
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAqueous alkaline pH stable halide ((PEA)2CoCl4) perovskite for oxygen reaction electrocatalysis†
Pranjit
Barman
a,
Vijay
Kumar
bc,
Kotaro
Takeyasu
 d and
Santosh K.
Singh
*a
d and
Santosh K.
Singh
*a
aElectrochemical Energy Research Laboratory, Department of Chemistry, School of Natural Sciences, Shiv Nadar Institution of Eminence (SNIoE), Dadri, Uttar Pradesh, India-201314. E-mail: santoshk.singh@snu.edu
bCenter for Informatics, School of Natural Sciences, Shiv Nadar Institution of Eminence (SNIoE), Dadri, Uttar Pradesh-201314, India
cDr Vijay Kumar Foundation, 1969 Sector 4, Gurgaon 122001, Haryana, India
dInstitute of Catalysis, Hokkaido University, Kita 21 Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan
First published on 12th May 2025
Abstract
Despite gaining significant importance as an exciting class of materials, the extremely low stability of metal halide perovskites (MHPs) in aqueous medium impedes their potential application beyond photovoltaics and photocatalysis. Herein, the stability of cobalt-based halide perovskite (PEA)2CoCl4 (PEA = phenylethylammonium) in an aqueous alkaline pH electrolyte has been enhanced by employing a hydrophobic organic cation as the A-site and further wrapping it with electronic conductive N-doped reduced graphene oxide (NrGO). The increased stability allows the developed perovskite composite to be applied to electrocatalysis, such as the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER). The effective surface passivation of (PEA)2CoCl4 results in controlled exposure of aqueous electrolytes to the active sites of perovskite during catalysis. (PEA)2CoCl4 coated with NrGO (NPCo) shows an ORR onset potential of 0.94 V at 0.1 mA cm−2 and an overpotential of 353 mV at 10 mA cm−2 towards the OER as compared to the state-of-the-art (Pt/C) (1.00 V) and RuO2 (346 mV) catalysts. Furthermore, the NPCo catalyst is used in rechargeable Zn–air batteries and shows a specific capacity of 760 mA h g−1 with a peak power density of 116 mW cm−2. The developed NPCo electrode materials are utilized to demonstrate flexible electronics. Density functional theory (DFT) and ab initio molecular dynamics (AIMD) simulations clarify the active role of NrGO coating in preventing the perovskite structure from flooding with water molecules and facilitating electrocatalysis.
Introduction
Metal halide perovskites (MHPs) with the chemical formula ABX3 or A2BX4 (B: divalent cations such as Pb, Sn, Bi, Co, and Mn; X: Cl−, Br−, and I−) consist of inorganic or organic ammonium cations as the A-site.1–4 However, A2BX4 is a 2D layered structure of ABX3 perovskite where long-chain organic molecules can occupy the A-site, providing better stability under moisture conditions.1–3 Over the last decade, MHPs have received significant attention for their application in photocatalysis and optoelectronic devices, such as solar cells,5,6 LEDs,7 lasers,8etc., due to their superior optoelectronic properties,9 quantum confinement effect,10 easy preparation,11 and bandgap tunability.12,13While MHPs have demonstrated great success in photocatalysis, their application in electrocatalysis faces significant challenges, mainly due to their rapid decomposition in aqueous environments.14,15 To address this issue, various strategies have been explored to improve MHP stability, including compositional modifications, ligand engineering, and surface encapsulation using dense layers of SiO2,16 Al2O3,17 TiO2,18 polymers (use of a PMMA layer),19 and two-dimensional hBN,20 and supporting over graphene.21 In addition to water stability, toxicity (Pb) in MHPs is another critical concern. Pb-based perovskites have shown superior efficiency as light absorbers in photovoltaics and photocatalysis,22,23 but their environmental and health risks limit broader applications.24–26 Therefore, developing Pb-free MHPs has become an important research direction.27,28 Recent studies have explored transition metal-based perovskites, including Mn2+, Fe2+, Co2+, Rh2+, Cu2+, Pd2+, and Pt2+, as well as post-transition metal-based perovskite-like compounds,29–36 for applications beyond photovoltaics.
Despite these advances, MHPs still face critical challenges in electrocatalysis, such as low electrical conductivity, insufficient stability, and a lack of exposed active sites, limiting their ability to achieve large current densities and long-term stability.37 While some studies have investigated MHPs as oxygen evolution reaction (OER) catalysts, the problem of low electrical conductivity remains unresolved. R. Babu et al. reported a lead-free, stable organic–inorganic perovskite (MA)2CoBr4 for water oxidation,38 while N. Navarro et al. demonstrated that (CH3NH3)2Zn1−xCoxBr4, particularly (MA)2Zn0.3Co0.7Br4, exhibited the highest activity for the OER.39 Recently, X. Ren et al. developed a MAPbX3@AlPO-5 host–guest composite to improve perovskite stability for water oxidation.40 However, these studies did not fully address the issue of low electrical conductivity. Furthermore, to our knowledge, the application of halide perovskites for the oxygen reduction reaction (ORR) remains largely unexplored.
Herein, we have designed an alkaline pH-stable organic–inorganic halide hybrid perovskite (PEA)2CoCl4, further stabilized by coating with N-doped reduced graphene oxide (NrGO) to form the NPCo catalyst. The NrGO coating not only enhances the conductivity of the NPCo catalyst but also improves the stability of perovskite by controlling its exposure to the aqueous electrolyte, thereby increasing surface utilization. The developed strategy is also useful for the stabilization and electronic conductivity improvement of other halide perovskites (ABX3, A2BX4, A4BX6,etc.) as well.41,42 X-ray photoelectron spectroscopy and electron paramagnetic resonance studies revealed a strong electronic interaction between N-doped graphene and perovskite materials. Furthermore, density functional theory (DFT) and ab initio molecular dynamics (AIMD) simulations demonstrated the role of NrGO in influencing the electronic interaction between explicit water molecules and perovskite stability. Extensive electrocatalytic activity investigations indicated that the N-doped rGO-coated NPCo catalyst significantly outperforms conventional catalytic materials in bifunctional ORR and OER activities. The well-defined Co active sites in NPCo serve as a model catalyst for electrocatalytic oxygen reactions. The developed NPCo catalyst exhibited an ORR onset potential of 0.94 V vs. RHE and reduced the OER overpotential from 454 mV to 353 mV at 10 mA cm−2 in an alkaline electrolyte. The bifunctional NPCo catalyst outperformed state-of-the-art catalysts in rechargeable and flexible zinc–air batteries. This work elucidates the design principles for highly stable and electrocatalytically active perovskite-based materials and identifies the key factors governing their ORR and OER mechanisms.
Results and discussion
Designing and physical characterization of water-stable perovskite
The synthesis of (PEA)2CoCl4 perovskite was carried out using the acid precipitation method, generating perovskite crystals. In brief, during the synthesis process, 2-phenylethylamine (PEA) gets protonated by dissolving in an aqueous HCl solution. Another solution was prepared by dissolving CoCl2·6H2O in aqueous HCl. Finally, both the solutions were mixed at 100 °C with vigorous stirring and allowed to cool down to room temperature slowly, resulting in the blue bulk perovskite (PCo) crystals. The in situ growth of (PEA)2CoCl4 crystals with N-doped reduced graphene oxide (NrGO) generated the NrGO-coated perovskite (NPCo) by a facile heating method, as shown in Fig. 1A. Furthermore, single-crystal X-ray diffraction (SCXRD) analysis was performed at room temperature, revealing that (PEA)2CoCl4 crystallizes in a monoclinic lattice with the space group P21/C. The lattice parameters of the monoclinic crystals are a = 7.453 Å, b = 24.649 Å, c = 11.187 Å, α = γ = 90°, and β = 91.786°. The asymmetric unit of (PEA)2CoCl4 contains two independent C6H5C2H4NH3+ (PEA) ions and one tetrachloride cobaltate(II) [CoCl4]2− dianion.43 Each of the [CoCl4]2− inorganic layers is separated by one layer of organic cations. The overall crystal structure and detailed crystal data are summarized in Fig. 1B, S1, and Table S1, ESI.† | ||
| Fig. 1 Schematic presentation of the material synthesis and aqueous medium stability test of the developed perovskite: (A) schematic presentation of the stepwise synthesis of the NPCo catalyst; (B) single crystal X-ray diffraction (SCXRD) analysis of the perovskite (PCo) crystal showing the molecular arrangement and the crystallographic details; (C) comparative powder XRD analysis of the as-synthesized and KOH treated PCo, showing similarity in diffraction peaks; (D) comparative liquid and solid state UV-visible spectra of PCo and KOH treated PCo, showing π–π*, n–π*, and ligand to metal charge transfer (LMCT) peaks; (E) FTIR spectra of PEACl, PCo, and PCo in KOH, showing the resemblance in stretching frequencies of materials; (F) photoluminescence spectra of PCo and PCo in KOH, showing the similarity in emission spectra; (G) deconvoluted N 1s spectra of NPCo, NPCo(phy), and NrGO, where NPCo shows significant contributions from pyridinic-N (398.2 eV), graphitic-N (401.1 eV), and pyrrolic-N (399.8 eV), confirming successful nitrogen doping and its interaction with PCo; (H) Co 2p spectra for PCo, NPCo(phy), and NPCo showing Co2+ and Co3+ peaks at ∼780 eV and ∼796 eV, respectively, and a shift in NPCo peaks due to interaction with NrGO, indicating electronic structure modulation; (I) comparative electron paramagnetic resonance (EPR) spectra showing the hyperfine structure of NrGO, PCo, and NPCo. | ||
