
Halide vacancy passivation in cesium lead halide perovskite nanocrystals with mixed halide compositions: the impact of prolonged reaction time†
Yeongcheol Kim‡
 ab,
Seongwoo Cho‡ac,
Seohee Parkac,
Min Ju Kimad,
Younghoon Kime,
Gui-Min Kimf,
Doh C. Leef,
Sung Nam Limabj,
Shin Ae Songab,
Cheolsang Young,
Seongho Leebi,
Seong-Yong Cho*bd,
Sohee Jeong*c,
Seunghyun Lee*bh and
Ju Young Woo*abj
ab,
Seongwoo Cho‡ac,
Seohee Parkac,
Min Ju Kimad,
Younghoon Kime,
Gui-Min Kimf,
Doh C. Leef,
Sung Nam Limabj,
Shin Ae Songab,
Cheolsang Young,
Seongho Leebi,
Seong-Yong Cho*bd,
Sohee Jeong*c,
Seunghyun Lee*bh and
Ju Young Woo*abj
aAutonomous Manufacturing & Process R&D Department, Korea Institute of Industrial Technology (KITECH), Ansan 15588, Republic of Korea. E-mail: jywoo@kitech.re.kr
bHYU-KITECH Joint Department, Hanyang University, Ansan 15588, Republic of Korea. E-mail: leeshyun@hanyang.ac.kr; seongyongcho@hanyang.ac.kr
cDepartment of Energy Science (DOES), Center for Artificial Atoms, Institute of Energy Science and Technology (SIEST), Sungkyunkwan University (SKKU), Suwon 16419, Republic of Korea. E-mail: s.jeong@skku.edu
dDepartment of Photonics and Nanoelectronics, Hanyang University ERICA, Ansan 15588, Republic of Korea
eDepartment of Chemistry, Kookmin University, Seoul 02707, Republic of Korea
fDepartment of Chemical and Biomolecular Engineering, KAIST Institute for the NanoCentury, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea
gReliability Research Center, FITI Testing & Research Institute, 79, Magokjungang 8-ro 3-gil, Gangseo-gu, Seoul, Republic of Korea
hDepartment of Energy and Bio Sciences, Department of Applied Chemistry, Hanyang University ERICA, Ansan 15588, Republic of Korea
iDepartment of Applied Physics, Hanyang University, Ansan, 15588, Republic of Korea
jSchool of Integrative Engineering, Chung-Ang University, Seoul 06974, Republic of Korea
First published on 15th May 2025
Abstract
Perovskite nanocrystals offer a significant advantage in covering the entire visible spectrum with outstanding color purity by simply tuning their halide compositions, making them highly desirable for light-emitting diode applications with excellent color reproducibility. However, mixed halide perovskite nanocrystals often suffer from instability issues, such as halide segregation under various stimuli. In this study, we present a straightforward synthetic approach to produce stable blue-emitting perovskite nanocrystals with mixed halide compositions. Specifically, we synthesized blue-emitting CsPbBrxCl3−x nanocrystals in the presence of impurity metal halides, such as ZnBr2, as halide sources. Our findings show that the reaction can be prolonged for over 90 min at elevated temperature when ZnBr2 is added as a bromine source, while the addition of PbBr2 fails to extend the reaction. In CsPbBrxCl3−x nanocrystals synthesized in the presence of ZnBr2, it turned out that the photoluminescence quantum yield gradually increases due to effective passivation of halide vacancies over time during the reaction. In addition, air and thermal stabilities, which are strongly associated with the compositions, were significantly enhanced. Finally, we demonstrate blue light-emitting diodes based on these nanocrystals, exhibiting greatly suppressed phase segregation even under high operating voltage.
Introduction
All-inorganic cesium lead halide perovskite NCs exhibit remarkable optical properties, such as high photoluminescence quantum yield (PL QY), narrow emission linewidth, defect tolerance, and easy solution processability. Additionally, their widely tunable spectra, which can be achieved through engineering the halide compositions, make them ideal for display applications requiring high color purity.1–3For electroluminescent light-emitting diodes (LEDs), it is essential to achieve efficient emission of the three primary colors: red, green, and blue. Among these, the development of blue-emitting materials has posed the greatest challenge, both for conventional semiconductor quantum dots and for perovskite NCs.4,5 While green and red perovskite NCs have achieved exceptionally high PL QY (∼100%) and external quantum efficiency (EQE) exceeding 20% in devices, blue-emitting perovskite NCs have lagged behind, displaying lower PL QY and poorer device EQE.6–8 This limitation is mainly attributed to the defect intolerance of chloride-based compositions compared to bromide and iodide, and insufficient passivation strategies targeting halide defects in blue-emitting perovskite NCs.9–13 Additionally, instability arising from phase segregation remains a significant issue, since mixed-halide compositions, which are vulnerable to phase segregation, are typically required to achieve blue emission in all-inorganic perovskite NCs.14–19
Recent advancements have been made in blue-emitting perovskite NCs through post-synthetic modifications. For instance, Gao et al. demonstrated effective passivation of blue-emitting perovskite NCs by treating CsPbCl3 NCs with octylammonium hydrobromide, resulting in CsPbBrxCl3−x NCs with a PL QY exceeding 90%. Efficient operation of blue-emitting LEDs based on these NCs was also achieved.20 Similarly, the incorporation of polyethylene glycol dimethacrylate into post-treatment solutions has proven to be an effective dual strategy for defect passivation in perovskite films. This method resulted in enhanced performance of blue 3D mixed-halide perovskite LEDs with high EQE. Additionally, device lifetime saw a significant improvement, achieving a duration five times longer than devices without post-synthetic defect passivation.21 Zou et al. reported notable CIE stability in blue-emitting perovskite NCs achieved through a post-treatment process utilizing a functional organotrichlorosilane. This methodology effectively enhances the Cl− concentration derived from HCl within the perovskite lattice, leading to a spectral blueshift with mitigation of ion migration, which contributes to improved device stability.22 Although post-synthetic processes somewhat enhance the performance and stability of blue-emitting perovskite NCs, they often involve complex steps that can lead to challenges such as irreversible degradation.23 In this respect, a simpler synthetic protocol without employing post-synthetic treatment has been desired.24
In this study, we developed a simple and novel in situ approach to synthesize high-quality blue-emitting perovskite NCs with mixed halide compositions (Cl and Br) by simply extending the reaction time—a straightforward and effective variable that has often been overlooked, particularly in the synthesis of perovskite NCs. Conventional synthesis of blue-emitting CsPbBrxCl3−x NCs using the mixture of PbCl2 and PbBr2 could not sustain prolonged reaction times at high temperature. However, we discovered that adding impurity metal halides, such as ZnBr2 in place of PbBr2, enabled the synthesis of CsPbBrxCl3−x NCs with extended reaction times over 90 min. The resulting NCs exhibited significantly improved PL QY, air stability, and thermal stability compared to control samples. Finally, we fabricated LEDs using the blue-emitting NCs synthesized with prolonged reaction times and without post-treatment, demonstrating greatly reduced phase segregation even under high operating voltages, because of effective passivation of halide vacancies.
Experimental
Materials
Cesium carbonate (Cs2CO3, 99%, Thermo Fisher Scientific), zinc bromide (ZnBr2, 99.9%, Alfa Aesar), lead bromide (PbBr2, ≥98%, Alfa Aesar), lead chloride (PbCl2, 99%, Thermo Fisher Scientific), oleic acid (OA, technical grade, Sigma-Aldrich), 1-octadecene (ODE, ≥91.5%, UNIAM), and oleylamine (OLA, >50%, Tokyo Chemical Industry) were purchased.Preparation of Cs-oleate solution
The original synthetic method for Cs-oleate was optimized from a procedure reported in 2015 with some modifications.25 Specifically, 0.326 g of Cs2CO3, 1 mL of OA, and 8 mL of ODE were loaded into a 100 mL three-neck flask and degassed for 1 hour at 110 °C. After degassing, the solution was further heated to 150 °C under an argon (Ar) flow and maintained at this temperature.Synthesis of PbBr2-CsPbBrxCl3−x NCs
The original PbBr2-CsPbBrxCl3−x NCs were optimized from a conventional hot injection method reported in 2015 with some modifications.25 For the synthesis of pristine PbBr2-CsPbBrxCl3−x NCs, 0.93 g of PbBr2 (2.532 mmol), 0.624 g of PbCl2 (2.256 mmol), 6 mL of OA, 6 mL of OLA, and 60 mL of ODE were loaded into a 100 mL three-neck flask. The mixture was degassed at 110 °C for 1 hour. After degassing, the temperature of the solution was increased to 200 °C under Ar, and 2.4 mL of Cs-oleate solution (preheated to 150 °C) was swiftly injected into the precursor solution using a glass syringe. After the injection of Cs-oleate, the reaction proceeded for a controlled duration before the flask was quenched to room temperature in an ice-water bath.Synthesis of ZnBr2-CsPbBrxCl3−x NCs
For the synthesis of ZnBr2-CsPbBrxCl3−x NCs, 0.57 g of ZnBr2 (2.532 mmol), 0.624 g of PbCl2 (2.256 mmol), 6 mL of OA, 6 mL of OLA, and 60 mL of ODE were loaded into a 100 mL three-neck flask. The mixture was degassed at 110 °C for 1 hour. After degassing, the temperature of the solution was increased to 200 °C under Ar, and 2.4 mL of Cs-oleate solution (preheated to 150 °C) was swiftly injected into the precursor solution using a glass syringe. After the injection of Cs-oleate, the reaction proceeded for a controlled duration before the flask was quenched to room temperature in an ice-water bath.Purification of PbBr2- and ZnBr2-CsPbBrxCl3−x NCs
Samples of 5 mL of NCs were extracted at various reaction times using a glass syringe and subjected to a series of purification steps. Initially, the samples were centrifuged for 10 min at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. After centrifugation, the supernatant was discarded, and the precipitate was dried. The dried precipitate was then redispersed in 4 mL of toluene and vigorously mixed. The resulting solution was centrifuged again for 10 min at 13000 rpm. Following this centrifugation, the supernatant was discarded, and the precipitate was dried once more. Subsequently, the precipitate was redispersed in 3 mL of hexane and thoroughly mixed. The solution underwent a final centrifugation for 10 min at 6000 rpm. After this step, only the supernatant was collected and filtered using a 0.45 μm pore size filter. The temperature was set to 22 °C during centrifugations. All processes were carried out under ambient conditions.
000 rpm. After centrifugation, the supernatant was discarded, and the precipitate was dried. The dried precipitate was then redispersed in 4 mL of toluene and vigorously mixed. The resulting solution was centrifuged again for 10 min at 13000 rpm. Following this centrifugation, the supernatant was discarded, and the precipitate was dried once more. Subsequently, the precipitate was redispersed in 3 mL of hexane and thoroughly mixed. The solution underwent a final centrifugation for 10 min at 6000 rpm. After this step, only the supernatant was collected and filtered using a 0.45 μm pore size filter. The temperature was set to 22 °C during centrifugations. All processes were carried out under ambient conditions.
Air stability test under ambient conditions
The glass substrates were coated with NCs under ambient conditions. Subsequently, the samples were stored in ambient air at room temperature. Characterization studies, including PL QY, were performed over time to assess the stability and performance of the NCs.Thermal stability test under ambient conditions
NCs were coated onto glass substrates via drop casting. All processes were conducted under ambient conditions. Subsequently, the samples were heated on a hot plate set to 70 °C, and the PLQY was measured over time.Materials characterization
Photoluminescence quantum yield measurements were performed using an integrating sphere (Quantaurus, Hamamatsu Photonics). The crystal structure of the perovskites was determined by X-ray diffraction using a diffractometer (Miniflex 600, Rigaku). The samples for X-ray diffraction analysis were prepared by drop-casting the NC solutions onto conductive Au/Cu-deposited silicon wafer substrates under ambient conditions. X-ray photoelectron spectroscopy measurements were conducted using an X-ray photoelectron spectrometer (ESCALAB 250Xi). For X-ray photoelectron spectroscopy analysis, the samples were prepared by drop-casting the NC solutions onto indium tin oxide substrates under ambient conditions. Transient photoluminescence was measured with 0.01 mW pump power, 0.1 mm beam diameter, and 310 nm wavelength. The pump beam was generated using an optical parametric amplifier (ORPHEUS, LIGHT CONVERSION) with a 1030 nm seed beam (PHAROS, LIGHT CONVERSION), which has a 200 fs pulse duration and a 200 kHz repetition rate. Photoluminescence from the sample was measured using a photon counting detector (PDM, Micro Photon Devices) and a time-correlated single-photon counting module (PicoHarp 300, PicoQuant). Transmission electron microscopy analysis to obtain nanoscale crystalline information of the perovskites was carried out using a Talos L120C.Device fabrication
Patterned ITO glass was ultrasonically cleaned with isopropyl alcohol (IPA) for 5 min. Then, the substrate was treated with oxygen plasma for 3 min. Poly(3,4-ethylenedioxythiophene) (PEDOT:PSS) was spin-coated at 4000 rpm for 30 s and annealed at 120 °C for 5 min in air. The substrate was placed into a N2-filled glove box and annealed at 180 °C for 10 min. Then, poly[(9,9-dioctylfluorenyl-2,7-diyl) co-(4,4′-(N-(4-sec-butylphenyl))diphenylamine)] (TFB) (5 mg mL−1) and 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) (1 mg mL−1) were dissolved in m-xylene, spin-coated at 3000 rpm for 30 s and annealed at 180 °C for 30 min. Perovskite NCs (25 mg mL−1 in octane) were spin-coated at 2000 rpm for 30 s and annealed at 80 °C for 15 min. Then, 2,2′,2′′-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole) (TPBi) (40 nm) and LiF/Al electrodes (1 nm/100 nm) were deposited using thermal evaporation. The LED device was encapsulated by sealing with Norland Optical Adhesive (NOA) 8610 B epoxy and a cover glass.Device characterization
The EL spectra and luminance–current density–voltage curves of the device were measured using a Keithley 2400B sourcemeter and a CS-2000 spectroradiometer (Konica Minolta).Results and discussion
We synthesized all-inorganic CsPbBrxCl3−x perovskite nanocrystals (NCs) with mixed halide compositions using a modified approach.25 Initially, a mixture containing PbBr2, oleic acid, oleylamine, and 1-octadecene (ODE) was prepared. The mixture solution was degassed at 110 °C and then heated to 200 °C under an inert atmosphere for the synthesis of NCs. Subsequently, a preheated Cs-oleate solution was injected into the lead halide mixture solution, allowing the reaction to proceed for a controlled duration (denoted hereafter as PbBr2-CsPbBrxCl3−x NCs). Previously, the effect of impurity metal halides in perovskite NCs with homogeneous halide compositions has been widely studied.26 However, the effect of such impurities in mixed halide systems has been nearly lacking. Therefore, to investigate the effect of different metal halides, we replaced PbBr2 with other metal halides. For example, ZnBr2 was added in place of PbBr2, and the CsPbBrxCl3−x NCs were synthesized following the same procedure as for the PbBr2-CsPbBrxCl3−x NCs (denoted hereafter as ZnBr2-CsPbBrxCl3−x NCs). The synthetic procedures for each type of perovskite NC are schematically shown in Scheme S1.†While previous studies on perovskite NCs typically employed very short reaction times (∼10 s), other types of NCs have shown that prolonged reaction times can result in higher-quality NCs.27–29 Based on this insight, we hypothesized that extending the reaction time might similarly improve the quality of perovskite NCs. For example, the passivation of NCs might be improved by allowing enough reaction time for crystal formation. Based on the hypothesis, we explored the effect of prolonged reaction times on the properties of the synthesized perovskite NCs.
Fig. 1(a) presents the PL peak wavelength data of PbBr2-CsPbBrxCl3−x and ZnBr2-CsPbBrxCl3−x NCs as a function of reaction time. For the PbBr2-CsPbBrxCl3−x NCs, a redshift in the PL peak was observed until the reaction time reached 20 min. However, extending the reaction time beyond 20 min resulted in the permanent precipitation of the NCs. In contrast, the ZnBr2-CsPbBrxCl3−x NCs showed a continuous redshift in the PL peak up to 90 minutes without any signs of precipitation. Notably, the PL QY of ZnBr2-CsPbBrxCl3−x NCs was markedly increased with prolonged reaction time, whereas the PL QY of PbBr2-CsPbBrxCl3−x NCs remained largely unchanged across different reaction times (Fig. 1(b)).
 | ||
| Fig. 1 (a) PL peak wavelength of ZnBr2- and PbBr2-CsPbBrxCl3−x NCs as a function of reaction time. (b) PL QYs of ZnBr2- and PbBr2-CsPbBrxCl3−x NCs depending on reaction time. | ||
To explore the effect of reaction time on the size and morphology of CsPbBrxCl3−x NCs, TEM analysis was carried out. Fig. 2(a) and (b) display the TEM images of PbBr2-CsPbBrxCl3−x NCs at reaction times of 10 s and 20 min, respectively. The morphology remained unchanged as the reaction time increased, and notably, the size of the PbBr2-CsPbBrxCl3−x NCs also showed no significant variation despite different reaction times. A similar trend was observed for the ZnBr2-CsPbBrxCl3−x NCs (Fig. 2(c) and (d)), where the PL spectra exhibited a pronounced redshift with increasing reaction time, yet the size of the NCs remained constant throughout the reactions (Table S1 and Fig. S1†). These findings strongly suggest that the observed spectral redshift of both PbBr2- and ZnBr2-CsPbBrxCl3−x NCs (Fig. 1(a)) was primarily due to anion exchange from Cl to Br, rather than a weakened quantum confinement effect due to an increase in particle size as the reaction time was extended.
 | ||
| Fig. 2 TEM images of PbBr2-CsPbBrxCl3−x NCs with the (a) shortest (10 s) and the (b) longest (20 min) reaction times. TEM images of ZnBr2-CsPbBrxCl3−x NCs with the (c) shortest (10 s) and the (d) longest (90 min) reaction times. The scale bars are 20 nm. | ||
Fig. 3(a) shows the X-ray diffraction (XRD) patterns of ZnBr2-CsPbBrxCl3−x NCs. As the reaction time increased, the XRD peaks gradually shifted closer to the reference pattern of CsPbBr3. The results clearly indicate that anion exchange reaction from Cl to Br occurs during the prolonged reaction and are consistent with the data shown in Fig. 1(a). XRD patterns of PbBr2-CsPbBrxCl3−x NCs displayed a similar trend to those of ZnBr2-CsPbBrxCl3−x NCs, indicating the progress of the anion exchange reaction from Cl to Br (Fig. S2†). However, quantitative elemental analysis revealed distinctly different behavior between the two systems.
 | ||
| Fig. 3 (a) XRD patterns of ZnBr2-CsPbBrxCl3−x NCs with various reaction times (reference patterns: JCPDS PDF#54-0752 and JCPDS PDF#84-0438 for CsPbBr3 NCs and CsPbCl3 NCs, respectively). (b) Anion (Br + Cl) to cation (Cs + Pb) atomic ratios of ZnBr2- and PbBr2-CsPbBrxCl3−x NCs. | ||
Quantitative composition analysis data obtained from X-ray photoelectron spectroscopy (XPS) are shown in Fig. 3(b). For the PbBr2-CsPbBrxCl3−x NCs, the total anionic compositions remained largely unchanged with varying reaction times, indicating that the anion exchange reaction exclusively occurred without effectively filling halide vacancies. This lack of halide vacancy passivation is crucial, as it explains why there was no significant increase in PL QY during the reaction from 10 s to 20 min. In contrast, the ZnBr2-CsPbBrxCl3−x NCs showed a substantial increase in the total halide content over time, suggesting that halide supplementation to the NCs occurred in addition to the exchange of Cl for Br. It is noteworthy that the distinct interaction mechanism of ZnBr2 with the NCs, compared to PbBr2, may lead to additional halide incorporation into the NCs. This halide enrichment contributed to the effective passivation of halide vacancies, which in turn increased the PL QY of the NCs. As a result, the ZnBr2-CsPbBrxCl3−x NCs achieved significantly higher PL QY with prolonged reaction times. Previous studies suggest that substituting the B site cation with a smaller cation can enhance structural stability by forming shorter bond lengths that can suppress octahedral rotation and tilting.30–33 We indeed observed partial substitution of Pb to Zn in ZnBr2-CsPbBrxCl3−x NCs, possibly due to lattice compatibility between the CsPbBrxCl3−x host lattice and Zn2+ ions (Fig. S4 and S5†). This substitution should result in a more stable octahedral structure that could persist under prolonged reactions at high temperature, facilitating halide vacancy passivation during reactions.
We conducted nuclear magnetic resonance (NMR) spectroscopy to reveal the surface of CsPbBrxCl3−x NCs. To better understand the surface structures, we employed a selective ligand exchange with methyl acetate.34 In previous studies, a selective ligand exchange strategy using methyl acetate was employed to distinguish surface ligands, as methyl acetate molecules selectively replace oleate ligands through a hydrolysis reaction. In the NMR spectra of the as-prepared PbBr2-CsPbBrxCl3−x NCs, a broad alkene resonance around 5.6 ppm was observed (Fig. 4(a)). However, this resonance disappeared completely after selective ligand exchange with methyl acetate (Fig. 4(b)), indicating that the surface termination of PbBr2-CsPbBrxCl3−x NCs should be predominantly cesium oleate. In contrast, for the ZnBr2-CsPbBrxCl3−x NCs, the broad resonance at 5.6 ppm persisted even after repeated selective ligand exchange with methyl acetate (Fig. 4(c) and (d)), suggesting that the surface termination consisted of oleylammonium halide. Additionally, broad resonances at 7.5 ppm and 4.1 ppm were detected, corresponding to α and β protons of oleylammonium molecules, respectively.35–37 These findings indicate that the surface termination of ZnBr2-CsPbBrxCl3−x NCs is anion-rich oleylammonium. This anion-rich surface may contribute to effective passivation of halide defects at the surface of perovskite NCs.
 | ||
| Fig. 4 1H NMR spectra of PbBr2-CsPbBrxCl3−x NCs purified (a) without methyl acetate and purified (b) with methyl acetate. 1H NMR results of ZnBr2-CsPbBrxCl3−x NCs purified (c) without methyl acetate and purified (d) with methyl acetate. | ||
To understand the impact of reaction time on carrier dynamics in perovskite NCs, we conducted time-resolved photoluminescence spectroscopy (TRPL) using time-correlated single-photon counting. The resulting decay curves were fitted with a tri-exponential decay function,38 and the corresponding data are summarized in Table S2 and Fig. S3.† Each decay time component corresponds to a specific recombination process: Auger recombination or trap-assisted charge recombination (τ1), bi-molecular radiative recombination (τ2), and a slow radiative recombination process (τ3). Additionally, A1, A2, and A3 represent the fractional contributions of the fast (A1), intermediate (A2), and slow (A3) components, respectively.39
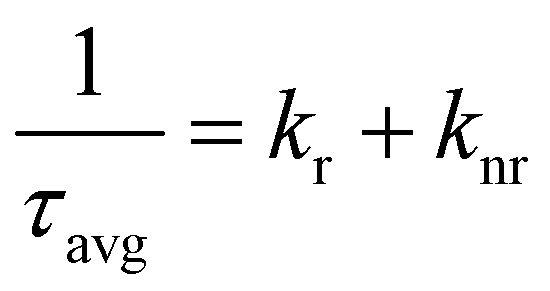 | (1) |
Overall, the average PL lifetime of ZnBr2-CsPbClxBr3−x NCs was markedly longer compared to the average PL lifetime of PbBr2-CsPbBrxCl3−x NCs. This fact likely indicates a much lower defect density in the ZnBr2-CsPbBrxCl3−x NCs, leading to more efficient radiative recombination.40–42 To understand the carrier dynamics more deeply, we extracted the radiative recombination rate (kr) and non-radiative recombination rate (knr) from the data. The average lifetime (τavg) reflects the contributions of both kr and knr, and related parameters can be calculated using the following equation:43,44
 | (2) |
The PL QY can be calculated from the ratio of kr to the total recombination rate. From eqn (1) and (2), kr can be derived as:
 | (3) |
The parameters extracted from TRPL data are summarized in Table 1. We compare the relaxation ratio (kr/knr) to investigate the carrier dynamics of NCs. In PbBr2-CsPbBrxCl3−x NCs, both kr and knr increased when the reaction time was increased from 10 s to 20 min. Therefore, the relaxation ratio, kr/knr, of PbBr2-CsPbBrxCl3−x NCs exhibited only a slight difference, resulting in similar PL QY even at different reaction times (10 s and 20 min), as shown in Fig. 1(a). In stark contrast, in ZnBr2-CsPbBrxCl3−x NCs, knr noticeably decreased, whereas kr increased as the reaction time was extended, possibly due to effective halide passivation in ZnBr2-CsPbBrxCl3−x NCs, which underwent longer reactions (Fig. 3(b)). As a result, we observed a considerable increase in kr/knr, and thus enhanced PL QY as the reaction time increases in ZnBr2-CsPbBrxCl3−x NCs.
| Perovskite NCs | τavg [ns] | kr [μs−1] | knr [μs−1] | kr/knr |
|---|---|---|---|---|
| PbBr2-CsPbBrxCl3−x 10 s | 11.5 | 15.6 | 71.3 | 0.22 |
| PbBr2-CsPbBrxCl3−x 20 min | 6.9 | 39.0 | 105.0 | 0.37 |
| ZnBr2-CsPbBrxCl3−x 10 s | 18.7 | 12.8 | 40.6 | 0.32 |
| ZnBr2-CsPbBrxCl3−x 20 min | 19.8 | 19.2 | 31.4 | 0.61 |
| ZnBr2-CsPbBrxCl3−x 50 min | 19.9 | 21.1 | 29.2 | 0.72 |
| ZnBr2-CsPbBrxCl3−x 90 min | 18.3 | 28.0 | 26.8 | 1.04 |
Previously, it was very well known that perovskite NCs are vulnerable to the ambient environment due to their ionic nature and dynamic surfaces. This vulnerability usually leads to shape and phase transformation when the perovskite NCs are exposed to ambient conditions, finally deteriorating optoelectronic properties.45–48 Therefore, we investigated the stability of our perovskite NCs under both thermal and ambient conditions. The perovskite NCs were exposed to either ambient conditions or heat as films, and then PL quenching was measured. When the PbBr2-CsPbBrxCl3−x NCs were exposed to elevated temperature around 70 °C, the PL QY was quenched nearly 80% compared to the initial value, accompanied by the emergence of a shoulder peak around 500–510 nm attributed to phase segregation, indicating their high susceptibility to heat (Fig. 5(a)). In the case of ZnBr2-CsPbBrxCl3−x NCs, the sample showed just around 20% quenching compared with the initial value when stored at 70 °C, demonstrating much greater thermal stability compared to PbBr2-CsPbBrxCl3−x NCs (Fig. 5(b)). The air stability also shows a marked contrast between the samples. While PbBr2-CsPbBrxCl3−x NCs exhibited a decrease in PL QY around 60% compared to the initial value (Fig. 5(c)), the ZnBr2-CsPbBrxCl3−x NCs showed nearly comparable PL QY after air storage for 2 weeks (Fig. 5(d)). In addition, a slight redshift resulting from phase segregation was also observed in PbBr2-CsPbBrxCl3−x NCs, whereas ZnBr2-CsPbBrxCl3−x NCs showed no sign of phase segregation after 2 weeks of air exposure. All the stability data shown in Fig. S6† also point to significantly superior properties of ZnBr2-CsPbBrxCl3−x NCs, possibly due to effective passivation of halide vacancies and thus stable structures of ZnBr2-CsPbBrxCl3−x NCs. Partial substitution of B sites from Pb to Zn may also contribute to enhanced stability (Fig. S4†).30
 | ||
| Fig. 5 Thermal stability data of (a) PbBr2-CsPbBrxCl3−x NCs and (b) ZnBr2-CsPbBrxCl3−x NCs. The samples were kept at 70 °C for 2 h. Air stability data of (c) PbBr2-CsPbBrxCl3−x NCs and (d) ZnBr2-CsPbBrxCl3−x NCs. For the air stability test, the samples were stored under ambient conditions for 2 weeks. | ||
We fabricated LEDs employing PbBr2- and ZnBr2-CsPbBrxCl3−x NCs, and the device performance was investigated (Fig. 6 and Scheme S2†). In LEDs based on perovskite NCs with mixed halide compositions, it has been frequently reported that the presence of halide vacancies leads to halide ion migration within the device, causing phase segregation, which finally results in lower energy emissions.49,50 In our devices, ZnBr2-CsPbBrxCl3−x NCs synthesized with a relatively short reaction time exhibited significant electroluminescent spectral shift as the applied voltage increased, due of the high density of halide vacancies (Fig. 6(a)). However, as the reaction time increases from 20 min to 90 min (Fig. 6(b)–(d)), the electroluminescent spectral shift was significantly mitigated. Finally, we observed just little spectral shift (∼3 nm) for the LEDs based on the ZnBr2-CsPbBrxCl3−x NCs synthesized with the longest reaction time (90 min), as shown in Fig. 6(d). We believe the considerably suppressed spectral shift resulted from the effective passivation of halide vacancies in ZnBr2-CsPbBrxCl3−x NCs due to prolonged reaction time.
 | ||
| Fig. 6 Normalized EL spectra of ZnBr2-CsPbBrxCl3−x NCs with the reactions of (a) 10 s, (b) 20 min, (c) 50 min, and (d) 90 min, respectively. | ||
In contrast, importantly, the devices using PbBr2-CsPbBrxCl3−x NCs showed significant electroluminescent spectral shift even in NCs with prolonged reaction time (Fig. S7†). The results are in line with the elemental analysis data (Fig. 3(b)), demonstrating the presence of considerable halide vacancies even at the extended reaction in PbBr2-CsPbBrxCl3−x NCs. Also, it is important to note that LEDs fabricated with PbBr2-CsPbBrxCl3−x NCs exhibited marked TFB emission peaks (Fig. S7†). The observed TFB peaks can be attributed to recombination at the interface between the hole transport layer (HTL) and the emissive layer, due to excessive electron accumulation at the interface.51 In mixed halide perovskite LEDs, phase segregation of narrow bandgap domains occurs near the HTL under the electric field.52 This phenomenon can lead to the accumulation of electrons at narrow bandgap domains near the HTL.53 Consequently, HTL emission is readily observed in PbBr2-CsPbBrxCl3−x NCs, indicating that this behavior is a result of significant phase segregation. And the result is also a direct sign of high density of halide vacancies in PbBr2-CsPbBrxCl3−x NCs. We note that previous studies performing halide defect suppression also reported similar results of contrasting TFB emission.54 This implies that halide defects in PbBr2-CsPbBrxCl3−x NCs can trigger excessive charge imbalance. Therefore, we believe the reduction of surface defects in the NCs and thus suppressed phase segregation contributed to the enhanced electroluminescent spectral stability of ZnBr2-CsPbBrxCl3−x NC LEDs compared to their PbBr2-CsPbBrxCl3−x counterparts.
The device performance of ZnBr2-CsPbBrxCl3−x NCs reached a significantly improved maximum EQE of 0.02% compared to a maximum EQE of 0.004% in PbBr2-CsPbBrxCl3−x NCs (Fig. S8†). Importantly, ZnBr2-CsPbBrxCl3−x NCs exhibited an increase in maximum luminance and EQE with prolonged reaction time, which can be attributed to effective halide passivation and thus improved carrier injection balance. The results strongly suggest the potential for fabricating stable and high-performance blue-emitting LEDs based on mixed halide perovskite NCs.
Conclusions
In conclusion, we have developed a straightforward and effective in situ approach for synthesizing bright and stable all-inorganic perovskite NCs with mixed halide compositions by simply extending the reaction time – an essential but previously underexplored experimental variable in the synthesis of perovskite NCs. Partial replacement of B sites from Pb to Zn by employing impurity metal halides, such as ZnBr2, stabilizes the CsPbClxBr3−x NCs over longer reaction periods at high temperature, preventing precipitation that would otherwise occur without the impurity metal halides. Our findings reveal that CsPbClxBr3−x NCs synthesized with extended reaction times are halide-rich, suggesting effective passivation of halide vacancies during prolonged reactions. This passivation significantly enhances the optical properties, as demonstrated by static PL QY studies and carrier dynamics investigations depending on reaction times. Additionally, the CsPbClxBr3−x NCs synthesized with the prolonged reaction time exhibit improved air and thermal stability. Notably, LEDs fabricated using ZnBr2-CsPbClxBr3−x NCs synthesized with extended reaction times demonstrated superior spectral stability during operation, which should be attributed to suppressed phase segregation due to effective passivation of halide vacancies. This synthetic method offers valuable insights for producing high-quality perovskite NCs by simply adjusting the basic reaction variable (reaction time), thereby avoiding the need for complex post-synthetic processes.Data availability
The data supporting this finding are available in this article and included as part of the ESI.† Raw data are available from the corresponding author upon reasonable request.Author contributions
Y. K.: investigation, data curation, formal analysis, writing – original draft. S. C.: investigation, data curation, formal analysis, writing – original draft. S. P.: investigation, data curation, formal analysis. M. J. K.: investigation, data curation, formal analysis. Y. K.: methodology, writing – review & editing. G.-M. K.: data curation, formal analysis. D. C. L.: methodology, writing – review & editing. S. N. L.: formal analysis. S. A. S.: formal analysis. C. Y.: data curation, formal analysis. S. L.: data curation. S.-Y. C.: supervision, methodology, writing – review & editing. S. J.: supervision, methodology, writing – review & editing. S. L.: supervision, methodology, writing – review & editing. J. Y. W.: conceptualization, methodology, supervision, project administration, writing – original draft, writing – review & editing. All authors have critically reviewed the manuscript for intellectual content, provided final approval for publication, and agreed to be accountable for all aspects of the work to ensure its accuracy and integrity.Conflicts of interest
The authors declare that they have no financial or personal conflicts of interest that could have influenced the results presented in this study.Acknowledgements
This study was supported by grants funded by the Ministry of Trade, Industry, and Energy (RS-2022-00144108, RS-2025-02315917 and RS-2024-00423271) of the Korean Government. This research was also supported by grants from the National Research Foundation of Korea (RS-2024-00339674) funded by the Ministry of Science and ICT, Republic of Korea.References
- Y. Hong, C. Yu, H. Je, J. Y. Park, T. Kim, H. Baik, G. M. Tomboc, Y. Kim, J. M. Ha, J. Joo, C. W. Kim, H. Y. Woo, S. Park, D. H. Choi and K. Lee, Adv. Sci., 2023, 10, 2302906 CrossRef CAS PubMed.
- S. A. Veldhuis, P. P. Boix, N. Yantara, M. Li, T. C. Sum, N. Mathews and S. G. Mhaisalkar, Adv. Mater., 2016, 28, 6804–6834 CrossRef CAS PubMed.
- X. Zhao, J. D. A. Ng, R. H. Friend and Z.-K. Tan, ACS Photonics, 2018, 5, 3866–3875 CrossRef CAS.
- S. Cho, Y. Kim, S. Lee and J. Y. Woo, Korean J. Chem. Eng., 2024, 41, 3359–3370 CrossRef CAS.
- D. Kim, G. Cho, Y. J. Kim, J. H. Kwon, Y. Oh, M. Yang, S. Jee, I. S. Lee, M.-J. Si, Y. Jung, H. Y. Yang, Y. Ahn, B.-K. Kim, C. Kim, H. S. Kim and S.-W. Baek, Adv. Energy Mater., 2024, 14, 2302579 CrossRef CAS.
- A. Fakharuddin, M. K. Gangishetty, M. Abdi-Jalebi, S.-H. Chin, A. R. bin Mohd Yusoff, D. N. Congreve, W. Tress, F. Deschler, M. Vasilopoulou and H. J. Bolink, Nat. Electron., 2022, 5, 203–216 CrossRef CAS.
- Q. Shan, Y. Dong, H. Xiang, D. Yan, T. Hu, B. Yuan, H. Zhu, Y. Wang and H. Zeng, Adv. Funct. Mater., 2024, 34, 2401284 CrossRef CAS.
- Y. Hu, S. Cao, P. Qiu, M. Yu and H. Wei, Nanomaterials, 2022, 12, 4372 CrossRef CAS PubMed.
- D. P. Nenon, K. Pressler, J. Kang, B. A. Koscher, J. H. Olshansky, W. T. Osowiecki, M. A. Koc, L. W. Wang and A. P. Alivisatos, J. Am. Chem. Soc., 2018, 140, 17760–17772 CrossRef CAS PubMed.
- X. Zheng, S. Yuan, J. Liu, J. Yin, F. Yuan, W.-S. Shen, K. Yao, M. Wei, C. Zhou, K. Song, B.-B. Zhang, Y. Lin, M. N. Hedhili, N. Wehbe, Y. Han, H.-T. Sun, Z.-H. Lu, T. D. Anthopoulos, O. F. Mohammed, E. H. Sargent, L.-S. Liao and O. M. Bakr, ACS Energy Lett., 2020, 5, 793–798 CrossRef CAS.
- D. Li, C. Zhao, X. Zhang, X. Zhao, H. Huang, H. Li, F. Li and J. Yuan, Adv. Mater., 2025, 37, 2417346 CrossRef CAS PubMed.
- X. Zhang, H. Huang, C. Zhao and J. Yuan, Chem. Soc. Rev., 2025, 54, 3017–3060 RSC.
- C. Zhao, C. Cazorla, X. Zhang, H. Huang, X. Zhao, D. Li, J. Shi, Q. Zhao, W. Ma and J. Yuan, J. Am. Chem. Soc., 2024, 146, 4913–4921 CrossRef CAS PubMed.
- H. Yu, H. Wang, T. Zhang, C. Yi, G. Zheng, C. Yin, M. Karlsson, J. Qin, J. Wang, X. K. Liu and F. Gao, J. Phys. Chem. Lett., 2021, 12, 6041–6047 CrossRef CAS PubMed.
- L. Cao, Y. Shen, B. Lei, W. Zhou, S. Li, Y. Liu, Y. Lu, K. Zhang, H. Ren, Y. Q. Li, J. X. Tang and W. Wang, Laser Photonics Rev., 2023, 17, 2200861 CrossRef CAS.
- S.-Q. Sun, C. Liu, M. Zhu, Y.-L. Xu, W. He, D.-D. Feng, C.-C. Huang, Q. Sun, Y.-M. Xie, Y.-Y. Li and M.-K. Fung, Mater. Today Energy, 2022, 29, 101139 CrossRef CAS.
- G. Li, F. W. Rivarola, N. J. Davis, S. Bai, T. C. Jellicoe, F. de la Pena, S. Hou, C. Ducati, F. Gao, R. H. Friend, N. C. Greenham and Z. K. Tan, Adv. Mater., 2016, 28, 3528–3534 CrossRef CAS PubMed.
- S. Sun, M. Lu, Y. Zhong, P. Lu, F. Qin, Y. Gao, X. Bai, Z. Wu and Y. Zhang, ACS Energy Lett., 2022, 7, 3974–3981 CrossRef CAS.
- D. Lee, S.-J. Lee, J. H. Kim, J. Park, Y.-C. Kang, M. Song, H. W. Lee, H. S. Kim and J. W. Choi, Adv. Funct. Mater., 2022, 32, 2202207 CrossRef CAS.
- L. Gao, T. Cheng, L. Gou, Y. Zhang, Y. Liu, L. Yuan, X. Zhang, Y. Wang, F. Meng and J. Zhang, ACS Appl. Mater. Interfaces, 2023, 15, 18125–18133 CrossRef CAS PubMed.
- X. Bi, X. Yang, S. Xu, Y. Tong, X. Liang, Y. Nan, L. Zhu, N. Wang and J. Wang, J. Mater. Chem. C, 2023, 11, 10404–10408 RSC.
- G. Zou, Z. Li, Z. Chen, L. Chu, H. L. Yip and Y. Cao, Adv. Funct. Mater., 2021, 31, 2103219 CrossRef CAS.
- M. Li, Z. Zhu, Z. Wang, W. Pan, X. Cao, G. Wu and R. Chen, Adv. Mater., 2024, 36, e2309428 CrossRef PubMed.
- H. S. Kim, Y. Kim, S. Cho, S. Jeong, S. N. Lim, H. S. Jung, Y. Choa, S. Lee, K. Park, W. Chae and J. Y. Woo, J. Phys. Chem. C, 2024, 8, 3343–3350 CrossRef.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- A. Swarnkar, V. K. Ravi and A. Nag, ACS Energy Lett., 2017, 2, 1089–1098 CrossRef CAS.
- A. Dutta, R. K. Behera, P. Pal, S. Baitalik and N. Pradhan, Angew. Chem., Int. Ed., 2019, 58, 5552–5556 CrossRef CAS PubMed.
- A. Dutta, R. K. Behera, S. K. Dutta, S. Das Adhikari and N. Pradhan, J. Phys. Chem. Lett., 2018, 9, 6599–6604 CrossRef CAS PubMed.
- W. Cui, J. Zhao, L. Wang, P. Lv, X. Li, Z. Yin, C. Yang and A. Tang, J. Phys. Chem. Lett., 2022, 13, 4856–4863 CrossRef CAS PubMed.
- A. Swarnkar, W. J. Mir and A. Nag, ACS Energy Lett., 2018, 3, 286–289 CrossRef CAS.
- Y. Hu, F. Bai, X. Liu, Q. Ji, X. Miao, T. Qiu and S. Zhang, ACS Energy Lett., 2017, 2, 2219–2227 CrossRef CAS.
- C. F. J. Lau, M. Zhang, X. Deng, J. Zheng, J. Bing, Q. Ma, J. Kim, L. Hu, M. A. Green, S. Huang and A. Ho-Baillie, ACS Energy Lett., 2017, 2, 2319–2325 CrossRef CAS.
- M. T. Klug, A. Osherov, A. A. Haghighirad, S. D. Stranks, P. R. Brown, S. Bai, J. T. W. Wang, X. Dang, V. Bulović, H. J. Snaith and A. M. Belcher, Energy Environ. Sci., 2017, 10, 236–246 RSC.
- L. M. Wheeler, E. M. Sanehira, A. R. Marshall, P. Schulz, M. Suri, N. C. Anderson, J. A. Christians, D. Nordlund, D. Sokaras, T. Kroll, S. P. Harvey, J. J. Berry, L. Y. Lin and J. M. Luther, J. Am. Chem. Soc., 2018, 140, 10504–10513 CrossRef CAS PubMed.
- J. Park, S. Park, S. Cho, Y. Kim, H. Kim, S. Jeong and J. Y. Woo, New J. Chem., 2022, 46, 19514–19522 RSC.
- D. Yoo, J. Y. Woo, Y. Kim, S. W. Kim, S. H. Wei, S. Jeong and Y. H. Kim, J. Phys. Chem. Lett., 2020, 11, 652–658 CrossRef CAS PubMed.
- M. J. Kim, M. S. Kim, J. Y. Woo and S. Y. Cho, J. Phys. Chem. Lett., 2024, 15, 11437–11444 CrossRef CAS PubMed.
- K. Zheng, K. Žídek, M. Abdellah, M. E. Messing, M. J. Al-Marri and T. Pullerits, J. Phys. Chem. C, 2016, 120, 3077–3084 CrossRef CAS.
- M. B. Johnston and L. M. Herz, Acc. Chem. Res., 2016, 49, 146–154 CrossRef CAS PubMed.
- Y. H. Zhou, Y. H. Lou, X. Q. Wang, K. L. Wang, J. Chen, C. H. Chen and Z. K. Wang, Adv. Opt. Mater., 2021, 10, 2101655 CrossRef.
- H.-C. V. Tran, B. Kim, H. Kim, S. Park, J. Y. Woo and S. Jeong, Chem. Mater., 2022, 34, 6402–6407 CrossRef CAS.
- Y. Shen, Y. Q. Li, K. Zhang, L. J. Zhang, F. M. Xie, L. Chen, X. Y. Cai, Y. Lu, H. Ren, X. Gao, H. Xie, H. Mao, S. Kera and J. X. Tang, Adv. Funct. Mater., 2022, 32, 2206574 CrossRef CAS.
- W. Cai, M. U. Ali, P. Liu, M. He, C. Zhao, Z. Chen, Y. Zang, M. C. Tang, H. Meng, H. Fu, G. Wei and H. L. Yip, Adv. Sci., 2022, 9, 2200393 CrossRef CAS PubMed.
- J. M. Richter, M. Abdi-Jalebi, A. Sadhanala, M. Tabachnyk, J. P. H. Rivett, L. M. Pazos-Outon, K. C. Godel, M. Price, F. Deschler and R. H. Friend, Nat. Commun., 2016, 7, 13941 CrossRef CAS PubMed.
- J. Y. Woo, Y. Kim, J. Bae, T. G. Kim, J. W. Kim, D. C. Lee and S. Jeong, Chem. Mater., 2017, 29, 7088–7092 CrossRef CAS.
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS PubMed.
- H. Zhang, Y. Wu, C. Shen, E. Li, C. Yan, W. Zhang, H. Tian, L. Han and W. H. Zhu, Adv. Energy Mater., 2019, 9, 1803573 CrossRef.
- M. Kazes, T. Udayabhaskararao, S. Dey and D. Oron, Acc. Chem. Res., 2021, 54, 1409–1418 CrossRef CAS PubMed.
- D. J. Slotcavage, H. I. Karunadasa and M. D. McGehee, ACS Energy Lett., 2016, 1, 1199–1205 CrossRef CAS.
- A. J. Knight, J. B. Patel, H. J. Snaith, M. B. Johnston and L. M. Herz, Adv. Energy Mater., 2020, 10, 1903488 CrossRef CAS.
- H.-S. Yun, K. Noh, J. Kim, S. H. Noh, G.-H. Kim, W. Lee, H. B. Na, T.-S. Yoon, J. Jang, Y. Kim and S.-Y. Cho, Phys. Status Solidi RRL, 2019, 14, 1900573 CrossRef.
- Y. Nah, O. Allam, H. S. Kim, J. I. Choi, I. S. Kim, J. Byun, S. O. Kim, S. S. Jang and D. H. Kim, ACS Nano, 2021, 15, 1486–1496 CrossRef CAS PubMed.
- S. G. Motti, J. B. Patel, R. D. J. Oliver, H. J. Snaith, M. B. Johnston and L. M. Herz, Nat. Commun., 2021, 12, 6955 CrossRef CAS PubMed.
- S. Sun, P. Huang, X.-g. Wu, C. Chen, X. Hu, Z. Bai, A. Pushkarev and H. Zhong, J. Phys. Chem. C, 2024, 128, 3602–3608 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01550b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |