
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic Fe,Pd diatomic sites anchored on porous nitrogen-doped carbon for efficient oxygen reduction in the entire pH range†
Yejung Choi‡
ab,
Byoung Joon Park‡c,
Yechan Leeb,
Kug-Seung Lee *d,
Jin Kon Kim*ab and
Jeong Woo Han*c
*d,
Jin Kon Kim*ab and
Jeong Woo Han*c
aNational Creative Research Initiative Center for Hybrid Nano Materials by High-level Architectural Design of Block Copolymer, Republic of Korea
bDepartment of Chemical Engineering, Pohang University of Science and Technology (POSTECH), Pohang, Gyeongbuk 37673, Republic of Korea. E-mail: jkkim@postech.ac.kr
cDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: jwhan98@snu.ac.kr
dBeamline Division, Pohang Accelerator Laboratory, Pohang, Gyeongbuk 37673, Republic of Korea. E-mail: lks3006@postech.ac.kr
First published on 6th May 2025
Abstract
Diatomic catalysts (DACs) have shown great potential for the oxygen reduction reaction (ORR). However, designing highly active and durable DACs that perform efficiently across the entire pH range remains a significant challenge. In this work, we introduce a novel Fe,Pd diatomic catalyst (Fe,Pd–NC), in which Pd serves as an effective electronic modulator adjacent to Fe, supported on a porous nitrogen-doped carbon matrix. By optimizing the electronic environment and enhancing the accessibility of Fe,Pd diatomic sites, Fe,Pd–NC demonstrates outstanding ORR activity across all pH conditions, achieving half-wave potentials of 0.884 V, 0.782 V, and 0.781 V in alkaline, neutral, and acidic media, respectively. Furthermore, Fe,Pd–NC exhibits exceptional durability across the pH spectrum. When incorporated into zinc–air batteries (ZABs) operating in alkaline and neutral electrolytes, the Fe,Pd–NC catalyst delivers superior power density, specific capacity, and long-term stability, outperforming conventional Pt/C-based ZABs. This study presents an effective design strategy for DACs with universal pH adaptability, offering promising applications in energy conversion and storage devices.
1. Introduction
The growing global energy demand and worsening environmental crisis have intensified research efforts toward sustainable energy conversion and storage technologies.1 The oxygen reduction reaction (ORR) plays a crucial role in electrochemical devices operating across diverse pH environments, including proton-exchange membrane fuel cells (PEMFCs, acidic medium), microbial fuel cells (MFCs, neutral medium), and zinc–air batteries (ZABs, alkaline medium).2–5 While platinum (Pt)-based catalysts have exhibited excellent ORR performance across a wide pH range, their high cost and scarcity hinder large-scale implementation.6,7 To overcome these limitations, significant progress has been made in developing metal–nitrogen-doped carbon (M–NC) catalysts, which offer advantages such as maximized atomic efficiency, uniform active sites, and high tunability in their coordination environment.8–10 Among them, Fe–NC catalysts have emerged as the most promising alternatives to Pt-based catalysts in alkaline media.11–13 However, their performance under neutral and acidic conditions is still low, due to poor activity and rapid deactivation.14–16 Therefore, enhancing the ORR activity and stability of Fe–NC catalysts across a broad pH range is highly desirable but remains a significant challenge.Morphological engineering has emerged as an effective strategy for enhancing catalytic performance.17,18 Although diverse porous structures have been introduced using hard templates and soft templates,19–21 these conventional methods need time-consuming processes and the use of corrosive solvents for template removal. To overcome these limitations, salt-assisted templating has garnered significant attention due to its non-toxicity, ease of removal, and low cost.22,23 Salts such as NaCl and ZnCl2 have been widely used as templates to create meso- and microporous structures in carbon substrates during high-temperature heat treatment.22–24 These porous structures increase the number of accessible Fe–Nx active sites and improve mass transport, thereby enhancing catalytic efficiency. However, despite these advancements, the intrinsic properties of Fe—particularly its strong binding affinity for oxygen intermediates—continue to pose challenges in further improving catalytic performance.25,26
A widely adopted strategy for tuning the intrinsic properties of Fe–NC catalysts is the construction of diatomic catalysts (DACs) by incorporating a secondary metal center.27,28 Adjacent secondary metals can modulate the local electronic environment of Fe through a long-range interaction (LRI), optimizing oxygen adsorption/desorption behavior and enhancing ORR activity.29–31 Recent efforts have focused on incorporating 3d transition metals such as Co, Cu, and Zn with Fe to develop efficient DACs for broad pH applications.32–34 However, due to their similar electronegativity to Fe, these metals offer limited electronic modulation, resulting in only marginal activity improvements, particularly in neutral media.35 In contrast, the 4d transition metal Pd exhibits a significantly higher electronegativity difference and possesses a fully filled 4d10 electronic configuration, allowing it to effectively modulate the local electronic environment around Fe sites and enhance ORR activity. Additionally, Pd demonstrates greater stability than 3d transition metals in acidic environments and exhibits strong resistance to impurities.36,37 Despite their promising attributes, Pd-based DACs remain largely unexplored. Therefore, incorporating Pd as a secondary metal presents a promising strategy for designing DACs with high activity and durability across the entire pH range.
In this work, we present a novel diatomic catalyst with atomically dispersed Fe and Pd sites on porous nitrogen-doped carbon (Fe,Pd–NC), synthesized using NaCl as a template. X-ray photoelectron spectroscopy (XPS) and X-ray absorption fine structure (XAFS) analyses confirm that adjacent Pd effectively modulates the electronic environment of Fe through LRI. As a result, the Fe,Pd–NC catalyst exhibits exceptional ORR activity across the entire pH spectrum, achieving half-wave potentials of 0.884 V, 0.782 V, and 0.781 V under alkaline, neutral, and acidic conditions, respectively. Furthermore, Fe,Pd–NC demonstrates remarkable durability in all electrolytes, surpassing Fe–NC in long-term stability. We also elucidate the role of Pd in enhancing the durability of Fe,Pd–NC. When incorporated into ZABs operating in alkaline and neutral electrolytes, Fe,Pd–NC delivers superior power density, specific capacity, and long-term durability compared to commercial Pt/C catalysts. These findings highlight Fe,Pd–NC as a promising candidate for energy conversion and storage applications across a broad pH range.
