
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen-modified supra-nanometer-sized RuPt for robust alkaline HER/HOR†
Youpeng Cao‡
 a,
Hongling Liu‡a,
Yuxuan Xiaoa,
Lun Lia,
Jiao Yanga,
Chunfa Liua,
Chengcheng Zhonga,
Wendi Zhanga,
Shuyang Penga,
Junge Yanga,
Zhichao Yua,
Weng Fai Ipb and
Hui Pan*ab
a,
Hongling Liu‡a,
Yuxuan Xiaoa,
Lun Lia,
Jiao Yanga,
Chunfa Liua,
Chengcheng Zhonga,
Wendi Zhanga,
Shuyang Penga,
Junge Yanga,
Zhichao Yua,
Weng Fai Ipb and
Hui Pan*ab
aInstitute of Applied Physics and Materials Engineering, University of Macau, Macao SAR 999078, China. E-mail: huipan@um.edu.mo; Tel: +86 853 88224427
bDepartment of Physics and Chemistry, Faculty of Science and Technology, University of Macau, Macao SAR 999078, China
First published on 30th May 2025
Abstract
Platinum (Pt) is a highly efficient catalyst for the hydrogen evolution reaction (HER) and the hydrogen oxidation reaction (HOR). However, the scarcity of Pt necessitates the development of catalysts with low Pt content while maintaining high activity. In this study, we report the synthesis of oxygen-modified supra-nanometer-sized RuPt nanocrystals with low Pt content by a simple air annealing method to achieve a catalyst with high activity. The annealed catalyst, RuPt/C-200, exhibits a very low overpotential of only 3.8 mV at 10 mA cm−2, a low Tafel slope of 16.6 mV dec−1, and a 2.8-fold increase in mass activity at an overpotential of 0.06 V for HER compared to the unannealed catalyst. Additionally, the HOR exchange current density increases 2.9-fold compared to the unannealed catalyst. AEMWE tests confirm the application potential of RuPt/C-200, while density-functional-theory (DFT) calculations reveal that oxygen modification, by optimizing the hydrogen binding energy, plays a crucial role in improving the catalytic performance of RuPt. This work highlights that combining the oxygen modification strategy with the nanoscale size effect could advance the development of high-performance RuPt electrocatalyst for both alkaline HER and HOR.
1. Introduction
As a clean energy source, hydrogen has the potential to replace fossil fuels such as oil and coal, thereby significantly reducing greenhouse gas emissions and playing a crucial role in the global energy transition.1–4 Technologies, such as anion exchange membrane water electrolysis (AEMWE) and the anion exchange membrane fuel cell (AEMFC), are important for achieving a hydrogen cycle due to their relatively lower corrosiveness to catalysts and lower equipment costs under alkaline conditions.5–7 However, both AEMWE and AEMFC face a significant drawback: the alkaline environment leads to low proton transport efficiency, which significantly reduces the kinetics of the hydrogen evolution reaction (HER) and hydrogen oxidation reaction (HOR).8–12 Therefore, developing efficient catalysts to enhance the HER of AEMWE and the HOR of AEMFC is very important.13,14Platinum (Pt) is a benchmark catalyst for alkaline HER and HOR due to its optimal d-band center and ideal adsorption/desorption energies for reactive intermediates, but its high cost hinders large-scale applications.15–17 Ruthenium (Ru)-based catalysts thus receive increasing attention as cost-effective alternatives due to the similar d-band center to Pt.18–20 Consequently, PtRu alloy catalysts are being explored to reduce Pt content and overall catalyst cost while maintaining high catalytic activity.17,21 However, the activity of Ru in hydrogen electrocatalysis still needs to be improved due to its relatively strong hydrogen (*H) adsorption energy, which is unbeneficial for efficient hydrogen desorption.22,23 To address this issue, two strategies are widely utilized to prepare Ru-based catalysts with high activity for HER and HOR. One is focusing on the morphology control for more exposure of electrochemical surface area (ECSA).24–27 Another one lies in the electronic structure optimization by heteroatom doping,16,27–30 heterojunction fabrication31 or vacancy engineering32–35 to reduce the adsorption energy of *H. Thus, it can be predicted that a combined optimization of both morphology and electronic structure could further enhance the performance of Ru-based catalysts, although it is still challenging to be realized through a simple synthetic strategy.
Herein, we report a facile two-step synthesis of oxygen-modified RuPt nanocrystals with low Pt content and high catalytic performance. Benefiting from the alloying effect of Ru and Pt, the size effect of supra-nanometer-sized RuPt nanocrystals and the optimized *H adsorption energy of Ru due to oxygen modification, the RuPt catalyst annealed at 200 °C (denoted as RuPt/C-200) exhibits a significantly improved catalytic activity for both HER and HOR. AEMWE testing validated the application potential of the catalyst, while DFT calculations confirm the performance enhancement mechanism through the optimization of hydrogen binding energy. Our work highlights that combining the oxygen modification strategy with the nanoscale size effect could advance the development of high-performance RuPt electrocatalyst for both alkaline HER and HOR.
2. Experimental section
2.1 Chemicals
Triruthenium dodecacarbonyl (Ru3(CO)12, >98%) and platinum(II) acetylacetonate (Pt(acac)2, 98%) were purchased from Bidepharm. Nafion perfluorinated resin solution (5 wt%) were purchased from Sigma Aldrich. Oleylamine (C18H37N, 80–90%) was purchased from Macklin. Commercial Pt/C (20 wt% Pt) was purchased from TANAKA. Ethanol (C2H6O, AR), hexamethylene (C6H12, AR), isopropanol (C3H8O, AR) and potassium hydroxide (KOH, AR) were purchased from Aladdin. Vulcan carbon XC-72R was purchased from CABOT. All the chemicals were used without further purification. The deionized water (18 MΩ cm−1) used in all the experiments was obtained by passing through an ultra-pure purification system (Aqua Solutions).2.2 Preparation of samples
2.3 Materials characterizations
The morphology and microstructure of the synthesized materials were examined by transmission electron microscopy (TEM) using a JEM2010-HR microscope. Raman spectra, revealing information about the vibrational modes and structure, were obtained from 200 to 1800 cm−1 using a LABHRev-UV Raman spectrometer. The crystal structure and phase purity of the samples were analyzed by X-ray diffraction (XRD) using a Rigaku SmartLab SE diffractometer with Cu Kα radiation (λ = 1.5418 Å). The data were collected over a 2θ range from 5° to 90°. Electron paramagnetic resonance (EPR) spectroscopy was used to investigate paramagnetic species at room temperature with a Magnettech MS 5000 EPR spectrometer operating at 9.86 GHz. The elemental composition was quantitatively determined by inductively coupled plasma-mass spectrometry (ICP-MS) using an Agilent 7850 instrument. Surface chemical composition and oxidation states were investigated by X-ray photoelectron spectroscopy (XPS) using a Thermo Scientific Escalab 250Xi spectrometer.2.4 Electrochemical measurements
All the electrochemical measurements were tested on the electrochemical station (CHI760, Shanghai Chenhua) with a three-electrode system. The working electrode is a glassy carbon disk electrode, and graphite rod and mercuric oxide electrode were used the counter electrode and the reference electrode, respectively. For the preparation of the working electrode, catalyst (2 mg), isopropanol (745 μL), deionized water (245 μL) and Nafion solution (10 μL, 5 wt%) were mixed and ultrasonicated for 1 h to form a homogeneous ink. Afterwards, 12 μL ink was dropped on the glassy carbon disk electrode for the HER tests. The equilibrium potential was determined by the zero point of HER/HOR using platinum sheet working electrode as working electrode at 1600 rpm in H2-saturated electrolyte. The linear sweep voltammetry (LSV) was performed at a scan rate of 5 mV s−1 with 100% ohmic drop compensation.38 Before each electrochemical test, the electrodes were swept with 25 cyclic voltammetry at 0.05 mV s−1 to stabilize the catalysts' double layers and ensure complete wetting. The working electrode for stability test of HER was prepared by coating 1 mg of the catalyst onto a carbon paper (1 cm2).HOR tests were conducted in a H2-saturated 0.1 M KOH aqueous solution at a rotating speed of 1600 rpm with a scanning rate of 5 mV s−1 and an 95% iR correction (i, current; R, solution resistance). The mass density of Ru and Pt was 20 μg cm−2 on the glassy carbon disk electrode.
The kinetic current (jk) was calculated according to the Koutecky–Levich equation:
 | (1) |
The exchange current density (j0) was acquired by fitting jk with the Butler–Volmer equation:
 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), α is the transfer coefficient and T is the temperature (298.15 K).
485 C mol−1), α is the transfer coefficient and T is the temperature (298.15 K).
Electrochemical impedance spectroscopy (EIS) measurements were conducted at a HER overpotential of 50 mV in the range from 100 kHz to 0.01 Hz. The stability of catalyst for HER was measured by drop-casting the catalyst on a carbon paper with a mass loading (RuPt/C-200) of 1 mg cm−2. For CO-stripping, CO was bubbled into aqueous KOH solution (1 mol L−1) for 15 min. After the electrolyte was saturated with N2 for 15 min, two cyclic voltammetry (CV) curves were collected at a scan rate of 20 mV s−1. For the Cu stripping experiments, a N2-saturated solution containing 50 mM CuSO4 and 0.5 M H2SO4 was used as the electrolyte. The potential was first held at 0.3 V versus RHE for 100 s to form a Cu deposition monolayer, and then CVs were obtained from 0.3 V to 1.0 V at 20 mV s−1.
2.5 Electrochemical measurements in AEMWE device
The AEMWE device was composed of the cathode (RuPt/C-200 and Pt/C coated on carbon paper), anode (NiFe@Ni felt), and commercial AEM membrane (X37-50, Dioxide Materials Sustainion). The membrane was immersed in 1 M KOH solution for at least 10 h prior to being employed as an electrolyte. The cathode was prepared by air-spraying catalyst ink onto the carbon paper. The Ru and Pt loading were controlled to be 0.15 mg cm−2 for the cathode. The MEA was prepared by integrating the cathode, membrane, and anode between two Ti bipolar plates with a torque of 10 N m to complete an AEMWE device. The active area of cell is 1 cm−2. 1 M KOH was circulated through the anodic side with a flow rate of 15 mL min−1 by a peristaltic pump (DIPump550-B253, Kamoer). The cell was activated at 50 mA cm−2 for 1 h prior to the test. The performance of AEMWE was evaluated by measuring polarization curves from 0.03 A cm−2 to 1.0 A cm−2 at 60 °C on a battery test system (ME-1S200, Kolibrik). The stability performance of AEMWE was evaluated by measuring at a current density of 0.5 A cm−2 at room temperature. All measurements in AEMWE were recorded without iR-correction.2.6 Calculational details
All of our first-principles calculations were carried out within the framework of the density functional theory (DFT) as implemented in the Vienna ab initio simulation package (VASP).39–41 Core-valence interactions were described using the projector augmented wave (PAW) method. The exchange-correlation energy functional of interacting electrons was described with the Perdew–Burke–Ernzerhof functional within the generalized gradient approximation (GGA).42 The cutoff energy for the plane-wave basis was set to 500 eV, and the convergence criterion was set to 10−5 eV and 0.02 eV Å−1 for energy and force, respectively. Grimme's DFT-D3 scheme of dispersion correction was adopted to consider the van der Waals (vdW) interaction in all systems.43 The 2 × 2 supercell of Pt (111) surface slab models were constructed with doping Ru (50 Ru atoms and 14 Pt atoms), which proportion is close to the experimental value. The bottom two layers of the slab were fixed at bulk lattice constant, the top two layers and adsorbates were allowed to relax. The oxidized RuPt catalyst was built by partially oxidizing the surface Ru with oxygen atoms. The Brillouin zone was sampled with a 3 × 3 × 1 Γ-centered Monkhorst–pack k point mesh. The periodic images in the z-direction were separated by about 15 Å of vacuum space. The free energy of the proton–electron pair is equal to that of a half of gaseous H2 under standard condition. The kinetic barrier for the decomposition of the absorbed H2O was calculated by AIMD simulation and slow growth method with explicit solution model.44 The Brillouin zone was sampled with a 1 × 1 × 1 Monkhorst–Pack k-point grid. The collective variable (CV) increment was set to 0.0005 Å, and the time step was set to 1 fs. The convergence criteria for the electronic step is set to 1 × 10−4 eV, and cutoff energy for the plane-wave basis was set to 400 eV. The POMASS in POTCAR of H is set to 2.The adsorption energies of the absorbates were calculated according to the following equations:
| Eads = Ecat-M − E*M − Ecat | (3) |
The Gibbs free energy change (ΔG) were acquired according to the computational hydrogen electrode (CHE) model proposed by Nørskov et al.:45
| ΔG = ΔE + ΔEZPE − TΔS | (4) |
The d band center (εd) of the systems in this work was calculated as:
 | (5) |
3. Results and discussion
3.1 Synthesis and characterizations
The RuPt/C-200 catalyst was prepared via a two-step route (Fig. 1a). The supra-nanometer-sized (<10 nm) RuPt nanocrystals were firstly obtained through a one-pot synthesis method by using triruthenium dodecacarbonyl (Ru3(CO)12) and platinum(II) acetylacetonate (Pt(acac)2) as metal precursors, and oleylamine (OAm) as solvent, surfactant and reducing agent. After ultrasonicated at 30 °C for 1.5 h to form a homogeneous solution, the mixture was then heated to 200 °C and held at the same temperature for 6 h in oven. When the reaction began, carbon monoxide was released from the Ru3(CO)12, and Ru and Pt precipitated from the solution. Afterwards, the RuPt/C-200 catalyst was synthesized by loading onto carbon black and annealing at 200 °C in air for one hour. The transmission electron microscopy (TEM), Raman scattering, electron paramagnetic resonance (EPR) spectroscopy, X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) were performed to characterize the morphology and intrinsic structure of the as-synthesized catalyst. The TEM characterization (Fig. 1b) shows that the synthesized RuPt alloy exhibits a supra-nanometer-sized morphology, with an average particle size of around 6 nm. Energy-dispersive X-ray spectroscopy (EDS) element mappings of RuPt alloy (Fig. S1, ESI†) confirm the uniform distributions of Ru and Pt. The TEM characterizations were also performed on the catalysts with reaction times of 0.5 h and 1.5 h (Fig. S2, ESI†), respectively. The average particle size of the RuPt nanocrystals synthesized with a reaction time of 0.5 h is around 5.2 nm. When the reaction time was prolonged to 1.5 h, the average particle size increases to around 5.6 nm. High resolution TEM (HRTEM) characterization (Fig. 1c and d) reveals that there is no significant change in the lattice spacing of RuPt/C before and after annealing at 200 °C, primarily showing the Pt (111) crystal plane.9 The Raman spectra of RuPt/C-200 (Fig. 1e) shows two main peaks at 505 cm−1 and 618 cm−1, which are corresponding to Eg (degenerate vibrational characteristic peak) and A1g (symmetric stretching vibrational characteristic peak) modes, respectively.47 RuPt/C without annealing shows no obvious Eg and A1g modes. The EDS mappings of RuPt/C-200 catalyst confirm the presence of oxygen in the catalyst and the uniform distributions of Ru and Pt on the carbon black after annealing (Fig. 1f). The RuPt/C-180 annealed at 180 °C (Fig. S3a, ESI†) exhibits the same morphology as RuPt/C-200. In contrast, the RuPt/C annealed at a high temperature of 220 °C (denoted as RuPt/C-220, see Fig. S3b for its TEM characterization, ESI†) shows a tendency to aggregate. The atomic ratio of the synthesized RuPt nanocrystals was measured by inductively coupled plasma mass spectrometry (ICP-MS) (Table S1, ESI†). Since the same initial sample was used for annealing, the Ru to Pt atomic ratio in samples RuPt/C, RuPt/C-180, RuPt/C-200 and RuPt/C-220 are all around 4:1, which confirms the low Pt content in the catalysts. In the RuPt alloy with a lower Pt content, the atomic ratio of Ru to Pt is approximately 84:16, designated as Ru84Pt16/C-200. Similarly, in the RuPt alloy with a lower Ru content, the atomic ratio of Ru to Pt is approximately 68:32, designated as Ru68Pt32/C-200.
 | ||
| Fig. 1 (a) Schematic illustration of the two-step synthesis process for oxygen-modified RuPt/C catalysts. (b) TEM image of supra-nanometer-sized RuPt nanocrystals. Size distribution diagram of the synthesized supra-nanometer-sized RuPt nanocrystals is described in the inset. HRTEM images of (c) RuPt nanocrystals and (d) RuPt/C-200 catalyst. (e) Raman spectra of RuPt/C and RuPt/C-200. (f) EDS mappings of the RuPt/C-200 catalyst. | ||
The crystal structure of catalyst was analyzed by XRD (Fig. 2a). All four samples display diffraction peaks at around 40.26° and 46.62°, assigned to the (111) and (200) planes of Pt.9,48 However, the (111) diffraction peak of RuPt/C is shifted to higher angle compared to that of the standard Pt (111) diffraction peak (JCPDS no. 04-0802), indicating a decrease in the lattice spacing of RuPt/C compared to Pt. This is attributed to the incorporation of Ru with smaller atomic radius into the Pt lattice to form a RuPt alloy. Based on the XRD results, we estimate a compression strain of 1.52% for RuPt/C-200 compared to Pt (111) (JCPDS no. 04-0802) (Table S2, ESI†). Combining the TEM and XRD characterizations confirm that Ru is incorporated into the crystal structure of Pt. Only the sample annealed at 220 °C shows the characteristic diffraction peaks at 28.01° and 35.05°, indicating the presence of crystalline RuO2 (JCPDS no. 40-1290). The oxygen vacancies are confirmed by the characteristic EPR signal at g-factor = 2.001 (Fig. 2b).32 Only the unannealed RuPt/C exhibits a distinct characteristic peak of oxygen vacancies, while all annealed samples do not show any significant peaks of oxygen vacancies.
 | ||
| Fig. 2 (a) XRD patterns, (b) EPR spectra, and high-resolution XPS spectra for (c) Pt 4f, (d) Ru 3d5/2, (e) Ru 3p, and (f) O 1s signals of RuPt/C, RuPt/C-180, RuPt/C-200 and RuPt/C-220. | ||
The full XPS surveys (Fig. S4, ESI†) confirm the existence of Ru, Pt, C and O in the samples. The electronic structures of Ru, Pt and O are further surveyed (Fig. 2c–f). From the XPS spectrum of Pt 4f (Fig. 2c), it can be observed that the binding energy (BE) of Pt 4f is significantly higher than the typical value of around 71.5 eV,9 which is caused by the high Ru content in the alloy leading to compressive strain on Pt.11,49,50 As the annealing temperature increases, the Pt 4f7/2 peaks of RuPt/C-180, RuPt/C-200 and RuPt/C-220 shift negatively by about 0.08, 0.19 and 0.22 eV compared with those of RuPt/C, respectively. The XPS spectra of Ru 3d5/2 (Fig. 2d) shows that the Ru4+/Ru0 atomic ratio for RuPt/C is 1.35, which means that the Ru4+ content is 57.45% and the Ru0 content is 42.55%.51 The Ru4+/Ru0 atomic ratio of RuPt/C-180 is 1.34 similar to that of RuPt/C because the oleylamine on the metal surface was removed during annealing at 180 °C. The Ru4+/Ru0 atomic ratio of RuPt/C-200 further increases to 2.14, with a Ru4+ content of 68.15% and a Ru0 content of 31.85%. In the RuPt/C-220 sample, the Ru4+/Ru0 atomic ratio is 5.80, indicating that a Ru4+ content is 85.29% and a Ru0 content is 14.71% (Table S3, ESI†). The analysis of the Ru 3d5/2 clearly shows an increase in Ru4+ content, which also confirms the increase in oxygen content in the metal catalysts. From the XPS spectra of Ru 3p (Fig. 2e), the BE of Ru 3p gradually increases with the increase in annealing temperature. The O 1s XPS spectra reveals three different O types, including the lattice O2− (at around 530.70 eV, OLat), the hydroxyl (HO) groups adsorbed to the metal atoms in the oxygen-deficient region (at around 532.80 eV, OV), and the adsorbed water (at around 534.30 eV, OAbs) (Fig. 2f).52 The OLat peaks of RuPt/C-180, RuPt/C-200 and RuPt/C-220 are negatively shifted by about 0.21, 0.23 and 0.67 eV compared with those of RuPt/C, respectively. The combined analysis of the decrease in the BE of Pt and OLat, and the increase in the BE of Ru indicates that air annealing regulates the distribution and content of electrophilic oxygen in the catalyst, thereby affecting the electron cloud density of OLat, Pt and Ru.
Clearly, the combined structural characterizations indicate that the RuPt alloy catalyst we synthesized exists in a crystal structure similar to Pt (111). After oxygen modification, the density of oxygen vacancies decreased and the content of oxygen in RuPt alloy increased. When a higher annealing temperatures (220 °C) was applied, crystalline RuO2 began to appear in the sample.
3.2 HER performance
The HER performances of the as-synthesized RuPt/C, RuPt/C-180, RuPt/C-200 and RuPt/C-220 and commercial Pt/C were investigated in 1 M KOH (Fig. 3a). The RuPt/C-200 exhibits the best HER activity performance with a low overpotential (η) of 3.8 mV and 33.9 mV at 10 and 100 mA cm−2, respectively, much lower than those of commercial Pt/C (28.3 mV at 10 mA cm−2), RuPt/C (9.8 mV at 10 mA cm−2), RuPt/C-180 (4.2 mV at 10 mA cm−2) and RuPt/C-220 (7.1 mV at 10 mA cm−2). The linear sweep voltammetry (LSV) curves without iR correction show the consistent results (Fig. S5, ESI†). The HER activities of catalysts with different Ru/Pt ratios were also tested (Fig. S6, ESI†). The mass activity of RuPt/C-200 (3.6 A mg−1) at the overpotential of 60 mV is 2.8 and 4.0 times as high as those of RuPt/C (1.3 A mg−1) and commercial Pt/C (0.9 A mg−1), respectively (Fig. 3b and d). The Tafel slopes of commercial Pt/C, RuPt/C, RuPt/C-180, RuPt/C-200 and RuPt/C-220 are 47.2, 23.6, 24.7, 16.6, and 20.2 mV dec−1, respectively (Fig. 3c). According to Tafel slope theory, a Tafel slope value below 30 mV dec−1 may be problematic. Therefore, we verified the Tafel slope values using the chronoamperometry method (Fig. S7, ESI†).53 The results showed that RuPt/C-200 still exhibited a very low Tafel slope, with values nearly consistent with those obtained from the LSV method. By varying the loading amounts of Pt/C and RuPt/C-200 catalysts and comparing the Tafel slope values of RuPt/C-200 with and without iR potential compensation, we found that the Tafel slope calculations were influenced by catalyst loading and iR potential compensation (Fig. S8, ESI†). Consequently, the calculated Tafel slopes cannot fully reflect the catalytic mechanism but only indicate that a small increase in overpotential drives a large increase in reaction rate, implying highly efficient charge transfer kinetics of RuPt/C-200. Compared to the current state-of-the-art alkaline HER catalysts (Fig. 3e), our RuPt/C-200 catalyst exhibits one of the best performances for both overpotential at 10 mA cm−2 and Tafel slope, which are two important performance indicators for alkaline HER (Table S5, ESI†). To further explore the HER kinetics, electrochemical impedance spectroscopy (EIS) was conducted (Fig. 3f). Nyquist plots for all samples exhibit near-semicircular profiles, which are effectively modeled using an equivalent circuit comprising solution resistance (Rs), charge transfer resistance (Rct), and constant phase element (CPE). The fitting results (Table S4, ESI†) show that solution resistances across all samples are approximately 5.6 Ω, while the charge transfer resistances are calculated as 15.34, 6.793, 4.729, 1.568, and 2.431 Ω for Pt/C, RuPt/C, RuPt/C-180, RuPt/C-200, and RuPt/C-220, respectively. Among these, RuPt/C-200 demonstrates the lowest charge transfer resistance, aligning with its superior HER performance. Furthermore, RuPt/C-200 exhibits excellent long-term stability, showing negligible performance degradation for 100 h at a current density of 10 mA cm−2 (Fig. 3g). Post-characterizations are crucial for assessing the structural stability of the catalyst. Therefore, XRD and XPS characterizations on the RuPt/C-200 catalyst after the stability test were done. XRD characterization was conducted on bare carbon paper, RuPt/C-200 loaded on carbon paper before the stability test, and RuPt/C-200 loaded on carbon paper after the stability test. The results show that the diffraction peaks (Fig. S9, ESI†) and the Ru 3p and Pt 4f XPS peaks of RuPt alloy on carbon paper (Fig. S10, ESI†) after stability test have no change compared to those of pristine ones, indicating a stable structure of the catalyst during the HER process.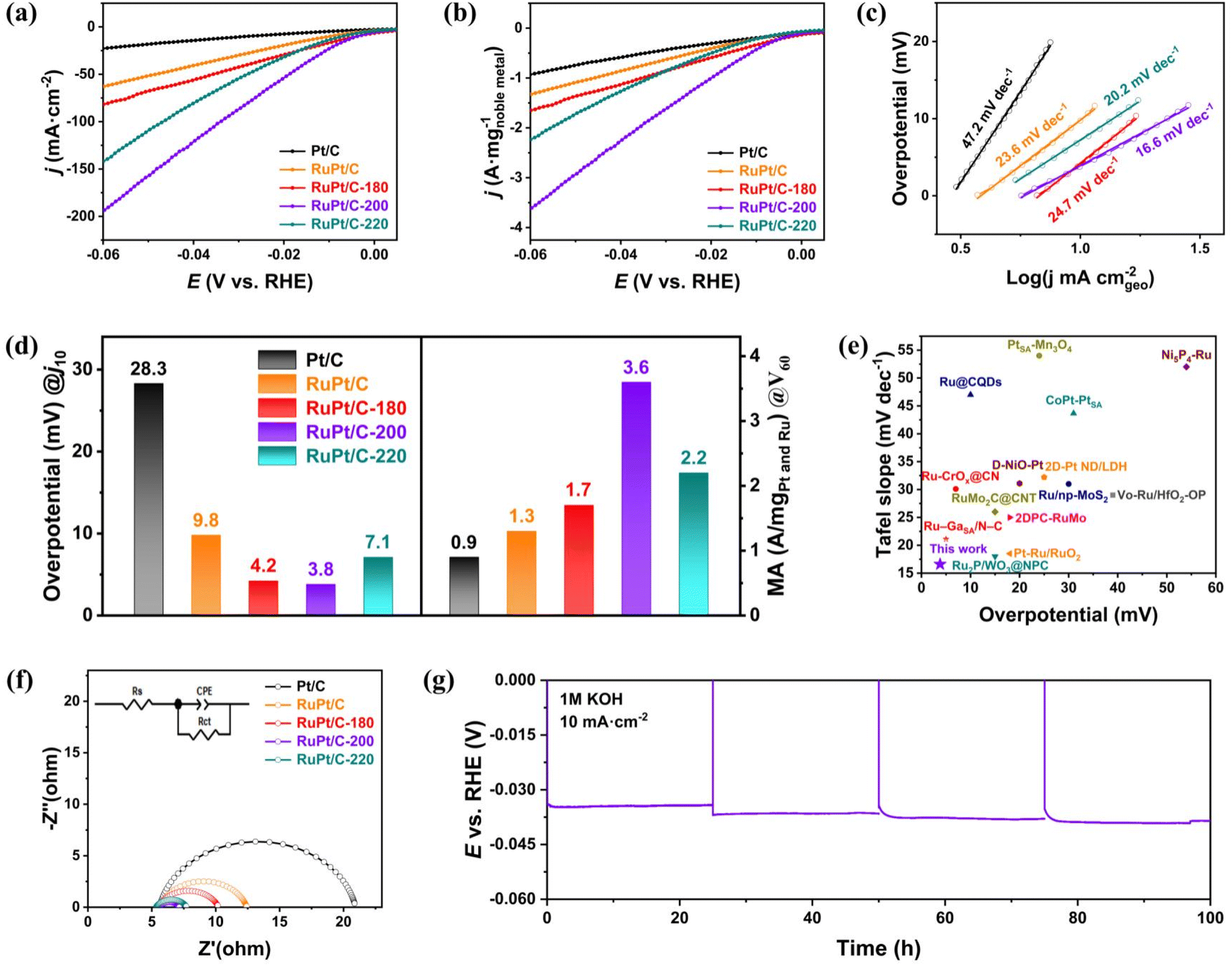 | ||
| Fig. 3 (a) HER polarization curves in a N2-saturated 1 M KOH electrolyte with a rotating speed of 1600 rpm. (b) Mass-normalized HER polarization curves of different catalysts. (c) Corresponding Tafel plot in (a). (d) Comparison of the overpotential of commercial Pt/C, RuPt/C, RuPt/C-180, RuPt/C-200, and RuPt/C-220 at the current density of 10 mA cm−2 and the mass activity at the overpotential of 60 mV. (e) Comparison of overpotential at 10 mA cm−2 and Tafel plots of different catalysts for alkaline HER. (f) EIS plots of commercial Pt/C, RuPt/C, RuPt/C-180, RuPt/C-200 and RuPt/C-220. X-Axis (Z′), real part; Y-axis (−Z′′), imaginary part. (g) Chronopotentiometric curves of RuPt/C-200 catalyst in 1 M KOH at 10 mA cm−2. | ||
CO-stripping, copper underpotential deposition (Cu-upd) and double-layer capacitance methods are the most commonly used techniques to measure the electrochemical active surface area (ECSA). However, since our catalyst is supported on carbon black, and carbon black itself does not serve as an active site, the double-layer capacitance method is not suitable for measuring ECSA of our catalysts. CO-stripping was employed to calculate the ECSA of the catalysts.54 From the CO-stripping tests (Fig. S11, ESI†), the peak potential positions follow the order of commercial Pt/C > RuPt/C ≈ RuPt/C-200 > RuPt/C-180 ≈ RuPt/C-220, indicating that all RuPt catalysts exhibit better resistance to CO poisoning compared to Pt/C. RuPt/C-180 shows a larger peak area compared to RuPt/C, because the 180 °C annealing process effectively removes oleylamine molecules from the surface of the RuPt. In contrast, the reduced peak areas observed for RuPt/C-200 and RuPt/C-220 may result from the diminished adsorption capacity of CO molecules on the metal oxide surfaces formed at higher annealing temperatures. In many studies, CO-stripping is not used to measure the ECSA of oxidized Ru.55 Therefore, the ECSA of oxygen-modified RuPt catalyst prepared in our work cannot be measured using the CO-stripping method. We then attempted to measure the ECSA using the Cu-upd method (Fig. S12, ESI†). Similarly, for the RuPt/C-200 and RuPt/C-220 samples, it was almost impossible to observe a distinct UPD peak, meaning that the Cu-upd method could not accurately measure the ECSA of the oxygen-modified RuPt catalysts either. It is worth noting that the peak appearing around 0.25 V is an overpotential deposition (OPD) peak, not UPD peak, and therefore cannot be used to calculate ECSA.56,57 In conclusion, although we know that measuring ECSA is very important for calculating the specific activity of catalysts, the most commonly used CO-stripping and Cu-upd methods cannot be applied to measure the ECSA of our oxygen-modified RuPt catalysts.
3.3 HOR performance
The catalytic performances of the electrocatalysts for alkaline HOR were also evaluated by rotating disk electrode (RDE) in H2-saturated 0.1 M KOH with a standard three-electrodes system. From the HOR polarization curves of different catalysts (Fig. 4a), it can be observed that the anodic current increases much faster on RuPt/C-200 than RuPt/C, RuPt/C-180, RuPt/C-220 and commercial Pt/C as the potential increases. The current density of RuPt/C-200 at an overpotential of 50 mV is 2.66 mA cm−2, which is much higher than those of RuPt/C (1.55 mA cm−2), RuPt/C-180 (1.99 mA cm−2), RuPt/C-220 (1.49 mA cm−2) and commercial Pt/C (1.70 mA cm−2). The calculated half-wave potential (E1/2) for RuPt/C-200 is 10 mV, which is also much lower than that of Pt/C (23 mV). The kinetic current density (jk) and exchange current density (j0) of HOR/HER are then calculated by the Koutecky–Levich equation9 and Butler–Volmer fitting.58 The Koutecky–Levich plot for RuPt/C-200 is derived from the polarization curves collected at different rotating speeds (Fig. 4b), which shows a slope of 3.92 cm2 mA−1 s−1/2. The Koutecky–Levich plots for the other four catalysts are also presented (Fig. S13, ESI†). The Tafel plots of the kinetic currents (jk) on RuPt/C-200, RuPt/C, RuPt/C-180, RuPt/C-220 and commercial Pt/C indicate that RuPt/C-200 displays the highest jk among these catalysts at various potentials (Fig. 4c and e). The micropolarization regions of the five catalysts (Fig. 4d) are depicted to calculate the exchange current density (j0) of these catalysts and we can find that RuPt/C-200 displays the highest j0 (Fig. 4e). These results indicate that the introduction of oxygen can remarkably boost the HOR activity. The HOR durability of RuPt/C-200 and commercial Pt/C was evaluated by performing a chronoamperometry test on RDE at an overpotential of 0.1 V with a continuous rotation speed of 1600 rpm. After a 3 hour stability test, the current density of RuPt/C-200 decreased from 2.17 mA cm−2 to 1.93 mA cm−2, and the current density of Pt/C decreased from 1.84 mA cm−2 to 1.56 mA cm−2 (Fig. 4f). RuPt/C-200 can retain approximately 89% of its original current density. In contrast, commercial Pt/C only maintains about 84.7% of its original current density (Fig. 4g). These results show that RuPt/C-200 are active and stable towards the alkaline HOR. | ||
| Fig. 4 (a) HOR polarization curves of commercial Pt/C, RuPt/C, RuPt/C-180, RuPt/C-200, and RuPt/C-220 in H2-saturated KOH solution (0.1 M) with a scanning rate of 5 mV s−1 at 1600 rpm. (b) HOR polarization curves of RuPt/C-200 at different rotating speeds (inset is Koutecky–Levich plot). (c) Calculated HOR kinetic current densities (jk) versus potential plots recorded from (a). (d) Micropolarization region of the five catalysts. (e) Comparison of exchange current densities (j0) and kinetic current density (jk) of different catalysts for alkaline HOR. (f) The current density–time chronoamperometry and (g) relative current–time chronoamperometry of commercial Pt/C and RuPt/C-200 recorded at 0.1 V vs. RHE in H2-saturated 0.1 M KOH electrolyte. | ||
3.4 AEMWE and mechanism investigation
To further verify the practical application potential of the catalyst and the mechanism of performance improvement, we conducted AEMWE device test for the catalyst, underpotentially deposited hydrogen tests, and DFT calculations.The RuPt/C-200 catalyst was integrated into a membrane electrode assembly (MEA) and evaluated under practical water electrolysis working conditions. An alkaline exchange membrane water electrolysis (AEMWE) device was constructed with RuPt/C-200 as the cathodic catalyst and NiFe layered double hydroxide (LDH) on Ni felt as the anode (Fig. 5a). The current–voltage curves for RuPt/C-200 and a commercial Pt/C-based AEMWE at 60 °C are presented (Fig. 5b). Notably, the RuPt/C-200-based electrolyzer achieves industrial-level current densities of 0.5 and 1.0 A cm−2 at low cell voltages of 1.64 and 1.74 V, respectively, which are significantly lower than that of commercial Pt/C (1.88 V @ 0.5 A cm−2 and 2.02 V @ 1.0 A cm−2). Under the same testing conditions, our catalyst exhibits the best device testing performance compared to catalysts from other studies59–61 (Fig. 5c). Moreover, remarkable stability is exhibited for AEMWE device with RuPt/C-200 as the cathode at room temperature, and it maintains a consistent voltage of approximately 1.74 V with no significant increase during 200 hours of continuous operation at an industrial current density of 0.5 A cm−2 (Fig. 5d). The reason we conducted the AEMWE stability test at room temperature is that the NiFe catalyst used at the anode undergoes significant iron leaching at 60 °C, which leads to a severe degradation of the device performance.62–64
 | ||
| Fig. 5 (a) Scheme diagram of the AEMWE device. (b) Polarization curves of the AEMWE using RuPt/C-200 and Pt/C as cathode catalysts without iR-correction at 60 °C. (c) Comparison of the cell voltage of RuPt/C-200-based AEMWE with other reported state-of-the-art Ru or Pt-based catalysts at current densities of 0.5 and 1.0 A cm−2. (d) Chronopotentiometric curves of RuPt/C-200-based AEMWE tested at 0.5 A cm−2 at room temperature. (e) CV polarization curves of RuPt/C, RuPt/C-180, RuPt/C-200, and RuPt/C-220 in N2-saturated KOH solution (1 M) with a scanning rate of 100 mV s−1 at 1600 rpm. (f) Bader charge analyses of the Ru active site on pristine RuPt and oxidized RuPt. The negative values represent loss of electrons. (g) Free energy changes of *H adsorption on RuPt and oxidized RuPt. (h) The projected densities of states (PDOSs) of RuPt and oxidized RuPt with the value of d-band center for Ru element. | ||
Based on the characterization of the catalyst structure and performance testing, we conclude that an increase in oxygen content can significantly enhance the catalytic activity of RuPt (RuPt/C-180 and RuPt/C-200). However, excessive oxygen and the formation of RuO2 crystals (RuPt/C-220) can lead to a decline in catalytic performance because RuO2 shows no activity for HER (Fig. S14, ESI†). To further elucidate the underlying mechanism for the enhanced HER activity of RuPt/C-200 in an alkaline electrolyte, the CV tests of these samples in N2-saturated 1 M KOH electrolyte at a scan rate of 100 mV s−1 were conducted (Fig. 5e). The peaks of the underpotentially deposited hydrogen (Hupd) for RuPt/C and RuPt/C-180 are significantly larger than those for RuPt/C-200 and RuPt/C-220, indicating a weaker but optimized Ru–H binding on RuPt/C-200. This is beneficial for both HER and HOR, in line with recent advancements in hydrogen binding energy mechanisms and hydrogen descriptors for alkaline HER and HOR.65,66
Density-functional-theory (DFT) calculations were employed as a complementary tool to further support this experimental conclusion.67 The pristine and oxidized RuPt (111) models were constructed (Fig. 5f and S15, ESI†). The differential charge density and Bader charge analyses indicate that the charge transfer between Ru and Pt remains basically unchanged after oxygen modifies RuPt (Fig. S16a and b, ESI†). However, surface Ru atoms lose more electrons to oxygen due to the significantly higher electronegativity of oxygen (3.44) than Ru (2.20) (Fig. S16c, ESI†). Thus, oxygen modification promotes electron redistribution on surface Ru sites, which is expected to modify the adsorption properties. The atomic structures of H2O adsorption and *H adsorption on RuPt and oxidized RuPt were simulated (Fig. S17 and S18, ESI†).68,69 The calculated water adsorption energy (ΔE) values for pristine and oxidized RuPt (111) surfaces are −0.63 eV and −0.84 eV, respectively, indicating stronger water molecule adsorption on the oxidized surface (Fig. S19, ESI†). It can be explained that the Ru surface is partially oxidized (Ru → Ruδ+), forming Lewis acid (Ruδ+)–Lewis base (Oδ−) dual functional sites. The coordination of Ruδ+ with the lone pair electrons of O in H2O promotes chemical adsorption, while the formation of hydrogen bonds between Oδ− and Hδ+ in H2O further enhances physical adsorption (Fig. S20b, ESI†). Bader charge analysis reveals that more electrons are transferred from the O atom of H2O to Ruδ+ to form coordination bonds, with a more pronounced electron accumulation region for oxidized RuPt with H2O molecules (Fig. S20, ESI†). It is well known that H2O serves as the proton source for HER in alkaline media. The water dissociation involves the broken of O–H bond to form *H and OH−, which is often the rate-limiting step with high energy barrier. Oxidized RuPt exhibits oxophilicity compared to pristine RuPt (stronger H2O adsorption), which is expected to stabilize OH− and thereby lower the energy barrier. Therefore, the AIMD simulation combined with the slow-growth approach was employed to evaluate the kinetic barriers for H2O decomposition. The kinetic barrier of water decomposition of oxidized RuPt (0.7 eV) is lower than that of pristine RuPt (1.24 eV), thus facilitating the HER process (Fig. S21, ESI†). In addition, the Gibbs free energy for hydrogen adsorption (ΔGH) is considered as a significant descriptor for the HER/HOR activity of electrocatalysts.65,66 (Fig. 5g) Compared with the pristine RuPt (−0.46 eV), the weakened *H adsorption free energy on oxidized RuPt (−0.19 eV) exhibits the improved alkaline HER/HOR activity, indicating that the oxygen modification strategy can effectively regulate ΔGH, thereby improving hydrogen electrocatalytic activity. In addition, the partial density of states (PDOS) plots (Fig. 5h) reveals that the d-band center of oxidized RuPt (111) (−1.863 eV) shows an obvious downshift relative to that of pristine RuPt (111) (−1.779 eV). Metallic Ru primarily relies on d-orbital electrons to interact with non-polar H. The downshift of the d-band center signifies a weakened metallicity for Ru, resulting in reduced interaction with non-polar H. Our DFT calculations support the conclusions obtained from experiments, demonstrating that the oxygen modification of RuPt optimizes the hydrogen adsorption energy, thereby significantly improving the hydrogen electrocatalytic activity.
4. Conclusion
In summary, we report the synthesis of supra-nanometer-sized RuPt nanocrystals with low Pt content and the application of an oxygen modification strategy to improve the electrocatalytic performance. The annealed catalyst, RuPt/C-200, exhibits remarkably improved activity in HER, including a significantly reduced overpotential of 3.8 mV at 10 mA cm−2, a low Tafel slope of 16.6 mV dec−1, and a 2.8-fold increase in mass activity at an overpotential of 0.06 V compared to the unannealed catalyst. Furthermore, the HOR exchange current density increases 2.9-fold compared to the unannealed catalyst. AEMWE tests confirm the application potential of RuPt/C-200, while DFT-assisted experiments reveal that the oxygen modification strategy, by optimizing the hydrogen binding energy, plays a crucial role in improving the catalytic performance of RuPt. XRD results indicate that as the annealing temperature increases, the inevitable formation of crystalline RuO2 lead to a decline in activity. This work highlights the advantages of combining the oxygen modification strategy with the nanoscale size effect in development of high-performance RuPt electrocatalyst for both alkaline HER and HOR.Data availability
All data supporting the findings of this study are available within the paper and ESI file.†Author contributions
Y. C. and H. L. conceptualized the study and designed the experiments, contributing equally to this work. Y. X. participated in data analysis and manuscript revision. L. L., C. L., and S. P. participated in AEMWE tests, XPS tests and analysis. J. Y. and W. Z. performed TEM characterization. C. Z. and J. Y. participated in electrochemical testing. Z. Y. participated in DFT analysis. W. F. I. participated in funding acquisition. H. P. participated in funding acquisition, experimental design, data analysis, and manuscript revision.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Science and Technology Development Fund (FDCT) from Macau SAR (0050/2023/RIB2, 0023/2023/AFJ, 006/2022/ALC, 0087/2024/AFJ and 0111/2022/A2) and Multi-Year Research Grants (MYRG-GRG2024-00038-IAPME, and MYRG-GRG2023-00010-IAPME) from the University of Macau.References
- W. A. Braff, J. M. Mueller and J. E. Trancik, Nat. Clim. Change, 2016, 6, 964–969 CrossRef.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- V. Stamenkovic, D. Strmcnik, P. Lopes and N. Markovic, Nat. Mater., 2017, 16, 57–69 CrossRef CAS PubMed.
- C. Wei, R. Rao, J. Peng, B. Huang, I. Stephens, M. Risch, Z. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef PubMed.
- J. Yang, K. An, Z. Yu, L. Qiao, Y. Cao, Y. Zhuang, C. Liu, L. Li, L. Peng and H. Pan, ACS Catal., 2024, 14, 17739–17747 CrossRef CAS.
- J. Yang, J. Feng, Y. Cao, Y. Xiao, L. Qiao, K. An, J. Yang, J. Peng, H. Pan and H. Cheng, Adv. Funct. Mater., 2024, 34, 2411081 CrossRef CAS.
- J. Durst, C. Simon, F. Hasché and H. A. Gasteiger, J. Electrochem. Soc., 2015, 162, F190–F203 CrossRef CAS.
- W. Ni, J. Meibom, N. Ul Hassan, M. Chang, Y. Chu, A. Krammer, S. Sun, Y. Zheng, L. Bai, W. Ma, S. Lee, S. Jin, J. Luterbacher, A. Schüler, H. Chen, W. Mustain and X. Hu, Nat. Catal., 2023, 6, 773–783 CrossRef CAS.
- W. Ni, A. Krammer, C. Hsu, H. Chen, A. Schüler and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 7445–7449 CrossRef CAS PubMed.
- F. Yang, Y. Wang, Y. Cui, X. Yang, Y. Zhu, C. Weiss, M. Li, G. Chen, Y. Yan, M. Gu and M. Shao, J. Am. Chem. Soc., 2023, 145, 27500–27511 CrossRef CAS PubMed.
- R. Liu, Z. Xu, F. Li, F. Chen, J. Yu, Y. Yan, Y. Chen and B. Xia, Chem. Soc. Rev., 2023, 52, 5652–5683 RSC.
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed.
- F. Xiao, Y. Wang, Z. Wu, G. Chen, F. Yang, S. Zhu, K. Siddharth, Z. Kong, A. Lu, J. Li, C. Zhong, Z. Zhou and M. Shao, Adv. Mater., 2021, 33, 2006292 CrossRef CAS PubMed.
- M. Shao, Q. Chang, J. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- Y. Xiao, J. Ying, J. Chen, X. Yang, G. Tian, J. Li, C. Janiak and X. Yang, Adv. Funct. Mater., 2024, 2418264 Search PubMed.
- C. Li and J. Baek, ACS Omega, 2020, 5, 31–40 CrossRef CAS PubMed.
- S. Han, Q. Yun, S. Tu, L. Zhu, W. Cao and Q. Lu, J. Mater. Chem. A, 2019, 7, 24691–24714 RSC.
- A. Salah, H. Ren, N. Al-Ansi, F. Yu, Z. Lang, H. Tan and Y. Li, J. Mater. Chem. A, 2021, 9, 20518–20529 Search PubMed.
- H. Shang, Z. Zhao, J. Pei, Z. Jiang, D. Zhou, A. Li, J. Dong, P. An, L. Zheng and W. Chen, J. Mater. Chem. A, 2020, 8, 22607–22612 RSC.
- W. Luo, Y. Wang and C. Cheng, Mater. Today Phys., 2020, 15, 100274 CrossRef.
- Y. Zheng, Y. Jiao, Y. Zhu, L. Li, Y. Han, Y. Chen, M. Jaroniec and S. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS PubMed.
- Y. Yang, Y. Yu, J. Li, Q. Chen, Y. Du, P. Rao, R. Li, C. Jia, Z. Kang, P. Deng, Y. Shen and X. Tian, Nano-Micro Lett., 2021, 13, 160 CrossRef CAS PubMed.
- G. Liu, W. Zhou, B. Chen, Q. Zhang, X. Cui, B. Li, Z. Lai, Y. Chen, Z. Zhang, L. Gu and H. Zhang, Nano Energy, 2019, 66, 104173 Search PubMed.
- J. Gu, L. Li, Y. Xie, B. Chen, F. Tian, Y. Wang, J. Zhong, J. Shen and J. Lu, Nat. Commun., 2023, 14, 5389 CrossRef CAS PubMed.
- C. Zhou, J. Shi, Z. Dong, L. Zeng, Y. Chen, Y. Han, L. Li, W. Zhang, Q. Zhang, L. Gu, F. Lv, M. Luo and S. Guo, Nat. Commun., 2024, 15, 6741 CrossRef CAS PubMed.
- K. Wang, J. Zhou, M. Sun, F. Lin, B. Huang, F. Lv, L. Zeng, Q. Zhang, L. Gu, M. Luo and S. Guo, Adv. Mater., 2023, 35, 2300980 CrossRef CAS PubMed.
- D. Zhang, H. Zhao, B. Huang, B. Li, H. Li, Y. Han, Z. Wang, X. Wu, Y. Pan, Y. Sun, X. Sun, J. Lai and L. Wang, ACS Cent. Sci., 2019, 5, 1991–1997 CrossRef CAS PubMed.
- J. Zhang, G. Ren, D. Li, Q. Kong, Z. Hu, Y. Xu, S. Wang, L. Wang, M. Cao and X. Huang, Sci. Bull., 2022, 67, 2103–2111 CrossRef CAS PubMed.
- D. Chen, R. Lu, Z. Pu, J. Zhu, H. Li, F. Liu, S. Hu, X. Luo, J. Wu, Y. Zhao and S. Mu, Appl. Catal., B, 2020, 279, 119396 Search PubMed.
- B. Zhang, J. Wang, G. Liu, C. Weiss, D. Liu, Y. Chen, L. Xia, P. Zhou, M. Gao, Y. Liu, J. Chen, Y. Yan, M. Shao, H. Pan and W. Sun, Nat. Catal., 2024, 7, 441–451 CrossRef CAS.
- J. Chen, C. Chen, M. Qin, B. Li, B. Lin, Q. Mao, H. Yang, B. Liu and Y. Wang, Nat. Commun., 2022, 13, 5382 CrossRef CAS PubMed.
- G. Li, H. Jang, S. Liu, Z. Li, M. Kim, Q. Qin, X. Liu and J. Cho, Nat. Commun., 2022, 13, 1270 CrossRef CAS PubMed.
- X. Liu, X. Wang, K. Li, J. Tang, J. Zhu, J. Chi, J. Lai and L. Wang, Angew. Chem., Int. Ed., 2024, 63, e202316319 CrossRef CAS PubMed.
- Q. Qin, H. Jang, X. Jiang, L. Wang, X. Wang, M. Kim, S. Liu, X. Liu and J. Cho, Angew. Chem., Int. Ed., 2024, 136, e202317622 CrossRef.
- Z. Lu, W. Xu, W. Zhu, Q. Yang, X. Lei, J. Liu, Y. Li, X. Sun and X. Duan, Chem. Commun., 2014, 50, 6479–6482 RSC.
- Q. Xiang, F. Li, W. Chen, Y. Ma, Y. Wu, X. Gu, Y. Qin, P. Tao, C. Song, W. Shang, H. Zhu, T. Deng and J. Wu, ACS Energy Lett., 2018, 3, 2357–2365 CrossRef CAS.
- S. Anantharaj and S. Noda, J. Mater. Chem. A, 2022, 10, 9348–9354 RSC.
- G. Kresse and J. Furthmuller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- W. K. P. Hohenberg, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- L. J. S. W. Kohn, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- T. Woo, P. Margl, P. Blochl and T. Ziegler, J. Phys. Chem. B, 1997, 101, 7877–7880 CrossRef CAS.
- J. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 Search PubMed.
- V. Wang, N. Xu, J. Liu, G. Tang and W. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- S. Y. Mar, C. S. Chen, Y. S. Huang and K. K. Tiong, Appl. Surf. Sci., 1995, 90, 497–504 CrossRef CAS.
- J. Gu, Y. Guo, Y. Jiang, W. Zhu, Y. Xu, Z. Zhao, J. Liu, W. Li, C. Jin, C. Yan and Y. Zhang, J. Phys. Chem. C, 2015, 119, 17697–17706 CrossRef CAS.
- K. Zhou, Z. Wang, C. Han, X. Ke, C. Wang, Y. Jin, Q. Zhang, J. Liu, H. Wang and H. Yan, Nat. Commun., 2021, 12, 3787 CrossRef PubMed.
- M. Wakisaka, S. Mitsui, Y. Hirose, K. Kawashima, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2006, 110, 23489–23496 CrossRef CAS PubMed.
- D. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- V. H. Do, P. Prabhu, V. Jose, T. Yoshida, Y. T. Zhou, H. Miwa, T. Kaneko, T. Uruga, Y. Iwasawa and J. M. Lee, Adv. Mater., 2023, 35, 2208860 CrossRef CAS PubMed.
- S. Roy, R. Madhu, A. Karmakar and S. Kundu, ACS Mater. Lett., 2024, 6, 3112–3123 CrossRef.
- M. C. Luo, Z. L. Zhao, Y. L. Zhang, Y. J. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. N. Qin, J. Y. Ma, F. Lin, D. Su, G. Lu and S. J. Guo, Nature, 2019, 574, 81–85 CrossRef CAS PubMed.
- L. Li, G. Zhang, C. Zhou, F. Lv, Y. Tan, Y. Han, H. Luo, D. Wang, Y. Liu, C. Shang, L. Zeng, Q. Huang, R. Zeng, N. Ye, M. Luo and S. Guo, Nat. Commun., 2024, 15, 4974 CrossRef CAS PubMed.
- L. Fan, T. Meng, Q. Li, D. Wang, Z. Xing, E. Wang and X. Yang, Catal. Sci. Technol., 2020, 10, 7302–7308 RSC.
- Y. Zhang, H. Huang, Y. Han, Y. Qin, N. Nie, W. Cai, X. Zhang, Z. Li, J. Lai and L. Wang, J. Mater. Chem. A, 2022, 10, 13241–13246 RSC.
- Y. Fang, C. Wei, Z. Bian, X. Yin, B. Liu, Z. Liu, P. Chi, J. Xiao, W. Song, S. Niu, C. Tang, J. Liu, X. Ge, T. Xu and G. Wang, Nat. Commun., 2024, 15, 1614 CrossRef CAS PubMed.
- L. Gao, F. Bao, X. Tan, M. Li, Z. Shen, X. Chen, Z. Tang, W. Lai, Y. Lu, P. Huang, C. Ma, S. Smith, Z. Ye, Z. Hu and H. Huang, Energy Environ. Sci., 2023, 16, 285–294 RSC.
- S. Park, T. Nguyen, D. Tran, V. Dinh, J. Lee and N. Kim, Energy Environ. Sci., 2023, 16, 4093–4104 Search PubMed.
- A. Majumdar, K. Tran, S. Prabhakaran, D. Kim, D. Tran, N. Kim and J. Lee, Adv. Funct. Mater., 2025, 2420517 CrossRef.
- X. Cui, T. Tang, F. Zhang, L. Sun and B. Zhang, Appl. Catal., B, 2025, 366, 125024 CrossRef CAS.
- Z. Li, G. Lin, L. Wang, H. Lee, J. Du, T. Tang, G. Ding, R. Ren, W. Li, X. Cao, S. Ding, W. Ye, W. Yang and L. Sun, Nat. Catal., 2024, 7, 944–952 CrossRef CAS.
- W. Sun, X. Zheng, X. Zheng, M. Gao, Y. Liu and H. Pan, Angew. Chem., Int. Ed., 2025, e202422062 Search PubMed.
- H. Li, C. Tsai, A. Koh, L. Cai, A. Contryman, A. Fragapane, J. Zhao, H. Han, H. Manoharan, F. Abild-Pedersen, J. Norskov and X. Zheng, Nat. Mater., 2016, 15, 48–53 CrossRef CAS PubMed.
- J. Zheng, W. Sheng, Z. Zhuang, B. Xu and Y. Yan, Sci. Adv., 2016, 2, e1501602 CrossRef PubMed.
- Y. P. Li, W. T. Wang, M. Y. Cheng, Y. F. Feng, X. Han, Q. Z. Qian, Y. Zhu and G. Q. Zhang, Adv. Mater., 2023, 35, 2206351 CrossRef CAS PubMed.
- L. Wei, W. Yan, Z. Huang, R. Li, Q. Kong, W. Huang, C. Pao, Z. Hu, H. Lin, N. Chen, Y. Xu, H. Geng and X. Huang, Energy Environ. Sci., 2024, 17, 5922–5930 RSC.
- S. Zhang, R. Ren, J. Cao, D. Zhang, J. Bai, C. Han, L. Xiao, L. Zhuang, P. Song and W. Xu, Adv. Energy Mater., 2024, 2404266 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta02064f |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |