
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vanadium-based oxyhalide photocatalysts for visible-light-driven Z-scheme water splitting: advancing conduction band engineering†
Hajime
Suzuki
 *a,
Ryuki
Tomita
a,
Yusuke
Ishii
a,
Osamu
Tomita
a,
Akinobu
Nakada
ab,
Akinori
Saeki
c and
Ryu
Abe
*a
*a,
Ryuki
Tomita
a,
Yusuke
Ishii
a,
Osamu
Tomita
a,
Akinobu
Nakada
ab,
Akinori
Saeki
c and
Ryu
Abe
*a
aDepartment of Energy and Hydrocarbon Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: suzuki.hajime.7x@kyoto-u.ac.jp; ryu-abe@scl.kyoto-u.ac.jp
bPrecursory Research for Embryonic Science and Technology (PRESTO), Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
cDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, 2-1 Yamadaoka, Suita, Osaka 565-0871, Japan
First published on 26th April 2025
Abstract
Z-scheme water-splitting systems have garnered significant attention as a promising technology for producing hydrogen cleanly from water using solar energy. Layered oxyhalides have emerged as efficient oxygen-evolving photocatalysts (OEPs) for these systems. However, the conduction band minimum (CBM) of these oxyhalides is excessively negative compared to the reduction potential of electron mediators. This limitation underscores the need for novel conduction band engineering approaches to narrow the band gap and enable the utilization of visible light across a broader spectrum. This study introduces vanadium-based oxyhalides as OEPs in the Z-scheme system. The physicochemical properties and photocatalytic activities of three lead-vanadium-based oxyhalides, Pb14(VO4)2O9Cl4, Pb5(VO4)3Cl, and PbVO3Cl, were comprehensively characterized. The CBMs of these materials were found to be more positive than those of conventional oxyhalide photocatalysts and displayed significant variation. PbVO3Cl exhibited the most positive CBM and the smallest band gap, enabling visible light absorption up to approximately 550 nm. Madelung site potential analysis of each vanadium cation highlighted the reasons for the significant difference in CBM positions among the Pb–V oxyhalides. Remarkably, PbVO3Cl exhibited oxygen evolution activity under visible light irradiation, marking the first instance of a vanadium-based oxyhalide as an OEP in a Z-scheme system. This exceptional activity of PbVO3Cl was attributed to the superior carrier transport properties, owing to the interconnected VO5 units, as revealed by time-resolved microwave conductivity (TRMC) measurements, as well as the extended visible light absorption.
Introduction
Water splitting using semiconductor photocatalysts has gained significant attention as a promising technology for producing hydrogen cleanly from water by harvesting abundant solar energy.1–8 To achieve practical (i.e., cost-effective) solar-to-hydrogen conversion efficiency, it is essential to efficiently utilize photons in the visible light region rather than just in the UV region. A Z-scheme water-splitting system has demonstrated great potential in capturing a broad range of visible light, and its efficiency has steadily improved in recent years.9–12 This system typically consists of a mixture of two different photocatalyst particles: a hydrogen-evolving photocatalyst (HEP) and an oxygen-evolving photocatalyst (OEP), along with a redox couple (Ox/Red), such as Fe3+/Fe2+, dissolved in an aqueous solution. By reducing the energy required to excite each photocatalyst, this system enables the utilization of various visible-light-responsive photocatalysts capable of absorbing light with long wavelengths. In other words, it allows semiconductors with water reduction or oxidation potentials suitable for one side of the system to be effectively employed. For example, visible-light-responsive oxides, such as tungsten trioxide (WO3), can function as OEPs if they can reduce the oxidant to a reductant.9 Similarly, non-oxide photocatalysts (e.g., (oxy)sulfides, (oxy)nitrides, and dyes) can act as HEPs if they can oxidize the reductant to an oxidant.13–18Recently, Sillén(-Aurivillius)-type layered oxyhalides such as Bi4NbO8Cl and PbBiO2Cl, have emerged as promising OEPs for visible-light-driven Z-scheme water splitting.19–21 The valence band maximum (VBM) of these oxyhalides is mainly composed of elevated O-2p orbitals, where photogenerated holes are preferentially consumed by water oxidation rather than self-oxidation (halogen oxidation), resulting in their substantially high stabilities as OEPs in Z-scheme water splitting systems.22,23 The conduction band minimum (CBM) of these oxyhalides is primarily composed of Bi-6p or Pb-6p orbitals and is sufficiently negative relative to the water reduction potential, enabling them to evolve hydrogen (H2) from water under light irradiation.19,24–27 However, such negative CBMs are not required when these materials are employed as OEPs in Z-scheme systems with redox mediators. In an ideal OEP in Z-scheme systems, the CBM should be somewhat more negative than the reduction potential of electron mediators. However, the CBM of the above oxyhalides (e.g., Bi4NbO8Cl) is excessively negative. This motivates the development of oxyhalides with CBMs positioned at more positive potentials, resulting in narrower bandgaps for harvesting photons in longer wavelength regions.
This study focused on pentavalent vanadium(V) cations. The V cation with high ionization energy is expected to form CBMs at more positive potentials. After exploring various Pb(Bi)–V-based oxyhalides, PbVO3Cl, Pb5(VO4)3Cl, and Pb14(VO4)2O9Cl4 (Fig. 1) were selected.28–30 Their optical properties, band structures, and photocatalytic activities were evaluated, and their relationships were investigated through various experimental and theoretical investigations.
 | ||
| Fig. 1 Crystal structures of Pb–V-based oxyhalides: PbVO3Cl, Pb5(VO4)3Cl, and Pb14(VO4)2O9Cl4. | ||
Experimental
Materials
Lead(II) oxide (PbO, 99.5%), vanadium(V) oxide (V2O5, 99.0%), sodium metavanadate(v) (NaVO3, 99.5%), boric acid (B(OH)3, 99.5%), sodium orthovanadate(V) (Na3VO4, 99.98%), sodium chloride (NaCl, 99.5%), lead(II) nitride (Pb(NO3)2, 99.9%), disodium hydrogenphosphate 12-water (Na2HPO4·12H2O, 99%), sodium dihydrogenphosphate 2-water (NaH2PO4·2H2O, 99%), iron(III) chloride hexahydrate (FeCl3·6H2O, 99%), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, 99%), iron(III) perchlorate n-hydrate (Fe(ClO4)3, 70% as anhydrous), iron(III) sulfate n-hydrate (Fe2(SO4)3, 60–80% as anhydrous), nitric acid (HNO3, 69% as assay), perchloric acid (HClO4, 70% as assay), hydrochloric acid (HCl, 35% as assay), sulfuric acid (H2SO4, 95% as assay), and ruthenium chloride n-hydrate (RuCl3·nH2O, 99.9%) were purchased from FUJIFILM Wako Chemicals. Lead(II) chloride (PbCl2, 99.999%) and sodium orthovanadate(V) (Na3VO4, 99.98%) were purchased from Sigma-Aldrich. Ammonium vanadate(V) (NH4VO3, 99.0%) was purchased from KANTO CHEMICAL.Synthesis of PbVO3Cl
PbVO3Cl was synthesized using solid-state and hydrothermal methods, following previously reported procedures.28,31 In the solid-state method, PbO, PbCl2, and V2O5 were used as starting materials, with masses of 0.37 g (1.65 mmol), 0.46 g (1.65 mmol), and 0.30 g (1.65 mmol), respectively. These components were mixed thoroughly for 15 minutes, pelletized, and sealed in a vacuum quartz tube. The sample was then calcined at 450 °C for 24 hours with heating and cooling rates of 3 °C min−1 to yield the desired product. For the hydrothermal method, PbCl2, NaVO3, and B(OH)3 were employed as raw materials, with masses of 3.7 g (13.2 mmol), 1.6 g (13.2 mmol), and 3.8 g (61.2 mmol), respectively. These reagents were added to 36 mL of deionized water, stirred, and transferred into a 100 mL Teflon-lined autoclave. The mixture was heated at 170 °C for 24 hours. The resulting product was washed with deionized water and dried overnight at 60 °C in a drying oven. The X-ray diffraction (XRD) patterns of the obtained samples are shown in Fig. S1.† The sample synthesized via the solid-state method contained impurities, including PbCl2 and Pb5(VO4)3Cl, whereas the hydrothermally synthesized sample was free of impurities. X-ray photoelectron spectroscopy (XPS) measurements were also performed for the hydrothermally synthesized sample, confirming the presence of the expected elements at the surface, predominantly in their intended oxidation states (Fig. S2†). Therefore, the hydrothermally synthesized sample was used for all investigations.Synthesis of Pb5(VO4)3Cl
Pb5(VO4)3Cl was synthesized via a precipitation method based on a procedure reported previously.32 Specifically, 0.55 g (3 mmol) of Na3VO4 and 0.058 g (1 mmol) of NaCl were dissolved in 50 mL of deionized water. To this solution, an aqueous solution containing 1.7 g (5 mmol) of Pb(NO3)2 dissolved in 50 mL of deionized water was slowly added dropwise. The resulting suspension was stirred for 1 hour, washed with deionized water, and dried overnight at 60 °C in a drying oven to obtain Pb5(VO4)3Cl (Fig. S3†).Synthesis of Pb14(VO4)2O9Cl4
Pb14(VO4)2O9Cl4 was synthesized via a solid-state reaction. PbO, PbCl2, and V2O5 were weighed precisely at 0.88 g (3.96 mmol), 0.18 g (0.66 mmol), and 0.060 g (0.33 mmol), respectively. These materials were mixed thoroughly for 15 minutes and then pelletized. The pellets were sealed in a vacuum quartz tube and subjected to heat treatment at 300–700 °C for 24 hours, with heating and cooling rates of 3 °C min−1.Electrochemical measurement
Mott–Schottky plots were recorded in a three-electrode electrochemical cell equipped with a platinum (Pt) wire counter-electrode and Ag/AgCl reference electrode in a phosphate-buffered solution (0.1 M, pH 6.0). Measurements were performed using an electrochemical analyzer (VersaSTAT 4, Princeton Applied Research) with an amplitude of 10 mV and a frequency of 1000 Hz. The Mott–Schottky plots for the V-based oxyhalides were obtained in the stable potential range determined from cyclic voltammetry measurements. The electrode was prepared using the squeegee method. A particulate sample was mixed with a small amount of water, coated on a fluorine-doped tin oxide (FTO) conductive substrate, and dried overnight at room temperature. The coated area was fixed at ca. 1.5 × 4.0 cm2. Photoelectrochemical measurements were also carried out using the same setup and electrode, under chopped visible light from a 300 W Xe lamp with a cutoff filter (L-42, HOYA).Characterization
Powder X-ray diffraction (XRD; MiniFlex II, Rigaku, Cu Kα), X-ray photoelectron spectroscopy (XPS; ESCA MT-5500, ULVAC-PHI, Mg Kα), scanning electron microscopy (SEM; NVision 40, Carl Zeiss-SIINT), and ultra-violet (UV)-vis diffuse reflectance spectroscopy (V-650, Jasco) were used to characterize the samples. The collected XRD data were analyzed using Jana2006.33 The XPS binding energies were corrected with reference to the C 1s peak at 284.8 eV. The VESTA program was used to draw crystal structures and calculate Madelung site potentials.34 The ionization energy was directly measured by PYS (BIP-KV201, Bunkoukeiki) in a vacuum (<5 × 10−2 Pa). Time-resolved microwave conductivity (TRMC) measurements were performed using the third harmonic generation (THG) at 355 nm (I0 = 4.6 × 1015 photons per cm2 per pulse) from a Nd:YAG laser (Continuum Inc., Surelite II, 5–8 ns pulse duration, 10 Hz) as the excitation source. The resonant frequency and microwave power were set to ∼9.1 GHz (X-band) and 3 mW, respectively.Calculation
The partial density of states (PDOS) was calculated using the Cambridge Serial Total Energy Package (CASTEP).35 The energy was calculated using the generalized gradient approximation (GGA) of density functional theory (DFT) as proposed by Perdew, Burke, and Ernzerhof (PBE). Electronic states were expanded using a plane-wave basis set with a cutoff value of 630 eV, and the k-point meshes were set as 5 × 5 × 5. Geometry optimization was performed before the PDOS calculations using the Broyden–Fletcher–Goldfarb–Shanno (BFGS) algorithm.Photocatalytic reactions
The loading of the Fe/Ru oxide ((Fe,Ru)Ox) cocatalyst was performed by simultaneous impregnation using FeCl3 and RuCl3 precursors corresponding to 0.5 and 0.9 wt% of Fe and Ru, respectively.36 The photocatalyst powder was dispersed in a portion of an aqueous solution containing FeCl3 and RuCl3, followed by evaporation of the solvent in a water bath with heating at 573 K for 1 h under an argon flow (20 mL min−1). As a HEP in Z-scheme water splitting, rhodium-doped strontium titanate (SrTiO3:Rh)37 was synthesized by a solid-state reaction. A mixture of TiO2, SrCO3, and Rh2O3, with a molar ratio of Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sr:Rh = 1:1.07:0.01, was calcined in an alumina crucible at 600 °C for 2 h and subsequently at 1000 °C for 10 h with intermediate grinding. A Ru-based cocatalyst was then deposited on SrTiO3:Rh by a photo-deposition method.38 SrTiO3:Rh was suspended in a 10 vol% methanol aqueous solution containing RuCl3 (0.7 wt% as Ru metal) and irradiated with visible light (400 < λ < 800 nm) for 2 h.
:Sr:Rh = 1:1.07:0.01, was calcined in an alumina crucible at 600 °C for 2 h and subsequently at 1000 °C for 10 h with intermediate grinding. A Ru-based cocatalyst was then deposited on SrTiO3:Rh by a photo-deposition method.38 SrTiO3:Rh was suspended in a 10 vol% methanol aqueous solution containing RuCl3 (0.7 wt% as Ru metal) and irradiated with visible light (400 < λ < 800 nm) for 2 h.
Photocatalytic reactions were carried out in a Pyrex glass reactor connected to a closed gas circulation system. For water oxidation with an electron acceptor, the photocatalyst powder (0.1 g) was suspended in 250 mL of an aqueous solution containing Fe(NO3)3, Fe(ClO4)3, FeCl3, or Fe2(SO4)3 (5 mM as Fe) in the reactor. A small amount of aqueous HNO3, HClO4, HCl, or H2SO4 solution was added to the respective aqueous solutions of Fe(NO3)3, Fe(ClO4)3, FeCl3, or Fe2(SO4)3, to adjust the pH to 2.4 before the reaction. For the Z-scheme water-splitting reaction with the Fe3+/Fe2+ redox couple, Ru-loaded SrTiO3:Rh (0.1 g) and (Fe,Ru)Ox-loaded PbVO3Cl (or unmodified PbVO3Cl) (0.1 g) were suspended together in an aqueous Fe(NO3)3 or Fe(ClO4)3 solution (5 mM, 250 mL) as the HEP and OEP, respectively. The suspensions were irradiated using a 300 W Xe lamp with a cutoff filter (L-42, HOYA) and a cold mirror (CM-1, Kenko) under magnetic stirring. The evolved gases were analyzed by using an online gas chromatograph (Shimadzu, GC-8A, thermal conductivity detector (TCD); 5 Å molecular sieve column packing; Ar carrier gas).
Results and discussion
Synthesis and characterization
PbVO3Cl and Pb5(VO4)3Cl particles were successfully synthesized as single phases using previously reported synthesis methods (Fig. S1 and S3†).28,32 Although the synthesis of Pb14(VO4)2O9Cl4 has not been reported, it is structurally characterized as a mineral.30 In this study, the synthesis was attempted via the conventional solid-state method. The XRD patterns of the synthesized samples are shown in Fig. 2. The XRD patterns of samples calcined at 550 °C and 600 °C are almost identical to the reported patterns. Furthermore, Le Bail analysis yielded good fits for the XRD patterns of both samples (Fig. S4†). These results confirm the successful synthesis of Pb14(VO4)2O9Cl4via solid-state synthesis at appropriate temperatures. The sample synthesized at 600 °C was used for further characterization. SEM images of the obtained PbVO3Cl, Pb5(VO4)3Cl, and Pb14(VO4)2O9Cl4 shown in Fig. S5† reveal large particles with sizes of several hundred nanometers to several micrometers. | ||
| Fig. 2 XRD patterns of the samples synthesized at various temperatures from precursor materials, aiming to synthesize Pb14(VO4)2O9Cl4. | ||
The diffuse reflectance spectra and Mott–Schottky plots for Pb5(VO4)3Cl, Pb14(VO4)2O9Cl4, and PbVO3Cl are shown in Fig. 3. The absorption edges of Pb5(VO4)3Cl, Pb14(VO4)2O9Cl4, and PbVO3Cl are approximately 415, 440, and 550 nm, respectively. The band gaps were estimated from Tauc plots (Fig. S6 and S7†). The flat-band potentials determined from the Mott–Schottky plots (vs. Ag/AgCl at pH 6) are −0.58 V, +0.10 V, and +0.23 V for Pb5(VO4)3Cl, Pb14(VO4)2O9Cl4, and PbVO3Cl, respectively.
 | ||
| Fig. 3 (a) Diffuse reflectance spectra and (b) Mott–Schottky plots of Pb5(VO4)3Cl, Pb14(VO4)2O9Cl4, and PbVO3Cl. The inset photographs are physical appearances of the oxyhalides. | ||
Based on the measurements, the estimated band positions of each Pb–V oxyhalide were compared with those of conventional layered oxyhalide photocatalysts (Fig. 4). The flat band potentials were assumed to be located just below the CBMs due to the n-type nature of these materials. Among the three Pb–V oxyhalides, the CBMs varied significantly, whereas the VBMs were close to each other. Notably, PbVO3Cl exhibited the most positive CBM, resulting in a narrower bandgap and enabling the broadest absorption in the visible region. Compared to conventional oxyhalide photocatalysts, all the Pb–V oxyhalides displayed more positive CBMs. The validity of this CBM position was further supported by PYS measurement (Fig. S8†).
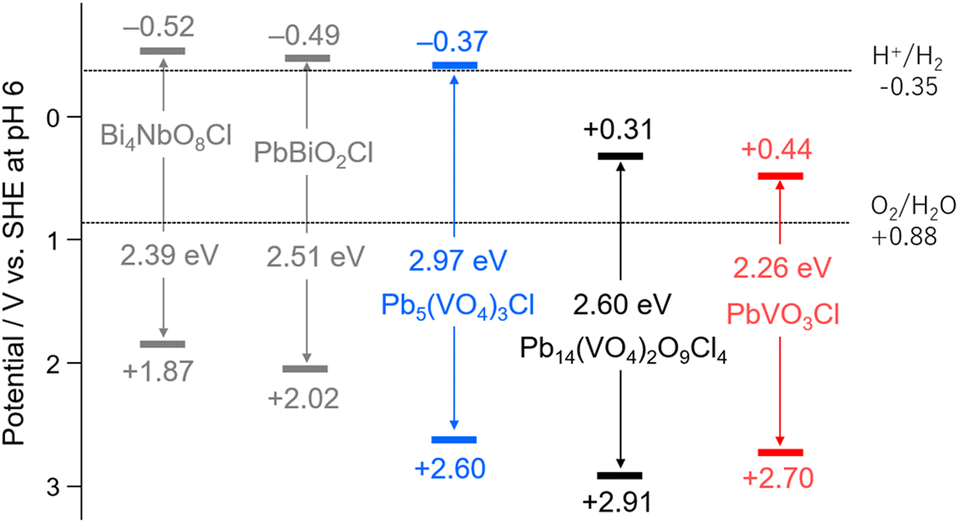 | ||
| Fig. 4 Band diagrams of Pb–V-based oxyhalides, along with those of conventional oxyhalide photocatalysts. The band levels of Bi4NbO8Cl and PbBiO2Cl were referred from previous studies.19,20 | ||
Band structures of Pb–V oxyhalides
The partial density of states (PDOS) calculated using DFT is shown in Fig. 5a–c. In all compounds, the VBM is predominantly composed of O-2p orbitals, with noticeable hybridization with Pb-6s orbitals, similar to what has been observed in other Pb-based oxyhalides such as PbBiO2Cl and PbBi3O4Cl3.20,39 Additionally, PbVO3Cl shows a distinct contribution from Cl-3p orbitals. The CBMs of all compounds are primarily composed of V-3d orbitals, with additional contributions from O-2p and Pb-6p orbitals. These results suggest that the more positive CBMs of Pb–V oxyhalides than that of conventional oxyhalide photocatalysts can be attributed to the introduction of the V5+ cation, which has a high ionization energy. Furthermore, the most positive CBM of PbVO3Cl consists almost entirely of V orbitals with a small contribution from O orbitals. This indicates that the variation in CBM positions among Pb–V oxyhalides arises not from changes in the orbital composition forming the CBM but from differences in the energy of the V-3d orbitals themselves.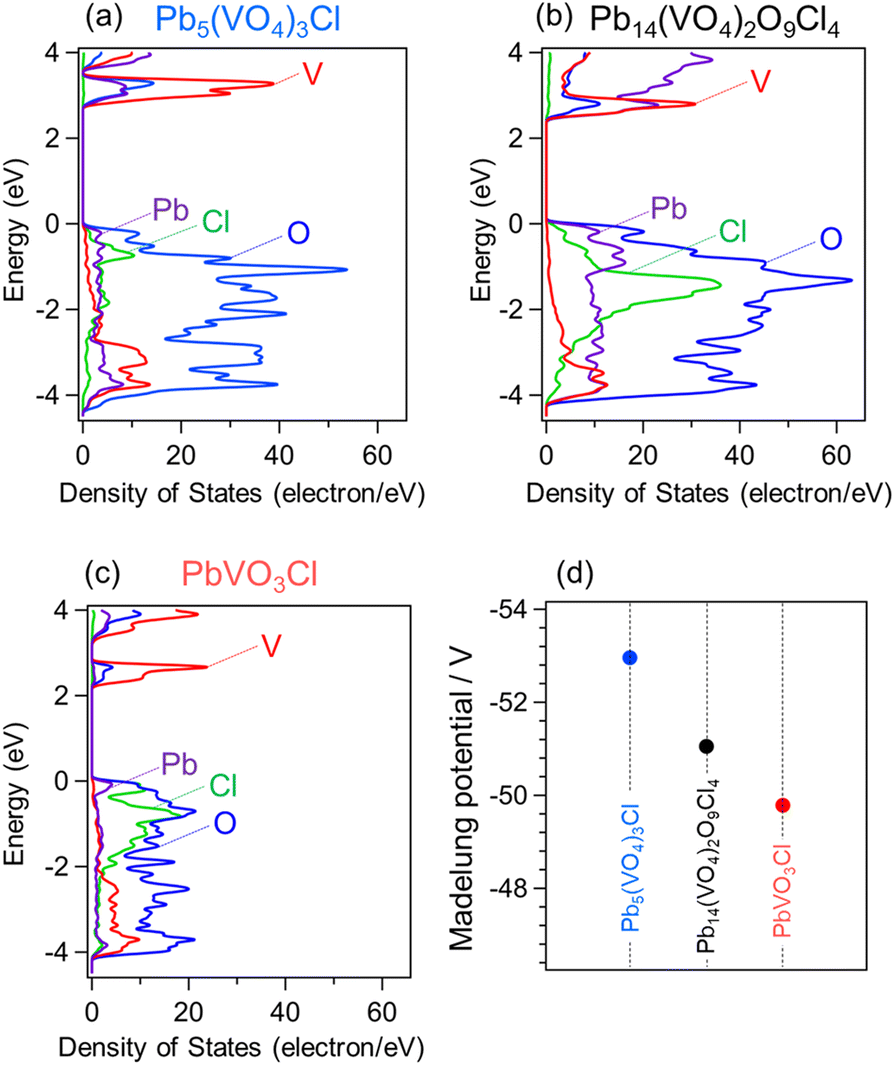 | ||
| Fig. 5 (a–c) Partial density of states of Pb5(VO4)3Cl, Pb14(VO4)2O9Cl4, and PbVO3Cl. (d) Madelung site potentials of vanadium in the Pb–V-based oxyhalides. | ||
P. A. Cox previously illustrated how bands in extended solids are formed from isolated atoms (Fig. S9†).40 The band structures can be determined by considering the following factors: (1) the electron affinity of anions or the ionization potential of cations, (2) the Madelung potential at each crystallographic site, (3) polarization effects, and (4) the bandwidth arising from orbital hybridizations. As discussed above, in the case of the Pb–V oxyhalide system, the energy levels of the V orbitals at crystallographic sites appear to play a significant role in determining the CBM positions. Fig. 5d shows the Madelung site potential of vanadium in each Pb–V oxyhalide. These values are significantly influenced by the crystal structures, and their sequence aligns closely with the CBM positions of the Pb–V oxyhalides. This correlation strongly suggests that the most positive Madelung site potential for V in PbVO3Cl substantially stabilizes its orbitals, resulting in the most positive CBM among that of the materials studied.
Photocatalytic O2 evolution
The Pb–V oxyhalides possess appropriate band levels suitable for reducing Fe3+(E0(Fe3+/Fe2+) = 0.77 V vs. NHE) and oxidizing water to O2. The O2 evolution activities of the three Pb–V oxyhalides were evaluated using Fe3+ as an electron acceptor under visible light irradiation. The (Fe,Ru)Ox cocatalyst was loaded to enhance the O2 evolution rate (Fig. S10†), as is often done with oxyhalide photocatalysts functioning as an OEP in Z-scheme systems with Fe3+/Fe2+ redox mediators.36Fig. 6a shows the time course of O2 evolution. Among the three oxyhalides, only PbVO3Cl exhibited O2 evolution activity under visible light, while Pb5(VO4)3Cl and Pb14(VO4)2O9Cl4 showed negligible activity. A similar trend was observed in the photocurrent response measurements (Fig. S11†). The XRD pattern of PbVO3Cl after the O2 evolution reaction revealed almost no changes compared to that of pristine PbVO3Cl, confirming that PbVO3Cl functioned stably as an OEP (Fig. S12†). The significantly higher O2 evolution activity of PbVO3Cl compared to other Pb–V oxyhalides can be attributed to two main factors: (1) the broader visible light absorption up to the longest wavelengths among the Pb–V oxyhalides as seen in Fig. 3a, and (2) the superior charge carrier transport properties. The second factor was evaluated using time-resolved microwave conductivity (TRMC) measurements,41–43 wherein the intensity of TRMC signals on oxyhalides (e.g., Bi4TaO8Cl and PbBiO2Cl) has been proven to strongly correlate with their photocatalytic activity.44,45Fig. 6b shows the TRMC kinetics for each oxyhalide. PbVO3Cl exhibited the highest photoconductivity (φΣμ) signal among the three oxyhalides. Fig. 6c highlights the structural features (VO4 or VO5 units) underlying this property. The VO4 units in Pb5(VO4)3Cl and Pb14(VO4)2O9Cl4 are isolated, while the VO5 units in PbVO3Cl are interconnected through edge-sharing. This interconnected feature facilitates superior electron transport, resulting in a higher TRMC signal. These findings reveal that the differences in crystal structures of Pb–V-based oxyhalides critically influence their light absorption and charge carrier transport properties, playing a crucial role in determining their photocatalytic activity.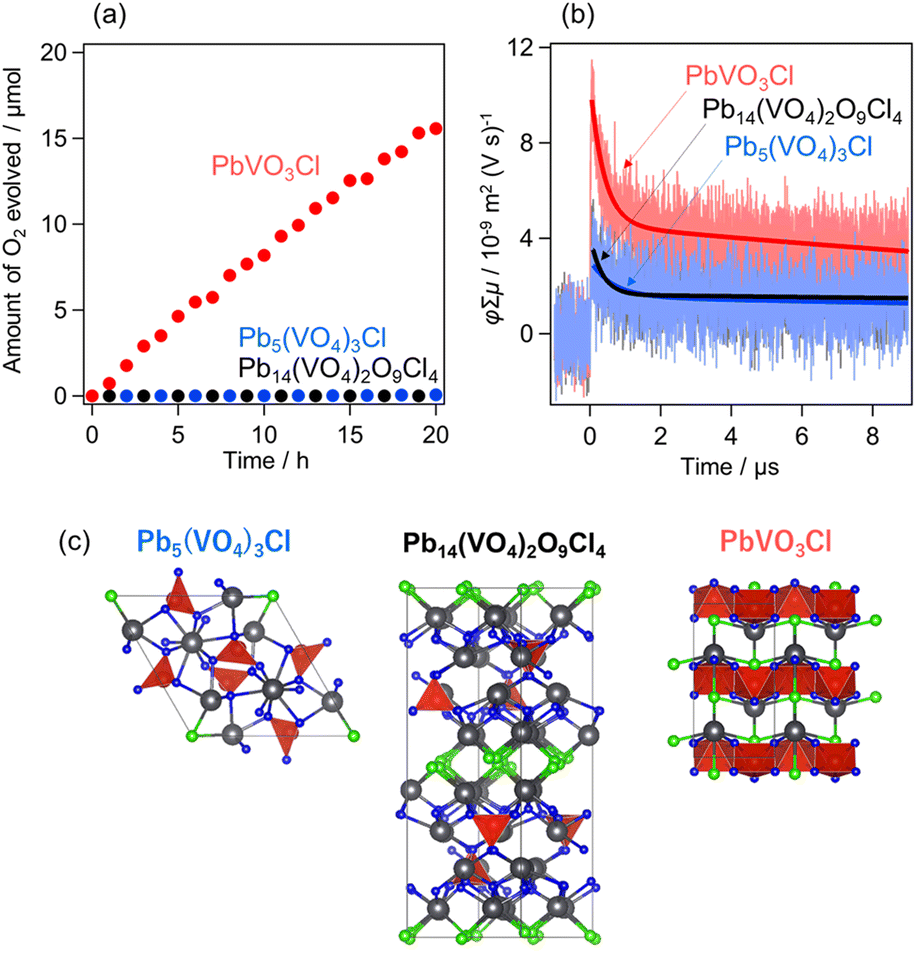 | ||
| Fig. 6 (a) Photocatalytic O2 evolution using Pb5(VO4)3Cl, Pb14(VO4)2O9Cl4, and PbVO3Cl in aqueous Fe(NO3)3 solution (5 mM, 250 mL, pH 2.4) under visible light irradiation (λ > 400 nm). (Fe,Ru)Ox was loaded as a cocatalyst. (b) TRMC transients (φΣμ) of Pb5(VO4)3Cl, Pb14(VO4)2O9Cl4, and PbVO3Cl after excitation at 355 nm. The solid line represents the curve obtained through double-exponential fitting. (c) Crystal structures highlight each material's VO4 or VO5 units. | ||
Z-scheme water-splitting
Finally, visible-light-driven Z-scheme water-splitting was attempted using PbVO3Cl as the OEP. The schematic image is illustrated in Fig. S13.† Before constructing the system, various Fe3+ species with different counter anions were tested to identify the optimal precursor of the Fe3+/Fe2+ redox mediator (Fig. 7a). The results revealed that Fe(NO3)3 and Fe(ClO4)3 were the most suitable, likely due to differences in the state of Fe aqua complexes, as suggested previously.46 Similar trends were also observed with other oxyhalides, such as PbBiO2Cl.20 Subsequently, Z-scheme water-splitting was attempted using SrTiO3:Rh as the HEP and Fe(ClO4)3 (or Fe(NO3)3) as the Fe3+/Fe 2+ redox mediator. When Fe(NO3)3 was used, gas production rates were very low, and H2 and O2 were not generated in a stoichiometric ratio (2:1) (Fig. S14†). In contrast, using Fe(ClO4)3 led to the relatively steady evolution of H2 and O2 in a 2:1 stoichiometric ratio, confirming successful Z-scheme water splitting under visible light (Fig. 7b). Since the O2 evolution activity of PbVO3Cl was comparable when using either Fe(ClO4)3 or Fe(NO3)3, the differences in gas evolution rates in the Z-scheme are likely attributed to the counter anion-dependent H2 evolution activity of SrTiO3:Rh.47 Notably, even when unmodified PbVO3Cl (i.e. without (Fe,Ru)Ox loading) was employed as the OEP, Z-scheme water splitting still proceeded, although the activity was moderately reduced (Fig. S15†). In conclusion, visible-light-driven Z-scheme water splitting was achieved for the first time using a V-based oxyhalide (PbVO3Cl) as an OEP.
 | ||
| Fig. 7 (a) O2 evolution over (Fe,Ru)Ox/PbVO3Cl from an aqueous Fe3+ solution (5 mM, 250 mL) with different counter anions (ClO4−, NO3−, Cl−, or SO42−) under visible-light irradiation (λ > 400 nm). (b) Z-scheme water splitting using (Fe,Ru)Ox/PbVO3Cl as an OEP and Ru/SrTiO3:Rh as a HEP under visible light (λ > 400 nm) in an aqueous Fe(ClO4)3 solution (5 mM, 250 mL, pH 2.4). | ||
Conclusions
We demonstrated the potential of V-based oxyhalides as OEPs in Z-scheme water-splitting systems. The Pb–V-based oxyhalides exhibited more positive CBMs compared to conventional oxyhalide photocatalysts, with PbVO3Cl showing the most positive CBM and the smallest band gap among the investigated Pb–V oxyhalides, attributed to the more positive Madelung site potential of vanadium. PbVO3Cl features a unique structure with interconnected VO5 units, significantly enhancing its carrier transport properties. The smaller band gap and superior carrier transport properties of PbVO3Cl enabled it to function as the first V-based oxyhalide OEP in a Z-scheme water-splitting system. While PbVO3Cl exhibits an ideal CBM position, its VBM remains excessively deep (positive), limiting its overall efficiency. The combination of vanadium introduction and valence band engineering could further narrow the band gap. Similar conduction band engineering could be achieved by introducing cations with high ionization energies other than vanadium.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS KAKENHI (JP20H00398 and JP23H02061) through Grant-in-Aid for Scientific Research (A) and (B), respectively. This study was also supported by the JSPS Core-to-Core Program (JPJSCCA20200004), the Kansai Research Foundation for Technology Promotion, and the ENEOS Tonengeneral Research/Development Encouragement & Scholarship Foundation. Computation time was provided by the SuperComputer System, Institute for Chemical Research, Kyoto University.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. J. Bard, Science, 1980, 207, 139–144 CrossRef CAS PubMed.
- K. Domen, J. N. Kondo, M. Hara and T. Takata, Bull. Chem. Soc. Jpn., 2000, 73, 1307–1331 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237–268 CrossRef CAS.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 Search PubMed.
- S. S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- K. Sayama, K. Mukasa, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2001, 23, 2416–2417 RSC.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- Y. O. Wang, H. Suzuki, J. J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. W. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS.
- W. Zhao, K. Maeda, F. X. Zhang, T. Hisatomi and K. Domen, Phys. Chem. Chem. Phys., 2014, 16, 12051–12056 RSC.
- T. Kato, Y. Hakari, S. Ikeda, Q. X. Jia, A. Iwase and A. Kudo, J. Phys. Chem. Lett., 2015, 6, 1042–1047 CrossRef CAS PubMed.
- R. Abe, T. Takata, H. Sugihara and K. Domen, Chem. Commun., 2005, 30, 3829–3831 RSC.
- M. Higashi, R. Abe, T. Takata and K. Domen, Chem. Mater., 2009, 21, 1543–1549 CrossRef CAS.
- D. J. Martin, P. J. T. Reardon, S. J. A. Moniz and J. W. Tang, J. Am. Chem. Soc., 2014, 136, 12568–12571 CrossRef CAS PubMed.
- R. Abe, K. Shinmei, N. Koumura, K. Hara and B. Ohtani, J. Am. Chem. Soc., 2013, 135, 16872–16884 CrossRef CAS.
- H. Fujito, H. Kunioku, D. Kato, H. Suzuki, M. Higashi, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2016, 138, 2082–2085 Search PubMed.
- H. Suzuki, H. Kunioku, M. Higashi, O. Tomita, D. Kato, H. Kageyama and R. Abe, Chem. Mater., 2018, 30, 5862–5869 Search PubMed.
- D. Kato, H. Suzuki, R. Abe and H. Kageyama, Chem. Sci., 2024, 15, 11719–11736 RSC.
- H. Kunioku, M. Higashi, O. Tomita, M. Yabuuchi, D. Kato, H. Fujito, H. Kageyama and R. Abe, J. Mater. Chem. A, 2018, 6, 3100–3107 RSC.
- D. Kato, K. Hongo, R. Maezono, M. Higashi, H. Kunioku, M. Yabuuchi, H. Suzuki, H. Okajima, C. C. Zhong, K. Nakano, R. Abe and H. Kageyama, J. Am. Chem. Soc., 2017, 139, 18725–18731 CrossRef CAS.
- K. Ogawa, R. Sakamoto, C. C. Zhong, H. Suzuki, K. Kato, O. Tomita, K. Nakashima, A. Yamakata, T. Tachikawa, A. Saeki, H. Kageyama and R. Abe, Chem. Sci., 2022, 13, 3118–3128 RSC.
- K. Ogawa, H. Suzuki, C. C. Zhong, R. Sakamoto, O. Tomita, A. Saeki, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2021, 143, 8446–8453 CrossRef CAS PubMed.
- D. Ozaki, H. Suzuki, K. Ogawa, R. Sakamoto, Y. Inaguma, K. Nakashima, O. Tomita, H. Kageyama and R. Abe, J. Mater. Chem. A, 2021, 9, 8332–8340 RSC.
- H. Kunioku, M. Higashi, C. Tassel, D. Kato, O. Tomita, H. Kageyama and R. Abe, Chem. Lett., 2017, 46, 583–586 CrossRef CAS.
- A. Sahin and M. Emirdag-Eanes, Z. Kristallogr. - New Cryst. Struct., 2007, 222, 159–160 CAS.
- J. B. Trotter and W. H. Barnes, Can. Mineral., 1958, 6, 161–173 CAS.
- M. Cooper and F. C. Hawthorne, Am. Mineral., 1994, 79, 550–554 CAS.
- U. Dang, W. Zaheer, W. Zhou, A. Kandel, M. Orr, R. W. Schwenz, G. Laurita, S. Banerjee and R. T. Macaluso, Chem. Mater., 2020, 32, 7404–7412 CrossRef CAS.
- M. Nakamura, K. Oqmhula, K. Utimula, M. Eguchi, K. Oka, K. Hongo, R. Maezono and K. Maeda, Chem.–Asian J., 2020, 15, 540–545 CrossRef CAS.
- V. Petricek, M. Dusek and L. Palatinus, Z. Kristallogr. - Cryst. Mater., 2014, 229, 345–352 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 Search PubMed.
- A. Nakada, H. Suzuki, J. J. M. Verizo, K. Ogawa, M. Higashi, A. Saeki, A. Yamakata, H. Kageyama and R. Abe, ACS Appl. Mater. Interfaces, 2019, 11, 45606–45611 CrossRef CAS.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992–8995 Search PubMed.
- Y. Sasaki, A. Iwase, H. Kato and A. Kudo, J. Catal., 2008, 259, 133–137 Search PubMed.
- H. Suzuki, M. Higashi, O. Tomita, Y. Ishii, T. Yamamoto, D. Kato, T. Kotani, D. Ozaki, S. Nozawa, K. Nakashima, K. Fujita, A. Saeki, H. Kageyama and R. Abe, Chem. Mater., 2021, 33, 9580–9587 CrossRef CAS.
- P. A. Cox, The Electronic Structure and Chemistry of Solids, Oxford Science Publications, Oxford, 1986, p. 146 Search PubMed.
- A. Saeki, S. Yoshikawa, M. Tsuji, Y. Koizumi, M. Ide, C. Vijayakumar and S. Seki, J. Am. Chem. Soc., 2012, 134, 19035–19042 Search PubMed.
- A. Saeki, M. Tsuji and S. Seki, Adv. Energy Mater., 2011, 1, 661–669 Search PubMed.
- A. Saeki, Y. Koizumi, T. Aida and S. Seki, Acc. Chem. Res., 2012, 45, 1193–1202 CrossRef CAS PubMed.
- H. Suzuki, M. Higashi, H. Kunioku, R. Abe and A. Saeki, ACS Energy Lett., 2019, 4, 1572–1578 CrossRef CAS.
- H. Suzuki, S. Kanno, M. Hada, R. Abe and A. Saeki, Chem. Mater., 2020, 32, 4166–4173 CrossRef CAS.
- Y. Miseki, H. Kusama, H. Sugihara and K. Sayama, Chem. Lett., 2010, 39, 846–847 CrossRef CAS.
- H. Kato, Y. Sasaki, A. Iwase and A. Kudo, Bull. Chem. Soc. Jpn., 2007, 80, 2457–2464 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00605h |
| This journal is © The Royal Society of Chemistry 2025 |