After the synthesis of pristine perovskite (PCo), it is imperative to assess its stability in an aqueous electrolyte since the structural decomposition of perovskite is likely to occur over the long run. To study the stability of the synthesized perovskite PCo in the catalytic environment, PCo was dispersed in 0.1 M KOH electrolyte solution and recovered as a powder (Fig. S2, ESI†). Various characterization techniques confirmed the structural integrity of the recovered PCo from an alkaline aqueous solution. The comparative powder X-ray diffraction (PXRD) spectra of pristine PCo and alkaline solution-treated PCo showed similarity in the diffraction peaks (Fig. 1C). This confirms that the perovskite has no structural degradation at alkaline pH. Fig. 1D shows the UV spectra of PCo, displaying absorption peaks at 280 nm, 315 nm, and 675 nm, attributed to π–π* and n–π* transitions and ligand to metal charge transfer occurring from the organic phenylethylammonium moiety to the cobalt transition metal, respectively.43–45 The comparative UV absorption spectra of PCo in the solid state, solution state, and after dispersion in 0.1 M KOH solution show no peak shifts, further indicating that the PCo structure remains intact, resulting in no structural degradation. In addition, comparative Fourier-transform infrared (FTIR) spectra of KOH solution-treated PCo, organic moiety PEACl, and pristine PCo are shown in Fig. 1E. Along with the peaks of the typical aromatic cation, pristine PCo shows vibrational peaks at 1650 cm−1 and 1130 cm−1, which are attributed to the NH3+ stretching and bending, respectively.45,46 The peaks of pristine PCo remain unaffected in the FTIR spectra of the PCo after the KOH solution treatment. However, an intense peak at 3560 cm−1 highlights the –OH stretch due to the presence of KOH solution. Furthermore, the photoluminescence (PL) spectra shown in Fig. 1F depict the stability of PCo in aqueous electrolytes. Pristine PCo shows a broad emission peak ranging from 300 to 650 nm upon excitation at 290 nm.47,48 The PL spectrum of KOH-treated PCo perovskite matches well with that of the pristine PCo. The PXRD, UV, FTIR, and PL analyses of the KOH-treated PCo confirm the stability of PCo in an aqueous alkaline electrolyte. The stability of PCo in aqueous electrolyte is mainly due to the presence of hydrophobic PEA, which has a strong van der Waals interaction that reduces the water adsorption on the perovskite surface. The PEA moieties create a close water-repellent environment that effectively penetrates water and increases stability.36,49–52
The composite catalyst (NPCo) was characterized to investigate the phase purity, stability to KOH, and interaction between NrGO and PCo. The PXRD pattern of the as-synthesized PCo matches well with the simulated one, confirming the successful formation of PCo perovskite in the composite NPCo material (Fig. S3, ESI†). The XRD pattern of NrGO exhibits broad peaks at 2θ values of 26° and 43°, indicating the graphitic carbon (002) and (100) planes of NrGO with interplanar spacings of 0.34 and 0.21 nm, respectively.53 The comparative PXRD spectra of NPCo reveal the appearance of PCo in the structure with a slight negative shift compared to the pristine perovskite due to the interaction between PCo and NrGO. Furthermore, the comparative Raman spectra of NrGO, PCo, and NPCo are shown in Fig. S4, ESI.† The pristine PCo peaks at 110 and 265 cm−1 indicate that cobalt exists in a tetrahedral environment with the chloride ion, consistent with the single-crystal XRD structure.39,54 The peaks at 1000 and 1600 cm−1 are attributed to C–N stretching and –NH3+ bending vibration, respectively, confirming the presence of PEA+ cation in the perovskite structure.39 In the Raman spectra, the typical defective (D) peak that appeared at 1350 cm−1 corresponds to the graphitic lattice vibration mode with A1g symmetry, while the graphitic (G) peak centered at 1589 cm−1 corresponds to the graphitic lattice vibration mode with E2g symmetry that appeared in NrGO and NPCo.55 The higher AD/AG ratio of NrGO (1.94) compared to NPCo indicates the incorporation of perovskite in NPCo, suggesting that PCo is successfully embedded in NrGO. In addition, a shift of the D and G peaks to a higher frequency in the case of NPCo compared to NrGO indicates the electronic interaction between NrGO and PCo. The thermogravimetric analysis shows the thermal stability and loading of the perovskite over NrGO, as displayed in Fig. S5, ESI.† NPCo shows the loading of cobalt in a meager amount. The pristine (PEA)2CoCl4 and physically mixed NPCo(phy) show the same cobalt loading. In addition, the onset of thermal degradation at lower temperatures observed for NPCo(phy) indicates that in physical mixing, no electronic interaction occurs between PCo and NrGO as they are separated. However, the in situ growth of PCo in the NrGO matrix leads to interaction with each other.
The survey scan X-ray photoelectron spectroscopy (XPS) analysis shows the presence of elements such as C, N, O, Co, and Cl in the corresponding catalysts (Fig. S6, ESI†). The elemental atomic percentage (C: 49.58%, N: 5.95%, Co: 4.66%, and Cl: 38.87%) confirms the structural composition of the pristine PCo. In addition, NrGO contains ∼7% of the doped-N with pyridinic-N (pyri-N), pyrrolic-N (pyrr-N), and graphitic-N (grap-N) at the binding energies (BE) of 397.22, 399.45, and 401.50 eV, respectively.56 The differences in the deconvoluted N 1s spectra of NrGO, NPCo(phy), and NPCo indicate the extent of interaction between the NrGO and perovskite materials (Fig. 1G). An observable shift in the BE of Pyri-N peaks from 397.22 eV to 398.21 eV in the case of pristine NrGO and NPCo evidences the electronic interaction between doped-N and PCo. In contrast, no observable changes have been evidenced in the physically mixed NPCo(phy) catalyst. Similarly, the deconvoluted Co 2p spectra of PCo, NPCo(phy), and NPCo, shown in Fig. 1H, display the characteristic Co 2p3/2 and Co 2p1/2 peaks at ∼780 and ∼796 eV, respectively.56 A remarkable shift in the Co2+ peak in the case of NPCo compared to PCo further evidences the electronic interaction between NrGO and PCo as observed in the N 1s spectra. The Co 2p spectra of NPCo show the existence of Co in two forms, where the peak at the low binding energy corresponds to Co2+, similar to the pristine PCo with a slight shift. However, the new Co 2p peak at a higher BE is due to the interaction between the oxy functional groups of NrGO and PCo. The deconvoluted C 1s, Cl 2p, and O1 spectra of the catalysts also show an observable shift in the BE (Fig. S7 and S8, ESI†). The observed Co 2p and N 1s peaks shift to a higher BE and those of Cl 2p and O 1s shift towards a lower BE in the case of NPCo, which indicate the mutual electron transfer between NrGO and PCo. The comprehensive XPS analysis confirmed the electronic interaction between NrGO and perovskite PCo materials. The electron paramagnetic resonance (EPR) studies explain the individual vibronic states of the metal center based on their environment in the lattice. Fig. 1I shows the EPR spectra of PCo, displaying a partially resolved peak averaging out the hyperfine splitting of cobalt. The EPR spectra of NrGO show a characteristic singlet peak due to the localized electrons. However, the interaction between the electronic spin and nuclear spin, known as hyperfine interaction, alters the energy levels, resulting in observable splitting patterns in the EPR spectra. The well-resolved EPR signals in NPCo indicate the hyperfine interaction of cobalt with NrGO, particularly the coupling with the N atom., which can be correlated with the XPS data. The splitting of peaks further indicates the dilution of the CoCl4− tetrahedron in the lattice due to NrGO sheets from the bulk structure in PCo. A similar interaction effect between NrGO and PCo was observed in the FTIR analysis, which is evidenced by a shift in stretching frequencies (Fig. S9, ESI†).
Density functional theory and ab initio molecular dynamics calculations
To check the effect of N-doped graphene coverage over the perovskite layer and its impact on the prevention of water molecules interacting with the perovskite material, a layer of N-doped graphene was introduced between the perovskite and water layers. The structure optimization using density functional theory (DFT) and ab initio molecular dynamics (AIMD) simulation studies was performed by considering the two layers of perovskite molecules representing the bulk structure. A water film was introduced on the top of the perovskite layer in a supercell. Furthermore, to simulate the aqueous alkaline electrolyte, potassium hydroxide (K+OH−) ions were introduced to the water film (calculation details in the ESI†). The pristine PCo supercell structure (with a vacuum of 12 Å) without water was optimized, and the resultant lattice parameters are a = 18.824 Å, b = 23.043 Å, and c = 11.436 Å. Then, aq. KOH electrolyte was introduced in the vacuum region. The optimized structure of the perovskite layer with aq. KOH electrolyte (PCo + aq. KOH) resulted in the lattice parameters a = 19.504 Å, b = 22.439 Å, and c = 11.349 Å. A substantial change in the lattice parameters after adding water molecules to the perovskite layer indicates their strong interaction. When a graphene layer was introduced between the perovskite and water layers (PCo/rGO + aq. KOH), the optimized structure shows the lattice parameters a = 23.727 Å, b = 22.775 Å, and c = 12.257 Å, which show that the structure of the perovskite layer is closely restored to the PCo structure without electrolyte. This is because there is no direct interaction between the perovskite and water layers. Also, a weak interaction between the perovskite and graphene layers is possible due to the pi–pi interaction of PEA and graphene layers. To improve the interaction between the shielding carbon layer and perovskite, nitrogen-doped reduced graphene (NrGO) was considered. In this case, the lattice parameters of NPCo + aq. KOH were a = 23.286 Å, b = 23.628 Å, and c = 12.035 Å, showing a slight change in the lattice parameters compared to the graphene-separated system. However, in this case, the water molecules and perovskites are expected to come closer to the NrGO due to the electronic interactions. To understand the dynamics of the systems, further calculations were carried out by performing the ab initio molecular dynamics (AIMD) simulations.Stability analysis of N-doped graphene–perovskite composite
Theoretical study was performed to elucidate the role of NrGO towards the stabilization of the perovskite layers with aqueous electrolyte for upholding accessible space to the reactant molecules for the catalytic reaction. A balanced interaction between NrGO and perovskite allowed NrGO to extend the movement between the H2O and perovskite layers. AIMD simulation studies of the perovskite (PCo) and derivative materials (PCo/rGO and NPCo) were performed using the periodic density functional theory (DFT) program Vienna Ab Initio Simulation Package (VASP). Fig. 2A shows the simulated final structure of perovskite (PCo) with an aqueous electrolyte, indicating the diffusion of H2O molecules and its contact with perovskite. The atomic distance variation studies of Co (1), Cl (5, 13), and N (366) with the nearest water molecule's oxygen (O) (1_O, 5_O, 13_O, and N366_O) reveal that there is a strong interaction between solvent water molecules and the perovskite structure (Fig. 2B). The average atomic distance of ∼3.8 Å was observed between the nearest water oxygen atom and the perovskite atoms. However, the separation between the water molecules and perovskite significantly increased after the graphene insertion (PCo/rGO + aq. KOH). The graphene layer created a barrier between the two layers (Fig. 2C). The average atomic distances between the atoms (1_O, 5_O, 13_O, and N487_O) are observed to be increased to ∼8.2 Å. Also, a slight variation in the atomic distances can be realized with the simulation time, indicating the stable structure of the PCo/rGO + aq. KOH system (Fig. 2D). In addition, the perovskite coating with NrGO stabilized the structure by protecting the perovskite layer, which is visualized in Fig. 2E. The doping of nitrogen to the graphene layer introduces defects in the carbon framework responsible for the initialization of its interaction with water molecules as well as perovskite layers, as observed in XPS analysis (Fig. 2F) and AIMD simulation (Video VS3, ESI†). The defect in the coated NrGO sheet causes a breathing movement of NrGO between the perovskite and H2O layers due to their dynamic interaction. In comparison to PCo/rGO, the NrGO layer in the NPCo catalyst will allow the diffusion of reactant O2 molecules and the ions (OH− and H+) towards the active sites during the catalytic process. The breathing phenomena of NrGO in the case of NPCo + aq. KOH can be well realized in Fig. 3F, where the atomic distances (1_O, 5_O, 13_O, and N480_O) vary in the form of a wave with average atomic distances of ∼4–10 Å. The comparative simulation videos (VS1, VS2, and VS3, ESI†) of PCo + aq. KOH, PCo/rGO + aq. KOH, and NPCo + aq. KOH show the movement and mixing of the different components with simulation time. The breathing motion of the NrGO layer can effectively introduce the reactant molecules towards the active sites. | ||
| Fig. 2 Ab initio molecular dynamics (AIMD) simulation of PCo and its interaction with water clusters on different surfaces: (A) structural configuration of the PCo + aq. KOH system; (B) atomic distance variation of selected atoms with the nearest oxygen atoms (1_O, 5_O, 13_O, and N366_O) over a simulation time of 0–5 ps in the PCo + aq. KOH system; (C) PCo/rGO + aq. KOH structural configuration; (D) atomic distance variation of selected atoms and the nearest oxygen atoms (1_O, 5_O, 13_O, and N487_O) over a 0–5 ps simulation time in the PCo/rGO + aq. KOH; (E) structural configuration of the NPCo + aq. KOH system; (F) atomic distance variation of selected atoms with the nearest oxygen atoms (1_O, 5_O, 13_O, and N480_O) over a simulation time of 0–5 ps in the NPCo + aq. KOH system. The atomic distance variations are represented in angstrom (Å), indicating the stability and interaction dynamics of water molecules with the catalyst surface during the simulation period. | ||
 | ||
| Fig. 3 Imaging analysis of NrGO, PCo, NPCo(phy), NPCo catalyst solution stability, surface wettability, and electrical conductivity: (A) SEM image showing PCo particles with an irregular surface; (B) SEM image of NPCo, revealing uniform NrGO coverage over PCo; (C–H) SEM images of NPCo and elemental mapping showing the uniform distribution of carbon (green), oxygen (red), nitrogen (blue), and cobalt (cyan), confirming the successful integration of NrGO and PCo; (I) low-magnification transmission electron microscopy (TEM) image of NPCo showing NrGO sheets intertwined with PCo; (J) high-resolution TEM (HRTEM) image of NPCo, highlighting well-distributed NrGO layers and PCo particles; (K) HRTEM image showing lattice fringes corresponding to d-spacing values of 0.23 and 0.35 nm for NrGO and PCo, confirming the crystalline structure; (L) selected area electron diffraction (SAED) pattern of NPCo showing distinct diffraction rings, corresponding to the crystalline planes of perovskite PCo and NrGO; (M) and (N) atomic force microscopy (AFM) image and height profile of NPCo, indicating particle dimensions of ∼70 nm height and ∼1.1 μm width. (O) and (P) AFM image and height profile of NrGO, showing a thickness of ∼35 nm and a lateral dimension of ∼1.3 μm; (Q) UV-vis absorption spectra of PCo and NPCo catalyst inks, demonstrating enhanced dispersion stability for NPCo. The NPCo catalyst ink shows significantly higher absorption intensity in the 400–700 nm range, indicating improved stability; (R) contact angle measurements showing wettability differences: PCo (55°), NrGO (130°), NPCo(Phy) (64°), and NPCo (118°), NPCo showing the introduction of hydrophobicity with the introduction of NrGO to PCo; (S) four-probe electrical conductivity and sheet resistance of materials showing that NPCo achieves a high conductivity of 245 S m−1 and a low sheet resistance of 0.00028 Ω m−1, demonstrating superior electrical properties compared to PCo and NrGO. | ||
The surface morphology and coverage of NrGO over PCo were evaluated using field emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM), and atomic force microscopy (AFM) analyses. Fig. 3A shows the bulk morphology of the pristine perovskite (PCo) with a rough surface, with a size range of 2–3 μm. After in situ growth of the PCo crystals with NrGO, the particles' overall size decreased (1.5–2.5 μm) compared to pristine PCo, reflecting the effect of nucleation sites available in NrGO. During the in situ growth process of PCo, the doped-N in NrGO acts as nucleation sites. Due to the electronegativity difference between Co2+ and doped-N, there will be strong interaction between growing PCo and doped-N of NrGO, causing the wrapping of NrGO over the PCo, resulting in the generation of NrGO-wrapped PCo materials. Fig. 3B displays the FESEM image of NrGO-coated PCo (NPCo) where NrGO is exposed outside, where PCo is coated and not exposed directly. Fig. 3C–H show the elemental mapping of the NPCo observed in Fig. 3G, displaying the overlay elements and distribution of C, N, O, and Co. EDX-SEM mapping of elements shows the uniform distribution of C, N, Co, and O in NPCo. In contrast, Co and Cl are missing, indicating the coverage of NrGO over the surface PCo. In contrast, the FESEM analysis of the pristine PCo and physically mixed NrGO + PCo catalyst (NPCo(phy)) displayed a non-uniform distribution of NrGO and other elements with exposed Co metal ion (Fig. S10 and S11, ESI†). High-resolution transmission electron microscopy (HR-TEM) analysis of NPCo reveals the coating of thin and flexed sheets of NrGO over the micro-structured perovskite (PCo), which leads to the controlled exposure of perovskite to the electrolyte (Fig. 3I–J and S12, ESI†). Fig. 3K shows the 2.22 and 3.5 Å lattice fringes, which are attributed to the (002) and (100) facets of NrGO and PCo, respectively.55 The incorporation of PCo enhances the d-spacings of NrGO by lattice distortion. The selected area electron diffraction (SAED) pattern shown in Fig. 3L indicates the presence of polycrystalline planes of perovskite and NrGO. Fig. 3M–P show the AFM images and height profiles of NrGO and NPCo. The topographic images of NrGO show the NrGO sheets with rough surfaces having some porosity; however, NPCo displays a microstructure with agglomerated NrGO sheets. The height profile of NrGO represents a layer structure with few thin and flex sheets resulting in the height of nm size, which contains 100 layers. However, 3D NPCo with a micrometer height represents the incorporation of perovskite.
To confirm that the perovskite is not leaching out from NrGO in the NPCo catalyst ink, the UV absorption spectra were recorded (Fig. 3Q). The PCo shows all the absorption peaks; however, the NPCo catalyst ink shows no absorption peaks, indicating that PCo is not entirely exposed to the electrolyte and remains stable in the organic–inorganic solvent. Furthermore, the contact angle (CA) measurement shown in Fig. 3R indicates that NrGO (θ = 130°) increases the surface hydrophobicity of the composite NPCo catalyst. The PCo shows a macroscopic hydrophilic surface (CA = 55°). However, after coating with hydrophobic NrGO (CA = 130°), the composite material, displays hydrophobicity (CA = 118°). On the other hand, NPCo(phy) (CA = 65°), where NrGO has no electronic interaction with PCo and is separated from each other, is still hydrophilic in nature. Typically, the effective electronic interaction between PCo and NrGO should also be reflected in the electronic conductivity. Fig. 3S† shows that the coating of PCo with highly conductive NrGO eradicates the conductivity issue of perovskite in NPCo, consistent with the conductivity measurement data of the materials using four-probe conductivity measurements. The observed trend of electronic conductivity/resistance, evidencing the in situ growth of PCo particles with NrGO, establishes that the electronic interaction between the two is responsible for the improved conductivity. However, the physically mixed sample (NPCo(phy)) with the least interaction shows less conductivity than NPCo. The in situ inclusion of highly conductive NrGO in the NPCo catalyst increases the overall stability, conductivity, and active site electronic property modulation, which improves the activity of PCo towards ORR/OER electrocatalysis. Such improvements result from the electronic effect induced by the interaction between PCo and NrGO. The composite NPCo effectively transfers electrons to the organic, inorganic perovskite, and NrGO interfaces. The specific surface area of PCo and NPCo was determined by using N2 adsorption/desorption studies (Fig. S13, ESI†). The specific surface area of the pristine (PEA)2CoCl4 (PCo) was observed as 9.67 m2 g−1, which increased to 165 m2 g−1 after the introduction of NrGO to PCo (NPCo). Similarly, the BJH pore size distribution revealed the presence of mesopores in PCo. The calculated pore volume for PCo at P/P0 = 0.99 is 0.008 cm3 g−1. However, NPCo contains hierarchical mesopores with a pore diameter of 3.8 nm, and the calculated pore volume for NPCo at P/P0 = 0.99 is 0.316.57 The increased specific surface area and porosity of NPCo are an indication of improved accessibility of reactant molecules.
Electrocatalytic activity analysis
The electrocatalytic activity of the materials towards oxygen reactions, i.e., the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), was studied using a conventional 3-electrode setup. The electrochemical setup contains a glassy carbon rotating disc (Pt-ring) electrode (RDE/RRDE) coated with the catalytic material (working electrode), Hg/HgO (reference electrode), and a graphite rod (counter electrode) using N2/O2 saturated 0.1 M KOH as an electrolyte. The recorded potentials w.r.t. Hg/HgO were converted to a reversible hydrogen electrode (RHE) (details in the Experimental section, Fig. S14, ESI†). Fig. 4A shows the comparative cyclic voltammogram (CV) recorded for the PCo, NrGO, NPCo(phy), and NPCo catalysts in N2/O2 saturated 0.1 M KOH electrolyte. The higher reduction current obtained in O2-saturated electrolyte is due to the ORR process attained effectively by an NPCo catalyst at a lower reduction potential. The differences in ORR characteristic peaks in terms of the ORR onset potential and current density are attributed to the variation in catalytic active sites, accessibility of the reactant, and conductivity of the catalysts. The linear sweep voltammogram (LSV) recorded at 1600 rpm in O2-saturated electrolyte displays a distinguishable onset potential (Eonset). The comparative LSV curve shown in Fig. 4B displays the Eonset at 0.1 mA cm−2 in the order of Pt/C (1.00 V), NPCo (0.94 V), NrGO (0.84 V), NPCo(phy) (0.83 V), and PCo (0.60 V) vs. RHE. A closer Eonset of NPCo to the state-of-the-art (Pt/C) catalyst indicates the activity enhancement effect of the modulated active sites towards the catalytic process. In addition, the difference in ORR activity between NPCo and NPCo(phy) reveals the impact of electronic interaction between NrGO and PCo. The observed significant difference in the catalytic ORR activity of NPCo compared to PCo, NPCo(phy), and NrGO suggests that it is due to the changes in the electronic and physical properties of the catalyst and active sites. A significant difference in the Tafel slope of the catalysts suggests that the catalysts follow different ORR pathways (Fig. 4C). The state-of-the-art (Pt/C) and NPCo catalysts showed Tafel slopes of 54 and 60 mV dec−1, respectively. In contrast, the PCo catalyst prepared without NrGO exhibited the highest Tafel slope of 132 mV dec−1, revealing the effective electron and ion transport in the case of the NPCo catalyst. Fig. 4D shows the electrocatalytic surface area (ECSA) estimated from the electrical double-layer capacitance. The electrical double-layer capacitance CV profiles of the respective catalysts are shown in Fig. S15–S17, ESI.† NrGO and NPCo catalysts exhibited comparable ECSAs of 4.4 and 3.7 mF cm−2, respectively, due to the capacitive nature of NrGO. However, the lower ECSA observed for PCo and NPCo(phy) indicates the importance of effective interaction between NrGO and PCo.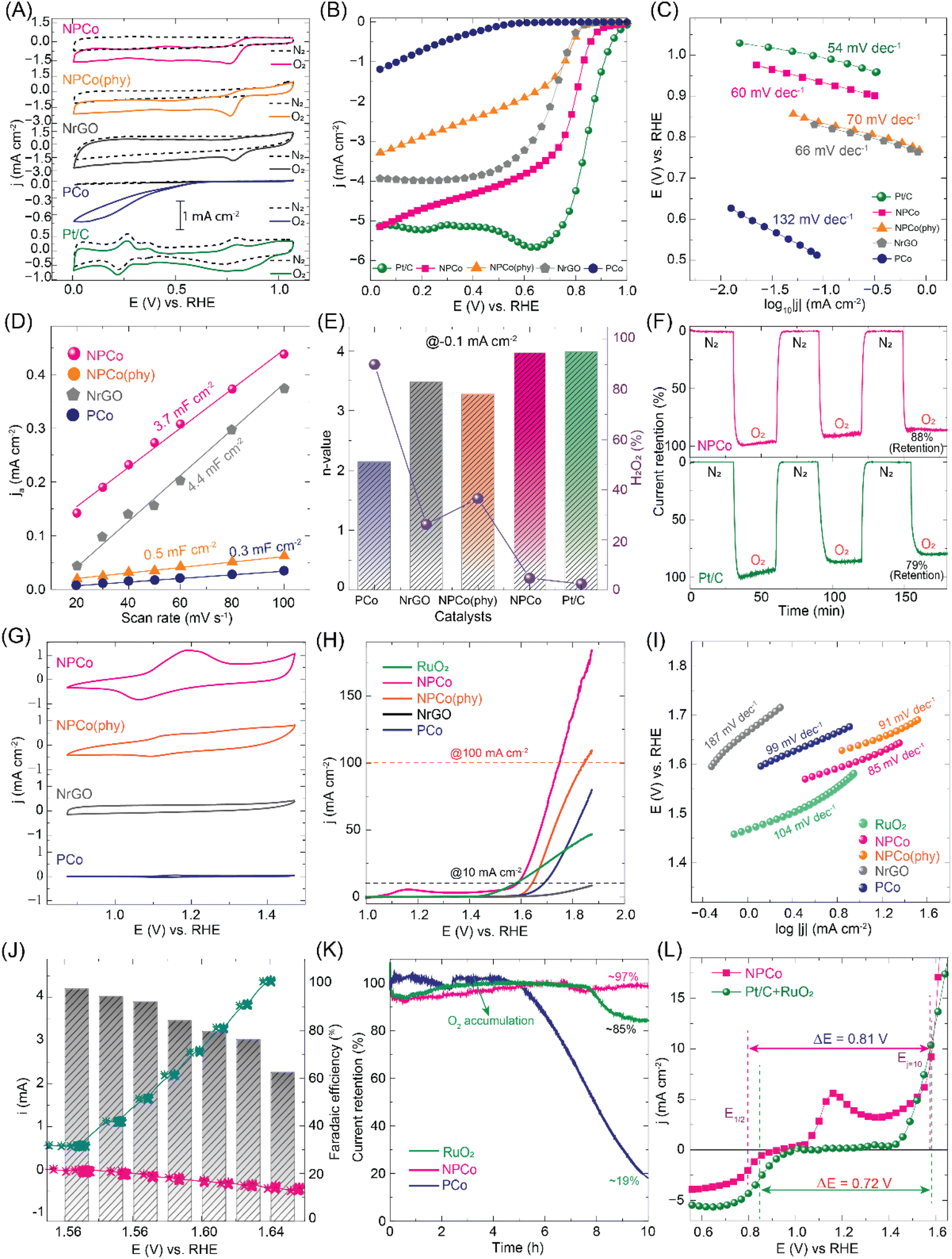 | ||
