
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFactors in the utilization of corrosive ruthenium in the oxygen evolution electrode of polymer electrolyte membrane water electrolysis†
Suji Leeab,
Chaeyeon Yangab,
Jihyun Choiab,
Jong Hyun Jangad,
Haneul Jin *c,
Yung-Eun Sung*b and
Hyun S. Park*ad
*c,
Yung-Eun Sung*b and
Hyun S. Park*ad
aCenter for Hydrogen and Fuel Cells, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea. E-mail: hspark@kist.re.kr
bSchool of Chemical and Biological Engineering, Seoul National University, Seoul 08826, Republic of Korea. E-mail: ysung@snu.ac.kr
cDepartment of Energy and Materials Engineering, Dongguk University, Seoul 04620, Republic of Korea. E-mail: hjin1@dongguk.edu
dDivision of Energy & Environment Technology, KIST School, University of Science and Technology (UST), Seoul 02792, Republic of Korea
First published on 24th April 2025
Abstract
The use of non-Pt materials in polymer electrolyte membrane water electrolysis (PEMWE) anodes is imperative to reduce green hydrogen production costs. Ru provides sufficient electrical and thermal conductivity, but rapidly over-oxidizes to RuO42− or H2RuO5 during PEMWE device operation. Therefore, this study involved using corrosive Ru instead of expensive Pt to form an interlayer between a Ti porous transport layer and the IrOx catalyst in PEMWE anodes, while enhancing the water-oxidation-catalyzing ability of IrOx. The optimized Ru-containing anode was comparable to the Pt-containing equivalent in OER performance (5.5 A cm−2 at 2.0 VCell; ∼80 μg cm−2 of Ir). Importantly, the Ru in the PEMWE anode functioned stably for >250 h at a practical current density (2.0 A cm−2) during current swing operation. Consequently, the Ru-interlayer-based device was comparable to the conventional Pt-interlayer-based counterpart in performance and durability. The relationship between Ru dissolution and local oxygen accumulation at electrode surfaces was clarified.
Introduction
The use of hydrogen as a clean energy vector and complementary system to intermittent renewable sources is vital for making industries carbon-neutral. Among the various techniques for producing green hydrogen, polymer electrolyte membrane water electrolysis (PEMWE) has received significant attention for permitting low-partial-load operations and high-pressure hydrogen production with renewable electricity.1 However, two economic and technical barriers hinder the widespread adoption of PEMWE. First, the scarcity of Ir is a significant impediment to the gigawatt-scale production of green hydrogen via PEMWE.2 Second, the capital cost of PEMWE is significantly higher than that of alkaline water electrolysis owing to the substantial use of expensive metals—such as Pt, Au, and Ti—in addition to the utilization of Ir in the anode. To boost the commercial viability of PEMWE, the aforementioned expensive metals should be used in a limited manner or replaced with other inexpensive materials.Electrode development strategies, such as engineering the interface between the catalyst and the Ti porous transport layer (Ti-PTL), have been devised to enhance the economic efficiency of PEMWE.3 The conventional OER electrode of PEMWE cells comprises an interlayer made of physically and chemically robust precious metals such as Pt and Au (1–2 mg cm−2) between the Ti-PTL and the Ir catalyst layer (1–3 mg cm−2).4,5 The Pt- or Au-based metal layer is applied onto the Ti-PTL to ensure sufficient electrical and thermal connections between the Ti-PTL and the IrOx catalyst and prevent severe Ti-PTL corrosion during water electrolysis, thereby averting the formation of an insulating TiO2 film on the Ti surface during operation.1 Recently, the Ti-PTL/Ir-catalyst interlayer has been utilized as an effective support, increasing the surface area and reducing catalyst consumption.6,7
The effectiveness of the precious metal layer between the OER catalyst and the Ti-PTL has been demonstrated through PEMWE device durability measurements conducted for hundreds of hours.8 To construct appropriate Ti-TPL surfaces and reduce the capital cost of PEMWE electrodes, the substitution of Pt or Au with less expensive materials has also been extensively studied.9,10 Among these materials, Ru is a feasible candidate because its electrical conductivity (1.4 × 107 S m−1) is comparable to that of Pt (9.4 × 106 S m−1) and Au (4.5 × 107 S m−1);11,12 moreover, it is more affordable than Pt and Au (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 750, 28887, and 67020 € per kg, respectively, as of February 2024).13 Additionally, Ru exhibits high thermal conductivity (120 W m−1 K−1; 71 and 320 W m−1 K−1 for Pt and Au, respectively),14 making it appropriate for high-current operations.
750, 28887, and 67020 € per kg, respectively, as of February 2024).13 Additionally, Ru exhibits high thermal conductivity (120 W m−1 K−1; 71 and 320 W m−1 K−1 for Pt and Au, respectively),14 making it appropriate for high-current operations.
However, Ru and RuOx dissolve rapidly under practical PEMWE operating conditions, making them unsuitable as interlayer materials or active OER catalysts.15 Ru and RuOx have also been extensively investigated as active OER catalysts in half-cells with acidic aqueous electrolytes;16,17 however, they lack the (electro)chemical stability required for PEMWE devices to function at practical operational densities (for example, >1.0 A cm−2). The dissolution of Ru can be mitigated by alloying it with other metals,18,19 which more readily resist corrosion and moderately suppress the over-oxidation of Ru. However, to our knowledge, PEMWE devices containing anodes with these Ru-based materials and exhibiting prolonged lifetimes have not been reported.
To utilize these corrosive materials in PEMWE anodes, factors that accelerate their dissolution kinetics—such as applied potential, device voltage, excess current density, and a harsh local chemical environment—should be understood and avoided in operations. For example, when the device is operated at high current densities for prolonged periods, the supersaturated dissolved oxygen produced at the OER electrode creates surface-insulating gas bubbles, increasing the local overpotential and producing hot spots to accelerate metal dissolution.20 A few researchers have investigated the effects of these bubbles on water electrolysis. For instance, Dukovic et al. theoretically determined that the hydrogen bubbles at electrodes double the local current densities and shift the potentials near the bubbles as they insulate the surface.21 Kellogg et al. studied bubble-induced hot spots on an alloy steel electrode for aqueous AWE. The electrode temperature was found to increase to the boiling point of water owing to bubble insulation under specific conditions.22 Despite the well-established fact that Ru dissolves when the applied potential exceeds 1.387 VRHE, forming RuO4 or H2RuO5,15–18 the factors contributing to Ru dissolution kinetics remain largely unexplored. Although the direct relationship between electrode dissolution and bubble accumulation has not yet been revealed, high overpotentials or temperatures are likely to affect the electrode degradation kinetics when corrosive metals are used in PEMWE anodes, given these facts.
In this study, a practical PEMWE anode was fabricated using Ru to create the interlayer between the Ti-PTL and IrOx. The Ru interlayer exhibited the requisite properties such as electrical conductivity, thermal conductivity, and large surface area to minimize the use of IrOx in the electrode. The performance of the PEMWE cell whose anode contained Ru and IrOx was comparable with that of its counterpart comprising Pt and IrOx, with the Ru content being one-tenth that of Pt (47 μgRu cm−2 and 378 μgPt cm−2, respectively). Importantly, the durability of the Ru-anode-based device was ensured under current swing (CS) operation, a technique that intermittently interrupts the current in a controlled manner during normal operation. CS operation consists of high current operation (HCO, 2.0 A cm−2) for hydrogen production and low current operation (LCO, 0.1 A cm−2) for gas removal. Implementing CS operation helps prevent the dissolution of the Ru interlayer during prolonged functioning of the PEMWE cell at a high current density (2.0 A cm−2 for >200 h). The beneficial effects of the PEMWE operation protocol on the stability of the electrode containing degradable materials were demonstrated under practical water electrolysis conditions. In the optimized operation protocol, the degradation rate of the Ru@Ir-based PEMWE cell decreased significantly, that is, from 950 to 314 μV h−1 (67%) at 2.0 A cm−2, which was comparable to that of the Pt@Ir-based cell under the same conditions. The Ru dissolution at the PEMWE anode was associated with the presence of over-oxidized Rux>+4, which was related to bubble accumulation during high-current operations.
Results and discussion
Ru@Ir and Pt@Ir fabrication
Electrodeposition of Ru was performed by layering different amounts of Ru on the Ti-PTL (denoted as Ru/Ti-PTL) at different Ru deposition times (1, 2, 5, and 10 min; Fig. S1a and b, ESI†), exhibiting thicknesses of 105–394 nm (Fig. S2, ESI†).23 For comparison, a Pt interlayer was also prepared on the Ti-PTL (denoted as Pt/Ti-PTL) according to a previously reported method (Fig. S1c, ESI†).6 The electrodeposited Pt particles were ∼500 nm in diameter and hemispherical with roughened surfaces (Fig. S3, ESI†), and the average crystallite size of Ru and Pt was 2.60 and 3.32 nm, respectively, as estimated around 45° in the XRD patterns (Fig. S4, ESI†). Small 3–5 nm Ru nanoparticles were also observed in the TEM images of the Ru/Ti-PTL film (Fig. S4 and S5, ESI†). The different growth mechanisms led to differences in crystallite size between Ru and Pt and also resulted in disparities in the ECSA of the electrode (Fig. S6, ESI†).24 The ECSA of Ru and Pt was estimated from the Hupd charge values determined by CV (Fig. S7, ESI†). The agglomeration of small Ru particles exhibited a higher ECSA than the Pt layer deposited on the Ti-PTL with a low loading. For example, the Ru/Ti-PTL had ∼40% more conducting surfaces than the Pt/Ti-PTL. Thereafter, as an active OER catalyst, a metallic Ir layer was constructed on the fabricated Ru/Ti-PTL and Pt/Ti-PTL systems via sequential pulse electrodeposition, which enabled facile control over both the position and amount of Ir deposited at the Ru/Ti-PTL and Pt/Ti-PTL surfaces. The Ir loading in Ru@Ir was controlled between 47 and 67 μg cmgeo−2 with 30 pulse deposition sequences (Fig. S1d, ESI†).25 As discussed above, although the Ru/Ti-PTL provided a larger surface area as a catalyst support compared to the Pt/Ti-PTL, the Ir layer in Ru@Ir was around three times thicker than that in Pt@Ir in experiments (∼180 and ∼50 nm, respectively), as indicated by the cross-sectional images of the electrodes (Fig. 1a–f). The results strongly suggest that the Ir film in Ru@Ir has a lower tap density (or higher film porosity) and/or a lower surface coverage than that in Pt@Ir. In fact, the measured Hupd surface area of Ru@Ir (38.1 cmmaterial2 cmgeo−2) was approximately 1.6 times higher than that of Pt@Ir despite having 20% less Ir deposited at the electrode (23 cmmaterial2 cmgeo−2) (Fig. S6, S7, and Table S1, ESI†). These results strongly suggest that Ru@Ir contained a porous film of small particles, and that the surface of the Ru/Ti-PTL was not entirely covered with the Ir film. In contrast, the Pt/Ti-PTL surface was completely covered with Ir, forming less porous Pt@Ir. These findings are also supported by the XPS, TPO analysis, intrinsic OER kinetic analyses in half cells. The Pt surface was not discernible in the XPS spectra of Ir-deposited Pt@Ir, whereas Ru was still detected in the spectrum of Ru@Ir, suggesting that the Pt surface was completely covered with Ir in Pt@Ir, which was not the case for Ru@Ir (Fig. S8, ESI†). In the TPO analysis, the oxidation of Ru in Ru@Ir was still observed, whereas that of Pt in Pt@Ir was barely detectable (Fig. S9, ESI†). In addition, RuOx and Ru@Ir exhibited an onset potential that was 80 mV more negative and an OER current density that was three times higher than those of IrOx and Pt@Ir in half cell tests (Fig. S10, ESI†). The effects of uncovered Ru in Ru@Ir on the long-term stability of the electrode are further discussed below. | ||
| Fig. 1 Comparison of surface morphology and OER performance (a) top-view FE-SEM image of Ru@Ir (the scale bar in the inset image on bottom-left: 1 μm), (b) cross-sectional FIB-SEM image of Ru@Ir, (c) cross-sectional elemental mapping image of Ru@Ir obtained via HAADF-STEM (yellowish green: Ir, sky b lue: Ru, and gray: Ti), (d) top-view FE-SEM image of Pt@Ir (the scale bar in the inset image on bottom-left: 1 μm), (e) cross-sectional FIB-SEM image of Pt@Ir, (f) cross-sectional elemental mapping image of Pr@Ir obtained via HAADF-STEM (yellowish green: Ir, navy: Pt, and gray: Ti). (g) LSV polarization curves, (h) current density at 2.0 V, and (i) overvoltage at 1.0 A cm−2 subdivided into ohmic (ηohm), kinetic (ηkin), and mass transfer (ηmass) components. | ||
Using Ru@Ir electrodes fabricated with different Ru deposition levels, a series of PEMWE devices were assembled to measure their initial performance and stability. Among these electrodes, Ru(47 μg cm−2)@Ir corresponding to 2 min of Ru deposition exhibited the highest activity and initial stability for 6 h, since the Ru film obtained via 2 min of deposition was more firmly attached to the Ti-PTL (Fig. S2 and S11, ESI†). Therefore, Ru(47 μg cm−2)@Ir—hereafter denoted as Ru@Ir—was selected as the representative electrode for subsequent investigations. After the initial assessments, the optimized Ru@Ir anode was compared with Pt@Ir and unsupported IrOx on the Ti-PTL in PEMWE devices. The Ir loading in the unsupported IrOx electrode was 0.5 mg cm−2. The OER performance of the optimized Ru@Ir anode was similar to that of Pt@Ir under device operation conditions (5.5 and 5.3 A cm−2, respectively, at 2.0 VCell) (Fig. 1g and h). Moreover, the initial performance of both Ru@Ir and Pt@Ir was 3.5 times higher than that of IrOx (1.5 A cm−2 at 2.0 VCell). Overvoltage analysis of Ru@Ir, Pt@Ir, and IrOx suggested that Ru@Ir and Pt@Ir exhibited similar kinetic overvoltages (308 and 309 mV, respectively, at 1.0 A cm−2), which were approximately 20% less than that of IrOx (382 mV). Furthermore, the ohmic and mass-transfer overvoltages of Ru@Ir and Pt@Ir decreased more prominently than those of IrOx. At 1.0 A cm−2, the ohmic overvoltage of IrOx (183 mV) was approximately 2.5 times higher than that of Ru@Ir (70 mV) and Pt@Ir (73 mV) (Fig. S12, ESI†). Last but not least, both Ru@Ir and Pt@Ir exhibited an insignificant mass-transfer overvoltage (<7.0 mV @ 1.0 A cm−2), whereas that of IrOx was considerable (91 mV) (Fig. 1i). Collectively, these results indicated that both Ru@Ir and Pt@Ir fully utilized the advantages of the interlayer—that is, the large surface area (Fig. S13, ESI†) and high electrical conductivity —thereby enhancing the PEMWE performance.
Overall, the effects of the Ru or Pt interlayer in the PEMWE anode were clearly demonstrated during device operation. The Ru- or Pt-interlayer-supported IrOx catalysts remarkably extended the surface area of the active electrode and decreased its mass-transfer resistance. Importantly, the analysis underscored the potential of inserting a Ru layer instead of Pt between the Ti-PTL and IrOx catalyst for fabricating PEMWE anodes.
Stability in constant-current operation
The initial assessment of Ru@Ir indicated that the Ru interlayer in Ru@Ir provided several beneficial features for replacing Pt in PEMWE anodes. However, as discussed above, the use of Ru in PEMWEs is generally avoided owing to its instability under practical operating conditions. Therefore, the operational stability of Ru@Ir had to be validated through long-term durability tests to determine the feasibility of employing Ru as a key Pt-replacing component of PEMWE anodes.The long-term OER performance of Ru@Ir was first evaluated by conducting chronopotentiometry (CP) for 80 h at a constant current (CC) density of 2.0 A cm−2 (Fig. 2a). As both reversible and irreversible degradation occurred in the durability tests, LSV curves of the electrodes were also recorded by excluding the significant reversible degradation occurring before and after the CP tests. As shown in Fig. 2b, Ru@Ir exhibited a 4.1 times higher degradation rate than that of Pt@Ir in the CP test performed at 2.0 A cm−2 (950 and 234 μV h−1, respectively). Furthermore, LSV analysis suggested that the current density of Ru@Ir at 2.0 VCell decreased by 11% from 2.6 to 2.3 A cm−2 in 80 h, whereas that of Pt@Ir decreased by only 3.8% from 2.6 to 2.5 A cm−2 in 100 h (Fig. 2c and d).
 | ||
| Fig. 2 Long-term durability test (LT) with constant current (CC) operation at 2.0 A cm−2 (a) CP curves of Ru@Ir and Pt@Ir, (b) Degradation rate calculated from voltage variations. (c) LSV polarization curves of Ru@Ir and Pt@Ir before and after LT, (d) current density at 2.0 V, (e) high-frequency resistance (HFR) at 10 kHz, and (f) line scanning study of cation distribution in the cross-section of MEA via electron probe micro analyzer (EPMA) analysis. | ||
The significantly higher degradation rate of Ru@Ir was primarily associated with the loss of the conductive interface between the Ti-PTL and the Ru (or Ir) catalyst, as indicated by the high-frequency resistance (HFR) measurements performed after the durability tests. The HFR of Ru@Ir increased by 0.23 mΩ cm2 h−1, that is, from 138 to 157 mΩ cm2 in 80 h, whereas that of Pt@Ir remained relatively uniform (Fig. 2e and S14, ESI†). The values corresponding to HFR degradation were 214 and 0 μV h−1 for Ru@Ir and Pt@Ir, respectively. This higher HFR degradation of Ru@Ir implied the possible destruction of its Ru interlayer during long-term operation, which did not occur in Pt@Ir. As a result, Ru@Ir also showed a remarkably higher voltage change rate compared to Pt@Ir, caused by changes in the charge-transfer resistance (736 and 234 μV h−1, respectively) (Fig. S15, ESI†). Additionally, the dissolved Ru ions generated via the degradation of Ru@Ir could poison the ion-conducting membrane of the PEMWE cell, thereby increasing the HFR; the presence of Ru and Ir at the membrane and cathode sides of the MEA was evident from the cross-sectional analysis of the electrodes after CC operation (Fig. 2f and S16, ESI†). The changes in the electrical resistance of the Ti-PTL and the ionic resistance of the membrane were not readily distinguished through EIS analysis, and the combined variations in the HFR of the MEA were used to evaluate the ohmic degradation.
The loss of the Ru interlayer in Ru@Ir was substantiated by changes in the ECSA of the electrodes determined through separate measurements (Fig. S17, ESI†). After a single LSV measurement from 1.3 to 2.0 VCell at 10 mV s−1, the Hupd area of Ru@Ir decreased by ∼64% from 38 to 14 cmmaterial2 cmgeo−2. The decrease in the Hupd area of Ru@Ir lessened in successive 1 h LSV tests, with only a 7% further reduction in the Hupd area from 14 to 13 cmmaterial2 cmgeo−2 in the subsequent measurement (Fig. S18 and S19, ESI†). Based on the changes in the Hupd area during repeated LSV tests, the Ru dissolution in Ru@Ir was speculated to occur in two stages. A large fraction of the uncovered Ru in Ru@Ir was dissolved in the initial few minutes of the LSV tests in water electrolysis. Subsequently, the residual Ru in Ru@Ir covered with IrOx slowly degraded in the repeated LSV tests. Consequently, the total loss of Hupd area for Ru@Ir was more than 66% in the repeated LSV tests. Stability measurements indicated that the steady increase in cell voltage during CC operation (Fig. 2a) was mainly caused by the slow surface passivation of the Ti-PTL and/or catalytic degradation of Ru@Ir. As the performance decrease was not prominent at the beginning of the water electrolysis, any membrane degradation caused by the initial Ru dissolution during long-term operation was not considered the primary reason for the performance decrease of Ru@Ir over 80 h.
In contrast, the Hupd loss of Pt@Ir was less than that of Ru@Ir, with ∼26% of the ECSA reduced in the 1 h LSV tests, that is, from 23 to 17 cmmaterial2 cmgeo−2 (Fig. S18, ESI†). The slow performance degradation of Pt@Ir (Fig. 2a and b) was related to catalytic deactivation, and no changes in HFR were observed via EIS analysis (Fig. 2e). This implied that both Ru@Ir and Pt@Ir suffered from steady loss of catalytic surfaces, with Ru@Ir undergoing an increase in HFR owing to Ru dissolution during operation. Therefore, a method was devised to preserve the Ru interlayer in Ru@Ir during long-term operation to practically employ Ru@Ir in PEMWE cells.
Stability in current swing operation
Ru@Ir exhibited limited stability in CC operation with a steady increase in HFR and catalytic degradation, or loss of ECSA. Moreover, its rate of performance degradation at 2.0 A cm−2 was four times higher that of Pt@Ir. However, the values could be lowered when the HFR changes in CC operation were excluded from the total degradation rate. Therefore, the destruction of corrosion-prone Ru in Ru@Ir had to be mitigated to enable its use in the PEMWE anode.As the destruction of Ru in Ru@Ir proceeded steadily over several tens of hours, a certain degradation route that was related to the increase in HFR resistance during water electrolysis had to be addressed to preserve the electrode structure. If the mechanism of corrosive Ru degradation in the anode can be avoided, it is likely related to any reversible degradation phenomenon associated with water electrolysis. Otherwise, it would be practically impossible to mitigate any degradation mechanism that intrinsically occurs in corrosive materials without addressing their fundamental properties or developing corrosion-resistant materials. In this context, operational variables that could modulate any reversible and long-term degradation routes during water electrolysis were investigated to utilize corrosive Ru and to preserve the initial performance of Ru@Ir, which was comparable to that of Pt@Ir.
Among the various causes of degradation, the continuous accumulation of oxygen gas bubbles at the anode of the water electrolyzer is a major factor on time-scales of several minutes to hours. When these bubbles are inefficiently removed from the electrode, the insulating gas bubbles trapped inside the catalyst layer temporarily inactivate the reaction sites before irreversibly damaging the electrode. However, continuous operation can be achieved if the gas bubbles are appropriately removed from the electrode before they permanently damage it.
In PEMWE operations, gas bubble trapping inside the electrode and the reversible degradation phenomena can be alleviated using methods such as manipulating the operating temperature, pressure, and/or water flow rate, thereby altering the fluid dynamics of the electrode.26 Current swing (CS) operation is an intuitive method for mitigating gas accumulation during water electrolysis. Essentially, water electrolysis is interrupted in a controlled manner in between normal operations, allowing the excess gas inside the electrode to diffuse out during the rest operation. For example, Rakousky et al. demonstrated that CS operation can achieve up to a 90% reduction in the degradation rate by effectively mitigating the corrosion of IrOx and Ti PTL,27 and further emphasized the importance of suppressing Ti PTL oxidation.28 Weiß et al. recommended using values higher than 1.3 VRHE as the lowest anode potential for CS operation; at potentials more negative than 1.3 VRHE, the CS operation evidently accelerates electrode degradation by reducing crystalline IrO2 to hydrous, less-conductive IrOOH with crossed-over H2.29
In this study, CS operation was also conducted to determine if the current interruption mode could curb the dissolution of corrosive Ru in the Ru@Ir anode of the water electrolyzer (Fig. 3a). Specifically, two different current densities—2.0 and 0.1 A cm−2—were chosen for high-current operation (HCO) and low-current operation (LCO), respectively, in the CS operations. The HCO corresponded to typical hydrogen production, whereas the LCO involved interrupting the water electrolysis and removing excess gas from the electrode. The duration of the LCO in between HCOs was varied (1, 3, and 6 h; denoted as CS-1, CS-3, and CS-6, respectively) to investigate the effects of current interruption on the electrode degradation rate. In each measurement, the total HCO duration was fixed at 100 h regardless of the LCO duration, and the total operation time was varied with the LCO mode.
 | ||
| Fig. 3 LT under CS operation, with 2.0 and 0.1 A cm−2 applied in high-current operation (HCO) and low-current operation (LCO), respectively. (a) Schematic illustration of CC and CS operation. CP curves of Ru@Ir in (b) CS-1, (c) CS-3, (d) CS-6, and (e) those of Pt@Ir in CS-6, (f) degradation rates of Ru@Ir and Pt@Ir calculated from voltage variation during HCO, and (g) HFR at 10 kHz. LSV polarization curves of Ru@Ir and Pt@Ir (h) before and (i) after LT, and (j) current density at 2.0 V before and after LT. | ||
The device stability and increase in HFR during the degradation of the interface in Ru@Ir depended on the duration of the low-current CS operation (Fig. 3b–g). As the LCO-related interruption increased from 0 to 6 h (CC and CS-6, respectively), the degradation rate at 2.0 A cm−2 decreased by 67% from 950 to 314 μV h−1 in the long-term stability test conducted for 80–250 h (Fig. 2b and 3f). Notably, the degradation rate or the change in HFR was calculated based on the duration of HCO, excluding that of LCO. Importantly, the HFR of Ru@Ir subjected to CS-6 was maintained at 135 mΩ cm2 for 250 h of operation (Fig. 3g, S20 and S21 ESI†). Consequently, both the performance and HFR changes of Ru@Ir became comparable to those of Pt@Ir in the stability test under CS-6 operation (234 μV h−1).
Additionally, the effectiveness of the CS operation depended considerably on the duration of LCO between the HCOs. When Ru@Ir was subjected to <6 h of LCO, the current interruption effect was significantly less prominent compared to that observed in CS-6. In CS-1 and CS-3, Ru@Ir exhibited performance degradation rates of 700 and 810 μV h−1, respectively; these values were more than 60% higher than that in CS-6 (Fig. 3f). The rate of HFR increase was also notably higher in CS-1 and CS-3 (Fig. 3g and S20, ESI†); therefore, the average ohmic voltage penalty associated with the increase in HFR was 166 and 285 μV h−1 for CS-1 and CS-3, respectively, but 0 μV h−1 for CS-6 (Fig. S22, ESI†). In contrast, the stability of Pt@Ir was not influenced by the use of LCO in the long-term measurements. In the presence and absence of LCO, the degradation rate of Pt@Ir changed by less than 1%, that is, 234 and 236 μV h−1, respectively, confirming the intactness of the Pt interlayer (Fig. 2 and 3e–g).
In the LSV measurements, the device current at 2.0 Vcell marginally increased for Ru@Ir after the long-term stability test in CS-6, as observed for Pt@Ir (Fig. 3h–j). The current density of Ru@Ir increased from 2.6 to 2.7 A cm−2 at 2.0 VCell after 250 h of CS-6, and was comparable with that of Pt@Ir (∼2.6 A cm−2), according to the LSV measurements conducted under appropriate operations. However, the LSV performance of Ru@Ir decreased by ∼16–20% when the LCO was performed for <6 h in the stability test. The current density at 2.0 VCell for Ru@Ir decreased from 2.6 to 2.2 A cm−2 and from 2.6 to 2.1 A cm−2, respectively, in CS-1 and CS-3.
In addition to the duration of LCO, the current applied in LCO was changed from 0.10 A cm−2 to 0.20 or 0.05 A cm−2 to ascertain the effect of current density or potential in CS operation. When the LCO duration was 1 h, no significant changes were observed in the adjusted LCO current, and the total degradation rate at 2.0 A cm−2 was between 610 and 650 μV h−1 regardless of the current level of LCO, with significant increases in HFR also observed (Fig. S23, ESI†). Overall, the stability analysis demonstrated the utility of CS operation with sufficiently long current interruption between normal operation events, thus permitting the use of corrosion-prone Ru in PEMWE anodes.
In addition to device performance, the effectiveness of CS operation on the conservation of the Ru@Ir structure was also explored using physical characterization techniques including cross-sectional EDS, XPS, and ECSA analyses. Cross-sectional HAADF-STEM and EDS images of Ru@Ir were obtained to compare the morphological and compositional changes after CC and CS-6 operations (Fig. 4a and b). Notably, only parts of Ru@Ir that remained on the Ti-PTL were analyzed by STEM, and part of the Ir catalyst layer was decal-transferred to the ion-conducting membrane during the disassembly of the MEA after the long-term stability test (Fig. S24, ESI†). The cross-sectional images of Ru@Ir indicated that the thickness of the Ru interlayer in Ru@Ir decreased after both CC and CS-6 operations, from ∼200 nm to less than 50 nm; this was not relevant to the variations in device performances or degradation rates. However, more cracks and pores in Ru and Ir layers were visible in Ru@Ir subjected to CC operation, whereas the structure remained comparatively undamaged after CS-6 operation (Fig. 4a, b and S25, ESI†). Importantly, the oxidation states of Ru and Ir after the long-term stability tests were significantly affected by the LCO, as revealed by XPS analysis (Fig. 4c, d and S26, ESI†). The amount of metallic Ru(0) decreased from 33% to 6% after CC operation at 2.0 A cm−2 for 80 h, whereas it gradually increased to 24% after CS-6 operation for 250 h. Similarly, catalytic IrOx exhibited a less oxidized state in CS operation; therefore, the content of metallic Ir(0) increased from 14% to 25% after CC and CS-6 operations, respectively. This tendency was corroborated by ECSA analysis, which accounted for the remaining metallic Ir (Fig. S27–32, ESI†).
 | ||
| Fig. 4 Physicochemical properties of the synthesized catalysts. HAADF-STEM image and elemental mapping image of the Ru@Ir electrode after (a) CC and (b) CS-6 operations (regions marked with dashed lines indicate holes in the catalyst layer; yellowish green: Ir, sky blue: Ru, and gray: Ti). XPS-derived deconvoluted area ratio of (c) Ru and (d) Ir in Ru@Ir, and (e) that of Ir in Pt@Ir. All data were obtained from the Ti-PTL side detached from the MEA. | ||
The results of the STEM, XPS, and ECSA analyses suggested that LCO for 6 h effectively reduced the oxidation state of Ru and Ir in the Ru@Ir electrodes under CS operation. Evidently, the reduced oxidation state of Ru was vital for explaining the structural integrity of stable Ru@Ir after CS-6 operation, whereas the oxidation state of Ir was irrelevant to the Ru interlayer stability or HFR changes after the stability tests. For example, the IrOx catalyst in Pt@Ir exhibited significantly different oxidation states owing to the effects of the LCO, similar to Ru@Ir (Fig. 4e and S33, ESI†); however, the degradation rate or stability was not influenced (Fig. 2, 3e and 3f). The dissolution of RuOx occurs via overoxidation to Ru(8+) at positive potentials above 1.387 VRHE, leading to the formation of soluble RuO4 (RuO2 + 2H2O → RuO4(aq) + 4H+ + 4e−).15 This implies that the high content of the Ru(4+) state in Ru@Ir increases the chance of its dissolution through electrochemical oxidation. The CS-6 operation reduced the oxidation state of Ru in Ru@Ir or mitigated the destruction of the Ru interlayer and the increase in HFR of Ru@Ir, via two routes. As the LCO interrupted the O2 evolution at the anode while allowing a certain amount of the H2 produced at the cathode to diffuse to the anode, the crossed-over H2 at the anode reduced the Ru in Ru@Ir during the LCO.29 Additionally, the degradation caused by the excessive accumulation of O2 bubbles at the anode was alleviated using appropriate LCOs, which allowed the bubbles to diffuse out of the cell (Fig. S34, ESI†). Given that the O2 bubbles insulated part of the electrode surface, the local overpotential and temperature around the area covered with the bubbles increased, accelerating the degradation reaction.20–22 Although it was difficult to separate the effects of the different Ru degradation mechanisms in CS operation, the physical and chemical changes observed in Ru@Ir demonstrated the appropriate use of CS operation to improve the stability of PEMWE devices with corrosion-prone Ru anodes.
Using a model electrode prepared on a plain Ti electrode, the relationship between the local degradation of Ru and Ir in Ru@Ir and bubble formation over the OER electrode was studied (Fig. 5, S35 and S36, ESI†). The local composition of Ru@Ir formed on a flat electrode measuring 1.5 × 2.0 mm2 was systematically examined by compartmentalizing the electrode into a 4 × 6 matrix (Fig. S37, ESI†). The atomic composition of Ru@Ir was quantified by EDS before and after 50 h of CP at a constant oxidation current of 5 mA cm−2.
 | ||
| Fig. 5 Model study on nonuniform catalyst dissolution due to stagnant bubbles. (a) Images of the Ru@Ir electrode deposited on Ti foil after 5, 10, 25, and 50 h of CP (dashed lines indicate areas where bubbles commonly existed over several tens of hours), and (b) 3D bar graphs showing the atomic percentage ratio of Ru and Ir, calculated by dividing the values obtained after the test by those before the test. | ||
During CP tests, transient bubbles generated and removed over the flat electrode were photographed to compare the local compositional changes in Ru@Ir and the area covered by the bubbles attached to the surface. The results showed that both Ru and Ir in Ru@Ir degraded severely after 50 h of CP. On average, 95% and 40% of Ru and Ir, respectively, dissolved after long-term electrolysis, similar to the dissolution observed at the Ti-PTL electrode. However, the degradation of Ru and Ir occurred nonuniformly over the electrode surface in the model study. As the OER proceeded at a constant current density for over 50 h, the produced O2 bubbles accumulated and stagnated mainly at the right and bottom edges (Fig. 5a and S38, ESI†). Interestingly, the area uncovered by the O2 bubbles showed more significant dissolution of both Ru and Ir, particularly around the left section of the compartmented matrix (Sections 1-1, 1-2, 1-3, 1-4, 2-1, 2-3, and 2-4); however, the right and bottom edges of the electrode covered by large O2 bubbles showed relatively higher portions of Ru and Ir retained through the electrolysis (Fig. 5b and S39, ESI†). These results indicated that the electrode degradation was related to the local O2 transport dynamics; essentially, the degradation was not directly accelerated by the attached O2 bubbles, but by the nonuniformly distributed electrode potential and/or temperature caused by the O2 bubbles. The potential and/or temperature of the active electrode around the inactive O2-covered surface could be increased using high local currents; therefore, the degradation of Ru and Ir can intensify at these spots.21,22 Essentially, CS operation, or sufficient LCOs in between HCOs, can decelerate the degradation of corrosion-prone metals in PEMWE anodes by generating uniformly distributed local potentials, current densities, and temperatures without excessive accumulation of gas bubbles. The model study of Ru@Ir strongly suggests using operational strategies to avoid the harsh oxidative environment at electrodes, enabling the use of inexpensive, corrosion-prone materials as electrode components of PEMWE cells.
Conclusions
To replace the expensive Pt interlayer in PEMWE anodes while enhancing the IrOx utilization for water oxidation, corrosive Ru was used in this study to form an interlayer between the Ti-TPL and the IrOx catalyst. The Ru interlayer in Ru@Ir provided sufficient electrical and thermal conductivity and a large surface area to efficiently support the IrOx catalyst for PEMWE. The optimized Ru@Ir specimen exhibited comparable OER performance to Pt@Ir in PEMWE device operation, that is, 5.5 A cm−2 at 2.0 VCell with less than 80 μg cm−2 of Ir usage. Furthermore, investigations were conducted to permit the use of Ru in PEMWE devices by addressing its tendency to rapidly over-oxidize to RuO42− or H2RuO5 in CC operation. To avoid the harsh oxidative environment at the electrodes and Ru degradation, CS operation was found to be effective for utilizing corrosion-prone Ru in Ru@Ir for PEMWE. In particular, LCO duration served as a critical factor in CS operation, likely due to its role in facilitating bubble removal during low-current intervals. Using the current interruption method in PEMWE device operation, the stability of Ru in the PEMWE anode was demonstrated for more than 250 h; moreover, the degradation rate at 2.0 A cm−2 was reduced by 67% from 950 to 314 μV h−1 in long-term stability tests conducted for 80–250 h. Consequently, the device with a Ru interlayer in the anode exhibited comparable performance and durability to a conventional PEMWE device with a Pt interlayer in the anode. These findings underscore the importance of developing strategies to enable the use of cost-effective but corrosion-prone materials as electrode components in future PEMWE systems. Given that corrosion-prone materials are more susceptible to dissolution in the OER environment, reducing the oxidation states of the catalyst through CS operation could be an effective strategy for enhancing durability. Moreover, the findings indicate that the accumulation of oxygen gas bubbles can accelerate Ru dissolution. Therefore, integrating CS operation with material modifications offers a promising approach to improving the durability of Ru-based cells. Additionally, exploring whether this method can enhance the stability of other highly corrosion-prone materials, such as Ni, Co, and Fe, would be a valuable direction for future research.Experimental section
Chemicals
Ru(III) chloride hydrate (RuCl3·xH2O; ≥99.98%), chloroplatinic acid hydrate (H2PtCl6·xH2O; ≥99.9%), potassium hexachloroiridate(III) (K3IrCl6; 35.5–38.1% Ir), sodium sulphate (Na2SO4; ≥99.0%), perchloric acid (HClO4; 69.0–72.0%), and sulfuric acid (95.0–98.0%) were purchased from Sigma-Aldrich. Iridium(IV) oxide (IrO2; 99.99% [metals basis], Ir 84.5% min, Alfa Aesar), isopropyl alcohol (IPA; 99.5%, Duksan), oxalic acid dihydrate (HO2CCO2H·2H2O; 99.5–100.2%, Daejung), and Pt/C powder (46.7 wt%, Tanaka K.K.)—all of which were analytical grade—were used as received without further purification.Electrode preparation
The electrode fabrication protocol was derived from those reported previously.6,23,25 To fabricate the anode, Ru, Pt, or Ir was electrodeposited on a 250 μm-thick Ti felt with 20-μm-diameter fibers (60% porosity, 2 GDL9N-025, Bekaert) in a three-electrode configuration. For electrodeposition, a Pt plate and a saturated calomel electrode (SCE) were used as the counter and reference electrodes, respectively. Prior to electrodeposition, the Ti felt was cleaned using 5 wt% oxalic acid at 60 °C for 30 min. The Ru interfacial layer in Ru@Ir was formed via electrodeposition in a 15 mM aqueous RuCl3 solution dissolved in 0.25 M perchloric acid. An electrochemical pulse sequence involving current densities of −55 mA cm−2 (0.05 s) and 20 mA cm−2 (0.05 s) was repeated 120 times (a total of 12 s) to create Ru seeds. Subsequently, a constant potential of −2.5 VSCE was applied for various deposition times (1, 2, 5, and 10 min) at room temperature. This was followed by Ir deposition in a 3 mM aqueous K3IrCl6 solution dissolved in 0.5 M Na2SO4 at pulsed potentials of 0.4 VSCE (2 s), −0.65 VSCE (10 s), and 0 VSCE (2 s), repeated for 30 cycles at 70 °C. To prepare the Pt@Ir anode, Pt was deposited on the Ti-PTL in a 20 mM aqueous H2PtCl6 solution dissolved in 0.5 M sulfuric acid at a constant current density of −10 mA cm−2 for 5 min at room temperature. The cathode for the HER was prepared by spraying 0.5 mg cm−2 Pt/C onto carbon paper (39BB, Sigracet). The catalyst ink slurry contained Pt/C powder (46.7 wt% Pt, Tanaka K.K.) suspended in a 5 wt% Nafion solution, deionized water, and IPA. For comparison, a commercial IrOx electrode was prepared by spraying commercial IrOx on a Ti felt using the catalyst powders suspended in a 5 wt% Nafion solution, deionized water, and IPA.Construction of membrane electrode assembly
A membrane electrode assembly (MEA) with a 1 cm2 geometric area was fabricated using a hot press (120 °C, 395 psi, 1 min) to create a sandwiched structure comprising proton-conducting membranes (50.8 μm-thick NR-212 or 127 μm-thick N-115, Dupont™) between the anode and cathode.Electrochemical analysis
Electrochemical measurements were performed at room temperature using a research-grade potentiostat (SP-300, BioLogic). Half-cell reactions were conducted in a conventional three-electrode system. For full-cell measurements, a home-built MEA was placed between two current collectors with a serpentine flow channel. Ti and graphite blocks were used as current collectors for the anode and cathode, respectively. A torque of 80 lb·in was applied to the cell assembly to prevent gas and electrolyte leakage through the layers. During water electrolysis, the cell temperature was maintained at 80 °C, and deionized water was supplied to the anode at a volumetric flow rate of 15 mL min−1. Linear sweep voltammetry (LSV) and electrochemical impedance spectroscopy (EIS) were performed using a high-current potentiostat (HCP-803, BioLogic). LSV curves were obtained by averaging the current density measured for 30 s at applied voltages of 1.3–2.0 Vcell with a 50 mV interval. EIS was performed at a cell voltage of 2.0 Vcell and AC frequencies of 50 kHz to 50 mHz.To acquire CV curves in full-cell settings, the cell voltage was controlled from 0.05 to 0.60 Vcell, or from 0.40 to 0.60 VCell, using the Pt/C electrode as the pseudo-reversible hydrogen electrode (pseudo-RHE) and counter electrode, with the Ir (or IrOx) electrode serving as the working electrode, at scan rates of 20, 50, 100, and 200 mV s−1 in a humidified hydrogen gas atmosphere (200 mL min−1) for the Pt/C electrode and in deionized water (15 mL min−1) for the IrOx electrode. From the CV data of the MEA, the electrochemically active surface area (ECSA) was estimated using the hydrogen underpotential deposition (Hupd) and double-layer charging current (Cdl). The Hupd area was calculated using eqn (1). In this equation, the first term represents the total charge of the Hupd peak, while the second term is a material-specific constant corresponding to the charge required for hydrogen adsorption per unit surface area. The Hupd charge density for polycrystalline Ru, Pt, and Ir was assumed to be 310, 210, and 218 μC cm−2, respectively.30,31 Cdl (mF) was simply calculated as the charging current difference in the forward and backward CV scans at 0.5 V (vs. pseudo-RHE). Cdl was used to compare the total conducting electrode area of the samples.
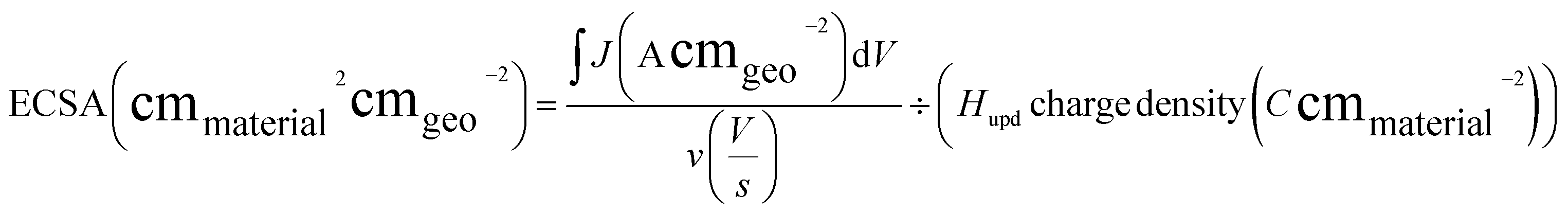 | (1) |
Characterization
The morphology and surface characteristics of the electrodes were investigated by field-emission scanning electron microscopy (FE-SEM; Teneo Volume Scope, Field Emission Inc.), focused ion beam-scanning electron microscopy (FIB-SEM; Hitachi-NX5000), high-resolution transmission electron microscopy (HRTEM; Talos, Titan), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), energy-dispersive X-ray spectrometry (EDS), X-ray diffractometry (XRD; Rigaku, 35 kV, 35 mA), and X-ray photoelectron spectroscopy (XPS; Thermo Scientific) with a soft X-ray source (Al Kα, 1486.6 eV). Electron probe microanalysis (EPMA) was conducted to examine the elements in a cross-section of the MEA after long-term OER performance tests, with the MEA cross-section obtained by embedding the electrode in a room-temperature-curing epoxy. The oxygen affinity of the electrodes was investigated by conducting temperature-programmed oxidation (TPO) and temperature-programmed desorption (TPD) experiments (Autochem 2920, Micromeritics). All samples were pretreated with Ar prior to the measurements. In the TPO analysis, oxygen was supplied as the temperature was gradually increased from 50 to 1000 °C. In the TPD analysis, oxygen was pre-adsorbed at 50 °C for 10 h and subsequently desorbed by increasing the temperature from 50 to 1000 °C. For the TPO and TPD experiments, the ramp rate was 10 °C min−1 and the flow rate of 5% O2 in Ar gas was maintained at 300 mL min−1. The catalyst composition was analyzed by inductively coupled plasma-optical emission spectroscopy (ICP-OES; Optima 7300DV, PerkinElmer) and inductively coupled plasma mass spectrometry (ICP-MS; NexION2000, PerkinElmer). All samples were dissolved via microwave digestion with aqua regia.Data availability
Crystallographic data for Ti, Ru, Ir, and Pt have been deposited in the JCPDS under accession numbers 44-1294, 06-0663, 46-1044, and 04-0802. Microscopy data, including SEM, TEM, and HAADF-STEM, have been provided as raw, unedited files in a high-resolution image format, available in both the manuscript and the ESI.† The data supporting this article have been included as part of the ESI.†Author contributions
S. L. contributed the concept and carried out the majority of the electrode fabrication, cell testing, characterization, and data analysis. C. Y., and J. C. conducted supplementary experiments. J. H. J assisted in the data analysis. H. N. J., Y.-E. S., and H. S. P. contributed to discussions about the data and played a significant role in revising the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Creative Materials Discovery Program through the National Research Foundation of Korea (RS-2024-00466554), the Ministry of Trade, Industry & Energy of Korea (MOTIE-20018989) and the Korea Institute of Science and Technology.Notes and references
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934, DOI:10.1016/j.ijhydene.2013.01.151.
- M. Clapp, C. M. Zalitis and M. Ryan, Catal. Today, 2023, 420, 114140, DOI:10.1016/j.cattod.2023.114140.
- C. V. Pham, D. Escalera-López, K. Mayrhofer, S. Cherevko and S. Thiele, Adv. Energy Mater., 2021, 11, 2101998, DOI:10.1002/aenm.202101998.
- S. H. Frensch, F. Fouda-Onana, G. Serre, D. Thoby, S. S. Araya and S. K. Kær, Int. J. Hydrogen Energy, 2019, 44, 29889–29898, DOI:10.1016/j.ijhydene.2019.09.169.
- M. Yasutake, Z. Noda, J. Matsuda, S. M. Lyth, M. Nishihara, K. Ito, A. Hayashi and K. Sasaki, J. Electrochem. Soc., 2023, 170, 124507, DOI:10.1149/1945-7111/ad1165.
- A. Lim, J. Kim, H. J. Lee, H.-J. Kim, S. J. Yoo, J. H. Jang, H. Y. Young Park, Y.-E. Sung and H. S. Park, Appl. Catal., B, 2020, 272, 118955, DOI:10.1016/j.apcatb.2020.118955.
- H.-Y. Jeong, J. Oh, G. S. Yi, H.-Y. Park, S. K. Cho, J. H. Jang, S. J. Yoo and H. S. Park, Appl. Catal., B, 2023, 330, 122596, DOI:10.1016/j.apcatb.2023.122596.
- C. Rakousky, U. Reimer, K. Wippermann, M. Carmo, W. Lueke and D. Stolten, J. Power Sources, 2016, 326, 120–128, DOI:10.1016/j.jpowsour.2016.06.082.
- J. Z. Y. Seow and T. D. Nguyen, Electrochim. Acta, 2020, 341, 136058, DOI:10.1016/j.electacta.2020.136058.
- N. F. Daudt, A. D. Schneider, E. R. Arnemann, C. J. Scheuer, L. S. Dorneles and L. F. Schelp, J. Mater. Eng. Perform., 2020, 29, 5174–5183, DOI:10.1007/s11665-020-05026-y.
- J. C. Bailar Jr, H. J. Emeleus, R. Nyholm and A. F. Trotman-Dickenson, Comprehensive Inorganic Chemistry, Pergamon Press, 1975 Search PubMed.
- R. A. Matula, J. Phys. Chem. Ref. Data, 1979, 8, 1147–1298, DOI:10.1063/1.555614.
- Umicore, Precious metals management, https://pmm.umicore.com/en/, accessed February 2024.
- R. W. Powell, R. P. Tye and M. J. Woodman, J. Less Common. Met., 1967, 12, 1–10, DOI:10.1016/0022-5088(67)90062-8.
- M. J. N. Pourbaix, J. Van Muylder and N. de Zoubov, Platin. Met. Rev., 1959, 3, 47–53, DOI:10.1595/003214059X324753.
- S. Hao, M. Liu, J. Pan, X. Liu, X. Tan, N. Xu, Y. He, L. Lei and X. Zhang, Nat. Commun., 2020, 11, 5368, DOI:10.1038/s41467-020-19212-y.
- L. An, F. Yang, C. Fu, X. Cai, S. Shen, G. Xia, J. Li, Y. Du, L. Luo and J. Zhang, Adv. Funct. Mater., 2022, 32, 2200131, DOI:10.1002/adfm.202200131.
- Y. Lin, Z. Tian, L. Zhang, J. Ma, Z. Jiang, B. J. Deibert, R. Ge and L. Chen, Nat. Commun., 2019, 10, 162, DOI:10.1038/s41467-018-08144-3.
- H. Jin, S. Choi, G. J. Bang, T. Kwon, H. S. Kim, S. J. Lee, Y. Hong, D. W. Lee, H. S. Park, H. Baik, Y. Jung, S. J. Yoo and K. Lee, Energy Environ. Sci., 2022, 15, 1119–1130, 10.1039/D1EE02636D.
- A. Angulo, P. van der Linde, H. Gardeniers, M. Modestino and D. F. Fernández Rivas, Joule, 2020, 4, 555–579, DOI:10.1016/j.joule.2020.01.005.
- J. Dukovic and C. W. Tobias, J. Electrochem. Soc., 1987, 134, 331–343, DOI:10.1149/1.2100456.
- H. H. Kellogg, J. Electrochem. Soc., 1950, 97, 133–142, DOI:10.1149/1.2777980.
- D. K. Oppedisano, L. A. Jones, T. Junk and S. K. Bhargava, J. Electrochem. Soc., 2014, 161, D489–D494, DOI:10.1149/2.0441410jes.
- S. H. Park and Y. J. Lee, J. Mater. Chem. A, 2021, 9, 1803–1811, 10.1039/D0TA10006D.
- H. Kim, J. Kim, J. Kim, G. H. Han, W. Guo, S. Hong, H. S. Park, H. W. Jang, S. Y. Kim and S. H. Ahn, Appl. Catal., B, 2021, 283, 119596, DOI:10.1016/j.apcatb.2020.119596.
- L. Guo, Y. Liu, P. Ran, G. Wang, J. Shan, X. Li, C. Liu and J. Li, Microsyst. Nanoeng., 2022, 8, 34, DOI:10.1038/s41378-022-00367-1.
- C. Rakousky, U. Reimer, K. Wippermann, S. Kuhri, M. Carmo, W. Lueke and D. Stolten, J. Power Sources, 2017, 342, 38–47, DOI:10.1016/j.jpowsour.2016.11.118.
- C. Rakousky, G. P. Keeley, K. Wippermann, M. Carmo and D. Stolten, Electrochim. Acta, 2018, 278, 324–331, DOI:10.1016/j.electacta.2018.04.154.
- A. Weiß, A. Siebel, M. Bernt, T.-H. Shen, V. Tileli and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, F487–F497, DOI:10.1149/2.0421908jes.
- D. Michell, D. A. J. Rand and R. Woods, J. Electroanal. Chem. Interfacial Electrochem., 1978, 89, 11–27, DOI:10.1016/S0022-0728(78)80027-8.
- J. Zheng, W. Sheng, Z. Zhuang, B. Xu and Y. Yan, Sci. Adv., 2016, 2, e1501602, DOI:10.1126/sciadv.1501602.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01168j |
| This journal is © The Royal Society of Chemistry 2025 |