
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Operando APXPS for direct probing of Li ion battery LCO electrode/electrolyte interface chemistry during lithiation/delithiation†
Qianhui Liu a,
Tove Ericsona,
Robert Tempertonb,
Ida Källquistc,
Fredrik Lindgrenac,
Laura Kinga,
Alenka Križande,
Katie L. Browningf,
Ethan J. Crumling,
Gabriel M. Veithf and
Maria Hahlin*ac
a,
Tove Ericsona,
Robert Tempertonb,
Ida Källquistc,
Fredrik Lindgrenac,
Laura Kinga,
Alenka Križande,
Katie L. Browningf,
Ethan J. Crumling,
Gabriel M. Veithf and
Maria Hahlin*ac
aDepartment of Chemistry - Ångström, Uppsala University, 751 20 Uppsala, Sweden. E-mail: maria.hahlin@kemi.uu.se
bMAX IV Laboratory, Lund University, 225 94 Lund, Sweden
cDepartment of Physics and Astronomy - Ångström, Uppsala University, 751 20 Uppsala, Sweden
dNational Institute of Chemistry, Hajdrihova 19, Ljubljana, Slovenia
eFaculty of Chemistry and Chemical Technology University of Ljubljana, Večna Pot 113, Ljubljana, Slovenia
fChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, 37831 Tennessee, USA
gAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
First published on 21st May 2025
Abstract
The real-time interface chemistry between the lithium cobalt oxide (LCO) working electrode and the LiClO4/propylene carbonate (PC) electrolyte is investigated during lithiation/delithiation using dip-and-pull ambient pressure photoelectron spectroscopy (APXPS). The APXPS results appear to exhibit the seldom discussed Co2+ state in the LCO structure, where the operando measurements indicate electron transfer among Co2+, Co3+, and Co4+ states. Specifically, the lithiation of LCO reduces the Co4+ state to both Co3+ and Co2+ states, where, as a function of voltage, reduction to the Co2+ state is initially more pronounced followed by Co3+ formation. In addition, a delay in surface delithiation is observed during the reverse potential steps. This is discussed in terms of overpotential at the interface measurement position as a consequence of the dip-and-pull setup for this experiment. Finally, the shifts in the apparent binding energies of the spectral features corresponding to the electrolyte and LCO at their interface show that the electrochemical potentials at delithiation voltage steps are different from the lithiation steps at the same applied voltages. This further explains the non-responsive delithiation. The BE shift observed from the LCO surface is argued to be dominantly due to the semi-conductive nature of the sample. Overall, this article shows the importance of operando APXPS for probing non-equilibrium states in battery electrodes for understanding electron transfer in the reactions.
Introduction
Since their first demonstration four decades ago, Li-ion batteries (LIBs) have revolutionized the portable energy storage market, mainly due to their high energy density and reversible charge/discharge properties.1 The development of the positive electrode has since drawn significant attention as it is often a capacity limiting factor. Lithium cobalt oxide (LCO), which for a long time was the most widely used positive electrode in commercial Li-ion batteries, has a practical capacity of roughly 140–150 mA h g−1 provided by the Li deintercalation/intercalation and electron transfer from/to the layered LixCoO2.2 In contrast to the higher theoretical capacity (273 mA h g−1), the suppressed practical capacity is due to the collapse of crystal structure when removing more than 50% of the lithium.3 During battery cycling it is reported that a phase transition occurs at high degree of delithiation, going from trigonal semi-conductive phase (LixCoO2 0.75 < x< 1) to monoclinic metallic phase (x < 0.75).4,5The change of the phase and the chemical states at the electrode due to lithiation/delithiation largely affect the battery performance in terms of capacity and reversibility.6,7 In the most simplistic models it is generally considered that the charge is fully transferred from/to Co during delithiation/lithiation, resulting in the transformation between Co3+ and Co4+ oxidation states.3,6–9 Although there are currently more complex models proposed that also include oxygen participation, what is still lacking is a clear picture of the interface chemistry during operation. This includes information on the transformation between chemical states at the electrode surface as well as changes in electrochemical potentials over the interface under during battery operation.
X-ray photoelectron spectroscopy (XPS) measures the kinetic energy of the photoelectrons emitted under X-ray illumination via the photoelectric effect, from which chemical information about the surface (and a few nm below surface) can be inferred. This makes XPS an efficient tool for ex situ characterization of the surface chemistry in LIBs after cycling.10 However, the surface chemistry of the battery electrode is sensitive to local environment, and is therefore dependent on real-time battery operation conditions. For example, volatile species in the interphase layers depletes under the ultra-high vacuum (UHV) condition most commonly used in XPS measurements.11 Additionally, intermediate states that exist only during battery operation or a non-equilibrated electrode surface are impossible to address using postmortem measurements. Operando surface/interface sensitive techniques, capable of measuring the electrode/electrolyte interface during battery cycling, are thus highly desirable. Ambient pressure X-ray photoelectron spectroscopy (APXPS) now routinely allows measurements in gas/vapor environments, in the mbar pressure range, opening up the possibility to maintain the liquid electrolyte phase which enables in situ/operando measurements of battery solid/liquid interfaces during redox reactions.12–17 One approach to access the solid/liquid electrochemical interfaces is the “dip-and-pull” method,12,17,18 which we apply here to a LIB. The setup involves suspending a three-electrode LIB system above a beaker of liquid electrolyte solution. After dipping into the liquid the electrodes are pulled up from the liquid whilst remaining in contact with the electrolyte solution to maintain a closed electric circuit. The dip-and-pull action forms a thin meniscus of electrolyte solution on a flat electrode surface, which then can be probed through with APXPS, and thus the electrode/electrolyte interface can be measured. For successful operando APXPS measurements, the meniscus needs to be thick enough as to conduct ions, evident from voltage dependence of the core level spectra binding energy from liquid species, and thin enough for photoelectrons to escape.19–21 Previous measurements performed by our group have shown that LIB electrolyte solutions can be successfully probed by operando dip-and-pull APXPS.16,19 Recently, Capone et al. probed the solid electrolyte interphase formation on a model glassy carbon electrode with APXPS, underscoring the effectiveness of APXPS for real-time interface probing in Li ion battery.22
In this study, the interface chemistry between a LCO thin film positive electrode and 1 M LiClO4/propylene carbonate (PC) electrolyte solution is probed by dip-and-pull operando APXPS during one cycle of lithiation/delithiation. Specifically, we are able to follow the complex transitions between Co2+/3+/4+ oxidation states during delithiation/lithiation, as well as monitor the electrochemical potential changes during operating conditions. Less change of Co oxidation states and an overall higher measured BE for both electrode and electrolyte peak during delithiation compared with lithiation reveals that extra overpotential is required to delithiate the sample. Overall, this article shows the importance of operando APXPS for developing a deeper understanding of the charge transfer over the electrode/electrolyte interface.
Results and discussion
Identification of oxidation states in the pristine LCO surface under ambient PC environment
The LCO electrode surface was investigated in a propylene carbonate vapor environment prior to being immersed in the liquid electrolyte solution. The resulting Co 2p, O 1s and C 1s spectra from the undipped electrode are presented in Fig. 1. The Co 2p spectra (Fig. 1a) is curve-fitted based on the raw data shape with the support from a set of theoretical and experimental studies on LCO and other cobalt compounds.23–25 It is noticed that in spite of the numerous XPS studies on LCO structure, a clear consistent interpretation of its Co 2p spectrum is still lacking.26 This stems from uncertainties in binding energy position, peak broadening and complex satellite structures, which make quantitative or accurate qualitative analysis of XPS spectra for Co chemical states challenging.6,27–31 Here, the asymmetric line shape of Co 2p3/2 spectra is fitted with three components (see fitting parameters in Table S1†). A sharp peak at 780 eV (red) is assigned to Co3+, and a peak at 781.8 eV (green) is assigned to Co4+.28 Between the Co3+ and Co4+ peaks, an additional contribution at ∼781 eV is assigned to Co2+ (yellow).4,23,30,32 Calculating the percentage of each oxidation state relative to the sum of the oxidation states results in 20% Co2+, 64% Co3+ and 16% Co4+, giving an average oxidation state of 3.0 at the surface of the uncycled electrode. Given that the relative weight of the satellite structures are different for these states,3,4 these values do not represent an actual distribution of state but rather serves as a reference value for the pristine LCO material when later comparing to the cycled sample.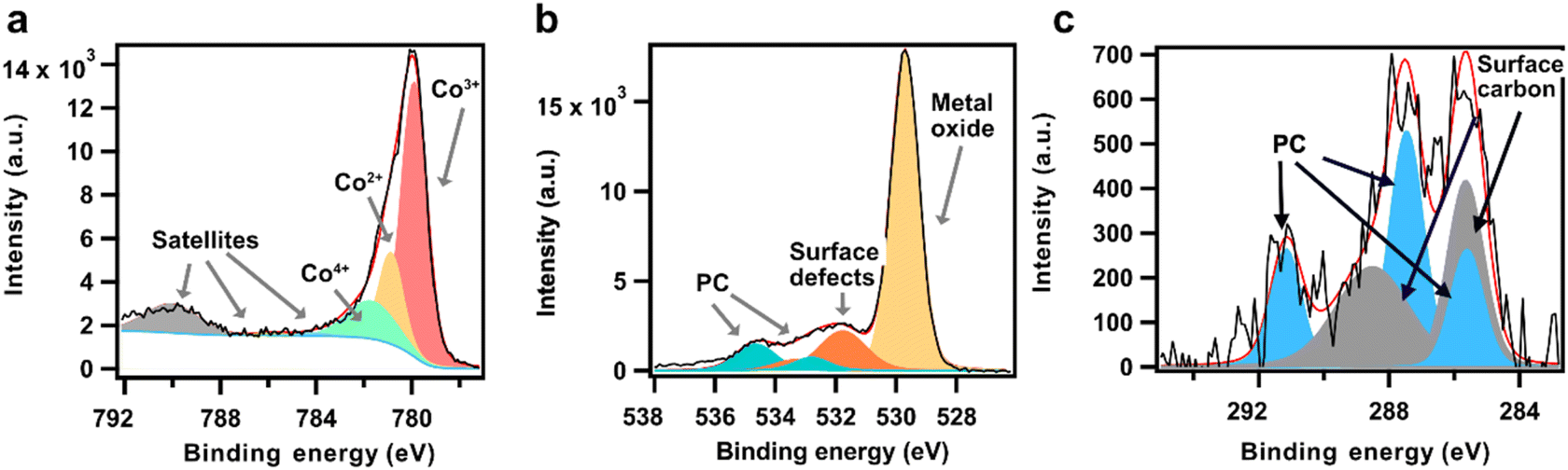 | ||
| Fig. 1 The (a) Co 2p, (b) O 1s, and (c) C 1s spectra of pristine LCO sample under the 7.9 mbar PC vapor and Ar atmosphere. | ||
The presence of Co2+ in LCO was less discussed in previous reports, despite of the significant contribution in the raw spectra.4,29,33,34 Some studies show that Co2+ is likely to show up due to local deficiency of oxygen,35 as well as due to surface degradation.36,37 Furthermore, heat treatment can remove the oxygen from the sample, or Ar ion sputter cleaning has also shown to cause Co ion reduction. In view of this, together with the lack of a general consistent interpretation of the Co 2p spectrum, a short motivation for our curve fitting model follows. In this work, by overlaying the two spectra acquired at lithiated state (3.2 V vs. Li+/Li) and delithiated state (4.0 V vs. Li+/Li) from the following operando APXPS measurement (Fig. S1†), two contributions at ∼781 eV and ∼783 eV are revealed at high-energy side of Co 2p main line. To model this behavior, a curve fit involving three peaks, assigned to Co3+, Co2+ and Co4+, is necessary. Furthermore, curve fitting tests excluding the Co2+ state resulted in a curve fit that lacks physical and chemical meaning (Fig. S2†). The binding energy of the Co2+ state with around 1 eV above that of Co3+ is further supported by previous studies.4,23,32 A higher BE position of Co2+ compared to that of Co3+ with higher oxidation state is a consequence of final state effect which occurs for some transition metal compounds with a localized open-d shell.26,38 A UHV HAXPES investigation on a replicate thin film LCO sample with high energy resolution substantiates the contribution from Co2+ (Fig. S3†). Thus, our data supports a curve fitting model using Co2+, Co3+, and Co4+ states.
The Co 2p shake-up satellite originates from photoelectrons with the energy loss from valence states excitation.29,39 The corresponding satellite peaks for Co3+ and Co4+ are assigned with energy spacing of 10 eV and 4.2 eV relative to the corresponding main peaks, respectively.4,25,28,29 The satellite position of Co2+ from LCO is hard to determine due to partially overlapped position with Co3+/Co4+ satellite peaks, and a lack of conclusive experimental or theoretical reports. Some studies on cobalt monoxide presented a broad satellite peak approximately 5–6 eV above the main peak of Co2+ with possibly several final states.23,32,40,41 However, since this satellite feature is due to the core hole screening effect by ligand O atom, the variation of band structure for different compounds will affect the satellite peak positions (see Fig. S4†).6,40,42 Considering a high relative weight of satellite for Co2+ state,3,35 the Co2+ satellite peak is expected to have a visible contribution.4 Here from the spectra, a signal at 3.4 eV above the Co2+ the main peak (4.5 eV above the spectra mainline) is presented, which we attribute to the corresponding satellite (blue peak).3,35 The relative ratio of it to the Co2+ main peak is on average 42% (Fig. S5†). We also note that due to the energy overlap between the Co 2p1/2 peak and Co 2p3/2 satellites it is not feasible to accurately determine the Shirley background offset.43 Thus, the satellite peak shape and intensity after background subtraction might to some degree be affected. Therefore, the components compositions and ratios presented below are determined solely from the main Co2p3/2 peaks.
The O 1s spectrum in Fig. 1b is dominated by the peak at 529.7 eV (yellow), assigned to the transition metal (TM) oxide from LCO.2,44 The two additional oxides with higher energy (orange) at 531.8 and 533.4 eV are attributed to the surface compounds typically seen in TM oxides, and have significantly lower intensity than the main oxide peak.45 The two components (green) at 534.7 and 532.8 eV are assigned to surface adsorbed PC. This is supported by the C 1s spectrum (Fig. 1c), where three components at 291.2, 287.5, and 285.6 eV with an atomic ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 (blue) are assigned to C
:2:1 (blue) are assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O and C–C from PC, respectively. The intensity ratio (total peak area ratio) of O 1s (IO) to C 1s (IC) from the same species PC here is 2.58, close to the theoretical intensity ratio (see method section). The rest of the contributions in the C 1s spectrum originate from surface carbon (grey peaks at 288.6 and 285.7 eV). The low C 1s intensity indicate that only minor amounts of carbonates or other surface species are present on the undipped LCO sample.
O, C–O and C–C from PC, respectively. The intensity ratio (total peak area ratio) of O 1s (IO) to C 1s (IC) from the same species PC here is 2.58, close to the theoretical intensity ratio (see method section). The rest of the contributions in the C 1s spectrum originate from surface carbon (grey peaks at 288.6 and 285.7 eV). The low C 1s intensity indicate that only minor amounts of carbonates or other surface species are present on the undipped LCO sample.
Oxidation state evolution during operando APXPS measurements
The electrodes were dipped into the electrolyte solution and the LCO working electrode was delithiated before the APXPS measurement by applying a voltage of 4.2 V vs. Li+/Li that was held for 200 min, (i.e. 2.65 V of potential differences between working electrode (WE) and LTO reference electrode, Fig. S6†), during which Li ions are deintercalated from the LCO working electrode and intercalated into the LTO counter electrode. The voltage over the cell was thereafter changed in steps from 4.1 V to 3.2 V vs. Li+/Li, to discharge the battery (i.e. lithiate the LCO working electrode), followed by the reversed steps back up to 4.1 V to charge the battery (i.e. delithiate the LCO working electrode). Voltage changes were applied when the electrode was at pulled-up position. APXPS measurements were conducted a few mm above the top of meniscus, since no signal from the electrode could be detected below this point. Fig. S7† exhibits changes in O 1s spectra during and after the potential ramping. The time-delayed core level shift seen for the electrolyte peaks, in contrast with the instant shift of TM oxide peak, shows individual voltage responses of the electrode and the electrolyte, and additionally that the ion redistribution time in the electrolyte is longer than the electronic response of the LCO. This also confirms that the probed area is electrochemically connected to the bulk electrolyte. APXPS measurements under close to steady-state conditions were acquired when the set potential value was reached and the current had decreased to a low value (below 15 μA). After the measurement at one voltage step, the electrodes were again dipped into the electrolyte before placed back in measurement position for the next voltage step to replenish the electrolyte. For some potentials an additional set of measurements was performed when observing a rising of current when dipping back to liquid (Fig. S8†). The Co 2p spectra at each voltage step in the discharge–charge cycle is presented in Fig. 2a–h. Fig. 2i shows the Co4+ % (blue dots and lines) given by the ratio of the Co4+ peak area to the sum of Co3+, Co2+ and Co4+ main peaks, and the average Co oxidation state (purple dots and lines) determined by the average contribution from all chemical states weighted by the corresponding valence. During lithiation the average Co state and the Co4+ % sees a continuous decrease. This suggests an active TM redox occurring during lithiation. The high level of correlation between Co4+ and Co average states trends (Fig. 2i) implies a continuous electron transfer to Co4+. | ||
| Fig. 2 Co 2p spectra measured during (a)–(d) lithiation and (e)–(h) delithiation of LCO after a pre-delithiation of material holding at 4.2 V. The spectra at each potential were measured when current converges to be stable (current below 15 μA) (i) Co4+ % (given by the Co4+ main peak area divided by the sum of all Co components main peaks area, same for Co3+ and Co2+ %) and averaged Co state (calculated by the relative weight of each Co state). And (j) Co3+ and Co2+ %. The data dots in (i) and (j) is from a summed Co 2p spectrum of 4 spectra due to low signal-to-noise ratio of individual spectrum. 20–25 spectra were acquired from Co 2p at each applied potential at 2 or 3 spots and (a)–(h) shows the full sum of these spectra. The dashed lines represent the mean value of the distributed dotted data. The solid lines are derived from curve fitting the summed Co 2p spectrum over the total 20–25 scans, corresponding to spectra (a)–(h). The overall trends of each components vs. voltages from both methods are well-aligned. Deviation at some voltages (e.g., 3.6 V delithiation) may result from a few outlier dotted points due to low signal-to-noise ratio of its corresponding spectrum. | ||
The evolution of the Co2+ and Co3+ population during lithiation/delithiation extracted from our curve fit model of the APXPS data is shown in Fig. 2j. Combined with the Co4+ population (Fig. 2i), the results reveals that all three components respond actively to the lithiation. Over the full voltage range during lithiation, the decrease of Co4+ % give rise to increases of both Co3+ and Co2+. It is further seen that the increase of Co2+% starts at an earlier stage of lithiation (from 4 V) with a change of higher magnitude (12% to 18%) compared to the small formation of new Co3+% states (49% to 51%). A few possible reaction pathways for the charge transfer compensation are plausible; (1) two electrons transfer to Co4+ yielding a one-step transition between Co4+ → Co2+ states, (2) two successive one-electron-transfer steps Co4+ → Co3+ → Co2+, 3) or a one-electron-transfer Co4+ → Co3+, followed by a fast disproportionation of two Co3+ states, 2 Co3+ → Co2+ + Co4+. The observed initial decrease of the Co3+ state can be argued to support the latter reaction pathway, while on the other hand the continuous decrease of the number of Co4+ states support the previous options. The data in these measurements are not sufficient to determine the exact reaction pathway, and further development of instrumentation with higher time resolution is most likely required to capture a possible intermediate Co3+ state. Up to now, the electron transfer mechanism during the Li ion intercalation/deintercalation to/from LCO is not fully understood. The expected behavior according to the simplistic “charge compensation” model is that the electron transfer occurs on Co, and specifically, through a transition between the Co4+ and Co3+ states.3,46 The presented data however implies the formation of Co2+ states at the surface of the LCO during lithiation, and indicates a more preferable state of Co2+ than Co3+ or Co4+ at the start of lithiation. Thus, we can conclude that to accurately describe the charge transfer processes at the LCO surface the models need to include the role of Co2+.
In recent years there has been much research emphasis placed on understanding the role of oxygen in the charge compensation during lithiation/delithiation.3,9,46,47 In the hybrid oxygen anion and Co cation redox model (HACR) an emerging XPS peak at ∼1 eV higher binding energy relative to the TM oxide peak in the O 1s spectra was linked to such oxygen redox in a voltage region >4.35V.8,9 However, Fantin et al. recently associated the presence of this peak with the rock-salt formation, attributing it to the unstable structure at high voltages.37 Meanwhile, the energy shift of the state formed from the oxygen redox, relative to the TM oxide, was calculated to be a minor 0.3 eV37. Such subtle shift is challenging to resolve with our limited energy resolution; however, a broadening of the TM-O peak may indicate its presence. In this work, the O 1s spectra have been curve fitted using a single TM-O peak, together with three peaks related to the electrolyte (Fig. 4). The slight variation of the full width half maximum of the TM-O peak shows no correlation to the material lithiation degree (Fig. S9†). Therefore, the data show no support for oxygen redox at the electrolyte/electrode interface. Furthermore, this work utilizes the stable cycling region for LCO, and thus cannot determine any oxygen loss as described in the HACR model.
Lack of surface delithiation-insufficient overpotential and ion transport resistance
At the end of the lithiation, LCO was held at 3.2 V for 2.5 hours under steady state lithiation current (−5 to −2 μA, see Fig. S10,† ∼0.11 C rate, calculated based on total capacity of 93 μA h−1) before starting the reverse potential steps. The following APXPS measurement points, i.e. the 3.2 to 3.6 V voltage step, show a continuous decrease of average Co state and Co4+ % due to the potential hold. The relative redistribution of the Co4+, Co3+, and Co2+ percentages further show a simultaneous transformation among the states during this time. However, on further increasing the WE voltage the values of average Co state is unreversed. This is at first thought surprising, but we attribute this to be a consequence of ineffective delithiation at the measurement position as outlined below.First of all, the delithiation of LCO material effectively occurs from 3.9 V, thus no major delithiation and/or Co states changes should be expected before reaching above 3.9 V (see Fig. S11† charge–discharge curve) as the driving force for redox reaction is very small in the lower voltage region. Furthermore, for a regular battery, the applied voltage often does not equal the driving potential for lithiation/delithiation and generally overpotentials are needed to overcome the internal resistances (e.g. iR-drop, activation polarization, concentration polarization) during operation.
Here, the dip-and-pull APXPS measurements are conducted on the sample 3 mm above the “dipped” position. This implies that lithiation/delithiation at the measurement spot is dependent on e.g. ionic transport through the electrolyte that resides in the 0.1–0.5 μm wide electrolyte channels that perforate the LCO material (Fig. 3a), and our hypothesis is that this may substantially increase the resistance between bulk electrolyte and the measurement position.48
 | ||
| Fig. 3 (a) The SEM image of LCO thin film surface. Cracks with a width of 0.1–0.5 μm are observed (see also Fig. S14† for cross section SEM), which here serves as electrolyte channels to transport electrolyte through capillary force, enabling the lithiation/delithation of the sample above where it was dipped into liquid. (b) The schematic illustration of limited Li ion transportation. | ||
To test this hypothesis APXPS measurement that probe vertically along the LCO surface while cycling at a pulled-up position at an applied voltage of 4.2 V was performed (HIPPIE beamline in MAXIV, Sweden, Fig. S12†). Both the solid and the liquid phase features are probed in the same O 1s spectrum. Shifts in the BE positions are observed for both the LCO surface and the electrolyte along the 500 μm vertical length, both below and above the probed-interface position. The LCO TM oxide signal shows a ∼0.4 eV shift from the “interface position” to the pure solid. This clearly demonstrates a non-uniform electrochemical potential distribution on the sample at different heights. Furthermore, the shift of the electrolyte peaks (0.3 eV) shows that the electrochemical potential of the electrolyte also varies depending on height. This means that there is a resistance present in the electrolyte solution. This effectively means that the driving potential at the interface measurement point is less than the set voltage, and that in this APXPS measurement geometry a higher applied voltage than 3.9 V is needed for delithiation. In addition, the hypothesis is supported by the electrochemical current rising following the re-dipping of the sample back into the electrolyte (see blue arrow in Fig. S10†).
In our experiment, the applied voltage window ranges between 4.1-3.2 V. From the cycling curve in Fig. S7† it is clear that for 4.1–3.9 V there are continuous reactions, while for 3.9–3.2 V very little is on-going. Assuming a voltage drop from the bulk electrolyte to the measurement point of 0.2 V, this would mean that 3.9 V would barely be reached when trying to delithiate with 4.1 V applied voltage. We would therefore still be operating at the “no-action” part of the curve, and thus no delithiation is expected. While for reverse voltage steps (lithiation) it would instead stay on the reaction-region below redox voltage.
Potentials at LCO electrolyte interface
The electrochemical potential changes over the solid/liquid interface can be investigated from operando APXPS spectra of the electrolyte solution for the case of a metallic WE.19 In this work, the interface potential between the thin film semi-conductive LCO electrode and liquid electrolyte solution, during electrode lithiation/delithiation, is directly probed from O 1s core level where the spectroscopic response from the electrode and electrolyte are probed simultaneously (Fig. 4). The curve fitted O 1s spectra exhibit two peaks (green) at binding energies 533.1 and 531.2 eV (at 4.1 V in Fig. 4a), with a relative area ratio of 2:1, which are attributed to the C–O–C and CO from PC, respectively.14 The peak (pink) at 531.6 eV is assigned to the salt LiClO4, for which the fitted peak position is constrained to be 0.42 eV above that of the minor PC peak (CO). The contribution to the lowest BE peak is from the LCO TM oxide (MO, yellow peak at 528.6 eV). Fig. 5a and b gives the extracted binding energies of each O components from electrolyte and TM oxide as a function of applied voltage, where each scan/spectrum was individually fitted and are included as single dots to show the spread in data.
 | ||
| Fig. 4 O 1s spectra from interface during (a)–(d) lithiation of LCO (from 4.1 V to 3.2 V vs. Li) and (e)–(h) delithiation of LCO (from 3.6 V to 4.1 V vs. Li+/Li). The blue double peaks with 1.8 eV energy separation refer to the electrolyte solvent PC, and the pink peak is attributed from theLiClO4 salt. The yellow peak is assigned to the TM oxide. | ||
 | ||
| Fig. 5 Measured binding energy of (a) PC CO contribution from O 1s, (b) metal oxide peak from O 1s, and (c) Co3+ peak position from Co 2p spectra during lithiation and delithiation. The blue line in each graph is 1:1 to potential as a reference. The data points in the O 1s at an applied potential are from 10–15 spectra acquired from 2–3 measurement spots on the sample. Each data point is a fitted BE position from one spectrum. Each data point of Co 2p is from a summed spectrum of 4 spectra due to low signal to noise ratio. 20–25 spectra were acquired from Co 2p at each applied potential at 2 or 3 measurement spots. | ||
The voltage stepping down from 4.1 to 3.2 V (lithiation voltage steps) shifts the BE for all components to higher BE values, and this is reversible during the following delithiation voltage steps. A nearly 1 eV/1 V (Fig. 5a) response of the PC solvent oxygen peak shifts relative to the applied voltages is observed over the full voltage range (see also C 1s BE shift in Fig. S13†). The O 1s binding energies from LCO electrode TM oxide (Fig. 5b) also shift with respect to the corresponding voltages, but with an overall smaller magnitude Fig. 5b) than the electrolyte peak shift. This BE shift can also be observed for the Co 2p spectra (Fig. 5c) which follows the same trends as the LCO O 1s peak. Finally, for both electrolyte and electrode spectra, significantly higher binding energies are seen during delithiation compared to lithiation at all applied voltages, yielding a hysteresis of BE energy vs. applied potential. It is challenging to fully determine the origin of these binding energy changes, however some suggestions to its origin follow.
Concerning the shift of the LCO O 1s and the Co 2p as a function of voltage: Firstly, as shown above (Fig. 2i and j), all the measurement points in the delithiation series are on essentially the same material, and thus no chemical shift in the core level spectra is expected. Secondly, for a semi-conductive WE, the alignment of the Fermi level with the spectrometer/metal current collector generally cause band bending and alignment barriers. Changing the voltage in such a system can cause an increase/decrease in band-bending which is seen as a shift in core level binding energy.20,49 In view of this, we argue that the observed binding energy changes in the LCO peaks on delithiation voltage are dominantly related to the semiconducting nature of the material. As the changes are similar for the lithiation, it is likely that it mainly has the same origin, however no firm conclusion can be made for the lithiation voltage steps since here also the material is changing as a function of voltage.
As the binding energy is referred to the Fermi level, which essentially reflects the electrochemical potential of electrons, the higher BE levels for the electrolyte solution at delithiation voltages steps compared to the equivalent lithiation steps reveals that the electrolyte is not electrochemically at the same energy level. A binding energy difference of around 0.2–0.3 eV is observed at the same applied potential when comparing delithiation to lithiation. The deviation is most probably due to sluggish ion mass transport in the meniscus electrolyte, which causes the electric potential in the meniscus to differ from the bulk electrolyte under operating conditions. This would essentially mean that an overpotential is needed to drive the reaction at the measurement point, which also agrees with the discussion above regarding the difficulty to delithiate the material.
As a final note, it should be acknowledged that with the challenges of resolving the full picture of interface potentials, more detailed operando APXPS measurements is vital. However, the direct probing of real-time electrode/electrolyte electrochemical potential, under varying cell voltage and chemical environment, clearly shows the importance of operando measurements for fully characterizing and understanding the interface functionality in LiBs.
Conclusions
A LCO/electrolyte interface was directly probed with operando APXPS. Curve fitting of the Co 2p spectra alludes to the presence of Co2+ in the LCO. An active Co redox during lithiation is observed from the changing relative ratios of the Co oxidation states. A well-aligned decrement of Co4+ % and averaged Co state, during lithiation from 4.1 to 3.2 V (vs. Li+/Li), reveals Co4+ as a main electron receiver during electron transfer. Correspondingly, both Co2+ and Co3+ respond to the lithiation, with an immediate increase of Co2+ from the beginning of lithiation compared to a slower response from Co3+.A constant Co chemical state from APXPS is observed at the surface of LCO during delithiation. This is attributed to a lack of overpotential resulting from the large electrolyte resistance between the interface probing position and the bulk liquid in this dip-and-pull experiment. This is also evidenced from the electrolyte/electrode core level BE positions, which shows overall higher values during delihitation voltage steps compared to those in lithiation at the same applied voltages.
This work presents a step forward in probing of LIB electrodes using operando APXPS, providing new insight into the reaction mechanisms during battery cycling. The demonstrated ability to resolve electrochemical potential distributions at the interface of real battery materials during operation opens up further studies to gain an atomic level understanding of functional battery materials and their interfaces.
Method
Materials
The investigated working electrodes (WEs) were thin films (1.8 μm thick, see Fig. S14 SEM and S15† EDS of LCO thin film cross section) of sputter deposited LCO (5 sccm Ar, 20 mTorr, 80 W, base pressure 7.7×10−7 mbar, deposition time 8 h 10 min). The substrate was alumina with a sputter deposited Pt layer on-top. The sample was transferred in air to an oven where it was annealed at 700 °C in O2 for 2 h, followed by a short transfer in air for packing.Operando APXPS setup
The dip-and-pull experiments were conducted at Beamline 9.3.1 tender APXPS beamline at the Advanced Light Source (ALS). A three-electrode cell setup was constructed in a glove-box and sealed in a bag before being transferred to the experimental chamber. An Ar glove bag was used to introduce the electrodes and electrolyte solution to the chamber. The counter electrode (CE) was a composite film comprising of lithium titanate oxide (LTO, Posco GS Materials), CMC (sodium carboxylate cellulose) and carbon black (Imerys graphite & carbon, C.NERGY SUPER C65) in 8:1:1 mass ratio coated on an aluminum substrate. An Al-wire with the LTO slurry dipped on the tip was used as a reference electrode (RE). The effective mass loading for the RE electrode was much smaller than for the CE. The RE was pre-cycled to the LTO voltage plateau (1.55 V vs. Li+/Li) so that the lithiation degree remained constant. The electrolyte used was 1 M LiClO4(Sigma Aldrich) in propylene carbonate (PC, Sigma Aldrich). The electrolyte was degassed for several hours in the experimental chamber. A specially designed plastic beaker was used as electrolyte container. All three electrodes were suspended from above the beaker using a 3-axis manipulator enabling the motion of the electrodes. The beaker was in a fixed position below the measurement position inside the chamber.
The PC's vapor pressure is around 0.3 mbar at room temperature but due to observed sample charging under X-ray illumination, additional Ar was introduced to the chamber. This was found to sufficiently minimize charging effects, where photoionized electrons from the gas compensate for the otherwise inherent buildup of positive charge in non-conductive samples (due to the photoemission process). For the measurement of the undipped samples the total chamber pressure for these measurements was around 8 mbar. For the dipped measurements no charging effects were observed so no Ar was needed.
A solid/liquid interface was created using the dip-and-pull method following similar methodology to our previous studies at the HIPPIE beamline at the MAX IV facility in Lund, Sweden.19,50 Using compact thin film electrodes successfully ensured that the wetting was limited (compared to porous electrodes). However, due to cracks in the thin film, capillary forces still transported the liquid several mm up the electrode. The major part of the meniscus was too thick for photoelectrons from the electrode to escape, but at approximately 3 mm above the dipped part of electrode, both the solid and liquid could be probed simultaneously. The electrical contact of the probed liquid was proven by the response of liquid core level spectra line position when ramping the applied potential (Fig. S7†). The O 1s core level spectra, which features components from both the TM oxide and the electrolyte solution, was used to identify the region of the meniscus where we could measure a solid/liquid interface. A photoelectron that is ejected from the electrode surface need to pass through the liquid layer atop with thickness d, and in this process the intensity of electron Is attenuates according to:
| Is=Is,0e−d/λL |
 | ||
| Fig. 6 (a) schematic illustration of LCO interface probing with O 1s at (b) solid, (c) interface, and (d) liquid. | ||
The sample was also moved sideways to ensure that the analyzed spot was fresh and free from radiation induced effects (we primarily observed beam induced growth of PC from the gas phase, which attenuated the electrode surface features long before the radiation induced any observable chemical changes to the surface). A photon energy of 5 keV was used for all APXPS measurements, with an energy bandwidth of 0.1% (double crystal monochromator Si (111)). The X-ray intrinsic energy resolution is estimated to be 675 meV. The Scienta HiPP-2 hemispherical analyzer was configured with a pass energy of 200 eV and a slit of 1.5 mm giving a theoretical analyzer resolution of 750 meV. The total overall measurement resolution is therefore estimated to be 1.0 eV
A BioLogic potentiostat was used in floating mode to run the electrochemical measurements. The working electrode was grounded directly to the hemispherical analyzer. Voltage was applied between WE and RE, initially set to 4.2 V (vs. Li+/Li, and 2.65 V vs. the RE). All the voltages declared in the paper are given vs. Li+/Li. At each subsequent voltage step the current was allowed to decay. When the current has decreased to a fairly stable value (below 15 μA), APXPS measurements were conducted. The total duration of the operando experiment was approximately 35 hours.
Spectra analysis
XPS spectra were processed with Igor Pro software. The binding energies for all spectra obtained are calibrated with a constant negative shift of 4.24 eV. This is based on alignment of pristine LCO O 1s metal oxide energy to be at 529.7 eV45 (the beamline energy was not changed throughout the measurements, which was proven to be stable through the entire dataset). A Shirley curve is used for the Co 2p spectra background subtraction, after a prior linear background level alignment based on the spectra shape at pre-peak region at low energy. Linear background substation are used for O 1s and C 1s. The curves were fitted with a pseudo-voigt function, where the Lorentzian function describes the natural broadening due to the life time of the core hole decay, while the Gaussian shape of the curve is due to the experimental broadening.The deconvoluted peak for each component from the probed core levels Co 2p, C 1s, and O 1s is considered symmetric. The FWHM for the same components/chemical state from one core level obtained at different potentials are constrained the same for curve fitting. For different chemical components contributing to one spectrum, the FWHM is not fitted exactly the same, as different broadenings are expected for different chemical states. The Lorentzian contribution giving the natural broadening from the life time of core hole is constrained to be similar for all components in one spectrum. The energy spacing between corresponding components are constraint to be the same for spectra from different applied voltages. See fitting parameters for Co 2p in Table S1.†
Peak intensity
The photoemission intensity is determined by51| I = nFσϕyATλ |
Given the signal from core levels, e.g. O 1s and C 1s from the same species PC in this study, the relation between the peak intensity IO1s and IC1s is

where the atomic ratio of O and C in PC is 3
 :4, and the photoionization cross section from O 1s and C 1s core levels at 5 k eV photon energy are 1.124 and 0.333.52
:4, and the photoionization cross section from O 1s and C 1s core levels at 5 k eV photon energy are 1.124 and 0.333.52
Scanning electron microscopy
Scanning electron microscopy (SEM) images was obtained with a Merlin from Zeiss (Germany) equipped with an energy-dispersive spectroscopy (EDS) system (X-Max 80 mm2 Silicon Drift Detector). AZtec (INCA energy) software was used for X-ray mapping and element analysis.Author contributions
M. H. and I.·K contributed to conceptualization. M. H. acquired funding, provide supervision to this project. I. K contributed to the method and model development of APXPS measurement on LCO. G. V contributed to providing resources for thin film LCO material growth. K. B., G. V., F. L., and T. E. contributed to material synthesis and sample preparation. T. E., M. H. and R. T. conducted the APXPS experiment at ALS. E. C. contributed to APXPS experiment support at ALS. Q. L., L. K. and A. K. conducted APXPS experiment at MAXIV. R. T. contributed to APXPS experiment support at MAXIV. Q. L. contributed to data analysis, data curation, validation and visualization. Q. L., T. E, and M. H. contributed to the interpretation of the results. Q. L., T. E, and M. H. created the original draft. All authors discussed the results, reviewed and edited the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Swedish research council (2020–04512_VR, 2022–06076_VR, 2018–06465_VR) for funding the presented research. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. For SEM images we acknowledge Myfab Uppsala for providing facilities and experimental support. Myfab is funded by the Swedish Research Council (2019-00207) as a national research infrastructure. A portion of this work (electrode growth, editing) was supported by the US Department of Energy’s Office of Energy Efficiency and Renewable Energy (EERE) Vehicle Technologies Office (Tien Duong Program Manager) as part of the US-DE program. This manuscript has been partially authored by UT-Battelle, LLC under Contract no. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a nonexclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doepublic-access-plan). RT is additionally grateful for funding from Lund University and MAX IV Laboratory. We acknowledge MAX IV Laboratory for preliminary and supporting measurements taken at the HIPPIE beamline50 under proposals 20180403 and 20230036. Research conducted at MAX IV, a Swedish national user facility, is supported by the Swedish Research council under contract 2018–07152, the Swedish Governmental Agency for Innovation Systems under contract 2018-04969, and Formas under contract 2019–02496. We acknowledge Diamond Light Source for the supporting XPS measurement at instrument I09 (proposal SI36581-1).References
- G. E. Blomgren, J. Electrochem. Soc., 2017, 164, A5019–A5025 CrossRef CAS.
- L. Dahéron, R. Dedryvère, H. Martinez, D. Flahaut, M. Méétrier, C. Delmas and D. Gonbeau, Chem. Mater., 2009, 21, 5607–5616 CrossRef.
- R. Fantin, A. van Roekeghem and A. Benayad, PRX Energy, 2023, 2, 043010 CrossRef.
- L. Dahéron, R. Dedryvère, H. Martinez, M. Ménétrier, C. Denage, C. Delmas and D. Gonbeau, Chem. Mater., 2008, 20, 583–590 CrossRef.
- Y. Shao-Horn, L. Croguennec, C. Delmas, E. C. Nelson and M. A. O’Keefe, Nat. Mater., 2003, 2, 464–467 CrossRef PubMed.
- Z. X. Yang, R. G. Li and Z. H. Deng, Sci. Rep., 2018, 8(1), 863 CrossRef PubMed.
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Mater. Res. Bull, 1980, 15, 783–789 CrossRef CAS.
- Z. Zhu, H. Wang, Y. Li, R. Gao, X. Xiao, Q. Yu, C. Wang, I. Waluyo, J. Ding, A. Hunt and J. Li, Adv. Mater., 2020, 32, 2005182 CrossRef CAS PubMed.
- A. T. S. Freiberg, S. Qian, J. Wandt, H. A. Gasteiger and E. J. Crumlin, ACS Appl. Mater. Interfaces, 2023, 15, 4743–4754 CrossRef CAS PubMed.
- Y. S. Jung, P. Lu, A. S. Cavanagh, C. Ban, G. H. Kim, S. H. Lee, S. M. George, S. J. Harris and A. C. Dillon, Adv. Energy Mater., 2013, 3, 213–219 CrossRef CAS.
- S. T. Oyakhire, H. Gong, Y. Cui, Z. Bao and S. F. Bent, ACS Energy Lett., 2022, 7, 2540–2546 CrossRef CAS.
- M. Favaro, F. Abdi, E. Crumlin, Z. Liu, R. van de Krol and D. Starr, Surfaces, 2019, 2, 78–99 CrossRef CAS.
- K. A. Stoerzinger, W. T. Hong, E. J. Crumlin, H. Bluhm and Y. Shao-Horn, Acc. Chem. Res., 2015, 48, 2976–2983 CrossRef CAS PubMed.
- J. Maibach, I. Källquist, M. Andersson, S. Urpelainen, K. Edström, H. Rensmo, H. Siegbahn and M. Hahlin, Nat. Commun., 2019, 10, 3080 CrossRef PubMed.
- R. Timm, A. R. Head, S. Yngman, J. V. Knutsson, M. Hjort, S. R. McKibbin, A. Troian, O. Persson, S. Urpelainen, J. Knudsen, J. Schnadt and A. Mikkelsen, Nat. Commun., 2018, 9, 1412 CrossRef PubMed.
- I. Källquist, T. Ericson, F. Lindgren, H. Chen, A. Shavorskiy, J. Maibach and M. Hahlin, ACS Appl. Mater. Interfaces, 2022, 14, 6465–6475 CrossRef PubMed.
- Y. Yu, A. Baskin, C. Valero-Vidal, N. T. Hahn, Q. Liu, K. R. Zavadil, B. W. Eichhorn, D. Prendergast and E. J. Crumlin, Chem. Mater., 2017, 29, 8504–8512 CrossRef CAS.
- S. Axnanda, E. J. Crumlin, B. Mao, S. Rani, R. Chang, P. G. Karlsson, M. O. M. Edwards, M. Lundqvist, R. Moberg, P. Ross, Z. Hussain and Z. Liu, Sci. Rep., 2015, 5, 9788 CrossRef CAS PubMed.
- I. Källquist, F. Lindgren, M. T. Lee, A. Shavorskiy, K. Edström, H. Rensmo, L. Nyholm, J. Maibach and M. Hahlin, ACS Appl. Mater. Interfaces, 2021, 13, 32989–32996 CrossRef PubMed.
- A. Shavorskiy, X. Ye, O. Karslloǧlu, A. D. Poletayev, M. Hartl, I. Zegkinoglou, L. Trotochaud, S. Nemšák, C. M. Schneider, E. J. Crumlin, S. Axnanda, Z. Liu, P. N. Ross, W. Chueh and H. Bluhm, J. Phys. Chem. Lett., 2017, 8, 5579–5586 CrossRef CAS PubMed.
- M. Favaro, B. Jeong, P. N. Ross, J. Yano, Z. Hussain, Z. Liu and E. J. Crumlin, Nat. Commun., 2016, 7, 12695 CrossRef CAS PubMed.
- F. G. Capone, J. Sottmann, V. Meunier, L. Pérez Ramírez, A. Grimaud, A. Iadecola, M. Scardamaglia, J. P. Rueff and R. Dedryvère, Energy Environ. Sci., 2024, 17, 1509–1519 RSC.
- K. S. Kim, Phys. Rev. B: Condens. Matter Mater. Phys., 1975, 11, 2177–2185 CrossRef CAS.
- J. P. P. Kemp and P. A. A. Cox, J. Phys.: Condens. Matter, 1990, 2, 9653–9667 CrossRef CAS.
- M. Oku, J. Solid State Chem., 1978, 23, 177–185 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- G. A. M Ali, K. Feng Chong and E. A. A Aboelazm, Chem. Adv. Mater., 2018, 3, 67–74 Search PubMed.
- J. C. Dupin, D. Gonbeau, H. Benqlilou-Moudden, P. Vinatier and A. Levasseur, Thin Solid Films, 2001, 384, 2332 CrossRef.
- M. Zarabian, M. Bartolini, P. Pereira-Almao and V. Thangadurai, J. Electrochem. Soc., 2017, 164, A1133–A1139 CrossRef CAS.
- J. Yu, J. Li, S. Zhang, F. Wei, Y. Liu and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, 1–9 Search PubMed.
- Q. Zuo, W. Liu, Y. Su, Y. Cao, K. Ren and Y. Wang, Scr. Mater., 2023, 233, 115511 CrossRef CAS.
- W. Zhou, J. Wu, C. Ouyang, Y. Gao, X. Xu and Z. Huang, J. Appl. Phys., 2014, 115, 093512 CrossRef.
- H. Kiuchi, K. Hikima, K. Shimizu, R. Kanno, F. Toshiharu and E. Matsubara, Electrochem. Commun., 2020, 118, 106790 Search PubMed.
- C. S. Lim, C. K. Chua, Z. Sofer, O. Jankovský and M. Pumera, Chem. Mater., 2014, 26, 4130–4136 CrossRef CAS.
- V. R. Galakhov, V. V. Karelina, D. G. Kellerman, V. S. Gorshkov, N. A. Ovechkina and M. Neumann, Phys. Solid State, 2002, 44, 257–264 CrossRef.
- N. Schulz, R. Hausbrand, L. Dimesso and W. Jaegermann, J. Electrochem. Soc., 2018, 165, A819–A832 CrossRef CAS.
- R. Fantin, PhD thesis, Université Grenoble Alpes, 2024.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B, 3, 943 Search PubMed.
- I. Källquist, PhD thesis, Uppsala University, 2021.
- Q. Lu, ACS Nano, 2024, 18, 13973–13982 CrossRef CAS PubMed.
- R. Dedryvère, S. Laruelle, S. Grugeon, P. Poizot, D. Gonbeau and J. M. Tarascon, Chem. Mater., 2004, 16, 1056–1061 CrossRef.
- S. Thapa, R. Paudel, M. D. Blanchet, P. T. Gemperline and R. B. Comes, J. Mater. Res., 2021, 36, 26–51 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- V. R. Galakhov, E. Z. Kurmaev, S. Uhlenbrock, M. Neumann, D. G. Kellerman and V. S. Gorshkov, Solid State Commun., 1996, 99, 221–224 CrossRef CAS.
- L. Dahéron, H. Martinez, R. Dedryvère, I. Baraille, M. Ménétrier, C. Denage, C. Delmas and D. Gonbeau, J. Phys. Chem. C, 2009, 113, 5843–5852 CrossRef.
- D. Ensling, G. Cherkashinin, S. Schmid, S. Bhuvaneswari, A. Thissen and W. Jaegermann, Chem. Mater., 2014, 26, 3948–3956 CrossRef CAS.
- A. Mikheenkova, S. Mukherjee, M. Hirsbrunner, P. Törnblom, C.-W. Tai, C. U. Segre, Y. Ding, W. Zhang, T. C. Asmara, Y. Wei, T. Schmitt, H. Rensmo, L. Duda and M. Hahlin, J. Mater. Chem. A, 2024, 12, 2465–2478 RSC.
- A. Križan, T. Ericson, L. King, Q. Liu, R. Temperton, R. Dominko, O. Vodeb, D. Strmčnik, M. Gaberšček and M. Hahlin, Phys. Chem. Chem. Phys., 2025, 27, 7456–7466 RSC.
- M. F. Lichterman, S. Hu, M. H. Richter, E. J. Crumlin, S. Axnanda, M. Favaro, W. Drisdell, Z. Hussain, T. Mayer, B. S. Brunschwig, N. S. Lewis, Z. Liu and H. J. Lewerenz, Energy Environ. Sci., 2015, 8, 2409–2416 RSC.
- S. Zhu, M. Scardamaglia, J. Kundsen, R. Sankari, H. Tarawneh, R. Temperton, L. Pickworth, F. Cavalca, C. Wang, H. Tissot, J. Weissenrieder, B. Hagman, J. Gustafson, S. Kaya, F. Lindgren, I. Kallquist, J. Maibach, M. Hahlin, V. Boix, T. Gallo, F. Rehman, G. D'Acunto, J. Schnadta and A. Shavorskiy, J. Synchrotron Radiat., 2021, 28, 624–636 Search PubMed.
- C. D. Wagner, L. E. Davis, M. V. Zeller, J. A. Taylor, R. H. Raymond and L. H. Gale, Surf. Interface Anal., 1981, 3, 211–225 CrossRef CAS.
- M. B. Trzhaskovskaya and V. G. Yarzhemsky, At. Data Nucl. Data Tables, 2018, 119, 99–174 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta01654a |
| This journal is © The Royal Society of Chemistry 2025 |