2. Results and discussion
Fig. 1a illustrates the schematic representation of the Fe,Pd–NC catalyst synthesis process. Initially, the Pd-based zeolitic imidazolate framework-8 (Pd-ZIF-8) precursor was synthesized by incorporating Pd2+ ions during the ZIF-8 formation. The Pd-ZIF-8 precursor was then physically mixed with NaCl, which served as a template to increase the accessible surface area of the catalyst. Subsequent pyrolysis at 900 °C under an Ar atmosphere converted Pd-ZIF-8 into porous Pd–NC. Above its melting point (∼800 °C), the molten NaCl penetrated into the Pd-ZIF-8 precursor through the capillary effect, facilitating the formation of meso- and macropores in the obtained Pd–NC.38–40 After removing residual NaCl, the porous Pd–NC was used as a host for Fe3+ ion impregnation. A final thermal activation at 900 °C induced the formation of Fe–N active sites, yielding the Fe,Pd–NC catalyst with highly accessible Fe and Pd diatomic sites. For comparison, Fe–NC and Pd–NC catalysts were synthesized following similar procedures but without the introduction of Pd and Fe, respectively.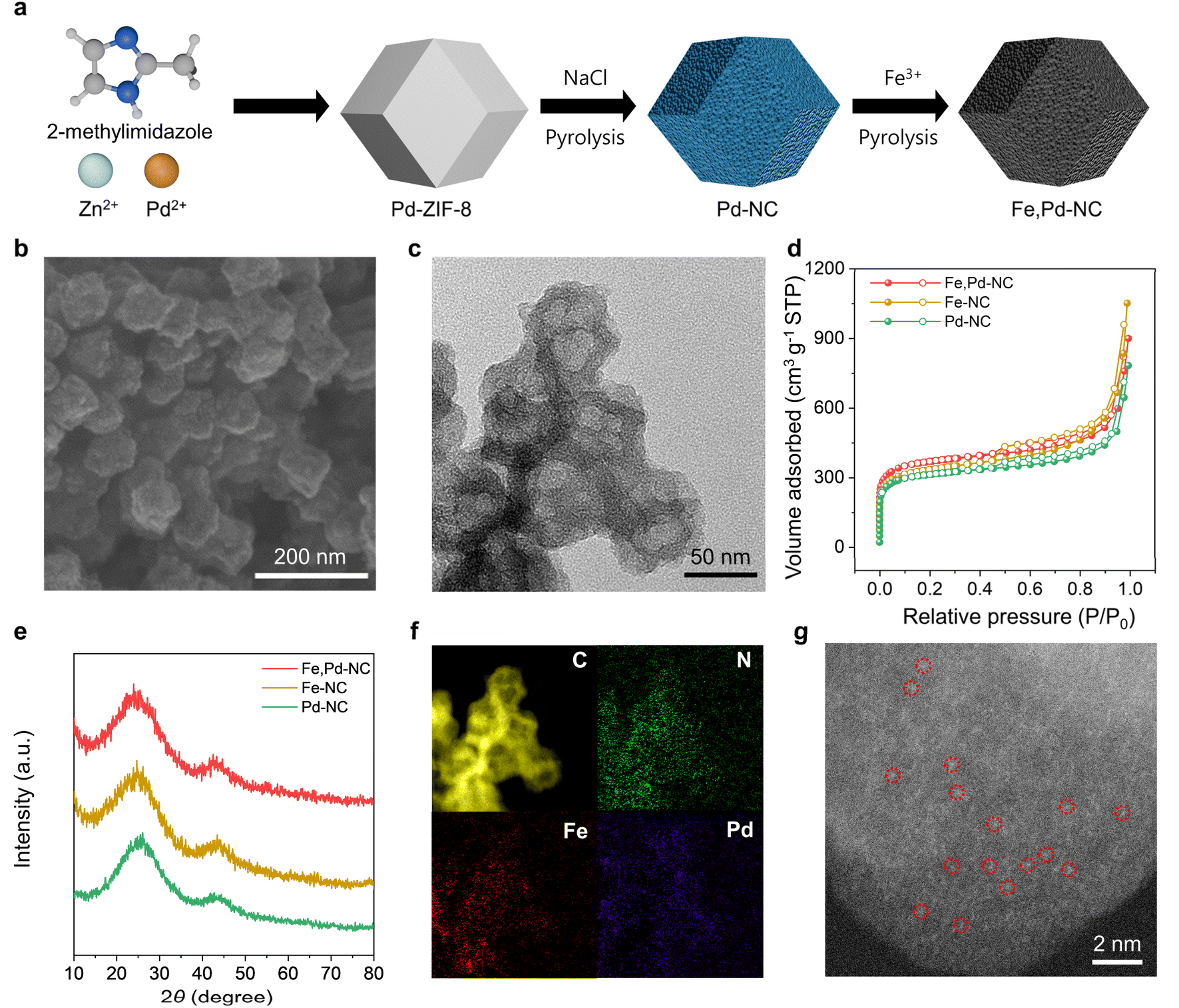 | ||
| Fig. 1 (a) Schematic of the synthesis process of Fe,Pd–NC. (b) FE-SEM and (c) HR-TEM images of Fe,Pd–NC. (d) Nitrogen adsorption–desorption isotherms and (e) XRD spectra of Fe,Pd–NC, Fe–NC, and Pd–NC. (f) EELS mapping and (g) HAADF-STEM image of Fe,Pd–NC. | ||
The morphological characteristics of the synthesized catalysts were examined using field emission scanning electron microscopy (FE-SEM) and high-resolution transmission electron microscopy (HR-TEM). All M–NC catalysts exhibited highly porous rhombic dodecahedral structures, confirming that the NaCl templates induced high porosity (Fig. 1b, c and S1†). To further analyze the porosity of the catalysts, N2 adsorption–desorption tests were performed. The M–NC catalysts demonstrated high BET surface areas ranging from 1150 to 1350 m2 g−1 and exhibited similar adsorption–desorption behavior (Fig. 1d and Table S1†). A sharp increase at low relative pressures (P/P0 = 0–0.015) indicated the presence of micropores.41–43 Additionally, the hysteresis loop observed at higher pressures (P/P0 = 0.4–0.8) and the sharp increase at P/P0 = 0.8–1.0 confirmed the existence of mesopores and macropores, respectively.41,44 From the pore size distribution, the M–NC catalysts possessed a hierarchical pore structure, with abundant meso/macropores (60–70% of the total pore volume) (Fig. S2†). Micropores are expected to enhance active site exposure by increasing the specific surface area, while mesopores and macropores facilitate the efficient mass transport of reactants and electrolytes.45,46 Raman spectroscopy was conducted to investigate the graphitization degree of the prepared catalysts (Fig. S3†). All M–NC catalysts exhibited a similar intensity ratio between the D peak at 1350 cm−1 and the G peak at 1590 cm−1, indicating comparable carbon disorder properties. XRD analysis revealed two broad peaks at 25° and 44°, corresponding to the (002) and (101) planes of amorphous carbon, suggesting that Fe and Pd did not form metallic aggregates but were instead atomically dispersed within the porous carbon matrix (Fig. 1e). Furthermore, electron energy loss spectroscopy (EELS) mapping (Fig. 1f) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images (Fig. 1g) confirmed the uniform distribution of single atomic Fe and Pd within the N-doped porous carbon framework of Fe,Pd–NC. The contents of Fe and Pd in Fe,Pd–NC were 0.48 wt% and 1.07 wt%, respectively, measured by inductively coupled plasma-atomic emission spectrometry (ICP-AES) (Table S2†).
The chemical states and composition of the Fe,Pd–NC catalysts were examined using X-ray photoelectron spectroscopy (XPS). As shown in Fig. S4a,† the XPS survey spectra confirmed the presence of C, N, O, Fe, and Pd elements in Fe,Pd–NC. The atomic contents of Fe and Pd were measured to be 0.10 and 0.13 at%, respectively, aligning well with the ICP-AES results (Table S3†). The Fe 2p and Pd 3d spectra revealed that Fe existed in a mixed oxidation state of Fe2+ and Fe3+ (Fig. 2a), while Pd was present in the Pd2+ state (Fig. 2b) following the formation of Fe–N and Pd–N bonds.47–49 Notably, the Fe 2p3/2 peak of Fe,Pd–NC shifted to a higher binding energy compared to Fe–NC, whereas the Pd 3d3/2 peak exhibited a shift toward a lower binding energy. This suggests electron transfer from Fe to Pd in Fe,Pd–NC due to the higher electronegativity of Pd, potentially modifying the electronic environment of the Fe sites. The N 1s spectra were deconvoluted into pyridinic (398.3 eV), pyrrolic (399.3 eV), graphitic (400.7 eV), and oxidized pyridinic (402.3 eV) nitrogen (Fig. S4b†).50,51 Among these, pyridinic N, one of the dominant species, plays a key role in facilitating the formation of Fe–N and Pd–N active sites.52 Additionally, no characteristic peaks of metal oxides or metal carbides were detected in the O 1s and C 1s spectra, confirming that Fe and Pd exist exclusively as single atoms coordinated with N (Fig. S4c and d†). Fe–NC and Pd–NC exhibited similar chemical states to Fe,Pd–NC, differing only in the metal species present. To further investigate the electronic structure and local coordination environments, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analyses were performed. In the Fe K-edge XANES spectra, the absorption edge of Fe,Pd–NC was positioned near that of Fe2O3, indicating that Fe atoms in Fe,Pd–NC have an oxidation state slightly lower than Fe3+ (Fig. 2c). The observed shift in the absorption edge of Fe,Pd–NC compared to Fe–NC suggests a slight increase in the oxidation state, consistent with the XPS findings. The k3-weighted Fourier transform (FT)-EXAFS spectra of the Fe K-edge displayed a single dominant peak at 1.49 Å, corresponding to Fe–N coordination (Fig. 2d).53 XANES and FT-EXAFS analysis of the Pd K-edge confirmed that Pd in Fe,Pd–NC remained in a Pd2+ oxidation state, with predominant Pd–N bonding (Fig. S5†).54 Importantly, no metal–metal bond peaks were detected in the higher R-region of the Fe and Pd K-edge FT-EXAFS spectra, confirming the absence of metallic aggregates. Wavelet transform (WT)-EXAFS analysis was conducted to distinguish the backscattering atoms using effective resolution in both k- and R-spaces (Fig. 2e). The WT-EXAFS contour plots of Fe,Pd–NC exhibited a primary peak near 5 Å−1, corresponding to Fe–N or Pd–N bonds.55–57 In contrast, distinct features observed in Fe2O3 and PdO further confirmed that Fe and Pd in Fe,Pd–NC exist exclusively as nitrogen-coordinated species. Moreover, quantitative EXAFS fitting was conducted to gain more insights into the local structure of the dual active sites (Fig. 2f), with the fitting parameters summarized in Table S4.† The best-fitting results revealed that the coordination number for both Fe and Pd centers is approximately 4.0, with an Fe–Pd bond distance of approximately 5.4 Å (Fig. 2g). Formation energy calculations confirmed the fitted model as the most stable structure among several constructed models (Fig. S6†). Additionally, the simulated XANES spectrum of the fitted model exhibited excellent agreement with the experimental result (Fig. S7†). Based on these findings, it can be concluded that Pd–N4 sites are positioned adjacent to Fe–N4 in Fe,Pd–NC, maintaining an interatomic distance of approximately 5.4 Å. This unique configuration is expected to efficiently modulate the electronic environment around Fe sites through long-range interactions, enhancing catalytic performance.
 | ||
| Fig. 2 (a) Fe 2p and (b) Pd 3d XPS spectra of the prepared catalysts. (c) Fe K-edge XANES spectra (inset: enlarged spectra) and (d) Fe K-edge k3-weighted FT-EXAFS spectra. (e) WT-EXAFS contour plots of Fe,Pd–NC, Fe2O3, and PdO. (f) Fitted EXAFS curves and (g) corresponding model of Fe,Pd dual active sites (gray, blue, orange, and turquoise represent C, N, Fe, and Pd atoms, respectively). | ||
As a result of the Fe,Pd dual active site formation and enhanced mass transport due to the highly porous structure, Fe,Pd–NC catalysts are expected to exhibit excellent ORR performance. The ORR electrocatalytic performance of the Fe,Pd–NC catalyst was first evaluated under alkaline conditions (0.1 M KOH, pH = 13) and compared with that of single atom catalysts (Fe–NC and Pd–NC) and commercial Pt/C. The cyclic voltammetry (CV) curves revealed distinct cathodic peaks in an O2-saturated electrolyte for all catalysts, confirming their catalytic activity toward the ORR (Fig. S8†). Further assessment using linear sweep voltammetry (LSV) (Fig. 3a) provided onset potential (E0) and half-wave potential (E1/2) values, summarized in Table S5.† The NaCl-templated porous structure of Fe,Pd–NC proved highly efficient in enhancing ORR catalytic activity (Fig. S9†). The Fe,Pd–NC catalyst achieved an E0 of 0.993 V, comparable to that of Pt/C (1.013 V), and outperformed Fe–NC (0.958 V) and Pd–NC (0.905 V). Notably, Fe,Pd–NC exhibited the highest E1/2 of 0.884 V in 0.1 M KOH, even surpassing Pt/C (0.852 V) and previously reported catalysts (Table S7†). The electrochemically active surface area (ECSA) was determined by calculating the double-layer capacitance (Cdl) from CV curves measured between 1.05 and 1.15 V (Fig. S10 and Table S6†). Fe,Pd–NC (270.5 cm2) and Fe–NC (275.5 cm2) displayed similar ECSA values, both exceeding Pd–NC (193.5 cm2). This suggests that the Fe sites are more effective than Pd sites in adsorbing and activating reactants.58 Despite their comparable ECSA values, the superior catalytic activity of Fe,Pd–NC over Fe–NC can be attributed to electronic environment optimization around Fe sites by the adjacent Pd, which has higher electronegativity. Fe,Pd–NC also exhibited the highest kinetic current densities (jk) of 16.9 mA cm−2 at 0.85 V (Fig. 3b) and the lowest Tafel slope of 77.2 mV dec−1 (Fig. 3c), indicating its fastest ORR kinetics. To gain insights into the ORR reaction pathway of Fe,Pd–NC, Koutecky–Levich (K–L) plots (Fig. 3d) were obtained from capacitive current corrected LSV curves at varying rotation speeds.59 The electron transfer number (n) for Fe,Pd–NC was determined to be approximately 4.1, confirming a predominantly 4e− ORR pathway. Furthermore, rotating ring-disk electrode (RRDE) measurements supported these findings, yielding an average n of approximately 3.97 over the 0.8 V to 0.2 V range (Fig. 3e). The slightly reduced H2O2 yield of Fe,Pd–NC compared to Fe–NC suggests that Pd incorporation enhances product selectivity toward the direct O2 to OH− reduction pathway. Stability, another key factor for the practical application of electrocatalysts, was evaluated using chronoamperometry measurements at 0.7 V in 0.1 M KOH (Fig. 3f). The Fe,Pd–NC catalyst exhibited only a 4.4% current attenuation after 12 hours of continuous operation, significantly lower than Pt/C (11.9%). Fe–NC showed a similar stability profile to Fe,Pd–NC, indicating that Pd incorporation has minimal impact on durability under alkaline conditions. Accelerated durability tests (ASTs) further confirmed the stability of Fe,Pd–NC, as it retained performance with only a 10 mV decrease in E1/2 after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 AST cycles (Fig. S11a†). In contrast, Pt/C exhibited a more pronounced 25 mV decline (Fig. S11b†). The methanol tolerance of Fe,Pd–NC was also examined by introducing 3 M methanol into the 0.1 M KOH electrolyte (Fig. S11c†). While Pt/C experienced a sharp current drop, Fe,Pd–NC maintained a stable current with negligible decay, demonstrating excellent resistance to methanol crossover effects. These findings highlight that the Fe,Pd dual atomic sites play a crucial role in imparting exceptional ORR activity, stability, and selectivity to Fe,Pd–NC in alkaline environments.
000 AST cycles (Fig. S11a†). In contrast, Pt/C exhibited a more pronounced 25 mV decline (Fig. S11b†). The methanol tolerance of Fe,Pd–NC was also examined by introducing 3 M methanol into the 0.1 M KOH electrolyte (Fig. S11c†). While Pt/C experienced a sharp current drop, Fe,Pd–NC maintained a stable current with negligible decay, demonstrating excellent resistance to methanol crossover effects. These findings highlight that the Fe,Pd dual atomic sites play a crucial role in imparting exceptional ORR activity, stability, and selectivity to Fe,Pd–NC in alkaline environments.
 | ||
| Fig. 3 (a) LSV curves, (b) kinetic current density, and (c) Tafel slopes of various catalysts in 0.1 M KOH solution. (d) Capacitive current (non-ORR current) corrected-LSV curves at various rotation speeds of Fe,Pd–NC (inset: corresponding K–L plot). (e) H2O2 yield and electron transfer number (n) of various catalysts. (f) Chronoamperometry curves of Fe,Pd–NC, Fe–NC, and Pt/C recorded at 0.7 V (vs. RHE) in 0.1 M KOH solution. | ||
The ORR performance of the Fe,Pd–NC catalyst was further evaluated in more challenging environments, including neutral (0.1 M PBS, pH = 7) and acidic (0.1 M HClO4, pH = 1) electrolytes, with results summarized in Table S5.† Under neutral conditions, Fe,Pd–NC exhibited significantly enhanced activity, achieving the highest E1/2 and jk values of 0.782 V and 7.8 mA cm−2, respectively, surpassing Pt/C (0.722 V and 2.1 mA cm−2) and previously reported catalysts (Fig. 4a, S12a and Table S8†). In acidic media, Fe,Pd–NC also demonstrated enhanced activity, with E1/2 and jk values of 0.781 V and 7.9 mA cm−2, respectively, outperforming Fe–NC and Pd–NC, while approaching the performance of Pt/C (0.813 V and 15.3 mA cm−2) and previously reported catalysts (Fig. 4d, S12b and Table S9†). These findings highlight the effectiveness of highly accessible Fe,Pd dual atomic sites in enhancing catalytic activity not only in alkaline solutions but also in neutral and acidic environments, demonstrating remarkable ORR performance across the entire pH spectrum. Notably, the ORR activity of Fe,Pd–NC is superior to that of most reported pH-universal catalysts, with particularly outstanding improvements in neutral media (Table S10†). This exceptional performance prompts further investigation into the intrinsic activity of each catalytic site. The turnover frequency (TOF) of Fe,Pd–NC is significantly higher than that of Fe–NC and Pd–NC across all electrolytes, confirming that the synergistic interaction between Fe and Pd effectively enhances intrinsic activity across a wide pH range (Table S11†). Furthermore, Fe,Pd–NC exhibited the lowest Tafel slopes of 94.2 mV dec−1 in 0.1 M PBS (Fig. 4b) and 82.8 mV dec−1 in 0.1 M HClO4 (Fig. 4e), demonstrating the fastest reaction kinetics under both neutral and acidic conditions. Long-term stability tests conducted over 12 hours revealed that Fe,Pd–NC exhibited significantly higher retention of catalytic performance compared to Pt/C in both neutral (Fig. 4c) and acidic (Fig. 4f) electrolytes. Interestingly, unlike under the alkaline conditions, Fe,Pd–NC showed a considerably lower attenuation rate compared to Fe–NC in acidic and neutral environments. The accelerated degradation of Fe–NC under lower pH conditions is primarily attributed to the increased efficiency of the Fenton reaction, in which H2O2, a byproduct of the ORR, interacts with Fe centers to generate ROS.60 Therefore, to explore the role of Pd sites in improving the durability of Fe,Pd–NC, it was hypothesized that Pd may mitigate ROS-related degradation by either facilitating the scavenging of ROS generated from H2O2 or accelerating the reduction of H2O2 to H2O.61 The ROS production level under different pH-conditions was assessed using 2,20-azinobis(3-ethylbenzthiazoline-6-sulfonate) (ABTS) as a probe molecule, which exhibits an absorbance change at 417 nm upon oxidation by ROS.62,63 In 0.1 M KOH, no significant absorbance changes were detected, indicating minimal ROS effects under alkaline conditions (Fig. 4g). However, in 0.1 M HClO4, a clear increase in absorbance was observed, with Fe,Pd–NC showing lower absorbance than Fe–NC, consistent with the durability trends. This suggests that the presence of Pd sites facilitates the scavenging of ROS generated from Fe–N4 sites under lower pH conditions (Fig. 4h). To further clarify the impact of Pd, the HPRR activities of Fe,Pd–NC and Fe–NC were compared across different pH conditions (Fig. S13†). As pH decreased, the HPRR activity of Fe,Pd–NC became lower than that of Fe–NC, suggesting that Pd does not primarily enhance durability by accelerating H2O2 reduction to H2O. Instead, the incorporated Pd sites effectively contribute to suppressing ROS-induced degradation in neutral and acidic environments, underscoring their crucial role in stabilizing Fe sites for pH-universal applications (Fig. 4i).
 | ||
| Fig. 4 (a) LSV curves, (b) Tafel slopes, and (c) chronoamperometry curves of various catalysts recorded at 0.7 V in 0.1 M PBS. (d) LSV curves, (e) Tafel slopes, and (f) chronoamperometry curves of various catalysts recorded at 0.7 V in 0.1 M HClO4. UV-vis spectra of Fe,Pd–NC + ABTS + H2O2, Fe–NC + ABTS + H2O2, and ABTS + H2O2 after 7 min reactions using (g) 0.1 M KOH and (h) 0.1 M HClO4. (i) Schematic illustration of the radical scavenging mechanism for Fe,Pd–NC. | ||
To investigate the origin of the enhanced catalytic activity in the Fe,Pd–NC catalyst and clarify the specific role of Pd, we performed comprehensive DFT calculations. The dual-site FeN4–PdN4 structure was modeled based on EXAFS and XANES results, as illustrated in Fig. 5a. For comparison, single-site FeN4 and PdN4 models were also constructed to represent Fe–NC and Pd–NC catalysts, respectively. Surface Pourbaix diagrams were calculated to identify the most stable surface species and corresponding active sites under realistic electrochemical conditions.64 At potentials between 0.8 and 1.0 V, which correspond to the onset potential region, the Fe sites in both FeN4 and FeN4–PdN4 structures are likely to be covered by *OH, as *OH exhibits the most stable free energy among the considered intermediates. In the FeN4(OH)–PdN4 configuration, where one side of the Fe site is already covered with *OH, further *OH adsorption on the opposite side is thermodynamically favorable, resulting in the FeN4(2OH)–PdN4 structure. This fully *OH-covered Fe site suggests that the ORR is more likely to occur at alternative sites, such as the adjacent Pd atom or neighboring carbon atoms. Unlike the Fe sites, the Pd sites remain in bare states without any intermediate coverage throughout the examined potential range, as shown in Fig. S14d–f.† To examine the electronic effects of Pd incorporation, we analyzed the Bader charges in the FeN4, PdN4, and FeN4–PdN4 models (Fig. S15†). In FeN4 and PdN4, carbon atoms bonded to the coordinating nitrogen atoms exhibit effective Bader charges of approximately +0.4, while the carbon atoms located in the third shell are nearly neutral. In the FeN4–PdN4 structure, the carbon atoms bridging between the two metal centers form bonds with two nitrogen atoms and display significantly higher Bader charges, around +1.0. This increased oxidation state suggests that these carbon atoms could potentially act as active sites for the ORR.65,66
 | ||
| Fig. 5 (a) Schematic illustrations of the considered active sites, including FeN4, PdN4, and FeN4–PdN4 configurations, as well as their OH pre-adsorbed structures: FeN4(OH), FeN4(OH)–PdN4, and FeN4(2OH)–PdN4, based on the surface Pourbaix diagram results shown in Fig. S14.† Gray, orange, blue, cyan-blue, red, and white spheres represent carbon, iron, nitrogen, palladium, oxygen, and hydrogen atoms, respectively. (b) ORR volcano plot of theoretical limiting potential versus ΔG*OH for Fe, Pd and C active sites. Gibbs free energy diagrams for the main process of Fenton reactions at (c) Fe sites, (d) Pd sites and (e) C sites. | ||
To investigate the ORR mechanism at different sites, we calculated the Gibbs free energy profiles for all considered active centers, including those with pre-adsorbed *OH. At all Fe sites, the potential-determining step (PDS) is the *OH removal step (Fig. S16†). The overpotentials for Fe@FeN4 and Fe@FeN4–PdN4 are 1.04 V and 0.99 V, respectively. For the *OH pre-adsorbed structures, Fe@FeN4(OH) and Fe@FeN4(OH)–PdN4, the overpotentials decrease to 0.63 V and 0.83 V, respectively, indicating slightly improved activity compared to the clean surfaces. In contrast, Pd@PdN4, Pd@FeN4(OH)–PdN4, and Pd@FeN4(2OH)–PdN4 exhibit O formation as the PDS, with overpotentials of 1.04 V, 1.08 V, and 2.03 V, respectively (Fig. S17†). The Pd@FeN4–PdN4 site shows *OOH formation as the PDS, with an overpotential of 1.52 V. These results indicate that Pd sites are not effective active centers for the ORR due to their large overpotentials exceeding 1.0 V. The overpotentials for carbon sites (C@FeN4–PdN4, C@FeN4(OH)–PdN4, and C@FeN4(2OH)–PdN4) are 0.86 V, 0.48 V, and 0.51 V, respectively, all with *OOH formation as the PDS. Notably, the overpotentials at C@FeN4(OH)–PdN4 and C@FeN4(2OH)–PdN4 are lower than those at Fe sites, confirming that these carbon sites are more active and can serve as dominant ORR active centers. Based on free energy calculations, we constructed a volcano plot to compare the intrinsic activities of different active sites (Fig. 5b). The carbon site in the FeN4(OH)–PdN4 structure is located near the top of the volcano. This result confirms that the carbon site exhibits the highest catalytic activity among Fe, Pd, and C sites bridging FeN4 and PdN4. Additionally, the results highlight the role of Pd in modulating the electronic structure of the carbon atoms located between FeN4 and PdN4, which are absent in single Fe–NC catalysts, thereby generating new active centers induced by the altered local environment around FeN4.
To compare the radical generation and scavenging behaviors of Fe, Pd, and C sites, we calculated the H2O2 decomposition energy profiles on Fe–N4, Pd–N4, and FeN4–PdN4 structures (Fig. 5c–e). DFT results indicate that all four Fe sites (Fe@FeN4, Fe@FeN4(OH), Fe@FeN4–PdN4, and Fe@FeN4(OH)–PdN4) strongly stabilize *O and *O2 intermediates. The free energy change (ΔG) for *O formation (* + 2H2O2 → *O + H2O + H2O2) ranges from −1.17 to −2.27 eV, and for O2 desorption (*O2 + 2H2O → * + O2 + 2H2O), it ranges from 0.55 to 1.04 eV (Fig. 5c). Such strong binding may hinder O2 release, leading to the accumulation of surface-bound oxygen species and an increased likelihood of ROS-related side reactions during H2O2 decomposition. In contrast, Pd sites (Pd@PdN4, Pd@FeN4–PdN4, Pd@FeN4(OH)–PdN4, and Pd@FeN4(2OH)–PdN4) exhibit significantly weaker *O binding, with ΔG values ranging from 0.97 to 1.14 eV. This suggests high energy barriers for *O formation and low H2O2 decomposition activity (Fig. 5d). As a result, Pd sites are unlikely to contribute to ROS generation. C sites (C@FeN4–PdN4, C@FeN4(OH)–PdN4, and C@FeN4(2OH)–PdN4) demonstrate thermodynamically favorable *O formation, with ΔG values from −0.52 to −1.10 eV, and relatively weak *O2 binding (ΔG from 0.03 to 0.07 eV) (Fig. 5e). These properties facilitate O2 desorption and efficient H2O2 decomposition, thereby minimizing the risk of ROS accumulation. Overall, Fe sites are highly active for H2O2 decomposition but prone to ROS generation due to strong oxygen intermediate stabilization. Pd sites are largely inactive and do not participate in ROS-generating pathways. On the other hand, C sites provide efficient and clean H2O2 conversion with low ROS generation potential, offering a favorable balance between activity and durability.
Based on the outstanding ORR performance of the Fe,Pd–NC catalyst, we assembled both alkaline and neutral ZABs to explore their potential in energy conversion and storage applications. For comparison, a ZAB using the commercial Pt/C catalyst was also evaluated. While the ZABs with Fe,Pd–NC and Pt/C catalysts exhibited similar open-circuit voltages of approximately 1.57 V in alkaline electrolyte, the open-circuit voltage of the ZAB with the Fe,Pd–NC catalyst (1.40 V) was higher than that of the ZAB with the Pt/C catalyst (1.38 V) in the neutral electrolyte (Fig. 6a). Additionally, at all overpotentials, ZABs incorporating Fe,Pd–NC demonstrated higher current densities compared to those with Pt/C (Fig. 6b). The peak power densities of ZABs using Fe,Pd–NC reached 74.1 mW cm−2 in alkaline electrolyte and 36.2 mW cm−2 in neutral electrolyte, significantly outperforming those of Pt/C-based ZABs (53.9 mW cm−2 and 24.0 mW cm−2, respectively) (Fig. 6c). Furthermore, Fe,Pd–NC exhibited superior specific capacities of 926.1 mA h g−1 in an alkaline electrolyte and 689.0 mA h g−1 in a neutral electrolyte, surpassing the corresponding values for Pt/C (873.2 mA h g−1 and 648.4 mA h g−1) (Fig. S19†). These results highlight the exceptional performance of Fe,Pd–NC as an air cathode material. To further assess the feasibility of Fe,Pd–NC for rechargeable ZAB applications, we evaluated long-term discharge/charge cycle stability using a composition air cathode comprising Fe,Pd–NC and RuO2 (Fig. 6d–e). Over a 100 hour cycling test, the ZABs with Fe,Pd–NC + RuO2 exhibited negligible increases in overpotential in both alkaline and neutral electrolytes. In contrast, the ZABs utilizing Pt/C + RuO2 showed a significant increase in overpotential after 60 hours in an alkaline electrolyte and 70 hours in a neutral electrolyte. These findings highlight the immense potential of the Fe,Pd–NC catalyst for real-world energy conversion applications, demonstrating its stability and high performance across a wide pH spectrum.
 | ||
| Fig. 6 (a) Open-circuit voltage, (b) discharge polarization curves, and (c) power density curves of alkaline ZABs and neutral ZABs. Long-term discharge/charge cycling tests of (d) alkaline ZABs at 5 mA cm−2 with each cycle for 20 min and (e) neutral ZABs at 2.5 mA cm−2 with each cycle for 20 min, using Fe,Pd–NC + RuO2 and Pt/C + RuO2 catalysts. | ||
3. Conclusion
In this study, Pd was introduced adjacent to Fe sites to modulate the electronic environment around Fe sites effectively. The NaCl-assisted ZIF-8 pyrolysis process generated a highly porous structure, ensuring optimal exposure of Fe,Pd diatomic sites and facilitating efficient mass transport. Consequently, Fe,Pd–NC exhibited exceptional ORR activity, achieving E1/2 of 0.884 V, 0.782 V, and 0.781 V under alkaline, neutral, and acidic conditions, respectively, significantly outperforming both Fe–NC and Pd–NC. Additionally, Fe,Pd–NC demonstrated superior durability compared to Fe–NC across all pH conditions. DFT results revealed that the introduced Pd enabled C sites between Fe–N4 and Pd–N4 serve as new active sites and contribute to ROS scavenging, thereby promoting both ORR activity and durability. When assembled into alkaline and neutral ZABs, Fe,Pd–NC surpassed Pt/C-based ZABs, delivering superior performance and long-term cycle stability. This work offers valuable insights into the rational design of highly active and durable diatomic catalysts for a wide range of applications across diverse pH environments.Data availability
All the data related to the current submission entitled “Synergistic Fe,Pd diatomic sites anchored on porous nitrogen-doped carbon for efficient oxygen reduction in the entire pH range” are available upon reasonable request directly from the authors via email to the corresponding authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study was supported by the National Creative Research Initiative Program supported by the National Research Foundation of Korea (No. 2022R1A3A3002149) (J. K. K.). This study was also supported by the National Research Foundation of Korea (NRF) (RS-2023-00235596) (J. W. H.). and (NRF-2019M3D1A1079309) (K.-S. L.).References
- Y. Yang, Y. Yang, Z. Pei, K.-H. Wu, C. Tan, H. Wang, L. Wei, A. Mahmood, C. Yan and J. Dong, Matter, 2020, 3, 1442–1476 CrossRef.
- Y. Deng, J. Luo, B. Chi, H. Tang, J. Li, X. Qiao, Y. Shen, Y. Yang, C. Jia and P. Rao, Adv. Energy Mater., 2021, 11, 2101222 CrossRef CAS.
- J. Pan, Y. Y. Xu, H. Yang, Z. Dong, H. Liu and B. Y. Xia, Adv. Sci., 2018, 5, 1700691 CrossRef.
- T. G. Vo, J. Gao and Y. Liu, Adv. Funct. Mater., 2024, 2314282 CrossRef CAS.
- L. Yang, J. Shui, L. Du, Y. Shao, J. Liu, L. Dai and Z. Hu, Adv. Mater., 2019, 31, 1804799 CrossRef PubMed.
- L. Huang, S. Zaman, X. Tian, Z. Wang, W. Fang and B. Y. Xia, Acc. Chem. Res., 2021, 54, 311–322 CrossRef CAS PubMed.
- J. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef CAS.
- J. Hu, W. Liu, C. Xin, J. Guo, X. Cheng, J. Wei, C. Hao, G. Zhang and Y. Shi, J. Mater. Chem. A, 2021, 9, 24803–24829 RSC.
- M. Humayun, M. Israr, A. Khan and M. Bououdina, Nano Energy, 2023, 113, 108570 CrossRef CAS.
- J. Zhang, H. Yang and B. Liu, Adv. Energy Mater., 2021, 11, 2002473 CrossRef CAS.
- U. Martinez, S. Komini Babu, E. F. Holby, H. T. Chung, X. Yin and P. Zelenay, Adv. Mater., 2019, 31, 1806545 CrossRef.
- F. L. Meng, Z. L. Wang, H. X. Zhong, J. Wang, J. M. Yan and X. B. Zhang, Adv. Mater., 2016, 28, 7948–7955 CrossRef CAS PubMed.
- C. Wang, Y. Liu, Z. Li, L. Wang, X. Niu and P. Sun, Chem. Eng. J., 2021, 411, 128492 CrossRef CAS.
- X. Wan and J. Shui, ACS Energy Lett., 2022, 7, 1696–1705 CrossRef CAS.
- X. X. Wang, V. Prabhakaran, Y. He, Y. Shao and G. Wu, Adv. Mater., 2019, 31, 1805126 CrossRef PubMed.
- T. G. Vo, J. Gao and Y. Liu, Adv. Funct. Mater., 2024, 34, 2314282 CrossRef CAS.
- J. Lian, J. Zhao, X. Wang and Q. Bai, Carbon, 2023, 213, 118257 CrossRef CAS.
- L. Zhang, D.-H. Wu, M. U. Haq, J.-J. Feng, F. Yang and A.-J. Wang, Appl. Catal., B, 2024, 351, 123991 CrossRef CAS.
- Z. Li, S. Ji, C. Xu, L. Leng, H. Liu, J. H. Horton, L. Du, J. Gao, C. He and X. Qi, Adv. Mater., 2023, 35, 2209644 CrossRef CAS PubMed.
- E. Zhu, C. Sun, C. Shi, J. Yu, X. Yang and M. Xu, Chem. Eng. J., 2023, 463, 142468 CrossRef CAS.
- X. Guan, Q. Wu, H. Li, R. Li, R. Li, S. Zeng, Q. Yao, H. Chen and K. Qu, Int. J. Hydrogen Energy, 2023, 48, 9659–9668 CrossRef CAS.
- Y. He, J. Yang, Y. Wang, Y. Jia, H. Li, Y. Liu, L. Liu and Q. Tan, ACS Appl. Mater. Interfaces, 2024, 16, 12398–12406 CrossRef CAS PubMed.
- X. Tan, J. Zhang, F. Cao, Y. Liu, H. Yang, Q. Zhou, X. Li, R. Wang, Z. Li and H. Hu, Adv. Sci., 2024, 11, 2306599 Search PubMed.
- W. Wang, W. Chen, P. Miao, J. Luo, Z. Wei and S. Chen, ACS Catal., 2017, 7, 6144–6149 Search PubMed.
- R. Liu, R. Tang, J. Feng and T. Meng, Chem. Eng. J., 2023, 470, 144261 Search PubMed.
- W. Wei, F. Lu, L. Cui, Y. Zhang, Y. Wei and L. Zong, Carbon, 2022, 197, 112–119 Search PubMed.
- Y. Yan, H. Cheng, Z. Qu, R. Yu, F. Liu, Q. Ma, S. Zhao, H. Hu, Y. Cheng and C. Yang, J. Mater. Chem. A, 2021, 9, 19489–19507 Search PubMed.
- W. Zhang, S. Yi, Y. Yu, H. Liu, A. Kucernak, J. Wu and S. Li, J. Mater. Chem. A, 2024, 12, 87–112 RSC.
- W. H. Li, J. Yang and D. Wang, Angew. Chem., 2022, 134, e202213318 CrossRef.
- K. Sun, K. Yu, J. Fang, Z. Zhuang, X. Tan, Y. Wu, L. Zeng, Z. Zhuang, Y. Pan and C. Chen, Adv. Mater., 2022, 34, 2206478 CrossRef CAS PubMed.
- M. Zhang, X. Cao, J. Dong, X. Zhu, Y. Zhu and L. Wang, Small, 2025, 21, 2409560 CrossRef CAS PubMed.
- L. Wang, Q. An, X. Sheng, Z. Mei, Q. Jing, X. Zhao, Q. Xu, L. Duan, X. Zou and H. Guo, Appl. Catal., B, 2024, 343, 123509 CrossRef CAS.
- L. Wu, Y. Chen, C. Shao, L. Wang and B. Li, Adv. Funct. Mater., 2024, 34, 2408257 Search PubMed.
- J. Xu, S. Lai, D. Qi, M. Hu, X. Peng, Y. Liu, W. Liu, G. Hu, H. Xu and F. Li, Nano Res., 2021, 14, 1374–1381 CrossRef CAS.
- O. Y. Bisen, A. K. Yadav, B. Pavithra and K. K. Nanda, Chem. Eng. J., 2022, 449, 137705 CrossRef.
- T. Wang, A. Chutia, D. J. Brett, P. R. Shearing, G. He, G. Chai and I. P. Parkin, Energy Environ. Sci., 2021, 14, 2639–2669 RSC.
- D. Duan, J. Huo, J. Chen, B. Chi, Z. Chen, S. Sun, Y. Zhao, H. Zhao, Z. Cui and S. Liao, Small, 2024, 2310491 CrossRef CAS PubMed.
- Y. Qian, T. An, K. E. Birgersson, Z. Liu and D. Zhao, Small, 2018, 14, 1704169 Search PubMed.
- C. Xue, W. Zhao, Q. Zhang, J. Wang, Y. Wei, K. Lv, T. Wu, Y. Lin, X. Li and X. Hao, J. Energy Storage, 2022, 51, 104421 CrossRef.
- J. Yan, X. Zheng, C. Wei, Z. Sun, K. Zeng, L. Shen, J. Sun, M. H. Rümmeli and R. Yang, Carbon, 2021, 171, 320–328 CrossRef CAS.
- F. Gao, C. Geng, N. Xiao, J. Qu and J. Qiu, Carbon, 2018, 139, 1085–1092 CrossRef CAS.
- M. Sevilla, G. A. Ferrero and A. B. Fuertes, Chem. Mater., 2017, 29, 6900–6907 Search PubMed.
- R. Song, H. Song, J. Zhou, X. Chen, B. Wu and H. Y. Yang, J. Mater. Chem., 2012, 22, 12369–12374 Search PubMed.
- C. Li, M. Wu and R. Liu, Appl. Catal., B, 2019, 244, 150–158 Search PubMed.
- X. Li, Y. Zhao, Y. Yang and S. Gao, Nano Energy, 2019, 62, 628–637 CrossRef CAS.
- Y. Zhu, Z. Zhang, Z. Lei, Y. Tan, W. Wu, S. Mu and N. Cheng, Carbon, 2020, 167, 188–195 Search PubMed.
- S. Huang, B. Hu, S. Zhao, S. Zhang, M. Wang, Q. Jia, L. He, Z. Zhang and M. Du, Chem. Eng. J., 2022, 430, 132933 Search PubMed.
- H. Liu, J. Fu, H. Li, J. Sun, X. Liu, Y. Qiu, X. Peng, Y. Liu, H. Bao and L. Zhuo, Appl. Catal., B, 2022, 306, 121029 CrossRef CAS.
- J. Tuo, Y. Lin, Y. Zhu, H. Jiang, Y. Li, L. Cheng, R. Pang, J. Shen, L. Song and C. Li, Appl. Catal., B, 2020, 272, 118960 CrossRef CAS.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai and R. Chen, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- R. Zhao, Z. Liang, S. Gao, C. Yang, B. Zhu, J. Zhao, C. Qu, R. Zou and Q. Xu, Angew. Chem., Int. Ed., 2019, 58, 1975–1979 Search PubMed.
- W. Tang, H. Zhang, X. Yang, Z. Dai, Y. Sun, H. Liu, Z. Hu and X. Zheng, Appl. Catal., B, 2023, 320, 121952 CrossRef CAS.
- L. Liu, T. Xiao, H. Fu, Z. Chen, X. Qu and S. Zheng, Appl. Catal., B, 2023, 323, 122181 CrossRef CAS.
- Q. He, J. H. Lee, D. Liu, Y. Liu, Z. Lin, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Adv. Funct. Mater., 2020, 30, 2000407 CrossRef CAS.
- F. Cao, Q. Zhao, D. Kong, X. Tan, X. Li, T. Liu, L. Zhi and M. Wu, Chem. Eng. J., 2023, 473, 145181 CrossRef CAS.
- J. Li, M. Lin, W. Huang, X. Liao, Y. Ma, L. Zhou, L. Mai and J. Lu, Small Methods, 2023, 7, 2201664 CrossRef CAS PubMed.
- Y. Liu, F. Tu, Z. Zhang, Z. Zhao, P. Guo, L. Shen, Y. Zhang, L. Zhao, G. Shao and Z. Wang, Appl. Catal., B, 2023, 324, 122209 CrossRef CAS.
- C. Chen, M. Sun, F. Zhang, H. Li, M. Sun, P. Fang, T. Song, W. Chen, J. Dong and B. Rosen, Energy Environ. Sci., 2023, 16, 1685–1696 RSC.
- J. Hu, X. Cai, J. Wu, C. Xin, J. Guo, Z. Liu, J. Wei, X. Cheng, C. Hao and H. Dong, Chem. Eng. J., 2022, 430, 133105 CrossRef CAS.
- G. Bae, M. W. Chung, S. G. Ji, F. Jaouen and C. H. Choi, ACS Catal., 2020, 10, 8485–8495 CrossRef CAS.
- Y. Li, M.-Y. Chen, B.-A. Lu, H.-R. Wu and J.-N. Zhang, Appl. Catal., B, 2024, 342, 123458 CrossRef CAS.
- Y. Chu, E. Luo, Y. Wei, S. Zhu, X. Wang, L. Yang, N. Gao, Y. Wang, Z. Jiang and C. Liu, Chem Catal., 2023, 3, 100532 CrossRef CAS.
- L. Ran, Y. Xu, X. Zhu, S. Chen and X. Qiu, ACS Nano, 2023, 18, 750–760 CrossRef PubMed.
- Y. Wang, Y.-J. Tang and K. Zhou, J. Am. Chem. Soc., 2019, 141, 14115–14119 CrossRef CAS PubMed.
- L. Yang, D. Cheng, H. Xu, X. Zeng, X. Wan, J. Shui, Z. Xiang and D. Cao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6626–6631 CrossRef CAS PubMed.
- C. Zhang, S. Qin, B. Li and P. Jin, J. Mater. Chem. A, 2022, 10, 8309–8323 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01442e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |