
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elucidating the phase transformations and grain growth behavior of O3-type sodium-ion layered oxide cathode materials during high temperature synthesis†
Anika Tabassum
Promi
 a,
Junyi
Yao
a,
Dawei
Xia
a,
Callum
Connor
a,
Afolabi Uthmon
Olayiwola
a,
Jianming
Bai
b,
Chengjun
Sun
c,
Dennis
Nordlund
d,
Kejie
Zhao
e and
Feng
Lin
*afg
a,
Junyi
Yao
a,
Dawei
Xia
a,
Callum
Connor
a,
Afolabi Uthmon
Olayiwola
a,
Jianming
Bai
b,
Chengjun
Sun
c,
Dennis
Nordlund
d,
Kejie
Zhao
e and
Feng
Lin
*afg
aDepartment of Chemistry, Virginia Tech, Blacksburg, Virginia 24061, USA. E-mail: fenglin@vt.edu
bNational Synchrotron Light Source II, Brookhaven National Laboratory, Upton, NY 11973, USA
cX-Ray Science Division, Argonne National Laboratory, Lemont, IL 60439, USA
dStanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Menlo Park, CA 94025, USA
eSchool of Mechanical Engineering, Purdue University, West Lafayette, IN 47907, USA
fDepartment of Materials Science and Engineering, Virginia Tech, Blacksburg, VA 24061, USA
gMacromolecules Innovation Institute, Virginia Tech, Blacksburg, VA 24061, USA
First published on 12th May 2025
Abstract
Understanding the formation mechanism of layered oxide cathodes via solid-state synthesis is imperative to achieving controllability over their materials properties and electrochemical behaviors. In this work, we investigate the phase and microstructure evolution during the synthesis of NaNi1/3Fe1/3Mn1/3O2, a model sodium-ion layered oxide cathode, using a combination of imaging, diffraction, and spectroscopic techniques. We unravel the synthetic mechanistic pathways involved in the high-temperature calcination reaction, as well as elaborate the synthesis-microstructure-performance relationship of this material. The formation of the final layered oxide phase involves a gradual transformation through a sodiated oxyhydroxide intermediate. During the reaction, the precursor dehydration reaction dominates at 250–550 °C, while the major sodiation reaction occurs at 550–850 °C. Alongside multiple stages of phase transformations, the final grain structure formation occurs through the continuous growth of the (003) and (104) facets. During the reaction, Mn acts as the charge-compensating element and exhibits depth-dependent characteristics. When the sodiation reaction dominates over dehydration, the reaction intermediates undergo gradual electronic structure changes with increasing temperature, as indicated by the spectral features of TM3d-O2p hybrid states. Calcination duration is also a critical parameter governing the microstructure, surface reactivity, phase fraction distribution and electrochemical performance of the material. The optimal calcination duration was determined to be 18 hours at 850 °C under the conditions evaluated here. Calcination beyond this duration was found to be detrimental to electrochemical performance due to Na and O loss and heterogeneous sodium distribution throughout the particles. Our work sheds light on the complex crystallographic-chemical-microstructural evolution of sodium ion layered oxide cathodes and provides insight into precisely tuning material properties which are intimately linked to battery performances.
Introduction
As the world is making progress toward ambitious carbon neutrality goals, advancements in battery technology are emerging as a powerful tool to decarbonize energy systems. Among these technologies, sodium-ion batteries (SIB) stand out as a promising alternative, and often a complementary option, to conventional lithium-ion batteries (LIB) for applications in short range electric vehicles and grid energy storage systems.1–3 Among the diverse cathode chemistries for sodium ion batteries, sodium ion layered oxide cathode materials offer promising electrochemical performance and ease of synthesis, making them suitable for industrial adoption.4–6 They also allow the exploration of more sustainable and low-cost compositions for battery materials by allowing the incorporation of several earth-abundant and non-critical transition metals into a layered oxide structure.5,7–9Despite the high practical value, layered oxides are sensitive to various synthetic parameters, accompanied by complex and heterogenous reactions. During high temperature calcination reaction, transition metal hydroxide precursor reacts with the alkali ion precursor in presence of O2 from the surrounding gaseous environment. In this reaction, multiple phase transitions occur driven by varying degrees of mass transport and reaction thermodynamics at increasingly high temperatures, leading to the formation of the final layered oxide cathode material.10 The phase propagation of the non-equilibrium reaction intermediates during this high temperature calcination reaction can govern the phase purity, strain development and structural defect formation, thereby controlling the electrochemical performance of these materials.10–13 Moreover, the high temperature calcination reactions also control the extent of grain growth of the primary particles, which in turn controls the ion transport properties of layered oxide cathode materials.14,15 Understanding the fundamental reaction pathways can inform better synthesis parameter tuning, leading to electrochemical performance optimization. This has led to the vast exploration of phase transformation kinetics and reaction mechanisms during calcination of the alkali-ion layered oxides.16 The literature highlights different reaction pathways based on the initial precursor compositions and chosen synthesis parameters. For example, for LiCoO2 material, a spinel-type intermediate leads to the formation of the final layered phase,17–19 whereas for LiNiO2 and quaternary Ni-rich layered oxide cathodes, the formation of layered oxide phase undergoes transformation through an intermediate rock-salt phase.10,20–22 Moreover, for quaternary Ni-rich compositions, other studies have reported conversion through an intermediate oxyhydroxide-to-spinel-to-layered phase,23 or direct conversion from layered-to-layered phases.24
Whereas many studies have focused on the phase transformation pathways during synthesis of lithium based layered oxide systems, sufficient literature on the sodium ion layered oxides is lacking. Sodium-based synthesis can be significantly more complicated than the lithium-based systems due to two reasons. One is the significantly larger ionic radius of sodium ions (1.02 Å) compared to lithium ions (0.76 Å), which can change the kinetic barrier and energy landscape of intermediate formation during synthesis. Another complexity arises from the vastly different precursor compositions for sodium ion layered oxide cathodes compared to their lithium counterparts. Many transition metal hydroxide precursors for sodium ion batteries are rich in Fe and Mn, rather than Ni, which can lead to the formation of different reaction intermediates and impurity phases compared to Ni-rich materials. Understanding the phase and chemical transformations during the high temperature sodiation process is crucial for optimizing reaction parameters and is essential for the successful commercialization of sodium ion layered oxide cathodes. For example, in a recent study, Zuo et al. demonstrated that structural defect generation and charge heterogeneity in a Ni-rich O3-type NaNMC811 cathode are influenced by phase transformation pathways during its formation process.12 They showed that control over these phenomena can be achieved by precise synthesis parameter tuning during calcination.
In this work, we have studied the reaction mechanism and calcination behavior of a popular O3-type cathode material for sodium-ion layered oxides, NaNi1/3Fe1/3Mn1/3O2 (NFM333), from a coprecipitated metal hydroxide precursor. We have utilized ensemble-averaged synchrotron X-ray diffraction and absorption techniques to study the global phase transformations and the concurrent local chemical changes occurring during different stages of calcination. Our study reveals that the precursor to layered oxide phase transformation occurs through an intermediate sodiated oxyhydroxide phase during calcination. Temperature ranges of precursor dehydration, onset of sodiation and layered phase formation are investigated and their correlation with the microstructure evolution of the material is studied. We also observe surface to bulk transition metal oxidation state heterogeneities and charge redistribution in the material at different stages of calcination due to the kinetically sluggish nature of the sodiation process. Finally, we study the effect of different calcination duration on the phase distribution, microstructure and the electrochemical performance of the material. We holistically establish crystallographic-chemical-microstructure relationships in the model layered oxide, which can inform more tailorable solid-state synthesis of multielement oxides in the future.
Experimental section
Material synthesis
The Ni1/3Fe1/3Mn1/3(OH)2+x precursor was synthesized by a coprecipitation method. First, a 2.5 M solution of metal sulfate salts was prepared by dissolving stoichiometric amounts of NiSO4·6H2O, FeSO4·7H2O and MnSO4·H2O in deionized water. Then this solution and 0.5 M NH4OH were fed to a continuous stirred tank reactor as two separate feeds at a flow rate of 0.4 mL min−1. At the same time NaOH was fed to the reactor to keep the reaction mixture at a constant pH of 10.8. The reactor was maintained at a constant temperature of 50 °C. The reaction mixture was agitated at a speed of 1100 rpm and was kept under continuous N2 inert gas protection. After 16.7 hours of reaction and 24 hours of ageing, the precipitate was collected, washed with deionized water and isopropyl alcohol, filtered and dried in a vacuum oven at 100 °C overnight.Ten calcination mixtures were prepared by mixing and grinding stoichiometric amounts of Ni1/3Fe1/3Mn1/3(OH)2+x precursor with Na2CO3. Grinding was done using a mortar and pestle. 2% excess Na2CO3 was used to compensate for the Na loss during calcination. Six of these mixtures were calcined in a box furnace at temperatures of 250 °C, 350 °C, 450 °C, 550 °C, 650 °C, 750 °C for a duration of one minute. The other four calcination mixtures were calcined in a box furnace (Lindberg, model: BF51894C-1) at 850 °C for four different durations of 6 hours, 12 hours, 18 hours and 24 hours. For all the calcinations, heating and cooling rates were 5 °C min−1. After calcination, all the ten calcined powders were stored in a glovebox to be used for characterization and electrode preparation.
Electrochemical characterization
To prepare the composite electrode slurry, the cathode powders obtained after calcination were ground with acetylene carbon black and then mixed thoroughly with a poly(vinylidene difluoride) (PVDF) binder dissolved in N-methyl-2-pyrrolidone solvent. The ratio of active material cathode powder, acetylene carbon and PVDF binder was 90 wt%![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 wt%:5 wt%. Then this slurry was cast on a carbon-coated aluminum foil and then dried in a vacuum oven at 130 °C overnight. The resultant dry electrode was then punched into disks of a diameter of 10 mm with an active material mass loading of ∼6 mg cm−2. To prepare the coin cells for electrochemical measurements, the prepared composite electrode was used as a cathode and Na foil was used as the anode with a Whatman glass fiber separator (Cytiva grade GF D microfiber prefilter, 675 μm thickness) between them. The coin cell assembly was done using CR2032 coin cell parts in an Ar-filled glove box. The electrolyte was 1.0 M sodium perchlorate (NaClO4) in propylene carbonate (PC) with 5% fluoroethylene carbonate (FEC). The galvanostatic discharge/charge of the coin cells were performed in the cutoff voltage range of 2.0–4.3 V at 22 °C and measured on the electrochemical workstation (Wuhan Land Company). 1C was defined as fully charging the cathode in 1 hour, which corresponded to the specific current rate of 120 mA g−1.
:5 wt%:5 wt%. Then this slurry was cast on a carbon-coated aluminum foil and then dried in a vacuum oven at 130 °C overnight. The resultant dry electrode was then punched into disks of a diameter of 10 mm with an active material mass loading of ∼6 mg cm−2. To prepare the coin cells for electrochemical measurements, the prepared composite electrode was used as a cathode and Na foil was used as the anode with a Whatman glass fiber separator (Cytiva grade GF D microfiber prefilter, 675 μm thickness) between them. The coin cell assembly was done using CR2032 coin cell parts in an Ar-filled glove box. The electrolyte was 1.0 M sodium perchlorate (NaClO4) in propylene carbonate (PC) with 5% fluoroethylene carbonate (FEC). The galvanostatic discharge/charge of the coin cells were performed in the cutoff voltage range of 2.0–4.3 V at 22 °C and measured on the electrochemical workstation (Wuhan Land Company). 1C was defined as fully charging the cathode in 1 hour, which corresponded to the specific current rate of 120 mA g−1.
GITT measurement details
Apparent diffusion co-efficient (D) values are obtained by the galvanostatic intermittent titration technique (GITT). In this method, the voltage response of the electrode was measured as a function of time after applying a short pulse of a fixed current. The electrode was then allowed to rest for three hours to allow the electrode potential to reach a steady state before applying another current pulse. The diffusion co-efficient (D) was then calculated using the equation:where τ is the pulse duration (which was 15 min), mB and MB are the active mass and molar mass of the active material, VM is the molar volume (for NaNi1/3Fe1/3Mn1/3O2, we employed 28.56 cm3 mol−1), and S is the active surface area of the electrode. ΔES and ΔEτ were obtained from the GITT curves.
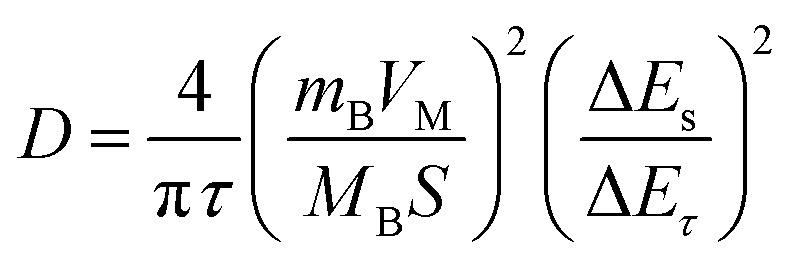
Material characterization
The morphologies of the materials were investigated using two scanning electron microscopes (SEM) (JEOL IT-500HR and LEO FESEM) at an accelerating voltage of 5 kV. The EDS results were conducted with the JEOL IT-500HR at an accelerating voltage of 75 kV. The STEM-EDS was performed using a JEOL JEM 2100 at an accelerating voltage of 200 kV. Synchrotron X-ray diffraction (XRD) of the calcined materials was collected at the 28-ID-2 beamline at National Synchrotron Light Source II (NSLS-II). The X-ray wavelength was 0.1814 Å. The samples were loaded in Kapton tubes. Rietveld refinements were performed with the GSAS-II software package. Hard X-ray absorption spectroscopy (XAS) measurements of Ni, Mn, and Fe K-edge X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) were conducted on the calcined powders in transmission mode at the 20-BM-B beamline of the advanced photon source (APS) at Argonne National Laboratory. The incident X-ray beam was monochromatized using a silicon (111) fixed-exit and double-crystal monochromator. For energy calibration, each spectrum was aligned by matching the first derivative peak of reference Ni, Mn, and Fe XANES spectra, which were collected simultaneously from metal foils in the reference channel. The Hard XAS data was analyzed using the Athena software package. Soft XAS measurements were performed at the 10-1 beamline at the Stanford Synchrotron Radiation Lightsource (SSRL). The energy resolution was 0.2 eV over a 1 mm2 beam spot. The data were collected under ultrahigh vacuum (10−9 Torr) at room temperature using total electron yield (TEY) and fluorescence yield (FY). The XAS samples were mounted on aluminum sample holders in an argon-filled glove box, then transferred to the load-lock chamber inside a double-contained container, using an argon-purged glove bag for safe transfer. The soft XAS data was analyzed using the PyMca software package. Thermogravimetric analysis (TGA) was performed in a Discovery SDT 650 thermogravimetric analyzer. Inductively coupled plasma mass spectrometry (ICP-MS) analysis was done using a Thermo Electron iCAP RQ spectrometer. BET surface area analysis was performed using a Micromeritics 3 plus instrument. For this measurement, degassing of the cathode powders was done at 200 °C for 12 hours.Results and discussion
Global phase progression and microstructure evolution as a function of calcination temperature
To study the structural changes and phase evolution during different stages of calcination, ex situ synchrotron XRD measurements were carried out. The transition metal hydroxide precursor consists of equimolar amounts of Ni, Fe, and Mn, uniformly distributed throughout the particles. Elemental quantification of the precursor is done with ICP-MS and the results are presented in ESI Table S1.†Fig. 1 shows the XRD patterns of the samples calcined at different temperatures in the range of 250 °C to 850 °C, along with the XRD patterns of the pristine uncalcined transition metal hydroxide precursor and precursor + sodium carbonate mixture. As previous literature report on the sintering of this material reported no obvious structural changes up to 200 °C except thermal expansion of the lattice,25 we chose a range of temperatures between 250 °C to 850 °C to study the structural evolution during calcination. The samples in the range of 250 °C to 750 °C were held at the final temperature for one minute to capture the nature of the reaction intermediates, whereas the sample calcined at 850 °C was held for different durations (in a range of 6 to 24 hours) for allowing complete transformation to layered oxide. Upon heating to 250 °C, the major peaks belonging to the transition metal hydroxide precursor disappear, suggesting precursor decomposition through dehydration. The reaction intermediate formed at this temperature consists of a transition metal oxyhydroxide structure (TMOOH), which was identified by the peaks at 2θ = 35.8°, 41°, 54.4° and 64.3°, using the crystal structure of Fe0.67Mn0.33OOH as a reference (Fig. S1†).25 This is further supported by the Fe-EXAFS of the sample calcined at 250 °C, which resembles the local coordination environment of a standard FeOOH sample (Fig. S2†). This oxyhydroxide intermediate has structural resemblance with a layered hexagonal δ-FeOOH structure, composed of octahedral FeO6 units.25,26 At the same time, major peaks belonging to Na2CO3 decrease in intensity at this temperature, suggesting its possible integration into the structure, forming a sodiated oxyhydroxide phase (see ESI Fig. S3†). In the work by Xie et al., where the authors have studied the sintering phase transformation of NaNi1/3Fe1/3Mn1/3O2 with in situ XRD, the first sodiated intermediate formation was reported to occur above 350 °C.25 However, our observations indicate that such an intermediate can form at a lower temperature of 250 °C. As the temperature increases up to 450 °C, the peaks belonging to the oxyhydroxide intermediate gradually shift to lower d-spacing, suggesting lattice shrinkage due to continuous dehydration and oxidation. At 550 °C, the nucleation of the layered phase is observed within the oxyhydroxide structure, as identified by the new peak formed at 2θ = 15.7°, adjacent to a peak belonging to Na2CO3 at 2θ = 16.5° (see Fig. S4†). The oxyhydroxide peaks also start shifting in the opposite direction towards higher d-spacing at this temperature. This is due to the insertion of a large amount of sodium ions in the structure, which leads to the expansion of the crystal lattice. As temperature increases above 550 °C, more sodium and oxygen are incorporated within the structure which leads to the gradual growth of the layered phase. At 650 °C, the layered peak becomes more pronounced, and the peaks belonging to the oxyhydroxide phase begin to diminish. As the crystal structure is transitioning between the oxyhydroxide and layered phase, we identify this phase as an intermediate layered oxide phase I. When the temperature has further increased to 750 °C, we observe the crystal structure undergoing another set of changes: (i) the peak belonging to the layered phase at 15.7° merges with its adjacent peak (belonging to Na2CO3) and become broader, (ii) the peaks at 2θ = 32.4° (100), 35.9° (101), 43.4° (012) and 63.3° (110) become more well defined and shift to higher d-spacing, and (iii) the nucleation of 104 facet at 2θ = 42°. These indicate structural transformation of the material due to a large volume of sodium insertion into the structure. We identify this phase as the intermediate layered oxide phase II. During the transformation to the intermediate layered phase I and II, atomic rearrangement and structural distortion are expected to occur, decreasing the transient atomic symmetry, causing low crystallinity of the samples.24 These two intermediate phases simultaneously underwent layered phase evolution and oxyhydroxide degradation. After the temperature has increased to 850 °C and held there for 12 hours, we observe the sharpening of all the evolving peaks, indicating the formation of a fully grown layered phase. The layered NaNi1/3Fe1/3Mn1/3O2 structure formed at 850 °C is identified by the characteristic (003), (006), (101), (012) and (104) facets in Fig. 1. Elemental distribution of the final layered oxide material is shown in Fig. S5† with EDS mapping, where the particles show uniform distribution of Na, Ni, Fe and Mn. | ||
| Fig. 1 Phase evolution of the NaNi1/3Fe1/3Mn1/3O2 material upon calcination studied with synchrotron X-ray diffraction. The precursor Ni1/3Fe1/3Mn1/3(OH)2+x undergoes transformation through a sodiated oxyhydroxide phase in the temperature range of 250–550 °C by concurrent precursor dehydration and sodiation. At 550 °C, the layered phase is nucleated, which then undergoes transformations through two intermediate layered oxide phases at 650 °C and 750 °C before forming the final O3 layered oxide phase at a temperature of 850 °C. The XRD pattern for (precursor + Na2CO3) was collected with lab X-ray of wavelength 1.5406 Å. All the other XRD patterns were collected with synchrotron X-ray of wavelength 0.1814 Å. The 2θ values of synchrotron XRD were converted to the equivalent lab XRD 2θ values using Bragg's equation. | ||
We also observe the grain growth behavior of the particles and its correlation with phase structure of the material at different stages of calcination, which can be seen in the SEM images in Fig. 2. The morphology of the precursor Ni1/3Fe1/3Mn1/3(OH)2+x is comprised of plate-like primary particles which aggregate together to form secondary particles with sizes ranging from 3 to 6 μm. When the precursor is heated with Na2CO3 to different temperatures, the calcined particles retain the spheroidal morphology of the precursor. Three distinct stages of grain growth can be observed with increased degree of sodiation: stage 1 (250–450 °C), stage 2 (550–750 °C) and stage 3 (upon holding at 850 °C for 12 hours). Upon heating the precursor with Na2CO3 at 250 °C (stage 1), growth of the primary particles occurs due to simultaneous dehydration and insertion of sodium. No further significant changes of the grain morphology are observed in the temperature range of 250 °C to 450 °C, where the material mostly maintains an oxyhydroxide crystal structure and undergoes continuous dehydration. Different particles were imaged at different temperatures in the ex situ SEM experiment which may lead to particle size variation in the images. Upon the nucleation of the layered phase at 550 °C, where bulk insertion of sodium begins, we can observe further growth of the grains occurring (stage 2), which continues as the temperature increases to 750 °C. This is also supported by the continuous growth of multiple facets in this temperature range (XRD pattern in Fig. 1). The growth of the particles with increasing temperature occurs from continued growth of multiple facets, particle fusion and recrystallization.14,15 After holding for 6 hours at the final temperature of 850 °C when the fully layered phase is formed, final stage of particle growth is observed (stage 3) through the continuous growth of the (003) and (104) planes (Fig. 1). At this temperature, as the crystal structure converts to a fully layered phase, we observe that many primary particles fuse together to form larger primary particles, which leads to the stabilization of the surface energy of the particles.
 | ||
| Fig. 2 Grain growth behavior of the particles during calcination observed with scanning electron microscopy. The precursor undergoes three stages of grain growth when calcined with Na2CO3 with increasing temperature. During the early stages of sodiation and precursor decomposition (250–450 °C), slight coarsening of the primary particles occurs. During the later stages of sodiation (550–750 °C) where nucleation and growth of the layered phase occurs, the particles exhibit rapid growth of the grains with increasing temperature. When the fully layered phase is formed, the particles exhibit further growth of the grains due to continuous sodiation. | ||
Surface and bulk level chemical changes during high temperature calcination reaction
To understand the calcination reaction mechanism and concurrent surface and bulk level chemical changes occurring during the phase transitions, we used a combination of soft and hard X-ray absorption spectroscopic techniques. Soft XAS allows us to probe the surface and sub-surface level ensemble-averaged electronic structure of the transition metals while hard XAS provides ensemble-averaged bulk electronic structure information.27 The combination of these two techniques, along with the phase structure information obtained from XRD, allows for estimating chemical reactions occurring at different stages of calcination.The soft XAS technique probes the 2p–3d transitions of transition metal L-edge, providing a distinct signature of the metal oxidations states. The spectra obtained from this technique shows two unique features due to spin–orbit coupling, namely the L3-edge feature at lower energy and L2-edge feature at higher energy, which arise from the transitions involving 2p3/2 and 2p1/2 states respectively.10,28,29 This technique allowed us to probe the oxidation states of the transition metals at different depths using different detection modes.29 We used the total electron yield (TEY) mode to probe the surface level (∼5 nm) oxidation states and the fluorescence yield (FY) mode to probe the subsurface level (∼50–100 nm) oxidation states. On the other hand, hard XAS measurements on the calcined samples provided information on the ensemble-averaged bulk oxidation states of the material by detecting the Ni, Fe and Mn K-edge absorption energy. Fig. 3 represents the soft XAS spectra (Fig. 3a), hard XAS spectra (Fig. 3c) and oxidation state quantification of Mn at the surface (Fig. 3b) and bulk (Fig. 3d) of the particles in the materials calcined at different calcination temperatures. Ni and Fe spectra for the same samples are provided in the ESI where Fig. S6 and S7† represent the Ni and Fe L-edge spectra, respectively, and Fig. S8 and S9† represent the Ni and Fe K-edge spectra, respectively.
 | ||
| Fig. 3 Surface, sub-surface and bulk oxidation state evolution of Mn upon calcination studied with soft and hard X-ray absorption spectroscopy (a) Mn L-edge spectra in the TEY mode (b) changes in Mn3+ and Mn4+ weight fractions at different calcination temperatures obtained through the linear combination fitting of the Mn soft XAS spectra (c) Mn K-edge XANES spectra at different stages of calcination (d) average oxidation state evolution of Mn as a function of temperature obtained from the linear combination fitting of the XAS data. | ||
The spectral shapes of Ni L-edge (Fig. S6†) and Fe L-edge spectra (Fig. S7†) are characteristic of Ni2+ and Fe3+ oxidation states respectively, exhibiting no significant changes occurring with increasing calcination temperature, both in the TEY and FY mode. Similarly, The Ni K-edge (Fig. S8†) and Fe K-edge spectra (Fig. S9†) show no significant shift of the absorption edge energy to indicate any significant change of bulk oxidation state. This suggests that nickel and iron do not participate in charge compensating redox reactions during the sodiation process and maintain homogenous oxidation state from the surface to bulk.
On the other hand, Mn shows continuous oxidation with increasing calcination temperature, both at surface and bulk levels, as can be seen by the Mn L-edge spectra (Fig. 3a) and Mn K-edge spectra (Fig. 3c). Due to the more stable and lower energy d electron configurations of Ni2+ and Fe3+ compared to Mn3+, Mn is more susceptible to oxidation and acts as the charge compensating element during calcination. Although it is not feasible to visualize Na/O incorporation in the oxyhydroxide structure through X-ray diffraction or absorption techniques, Mn oxidation state can be taken as a proxy to understand the extent of Na/O incorporation (e.g., sodiation). The extent of sodiation should increase with the extent of Mn oxidation to maintain charge neutrality during calcination. In the Mn L-edge spectra, a progressive change in spectral shape is evident, indicative of an increasing Mn oxidation state, as confirmed through comparison with reference Mn spectra. Using the linear combination fitting method, Mn3+ and Mn4+ oxidation state contributions to the spectral shapes were quantified as a function of increasing temperature (Fig. 3b). Fitting parameters for the Mn L-edge soft XAS spectra at different calcination temperatures can be seen in ESI Table S2.† The surface of the precursor Ni1/3Fe1/3Mn1/3(OH)2+x has an overall oxidation state closer to Mn3+. Compared to the precursor, when the calcination mixture is heated up to 250 °C, the Mn3+ fraction increases. In the temperature range of 250–550 °C, there is a sharp linear increase of Mn oxidation state, indicating continuous precursor dehydration and concurrent sodiation by Na/O incorporation. Beyond 550 °C, the increasing trend of Mn oxidation state at the surface slows down, and Mn4+ weight fraction reaches closer to 1 at 750 °C. Upon further heating to 850 °C, Mn oxidation states at the surface do not change significantly and remain closer to Mn4+. Similar changes are observed in the subsurface level of the particles, as can be seen by the Mn L-edge spectra in the FY mode (Fig. S10†). This indicates that most of the surface and subsurface conversions from precursor-to-oxyhydroxide-to layered oxide occur within 250–550 °C and the structural ordering processes reach completion between 550 and 750 °C.
The oxidation of Mn in the bulk occurs slower than the surface during calcination due to the sluggish kinetics of sodiation and mass transport limitations. Fig. 3c represents the Mn K-edge spectra, where a continuous shift of the absorption edge is observed with increasing temperature. Quantification of the Mn oxidation state of the calcined samples was done by a linear combination fitting method (Fig. 3d). Fitting parameters are provided in ESI Table S3.† Although the surface of the hydroxide precursor exhibits a Mn oxidation state closer to +3 (Fig. 3a), the bulk oxidation state is much lower than Mn3+ (Fig. 3c). In the temperature ranges of 250–450 °C, Mn oxidation occurs in the bulk of the particles due to concurrent precursor dehydration/oxidation and sodium insertion. After the nucleation of the layered phase at 550 °C, where major insertion of sodium starts to occur into the bulk of the crystal lattice, Mn oxidation state also increases concurrently. Mn oxidation continues in the temperature range of 550–850 °C and reaches completion at 850 °C, upon the formation of the final layered phase. It is evident that throughout the entire course of calcination reaction, mass transport limitation of the Na/O incorporation leads to surface to bulk oxidation state heterogeneities within the particles. The reaction reaches completion at the surface and subsurface of the particles much earlier than the bulk, as evident from the XAS data.
Fig. 4 shows the O K-edge soft XAS spectra in the pre-edge region, which provides information on the transition metal 3d – O 2p states. The spectral features A (∼529.9 eV) and B (∼532.4 eV) arise from the electron excitation from O 1s orbital to the unoccupied states of the hybridized TM3d – O2p orbitals. Since there are multiple transition metals in the material, deconvolution of the spectra to identify the contribution of each transition metal to the spectral feature can be very complicated.29 The possible contribution of multiple TM 3d – O 2p contributions to the same spectral feature is depicted in ESI Fig. S11.† In general, in a material with multiple transition metals, the center of the 3d electronic states, known as the d-band center, can be approximated by taking a weighted average of the density of states (DOS) of each transition metal's 3d bands. This approximation is based on the contribution each transition metal makes to the overall DOS, typically proportional to its concentration in the material.30 Since Mn is the only charge compensating transition metal during calcination, spectral changes can be correlated with changes in Mn oxidation state. In the temperature range of 250 °C to 550 °C, where the dehydration reaction dominates over the sodiation process, we see a gradual increase in intensity of spectral feature A and a gradual decrease in intensity of spectral feature B, as a function of Mn oxidation state increase. Beyond 550 °C, up to 750 °C, where the sodiation reaction dominates, we see the opposite behavior of spectral feature A and B with increasing Mn oxidation state. This could be due to a charge redistribution in the TM3d-O2p states accompanied by bulk insertion of sodium in the oxyhydroxide structure through a rapid Na+/H+ ion exchange reaction. Although a further detailed analysis of the O K-edge data is not feasible in our material, the different trends in the spectral features between 250 °C to 550 °C and 550 °C to 750 °C are consistent with Mn L-edge (Fig. 3a) and synchrotron XRD data (Fig. 1). Both spectral feature A and B decrease in intensity when the sodiation reaction is complete and a more stable layered oxide structure has formed at 850 °C.
 | ||
| Fig. 4 Evolution of TM 3d – O2p hybridization at different stages of calcination. (a) During the early stages of calcination (250–550 °C), O K-edge spectra show a gradually increasing trend of spectral feature A and a gradually decreasing trend of spectral feature B. (b) After nucleation of the layered phase at 550 °C, opposite behavior of the A and B feature is observed with increasing temperature, due to possible charge redistribution caused by rapid incorporation of sodium into the material. | ||
Analysis of the phase structure and transition metal oxidation state evolution with increasing temperature allows us to gain a deeper insight into the reaction intermediates and chemical reactions occurring during synthesis. In the early stages of calcination from room temperature up to 150 °C, we assume that dehydration of the precursor occurs leading to the removal of the surface adsorbed and interlayer water molecules form the lattice. This can also be seen in the thermogravimetric analysis (TGA) curve of the precursor and Na2CO3 mixture (Fig. S12†), where a 9.5% weight loss occurs. The bulk of the precursor transforms to an oxyhydroxide structure TMOOH at 250 °C and maintains the oxyhydroxide structure until 550 °C, while continuously undergoing concurrent sodiation and precursor decomposition. Based on the oxidation state of the transition metals obtained from XAS, we propose that this oxyhydroxide intermediate consists of Fe3+, Ni2+, and a mixture of Mn3+ and Mn4+ within the temperature ranges of 250–550 °C. We cannot rule out the presence of minor oxide phases at this stage. Then gradual sodium and oxygen insertion in this intermediate leads to the final layered oxide formation. From our findings from the combination of XRD and XAS, the reactions occurring during different stages of calcination can be represented as:
Precursor oxidation and dehydration:
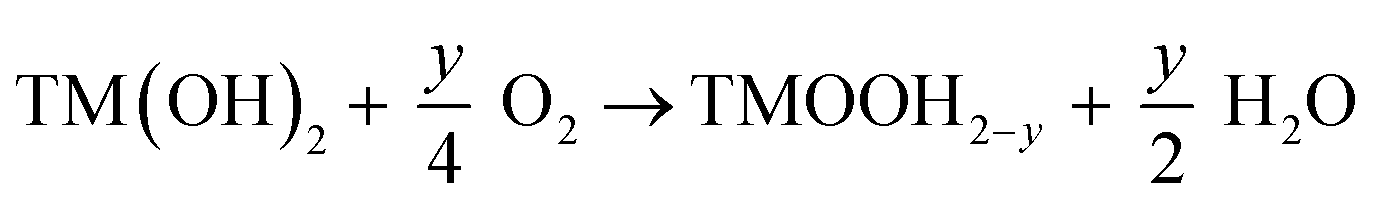 | (1) |
Sodiation of the oxyhydroxide intermediate via Na+/H+ exchange:
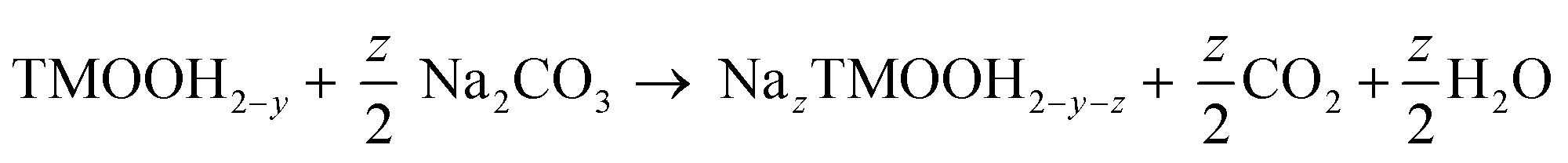 | (2) |
Oxyhydroxide to layered transformation:
 | (3) |
At the surface, these reactions occur faster compared to the bulk, within the temperature range of 250–550 °C, through concurrent sodiation and precursor dehydration (reaction (1)–(3)). This is due to higher accessibility of sodium and oxygen at the surface of the particles. At the bulk level, due to the sluggish nature of mass transport, the progression of these reactions occurs in two major phases: (i) 250–550 °C: where dehydration of the precursor dominates (reaction (1)) and (ii) 550–850 °C: where the sodiation process (reactions (2) and (3)) dominates over dehydration. The consequent weight loss during these reactions is quantified with TGA method (Fig. S12†).
Effects of calcination duration on microstructure, phase purity and electrochemical performance
To understand the effects of different calcination durations on the materials properties, we calcined the hydroxide precursor and Na2CO3 mixture at 850 °C for four different durations of 6 h, 12 h, 18 h and 24 h. We then investigated the effects of different calcination durations on the microstructure, phase purity and electrochemical performance of the material.Fig. 5a shows the microstructure of the cathode particles after calcination at 850 °C for different durations. The particles are composed of thick primary particle grains of sizes 200–500 nm which aggregate to form a secondary particle morphology. After calcining at 850 °C for 6 hours, a few small nanosized grains form on top of the primary particles, which have varying sizes in the range of 25–100 nm. As the calcination duration increases, the number of these smaller nanoparticles occupying the surface of the primary particles increases (Fig. 5b). The particles calcined for 24 hours at 850 °C show the highest number of nanoparticle formation, indicating a direct relationship between calcination duration and this unique microstructure evolution. This increased degree of nanoparticle formation with increased calcination duration has a direct effect on the surface reactivity of the material. These calcined particles were stored under ambient conditions for a week and then the O K-edge spectra were measured (Fig. 5c). The samples show an increased degree of surface carbonate formation with increasing calcination duration, which was identified by the carbonate peak signature at 534 eV in the O K-edge spectra.29,31,32 Increasing the calcination duration modifies the microstructure of the particles, which leads to their increased surface sensitivity to carbonate formation. In the literature, the surface of NaNi1/3Fe1/3Mn1/3O2 material has been found to be highly reactive due to the instability of surface sodium ions and high surface adsorption capacity towards air molecules.33 This high reactivity negatively impacts the material's storage stability and electrochemical performance.34 Our findings indicate that tuning the holding time at final calcination temperature can provide controllability of its surface reactivity.
 | ||
| Fig. 5 Effect of calcination duration on the microstructure and surface sensitivity of the material. (a) Scanning electron microscopy (SEM) images of the particles calcined at different durations of 6 h, 12 h, 18 h and 24 h. (b) Magnified SEM images of the particles calcined at different calcination durations, showing an increased number of nanoparticle formation on the surface of the particles with increasing calcination duration. Some of the nanoparticles are indicated with white arrows. (c) O K-edge soft X-ray absorption spectra of the samples calcined for different durations exhibit an increased degree of surface Na2CO3 formation as a function of increasing calcination duration after one week storage in ambient air. | ||
We further investigated the effect of calcination duration on the phase structure of the calcined materials through synchrotron X-ray diffraction. The calcined powders were stored in the glovebox (O2 < 0.5 ppm, H2O < 0.01 ppm) after calcination to minimize Na2CO3 formation through parasitic side reaction with air. Rietveld refinements of the XRD patterns of the samples calcined for different durations are shown in Fig. 6. Refinement results are given in Tables S4–S6.† Lattice parameters a and b decrease from 2.9821 Å to 2.9715 Å and lattice parameter c decreases from 16.1398 Å to 16.0768 Å when calcination duration is increased from 6 h to 12 h (Table S4†). Lattice parameters a and b do not show any significant change with further increase of calcination duration to 18 h and 24 h. However, lattice parameter c increases from 16.0768 Å to 16.1297 Å when the calcination duration is increased beyond 12 hours. Refinement results also show that the phase fraction of the O3 phase increases from 83.69% to 89.34% and the phase fraction of Na2CO3 decreases from 10.62% to 5.16% when calcination duration is increased from 6 h to 24 hours (Table S5†). This indicates that longer calcination duration can enhance the degree of sodiation. The results also show that the phase distribution of minor phases like Mn3O4 and NiO in the calcined materials can vary depending on the calcination duration. During calcination, although the major phase transformation occurs through an oxyhydroxide intermediate, it is possible that a minor NiO rocksalt phase forms at higher temperatures. The presence of a major rocksalt phase was not detected in the intermediate phases, but it might exist as a minor impurity phase with short-range order in the final calcined materials.
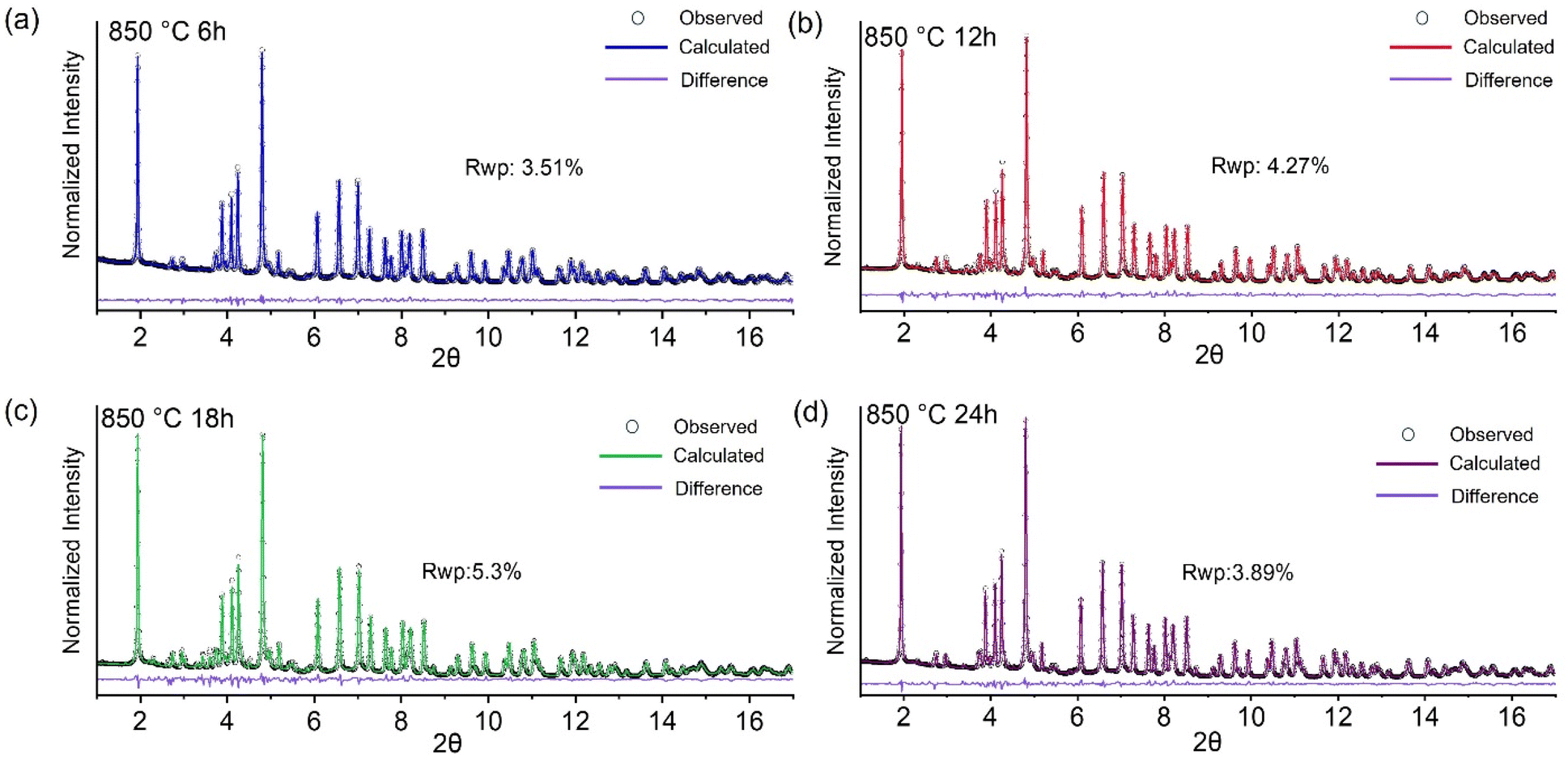 | ||
| Fig. 6 Synchrotron X-ray diffraction pattern and Rietveld refinement of the samples calcined at 850 °C for (a) 6 hours (b) 12 hours (c) 18 hours and (d) 24 hours. The X-ray wavelength is 0.1814 Å. | ||
We further evaluate the electrochemical performance of the NaNi1/3Fe1/3Mn1/3O2 material calcined at 850 °C for different calcination durations (Fig. 7). Fig. 7a shows the first cycle voltage profile of the materials, cycled at a rate of 0.1C in the voltage range of 2–4.3 V. The initial specific discharge capacity of the material calcined for 12 h (141 mA h g−1) and 18 h (143 mA h g−1) are similar, whereas the specific capacities for the materials calcined for 6 h (130 mA h g−1) and 24 h (120 mA h g−1) are lower. In terms of the capacity retention, all the materials show similar trends of fading behavior when cycled at 1C rate. Rate capability performance of the calcined materials is shown in Fig. 7c. All the materials show similar trend in their rate capability performance, with the sample calcined for 18 h showing the best rate capability behavior. The sample calcined for 24 hours shows poor electrochemical performance among all the other samples. We also employed the GITT method to estimate the apparent Na-ion diffusion coefficients of the cathode materials calcined for different durations (Fig. S13†). The material calcined for 18 hours shows the highest range of apparent diffusion coefficients values (1.36 × 10−9 to 6.68 × 10−10 cm2 s−1) and the material calcined for 24 hours shows the lowest range of apparent diffusion coefficients values (3.87 × 10−10 to 8.65 × 10−10 cm2 s−1). The samples calcined for 6 hours, and 12 hours show intermediate apparent diffusion coefficients values in the range of 4.68 × 10−10 to 1.16 × 10−9 and 6.21 × 10−10 to 1.26 × 10−9 cm2 s−1 respectively. Overall, with increasing calcination duration from 6 h to 18 h, a slight improvement of the electrochemical performance is observed, with the sample calcined for 18 hours showing the best electrochemical performance among all the samples.
 | ||
| Fig. 7 Electrochemical performance of the NaNi1/3Fe1/3Mn1/3O2 material at different calcination durations. (a) 1st cycle voltage profile at 0.1C cycling rate (b) capacity retention at 1C cycling rate (c) rate capability of the material at different calcination durations. All the electrochemical tests are done in a NaNFM333| Na half-cell configuration, with 1.0 M NaClO4 in PC with 5% FEC electrolyte, at a voltage range of 2.0–4.3 V. Average active material mass loading is 6 mg cm−2 and the testing temperature was 22 °C. | ||
We found that increasing the calcination duration beyond 18 hours (to up to 24 hours) has detrimental effects on electrochemical performance. This could be due to two reasons. One is due to Na/O loss from the material at high calcination duration, which is supported by the slight reduction of the Mn both at surface (Fig. 3b and S14a†) and bulk levels (Fig. 3d and S14b†). Although the 24 h calcined sample is structurally more phase-pure, it exhibits a slight chemical imbalance due to Na/O loss, resulting in average Mn reduction without the formation of new crystalline phases. Another is the unique microstructure evolution of the material with prolonged calcination duration. Although the O3 phase fraction is the highest at 24 hour calcination duration, the microstructure of this material is plagued by an increased number of nanoparticles which cover the surface of the primary particles. We performed BET surface area analysis of the cathode particles calcined at 850 °C for different durations to observe the effect of calcination duration on microstructure evolution. The results are presented in ESI Table S7.† With increasing calcination durations, two events are occurring. One is the thermally activated grain growth of the particles due to Ostwald ripening, which is reflected in the decreased surface area of the particles with increased duration (from 2.0 m2 g−1 at 6 h to 1.4 m2 g−1 at 18 h). However, at 24 hour holding, the surface area of the particles increases to 1.9 m2 g−1 due to excessive nanoparticle coverage, which outweighs the loss in surface area from Ostwald ripening. To look deeper into the effects of these nanoparticle formation, we performed BF-STEM EDS mapping of the particles calcined for 24 hours at 850 °C. We chose a total of fourteen spots on the particles and performed EDS mapping on them (Fig. S15†). The molar ratio of transition metal to sodium was found to be highly heterogenous throughout all fourteen spots. Nine of these spots were sodium deficient, and the rest of the five spots had a ratio greater than one. The elemental mapping results are presented in Table S8.† In our system, surface chemical instabilities driven by Na/O loss and local non-stoichiometry may trigger re-nucleation of smaller, sodium-deficient phases. The absence of a liquid phase and limited solid-state diffusion may introduce kinetic barriers that hinder ripening and freeze the microstructure in a nanoparticle-decorated state. Excessive nanoparticle formation can lead to a highly heterogeneous sodium distribution, potentially contributing to the material's poor electrochemical performance compared to samples calcined for shorter durations. This underscores the importance of tuning calcination parameters to control microstructure, optimize elemental distribution, and minimize Na/O loss.
Conclusion
Optimizing reaction parameters during material synthesis offers a straightforward approach to enhancing electrochemical performance, maintaining elemental simplicity, and avoiding complex material modification techniques. Gaining a deeper understanding of the mechanistic pathways involved in synthesis can further guide the precise tuning of these parameters. To summarize, the high temperature calcination reaction of NaNi1/3Fe1/3Mn1/3O2 material was studied to obtain insight into the microstructure, global phase evolution, and local chemical changes during different stages of synthesis using a range of synchrotron-based tools and supporting analytical methods. Specifically, our findings are as follows:(I) The precursor mixture undergoes multiple stages of phase transformation through dehydration and simultaneous sodiation reaction, where the major reaction intermediate consists of an oxyhydroxide structure. The dehydration reactions dominate in the temperature range of 250–550 °C and the sodiation reaction dominates in the temperature range of 500–850 °C. The phase transitions are accompanied by grain growth of the material along both the (003) and (104) directions, leading to its final microstructure.
(II) Mn serves as the major charge compensating element during sodiation and its surface to bulk oxidation state varies at different temperatures, reflecting upon the heterogenous sluggish nature of sodiation during synthesis.
(III) The results show that as the sodiation reaction starts to dominate over precursor dehydration reaction, the local chemical environment of the TM3d-O2p states undergo a charge redistribution.
(IV) Calcination duration also plays an important role in tuning the microstructure and electrochemical properties of the material. Calcining the material beyond 18 hours causes excessive nanoparticle formation on particle surface, which ultimately leads to the formation of sodium deficient regions in the individual particles and deterioration of electrochemical performance.
These findings provide deeper insight into the nature of the solid-state calcination reaction pathways in a sodium ion layered oxide system, which can be used to inform synthesis parameter tuning and optimization of electrochemical properties.
Data availability
The datasets supporting this article have been uploaded as part of ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work was supported by the National Science Foundation under DMR 2325464. K. Z. acknowledges the support from the National Science Foundation under DMR 2325463. The use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract no. DE-AC02-76SF00515. This research used resources of the Advanced Photon Source; a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract no. DE-AC02 06CH11357. This work used shared facilities at the Nanoscale Characterization and Fabrication Laboratory, which is funded and managed by Virginia Tech's Institute for Critical Technology and Applied Science. Additional support is provided by the Virginia Tech National Center for Earth and Environmental Nanotechnology Infrastructure (NanoEarth), a member of the National Nanotechnology Coordinated Infrastructure (NNCI), supported by NSF (ECCS 1542100 and ECCS 2025151). F.L. acknowledges the NCFL Faculty Fellowship program.References
- X. Pu, H. Wang, D. Zhao, H. Yang, X. Ai, S. Cao, Z. Chen and Y. Cao, Small, 2019, 15(32), 1805427 CrossRef.
- H. S. Hirsh, Y. Li, D. H. S. Tan, M. Zhang, E. Zhao and Y. S. Meng, Adv. Energy Mater., 2020, 10(32), 2001274 CrossRef CAS.
- J.-M. Tarascon, Joule, 2020, 4, 1616–1620 CrossRef.
- K. Kubota, N. Yabuuchi, H. Yoshida, M. Dahbi and S. Komaba, MRS Bull., 2014, 39, 416–422 CrossRef CAS.
- J. Lamb and A. Manthiram, Chem. Mater., 2020, 32, 8431–8441 CrossRef CAS.
- M. H. Han, E. Gonzalo, G. Singh and T. Rojo, Energy Environ. Sci., 2015, 8, 81–102 RSC.
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- M. M. Rahman, Y. Xu, H. Cheng, Q. Shi, R. Kou, L. Mu, Q. Liu, S. Xia, X. Xiao, C.-J. Sun, D. Sokaras, D. Nordlund, J.-C. Zheng, Y. Liu and F. Lin, Energy Environ. Sci., 2018, 11, 2496–2508 RSC.
- M. M. Rahman, Y. Zhang, S. Xia, W. H. Kan, M. Avdeev, L. Mu, D. Sokaras, T. Kroll, X.-W. Du, D. Nordlund, Y. Liu and F. Lin, J. Phys. Chem. C, 2019, 123, 11428–11435 CrossRef CAS.
- Z. Yang, L. Mu, D. Hou, M. M. Rahman, Z. Xu, J. Liu, D. Nordlund, C. Sun, X. Xiao and F. Lin, Adv. Energy Mater., 2021, 11(1), 2002719 CrossRef CAS.
- H. Park, H. Park, K. Song, S. H. Song, S. Kang, K.-H. Ko, D. Eum, Y. Jeon, J. Kim, W. M. Seong, H. Kim, J. Park and K. Kang, Nat. Chem., 2022, 14, 614–622 CrossRef CAS.
- W. Zuo, J. Gim, T. Li, D. Hou, Y. Gao, S. Zhou, C. Zhao, X. Jia, Z. Yang, Y. Liu, W. Xu, X. Xiao, G.-L. Xu and K. Amine, Nat. Nanotechnol., 2024, 1–10 CAS.
- X. Yang, S. Wang, H. Li, J. Peng, W.-J. Zeng, H.-J. Tsai, S.-F. Hung, S. Indris, F. Li and W. Hua, ACS Nano, 2023, 17, 18616–18628 CrossRef CAS PubMed.
- C.-K. Yang, L.-Y. Qi, Z. Zuo, R.-N. Wang, M. Ye, J. Lu and H.-H. Zhou, J. Power Sources, 2016, 331, 487–494 CrossRef CAS.
- L. Tang, X. Cheng, R. Wu, T. Cao, J. Lu, Y. Zhang and Z. Zhang, J. Energy Chem., 2022, 66, 9–15 CrossRef CAS.
- W. Hua, X. Yang, N. P. M. Casati, L. Liu, S. Wang, V. Baran, M. Knapp, H. Ehrenberg and S. Indris, eScience, 2022, 2, 183–191 CrossRef.
- S. A. Wicker and E. H. Walker, Inorg. Chem., 2013, 52, 1772–1779 CrossRef CAS PubMed.
- R. J. Gummow, D. Liles and M. Thackeray, Mater. Res. Bull., 1993, 28, 235–246 CrossRef CAS.
- Y. Kan, Y. Hu, Y. Ren, J. Bareño, I. Bloom, Y.-K. Sun, K. Amine and Z. Chen, J. Power Sources, 2014, 271, 97–103 CrossRef CAS.
- H. Park, H. Park, K. Song, S. H. Song, S. Kang, K.-H. Ko, D. Eum, Y. Jeon, J. Kim, W. M. Seong, H. Kim, J. Park and K. Kang, Nat. Chem., 2022, 14, 614–622 CrossRef CAS.
- S. Jo, J. Han, S. Seo, O. Kwon, S. Choi, J. Zhang, H. Hyun, J. Oh, J. Kim, J. Chung, H. Kim, J. Wang, J. Bae, J. Moon, Y. Park, M. Hong, M. Kim, Y. Liu, I. Sohn, K. Jung and J. Lim, Adv. Mater., 2023, 35(10), 2207076 CrossRef CAS.
- A. Tayal, P. Barai, H. Zhong, O. Kahvecioglu, X. Wang, K. Z. Pupek, L. Ma, S. N. Ehrlich, V. Srinivasan, X. Qu, J. Bai and F. Wang, Adv. Mater., 2024, 36(21), 2312027 CrossRef CAS.
- Y. Li, R. Xu, Y. Ren, J. Lu, H. Wu, L. Wang, D. J. Miller, Y.-K. Sun, K. Amine and Z. Chen, Nano Energy, 2016, 19, 522–531 CrossRef CAS.
- D. Wang, X. Zhang, G. Zhong, Y. Li, C. Hong, K. Dong, C. Chen and Y. Yang, J. Power Sources, 2022, 529, 231258 CrossRef CAS.
- Y. Xie, H. Gao, R. Harder, L. Li, J. Gim, H. Che, H. Wang, Y. Ren, X. Zhang, L. Li, Z. Chen, K. Amine and Z.-F. Ma, ACS Appl. Energy Mater., 2020, 3, 6107–6114 CrossRef CAS.
- D. Peddis, D. Rinaldi, G. Ennas, A. Scano, E. Agostinelli and D. Fiorani, Phys. Chem. Chem. Phys., 2012, 14, 3162 RSC.
- D. Hou, D. Xia, E. Gabriel, J. A. Russell, K. Graff, Y. Ren, C.-J. Sun, F. Lin, Y. Liu and H. Xiong, ACS Energy Lett., 2021, 6, 4023–4054 CrossRef CAS.
- L. Mu, X. Feng, R. Kou, Y. Zhang, H. Guo, C. Tian, C. Sun, X. Du, D. Nordlund, H. L. Xin and F. Lin, Adv. Energy Mater., 2018, 8, 1801975 CrossRef.
- C. Tian, D. Nordlund, H. L. Xin, Y. Xu, Y. Liu, D. Sokaras, F. Lin and M. M. Doeff, J. Electrochem. Soc., 2018, 165, A696–A704 CrossRef CAS.
- J. Suntivich, W. T. Hong, Y.-L. Lee, J. M. Rondinelli, W. Yang, J. B. Goodenough, B. Dabrowski, J. W. Freeland and Y. Shao-Horn, J. Phys. Chem. C, 2014, 118, 1856–1863 CrossRef CAS.
- F. Frati, M. O. J. Y. Hunault and F. M. F. de Groot, Chem. Rev., 2020, 120, 4056–4110 CrossRef CAS PubMed.
- L. Mu, Z. Yang, L. Tao, C. K. Waters, Z. Xu, L. Li, S. Sainio, Y. Du, H. L. Xin, D. Nordlund and F. Lin, J. Mater. Chem. A, 2020, 8, 17487–17497 RSC.
- X.-Q. Xu, J.-Y. Chen, Y. Jiang, B. Xu, X.-L. Li, C.-Y. Ouyang and J.-X. Zheng, J. Am. Chem. Soc., 2024, 146, 22374–22386 CrossRef CAS.
- Y. Sun, H. Wang, D. Meng, X. Li, X. Liao, H. Che, G. Cui, F. Yu, W. Yang, L. Li and Z.-F. Ma, ACS Appl. Energy Mater., 2021, 4, 2061–2067 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta00844a |
| This journal is © The Royal Society of Chemistry 2025 |