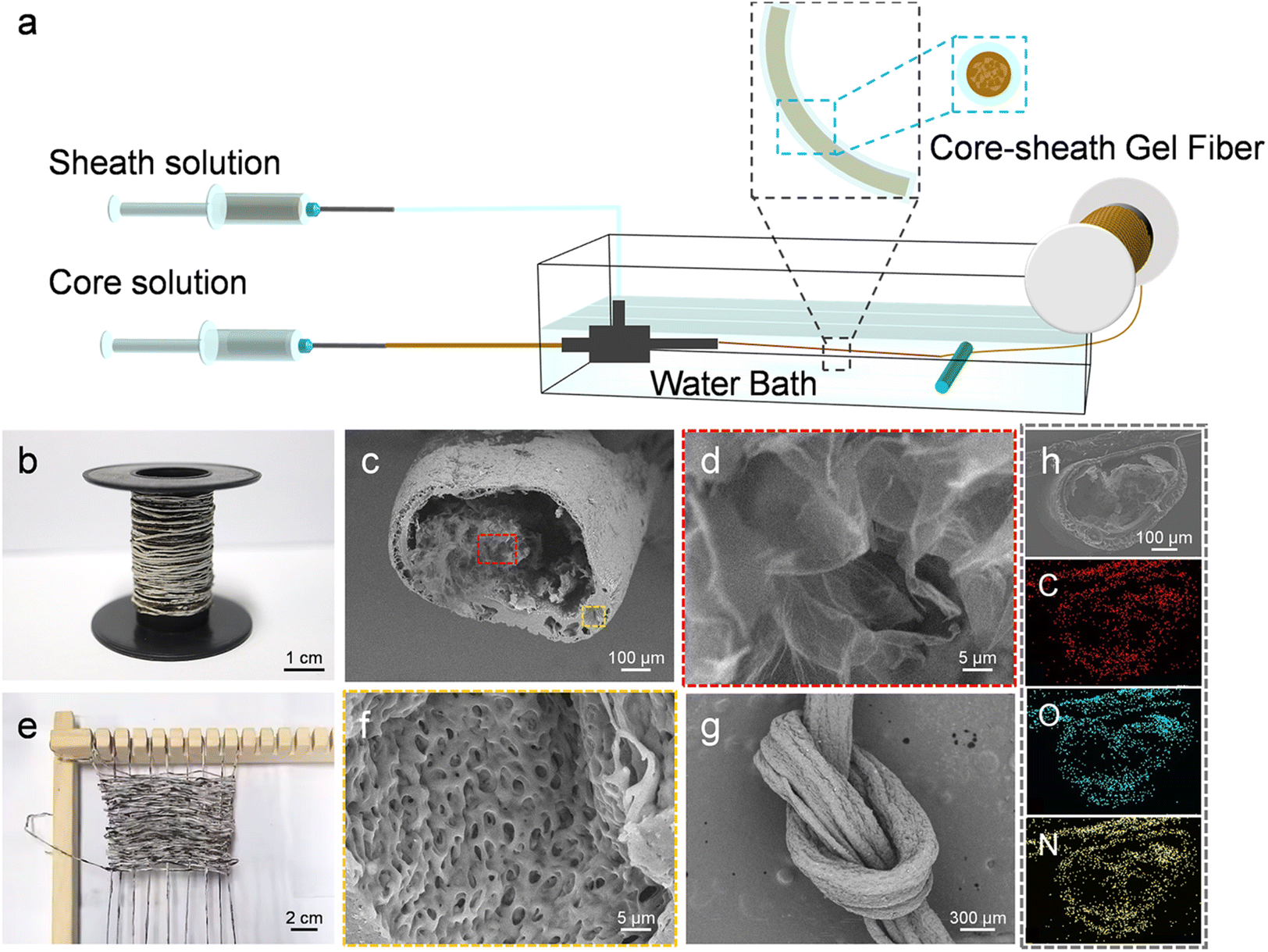 | ||
| Fig. 1 Schematic diagram of the preparation process, structural characterization and macroimages. (a) Schematic illustration of the coaxial wet spinning process and post-treatment process to produce GO/SF@TPU aerogel fibers. (b) Fiber collection display. (c) SEM image of the cross-section of GO/SF@TPU aerogel fibers. (d) and (f) Higher magnification SEM images of GO/SF@TPU aerogel fibers. Specifically, (d) shows a local magnification of the core region and (f) shows a local magnification of the shell region. (e) Piece of fabric woven from GO/SF@TPU aerogel fibers. (g) SEM images of knotted GO/SF@TPU aerogel fibers, and (h) EDS spectra of GO/SF@TPU composites. | ||
In practical applications, fibers are required to possess a certain level of mechanical properties. Fig. 2a presents the tensile stress–strain curve of the GO/SF@TPU fibers. During tensile testing, although the core structure may experience fractures, the cortex remains continuous, which ensures the overall integrity of the fiber structure and preserves its functional stability. The tensile strength of the GO/SF core-containing fibers is significantly higher compared to that of hollow TPU fibers, increasing from 7.75 MPa to 17.21 MPa. The comprehensive stress–strain curves for all samples are depicted in Fig. S5,† respectively. This marked improvement in tensile strength can be attributed to the thinner cortex observed in hollow fibers, which results from the compressive effect of water flow during their manufacturing process. In contrast, the core–shell structured fibers benefit from a more concentrated and less moisture-laden core solution, allowing the cortex to maintain its thickness during the gelation process. This structural advantage enhances the mechanical properties of the composite fibers. The hollow TPU aerogel fibers may experience stress concentration points after freeze-drying due to the presence of water in the core, making the fibers more prone to breaking under stress. In contrast, when the core is a GO/SF solution, the uniform structure and strong interface bonding reduce internal defects and stress concentration, leading to increased fracture strength. Additionally, the as-prepared GO/SF@TPU aerogel fibers demonstrate an impressive elongation rate of up to 1264%, with a single fiber easily supporting a load of 200 grams without fracturing, as shown in Fig. S6a,† and optical images of the fiber before and after lifting the weight are shown in Fig. S6b and c.† Moreover, our aerogel fibers exhibit excellent shape stability at different humidity levels, maintaining their integrity after twisting and stretching (Fig. S7a–c†). They also return to their original shape post-washing (Fig. S7d†). The tensile strength of these fibers is enhanced by over 80 times compared to the GO/SF aerogel fibers (Fig. S8†). Fig. S9† illustrates the effect of the inner flow rate on the tensile properties, with a constant outer flow rate of 300 μL min−1, revealing that the fiber exhibits optimal tensile performance at an inner flow rate of 800 μL min−1. As depicted in Fig. 2b, the toughness of the fibers increases from 57.7 MJ m−3 for the hollow TPU fibers to 136.8 MJ m−3, surpassing the majority of the aerogel fibers reported to date. The toughness was calculated using eqn (S1).† The mechanical properties are listed in detail in Table S1.† Concurrently, in order to evaluate the heat-shielding performance of GO/SF@TPU under ambient conditions, a GO/SF@TPU fabric was adhered to human skin for approximately 10 minutes. The infrared thermal image shows that the surface temperature of the GO/SF@TPU fabric is 24.2 °C, which is close to the background temperature of 18.6 °C. Compared with the wrist temperature of 31.6 °C, it represents a decrease of 7.6 °C, suggesting that GO/SF@TPU has a good heat – insulation effect (Fig. S10†). Furthermore, mercury intrusion porosimetry (MIP) was employed to characterize the pore structure of our GO/SF@TPU aerogel fibers. The test results show that the samples exhibit typical hierarchical pore structure characteristics, with the pore size distribution spanning from approximately 3 nm to 340 μm, covering the mesopore to macropore range. The total mercury intrusion volume amounts to 0.9219 mL g−1, and the porosity is 57.889%, indicating remarkable porous properties. This unique pore structure is advantageous for applications in fields like adsorption. The aerogel with a hierarchical pore structure holds broad application prospects in thermal insulation materials. High porosity and intricate pore diameters significantly hinder heat conduction, endowing the material with excellent heat-insulation capabilities. Moreover, high porosity is favorable for the infiltration of phase change materials (Fig. S11†).
 | ||
| Fig. 2 Characterization and properties of the GO/SF@TPU aerogel fiber. (a) Stress–strain curves of GO/SF@TPU aerogel fiber and hollow TPU aerogel fiber. (b) Corresponding toughness. (c) XRD patterns of SF, GO, TPU, and GO/SF@TPU composites. (d) FT-IR spectra of SF, GO, TPU and GO/SF@TPU composites. | ||
To further explore the crystal structural characteristics of the materials, X-ray diffraction (XRD) and Fourier transform infrared spectroscopy (FTIR) were utilized to analyze pure GO, SF, TPU, and the composite aerogel fiber GO/SF@TPU. The XRD pattern presented in Fig. 2c reveals distinct peaks for GO, featuring a sharp peak at 11.2° and a broad peak around 21.0°, which correspond to the (001) and (002) crystal planes of GO, respectively.44 SF displays a broad peak at 9.0°, while TPU exhibits broad peaks at 8.73° and 20.7°. The broad peaks of the composite material GO/SF@TPU at 8.81° and 21.1° suggest that the crystalline structures of the individual components are preserved without significant alteration during the formation of the composite. This indicates successful integration of the materials, maintaining their intrinsic crystalline properties. The Fourier transform infrared (FTIR) spectra shown in Fig. 2d further corroborate the XRD findings by demonstrating the absence of new absorption peaks in the GO/SF@TPU composite. This indicates that the characteristic peaks of GO, SF, and TPU remain unchanged, suggesting the stability of their crystalline structures during the composite formation process. In the FTIR spectrum of GO, distinct peaks are observed at 1717 cm−1 and 1637 cm−1, corresponding to carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and CC stretching vibrations, respectively. Additionally, a broad band at 1038 cm−1 is attributed to C–O–C stretching, and a broad band at 3169 cm−1 indicates the presence of hydroxyl (O–H) groups.45 For the SF spectrum, strong absorption peaks near 2873 cm−1 are associated with carbon–hydrogen stretching vibrations of methyl (–CH3) and methylene (–CH2–) groups. A peak at 1117 cm−1 corresponds to asymmetric C–O–C stretching.46 The FTIR spectrum of TPU displays several characteristic absorption peaks. An absorption peak at 3332 cm−1 is associated with amino groups (–NH–) in polyurethanes. A strong absorption peak at 1727 cm−1 results from the overlapping effects of ester bonds in both polyesters and polyurethanes. The range of 1600–1615 cm−1 corresponds to CC stretching vibrations, while the region from 1520 to 1560 cm−1 is attributed to N–H deformation vibrations. Absorption peaks between 1456 cm−1 and 1300 cm−1 indicate C–H deformation vibrations, and the range from 1217 to 1018 cm−1 corresponds to C–O–C ether bond vibrations. The peaks observed from 850 to 770 cm−1 suggest aromatic ring absorption, indicating that the isocyanates used in the polyurethane system contain aromatic rings.47 Notably, the FTIR spectrum of the composite material GO/SF@TPU retains the characteristic peaks of GO, SF, and TPU without the appearance of new peaks. This observation suggests that the components are interconnected through physical interactions, such as van der Waals forces and hydrogen bonding, rather than forming new chemical bonds. The stability of the characteristic peaks indicates that the crystalline structures of GO, SF, and TPU are preserved during the composite formation process. This is crucial for maintaining the desired material properties in the composite aerogel fiber, ensuring the integrity and functionality of the individual components within the composite matrix.
O) and CC stretching vibrations, respectively. Additionally, a broad band at 1038 cm−1 is attributed to C–O–C stretching, and a broad band at 3169 cm−1 indicates the presence of hydroxyl (O–H) groups.45 For the SF spectrum, strong absorption peaks near 2873 cm−1 are associated with carbon–hydrogen stretching vibrations of methyl (–CH3) and methylene (–CH2–) groups. A peak at 1117 cm−1 corresponds to asymmetric C–O–C stretching.46 The FTIR spectrum of TPU displays several characteristic absorption peaks. An absorption peak at 3332 cm−1 is associated with amino groups (–NH–) in polyurethanes. A strong absorption peak at 1727 cm−1 results from the overlapping effects of ester bonds in both polyesters and polyurethanes. The range of 1600–1615 cm−1 corresponds to CC stretching vibrations, while the region from 1520 to 1560 cm−1 is attributed to N–H deformation vibrations. Absorption peaks between 1456 cm−1 and 1300 cm−1 indicate C–H deformation vibrations, and the range from 1217 to 1018 cm−1 corresponds to C–O–C ether bond vibrations. The peaks observed from 850 to 770 cm−1 suggest aromatic ring absorption, indicating that the isocyanates used in the polyurethane system contain aromatic rings.47 Notably, the FTIR spectrum of the composite material GO/SF@TPU retains the characteristic peaks of GO, SF, and TPU without the appearance of new peaks. This observation suggests that the components are interconnected through physical interactions, such as van der Waals forces and hydrogen bonding, rather than forming new chemical bonds. The stability of the characteristic peaks indicates that the crystalline structures of GO, SF, and TPU are preserved during the composite formation process. This is crucial for maintaining the desired material properties in the composite aerogel fiber, ensuring the integrity and functionality of the individual components within the composite matrix.
The GO/SF@TPU aerogel fibers were impregnated with PEG to effectively utilize their porous structure. The fibers were subjected to vacuum infiltration for 2 h, followed by natural drying after excess PEG was removed from the surface. To preserve the flexibility of the fibers, a 50% PEG solution was employed in the preparation of the GO/SF@TPU-PEG composite material. Fig. 3a displays a SEM image of the fiber's cross-section after PEG impregnation, revealing that the small pores have been adequately filled, resulting in a denser and more compact porous structure. Furthermore, Fig. 3b and c offer magnified views of the core and shell regions of the fibers, clearly showing that PEG is uniformly distributed in the pores of both the core and the shell. This uniform distribution is crucial for enhancing the mechanical properties and functional stability of the composite material. The successful impregnation of PEG into the GO/SF@TPU aerogel fibers demonstrates their potential for various applications requiring a combination of porosity and flexibility. The presence of PEG not only reinforces the structural integrity but also imparts flexibility, making the composite suitable for a wide range of practical uses.
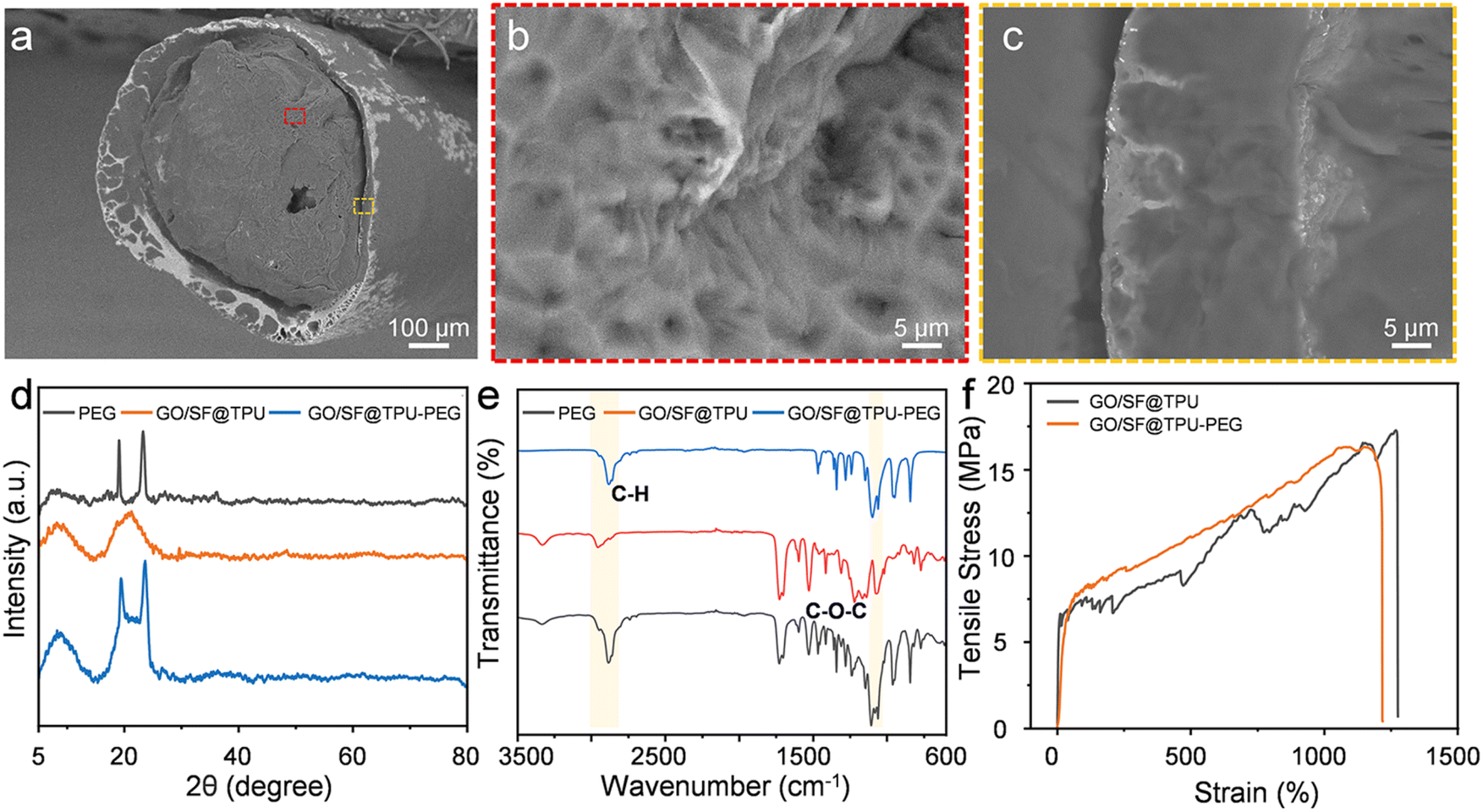 | ||
| Fig. 3 Characterization of the GO/SF@TPU-PEG fiber. (a) Cross-sectional SEM image of GO/SF@TPU-PEG aerogel fibers after PEG impregnation. (b) and (c) Higher magnification SEM images of GO/SF@TPU-PEG fiber. Specifically, (b) shows a local magnification of the core region and (c) shows a local magnification of the shell region. (d) XRD diffraction of PEG, GO/SF@TPU and GO/SF@TPU-PEG. (e) FT-IR spectra of PEG, GO/SF@TPU and GO/SF@TPU-PEG. (f) Stress–strain curves of GO/SF@TPU aerogel fibers and GO/SF@TPU-PEG fibers. | ||
As illustrated in Fig. 3d, pure PEG exhibits characteristic diffraction peaks at approximately 19.0° and 23.2°. The XRD pattern of GO/SF@TPU-PEG shows diffraction peaks near 8.84°, 19.4°, and 23.5°, similar to the individual components, indicating no new peaks. This observation suggests that the interaction between PEG and GO/SF@TPU is purely physical, without any chemical reaction. Fig. 3e presents the FTIR spectra of PEG, GO/SF@TPU, and GO/SF@TPU-PEG. In the spectrum of pure PEG, peaks at 962 cm−1 and 1061 cm−1 correspond to the stretching vibrations of C–O–C, with maximum absorption peaks at 1101 cm−1 and 1150 cm−1. Additionally, a stretching vibration peak of –CH2 is observed at 2882 cm−1. The FTIR spectrum of GO/SF@TPU-PEG reveals the characteristic peaks of both GO/SF@TPU and PEG, without the emergence of new peaks. The peak at 1466 cm−1 is attributed to the bending vibration of –CH2, and the region from 1360 cm−1 to 1240 cm−1 corresponds to the out-of-plane rocking vibration of –CH2. Peaks at 1150–1060 cm−1 are due to the stretching vibration of C–O, while peaks at 946 cm−1 and 841 cm−1 are related to the in-plane vibration of –CH2. These findings indicate that PEG is physically adsorbed onto the GO/SF@TPU aerogel fiber without undergoing any chemical reaction. The retention of characteristic peaks in both the XRD and FTIR spectra confirms that the integration of PEG into the GO/SF@TPU matrix is achieved through physical interactions, preserving the structural integrity and properties of the individual components within the composite. This physical bonding is crucial for maintaining the functional attributes of the composite material, ensuring its suitability for various applications requiring a combination of porosity, flexibility, and mechanical stability.
Fig. 3f displays the stress–strain curves of the fibers before and after PEG infusion. The maximum stress observed is 17.21 MPa before infusion and 16.07 MPa after infusion. The strain decreases from 1264% to 1071%, indicating that the PEG infusion does not significantly compromise the mechanical properties of the fibers. These results suggest that the infusion process maintains the overall structural integrity and flexibility of the fibers, making the composite material suitable for applications requiring both high mechanical strength and elasticity.
The shape stability of phase change materials is critical for their practical application. Fig. 4 presents photographs from leakage tests conducted on PEG and GO/SF@TPU-PEG composite samples at various time intervals. Two samples with initially identical shapes, along with pure PEG, were placed on hydrophobic paper and placed on a heating table at a constant temperature of 90 °C. Observations indicated that pure PEG began to liquefy after just 5 minutes of heating and was completely lost by 10 minutes. In stark contrast, the GO/SF@TPU-PEG composite material exhibited no signs of leakage under the same test conditions. This outcome demonstrates the high shape stability of GO/SF@TPU-PEG at elevated temperatures, highlighting its significant potential as an efficient phase change material. To evaluate the practical application potential of GO/SF@TPU-PEG fibers in fabric form, a series of tests, including bending and folding, were conducted. As illustrated in Fig. 5a, the results confirmed the fabric's excellent flexibility and shape stability. Thermal insulation properties were investigated by placing a fabric sample, approximately 2 cm × 2 cm in size and 0.5 mm thick, on a hot plate set at 100 °C. The temperature difference between the sample surface and the environment was monitored. Fig. 5b shows the variation in surface temperature of the fabric during heating and cooling cycles compared to the environmental temperature. At 100 °C, the maximum temperature difference between the fabric and the hot plate reached 26.4 °C. Even under extreme conditions of −30 °C, the fabric surface maintained a temperature of −13.7 °C, exhibiting a 16.3 °C difference. These data strongly indicate the superior thermal insulation performance of the fabric. Infrared thermography at different time points (0 min, 1 min, 3 min, 16 min, 18 min, and 20 min) is presented in Fig. 5c, visually reflecting the temperature changes of the fabric. The infrared images further validate the fabric's capacity to maintain its thermal properties over time, showcasing its effectiveness in thermal insulation applications.
 | ||
| Fig. 4 The shape stability test photographs of the pure PEG and GO/SF@TPU-PEG fabric. | ||
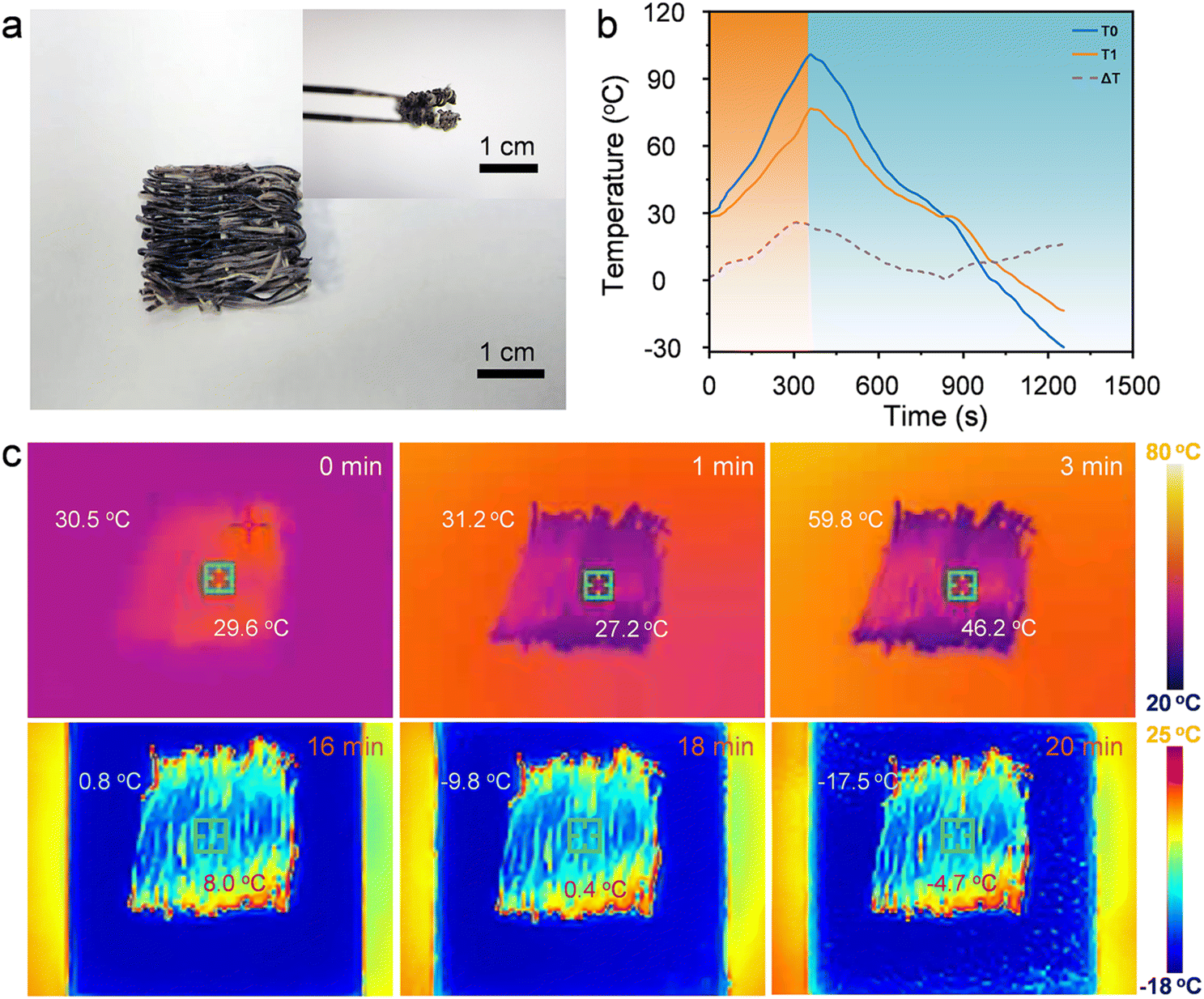 | ||
| Fig. 5 Shape stability and thermal insulation tests of GO/SF@TPU-PEG fabric. (a) The pictures of GAF-PW after the shape stability test. (b) The temperature–time curve for the hot plate (T0), sample surface (T1) and temperature difference (ΔT) between T0 and T1. (c) Thermal images of the fabric surface corresponding to the warming and cooling process. | ||
The thermal stability of pure PEG, TPU, and GO/SF@TPU-PEG was assessed using thermogravimetric analysis (TGA) and derivative thermogravimetry (DTG). As depicted in Fig. 6a and b, pure PEG exhibited a significant weight loss of 99.5% between 312 °C and 416 °C due to its decomposition. In contrast, GO/SF@TPU-PEG demonstrated a more intricate thermal degradation behavior, segmented into four distinct stages: (1) the initial stage (226–301 °C) showed a weight loss of 13.4%, indicating the commencement of TPU degradation (Fig. S12†); (2) the second stage (301–352 °C) exhibited a weight loss of 19.4%, corresponding to the decomposition of PEG and potential removal of oxygen-containing functional groups from GO, along with the initial thermal decomposition of SF. (3) The third stage (352–430 °C) recorded a weight loss of 55.3%, characterized by substantial TPU decomposition and further degradation of GO and SF, suggesting that TPU is the primary decomposing component at this stage. (4) The final stage (430–530 °C) with a weight loss of 3.9% represents the ultimate decomposition of the remaining thermally stable components within the composite. These observations indicate that GO/SF@TPU-PEG exhibits enhanced thermal stability at elevated temperatures, with the highest rate of thermal decomposition occurring at 392 °C. While PEG is almost entirely decomposed by 416 °C, GO/SF@TPU-PEG retains over 30% of its mass at this temperature. Additionally, its onset decomposition temperature is 226 °C, significantly exceeding the phase change temperature range of the composite. This demonstrates the high thermal stability of the GO/SF@TPU-PEG composite phase change materials under operational conditions. Detailed TGA data for the various materials are provided in Table S2.†
 | ||
| Fig. 6 Thermal properties of GO/SF@TPU-PEG. (a) TGA curves of PEG and GO/SF@TPU-PEG fibers. (b) DTG curves of PEG and GO/SF@TPU-PEG fibers. (c) DSC curves of PEG and GO/SF@TPU-PEG fibers. (d) Corresponding column chart of PEG and GO/SF@TPU-PEG fibers. (e) Thermal conductivity of PEG, GO/SF@TPU and GO/SF@TPU-PEG. (f) Comparison of toughness and thermal conductivity of our GO/SF@TPU-PEG with those reported in the literature. | ||
The phase change performance of pure PEG and GO/SF@TPU-PEG was evaluated using differential scanning calorimetry (DSC). The relevant data are summarized in Table S3.† The DSC curves for pure PEG and GO/SF@TPU-PEG composite phase change materials are presented in Fig. 6c, with the corresponding enthalpy values depicted in Fig. 6d. The loading rate (α), defined as the mass or volume fraction of PCM in the composite material, was determined to be 56.21% for PEG in the GO/SF@TPU-PEG composite fibers. The loading rate was calculated using eqn (S2).† The results indicate that pure PEG exhibits a melting enthalpy value of 198.47 J g−1 and a crystallization enthalpy value of 176.04 J g−1. In contrast, the GO/SF@TPU-PEG composite shows the corresponding values of 86.66 J g−1 and 69.84 J g−1. The encapsulation rate (R), defined as the ratio of the melting enthalpy of the CPCM to that of the pure PCMs, was calculated to be 43.7%. The encapsulation rate was calculated using eqn (S3).† The decreased melting and crystallization enthalpy values observed in GO/SF@TPU-PEG, along with the reduced melting temperature and increased crystallization temperature, may be attributed to the uniform distribution of PEG within the composite structure and its interaction with other components, such as GO, SF, and TPU. This uniform distribution weakens the intermolecular interactions in PEG, thereby affecting its phase transition characteristics. Specifically, the melting temperature of GO/SF@TPU-PEG decreases from 57.33 °C in pure PEG to 51.57 °C, and the crystallization temperature increases from 33.82 °C to 41.38 °C. This shift suggests that the presence of other components in the composite material alters the phase change behavior of PEG, providing additional nucleation sites that facilitate the crystallization process. The irregular peaks in the DSC curves likely reflect the crystallinity of TPU, which influences the overall phase change behavior (Fig. S13†). These findings elucidate the impact of the composite material structure on the properties of phase change materials and highlight the potential of GO/SF@TPU-PEG for temperature regulation and thermal energy storage. This is significant for the development of high-performance phase change materials and lays a foundation for further research and practical applications. The cycle stability was investigated through rapid heating–cooling tests conducted for 20 and 50 cycles. The phase-change properties of GO/SF@TPU-PEG composite phase-change fibers were measured by differential scanning calorimetry (DSC). As presented in the DSC curves in Fig. S14,† the characteristic values during the phase-change process were reduced. The melting and crystallization enthalpies listed in Table S3† differed slightly from those of the original GO/SF@TPU-PEG. Minor differences were also found in the phase-change temperature compared with the original one, suggesting that the phase-change properties during the cycle process demonstrated a certain degree of fluctuation. Nevertheless, it should be emphasized that the material exhibits excellent mechanical properties. In practical applications, the mechanical properties play a crucial role, which indicates that the overall performance of the material is still of great value. The thermal conductivity of pure PEG and the GO/SF@TPU-PEG fabric was assessed using a thermal constants analyzer. The results (Fig. 6e) indicate that the thermal conductivity of pure PEG is 0.2884 W m−1 K−1, whereas that of GO/SF@TPU is 0.0826 W m−1 K−1 and that of GO/SF@TPU-PEG is 0.0863 W m−1 K−1. Notably, the incorporation of PEG enhances the thermal conductivity of the composite material, as PEG fills pores and strengthens heat conduction pathways. Thermal conductivity, also known as the thermal conductivity coefficient, is a parameter that measures a substance's ability to conduct heat, defined as the amount of heat transmitted per unit area per unit time. Importantly, the duality of thermal conductivity in materials must be carefully considered; while high thermal conductivity aids in rapid heat transfer, excessive conductivity may lead to undesirable heat dissipation, affecting thermal storage performance.
In the realm of thermal management, particularly concerning insulating textiles, the importance of strong mechanical properties and low thermal conductivity is paramount. Fig. 6f compares the thermal conductivity and toughness of various materials, revealing that the GO/SF@TPU-PEG PCFs exhibit exceptional mechanical toughness-reaching up to 142.3 MJ m−3. Our GO/SF@TPU aerogel fibers and GO/SF@TPU-PEG PCFs outperform most reported aerogels and commercial textile materials. Additionally, they demonstrate moderate thermal conductivity, as detailed in Table S4,† which aids in heat conservation and further emphasizes their substantial applicability in practical scenarios.
Conclusions
In this study, we successfully engineered graphene oxide/silk protein core-sheath aerogel fibers for advanced thermal management applications. By employing a coaxial wet spinning technique, we integrated a graphene oxide/silk fibroin composite as the core, encapsulated with thermoplastic polyurethane (TPU) to form a robust core–shell structure. The resulting GO/SF@TPU aerogel fibers exhibited exceptional tensile strength and toughness, and retained shape stability over a wide range of humidity levels. All these characteristics are highly crucial and essential for wearable applications. Subsequent impregnation with polyethylene glycol (PEG), a phase change material, yielded the GO/SF@TPU-PEG composite fibers, which demonstrated remarkable thermal storage and insulation properties. A low thermal conductivity of 0.0863 W m−1 K−1 and a melting latent heat of 86.66 J g−1 underscore the fibers' efficacy in thermal regulation. Moreover, the fabrication process was both efficient and cost-effective, rendering the composite PCFs superior in insulation and energy storage performance. These findings not only substantiate the potential of the developed fibers in smart textile applications but also pave the way for the development of next-generation wearable devices with enhanced thermal management capabilities.Future research will optimize material composition and structure to enhance thermal conductivity while maintaining mechanical integrity. This will expand the applicability of aerogel fibers in diverse thermal management applications, advancing sustainable solutions for wearable technologies.
Data availability
The data that support the findings of this study are available upon reasonable request from the authors.Author contributions
W. J. X. and Z. W. contributed to the research design and analysis of the experimental results. W. J. X. carried out the experiments and analyzed the data. C. C. R. assisted in the mechanical testing of samples. W. J. X. and Y. Y. Z. wrote the manuscript. Y. Y. Z. and J. L. acquired funding and supervised the work. All authors discussed the results and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22279139 and 22465026), the Natural Science Foundation of Inner Mongolia Autonomous Region of China (20241Q06 and 2024MS05005), and the Program of Higher-level Talents of IMU (10000-23112101/173 and 10000-21311201/007).References
- L. T. Yeh, J. Electron. Packag., 1995, 117, 333–339 CrossRef.
- X. Zhang, H. Liu, Y. Kou, K. Sun, W. Han, Y. Zhao and Q. Shi, Mater. Today Sustain., 2024, 28, 100960 Search PubMed.
- R. Feng, P. Huang, Z. Tang, Y. He and Z. Bai, Energy Convers. Manage., 2022, 272, 116359 CrossRef.
- X. Shi, M. R. Yazdani, R. Ajdary and O. J. Rojas, Carbohydr. Polym., 2021, 254, 117279 CrossRef CAS PubMed.
- C. Yin, J. Lan, X. Wang, Y. Zhang, R. Ran and L.-Y. Shi, ACS Appl. Mater. Interfaces, 2021, 13, 21810–21821 Search PubMed.
- T. Zhicheng, S. Quan and L. Xin, in Phase Change Materials and Their Applications, ed. M. Mohsen, IntechOpen, Rijeka, 2018, ch. 5, DOI:10.5772/intechopen.78800.
- A. Al-Ahmed, M. A. J. Mazumder, B. Salhi, A. Sari, M. Afzaal and F. A. Al-Sulaiman, J. Energy Storage, 2021, 35, 102329 CrossRef.
- N. Sarier and E. Onder, Thermochim. Acta, 2012, 540, 7–60 CrossRef CAS.
- B. Zalba, J. M. Marín, L. F. Cabeza and H. Mehling, Appl. Therm. Eng., 2003, 23, 251–283 CrossRef CAS.
- S. Peng, A. Fuchs and R. A. Wirtz, J. Appl. Polym. Sci., 2004, 93, 1240–1251 CrossRef CAS.
- L. Feng, W. Zhao, J. Zheng, S. Frisco, P. Song and X. Li, Sol. Energy Mater. Sol. Cells, 2011, 95, 3550–3556 CrossRef CAS.
- L. Chen, R. Zou, W. Xia, Z. Liu, Y. Shang, J. Zhu, Y. Wang, J. Lin, D. Xia and A. Cao, ACS Nano, 2012, 6, 10884–10892 CrossRef CAS PubMed.
- S. Ramakrishnan, J. Sanjayan, X. Wang, M. Alam and J. Wilson, Appl. Energy, 2015, 157, 85–94 Search PubMed.
- X. Zhang, B. Wu, G. Chen, Y. Xu, T. Shi, Z. Huang, Y. Liu, M. Fang, X. Wu and X. Min, Energy Fuels, 2021, 35, 877–882 CrossRef CAS.
- Z. Liu, Y. Ran, J. Xi and J. Wang, Soft Matter, 2020, 16, 9160–9175 RSC.
- J. P. Randall, M. A. B. Meador and S. C. Jana, ACS Appl. Mater. Interfaces, 2011, 3, 613–626 CrossRef CAS PubMed.
- J. Lyu, Z. Liu, X. Wu, G. Li, D. Fang and X. Zhang, ACS Nano, 2019, 13, 2236–2245 CrossRef PubMed.
- B. Wang, G. Li, L. Xu, J. Liao and X. Zhang, ACS Nano, 2020, 14, 16590–16599 CrossRef CAS PubMed.
- L. Wang, J. Feng, Y. Luo, Y. Jiang, G. Zhang and J. Feng, Small Methods, 2022, 6, 2200045 CrossRef CAS PubMed.
- Y. Hou, Z. Sheng, C. Fu, J. Kong and X. Zhang, Nat. Commun., 2022, 13, 1227 CrossRef CAS PubMed.
- Y. Bao, J. Lyu, Z. Liu, Y. Ding and X. Zhang, ACS Nano, 2021, 15, 15180–15190 CrossRef CAS PubMed.
- M. Wu, Z. Shao, N. Zhao, R. Zhang, G. Yuan, L. Tian, Z. Zhang, W. Gao and H. Bai, Science, 2023, 382, 1379–1383 CrossRef CAS PubMed.
- T. Xue, C. Zhu, X. Feng, Q. Wali, W. Fan and T. Liu, Adv. Fiber Mater., 2022, 4, 1118–1128 CrossRef CAS.
- X. Li, G. Dong, Z. Liu and X. Zhang, ACS Nano, 2021, 15, 4759–4768 CrossRef CAS PubMed.
- Q. Ge, J. Chu, W. Cao, F. Yi, Z. Ran, Z. Jin, B. Mao, Z. Li and K. S. Novoselov, Adv. Funct. Mater., 2022, 32, 2205934 CrossRef CAS.
- A. A. Balandin, Nat. Mater., 2011, 10, 569–581 CrossRef CAS PubMed.
- H. Huang, H. Shi, P. Das, J. Qin, Y. Li, X. Wang, F. Su, P. Wen, S. Li, P. Lu, F. Liu, Y. Li, Y. Zhang, Y. Wang, Z.-S. Wu and H.-M. Cheng, Adv. Funct. Mater., 2020, 30, 1909035 CrossRef CAS.
- H. Zhang, A. F. Fonseca and K. Cho, J. Phys. Chem. C, 2014, 118, 1436–1442 CrossRef CAS.
- C. Wang, L. Feng, H. Yang, G. Xin, W. Li, J. Zheng, W. Tian and X. Li, Phys. Chem. Chem. Phys., 2012, 14, 13233–13238 RSC.
- G. Poggi, D. Chelazzi and M. Laurati, Colloids Surf., A, 2022, 636, 128121 CrossRef CAS.
- R. Rong, H. Li, X. Dong, L. Hu, X. Shi, Y. Du, H. Deng and Y. Sa, Int. J. Biol. Macromol., 2023, 233, 123501 CrossRef CAS PubMed.
- Z. Shao and F. Vollrath, Nature, 2002, 418, 741 CrossRef CAS PubMed.
- H. Yang, Z. Wang, Z. Liu, H. Cheng and C. Li, Polymers, 2019, 11, 1899 CrossRef CAS PubMed.
- Y. Shi, Y. Miao, L. Li, W. Li, X. Zheng, J. Zhao and Z. Liu, Mater. Today Phys., 2024, 40, 101323 CrossRef CAS.
- H. Maleki, L. Whitmore and N. Hüsing, J. Mater. Chem. A, 2018, 6, 12598–12612 RSC.
- Y. Okahisa, C. Narita and K. Yamada, Mater. Today Commun., 2020, 25, 101630 CrossRef CAS.
- A. Allahbakhsh and M. Arjmand, Carbon, 2019, 148, 441–480 Search PubMed.
- S. Wang, H. Ning, N. Hu, K. Huang, S. Weng, X. Wu, L. Wu and J. Liu, Alamusi, Composites, Part B, 2019, 163, 716–722 Search PubMed.
- M. Mehrali, S. T. Latibari, M. Mehrali, T. M. I. Mahlia, H. S. C. Metselaar, M. S. Naghavi, E. Sadeghinezhad and A. R. Akhiani, Appl. Therm. Eng., 2013, 61, 633–640 CrossRef CAS.
- L. Jiang, Y. Lei, Q. Liu and J. Lei, Energy, 2020, 193, 116802 CrossRef CAS.
- B. Tang, J. Cui, Y. Wang, C. Jia and S. Zhang, Sol. Energy, 2013, 97, 484–492 CrossRef CAS.
- C. S. Lee, J. Moon, J. T. Park and J. H. Kim, Chem. Eng. J., 2023, 453, 139818 CrossRef CAS.
- M. Choi, Y. Kim, S. Park, D. Ka, T. Kim, S. Lee, E.-H. Sohn, Y. Jin and J. Hong, Adv. Funct. Mater., 2021, 31, 2101511 CrossRef CAS.
- X. Zhang, A. Wang, X. Zhou, F. Chen and Q. Fu, Carbon, 2020, 165, 340–348 CrossRef CAS.
- Y. Zhang, Y. Li, P. Ming, Q. Zhang, T. Liu, L. Jiang and Q. Cheng, Adv. Mater., 2016, 28, 2834–2839 CrossRef CAS PubMed.
- C. Zhao, Y. Liu, Z. Lv, L. Cao, J. Ren, Z. Shao and S. Ling, Adv. Fiber Mater., 2022, 4, 1191–1208 CrossRef CAS.
- X. Wang, R. Xue, M. Li, X. Guo, B. Liu, W. Xu, Z. Wang, Y. Liu and G. Wang, Surf. Interfaces, 2022, 32, 102132 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00580a |
| This journal is © The Royal Society of Chemistry 2025 |