| Fig. 4 Electrocatalytic oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) performance analyses of PCo, NrGO, NPCo(phy), NPCo, and state-of-the-art catalysts: (A) comparative cyclic voltammogram (CV) of the catalysts recorded in N2/O2 saturated 0.1 M KOH electrolyte at a scan rate of 10 mV s−1; (B) linear sweep voltammograms of PCo, NrGO, NPCo(phy), NPCo and Pt/C catalysts recorded at a scan rate of 10 mV s−1 in O2 saturated electrolyte with the disc electrode rotation at 1600 rpm; (C) Tafel slope plot of the catalysts displaying the ORR kinetics; (D) double-layer capacitance (Cdl) as a function of scan rate, indicating the electrochemical surface area (ECSA) of NPCo, NPCo(phy), NrGO, and PCo; (E) rotating ring-disc electrode (RRDE) analysis showing the generated H2O2% and n-value during the ORR process observed at 0.1 mA cm−2 current density; (F) comparative durability analysis of the Pt/C and NPCo catalysts by conducting chronoamperometric analysis at the applied E1/2 potential in a N2–O2 saturated electrolyte solution; (G) CV curves of NPCo, NPCo(phy), NrGO, and PCo in 1 M KOH, highlighting their redox behaviour; (H) LSV curves of RuO2, NPCo, NPCo(phy), NrGO, and PCo, showing their OER activity (LSV data is 85% iR-compensated); (I) Tafel plots derived from LSV curves, comparing the OER kinetics of the catalysts; (J) faradaic efficiency and current density as a function of applied potential for NPCo; (K) chronopotentiometry stability tests of RuO2, NPCo, and PCo, indicating their current retention and oxygen accumulation over 10 hours; (L) the electrochemical potential required to carry out the bi-functional oxygen reactions (ORR and OER) by the Pt/C + RuO2 and NPCo catalysts. | ||
A rotating ring disc electrode (RRDE) analysis was carried out to estimate the generated H2O2% and the number of electron transfers (Experimental details in the ESI and Fig. S18†). Fig. 4E shows the H2O2% and n-value, suggesting that the NPCo catalyst followed a direct 4e− (∼3.8) electron pathway similar to the Pt/C catalyst at 0.85 V, estimated at 0.1 mA cm−2 current density. Consequently, these catalysts generate a lower H2O2% of ∼8–10% during the ORR process. In contrast, PCo and NPCo(phy) display the 2e− process at a lower potential, followed by ∼4e− transport at a higher reduction potential. Fig. 4F shows the chronoamperometric ORR durability of Pt/C and NPCo under N2/O2 switching conditions for 3 continuous cycles, showing current retention of 79% and 88%, respectively. The higher durability NPCo achieved is due to the integrated, stable structure of NrGO and PCo, as observed in the XPS studies. The observed stability of NPCo under operating conditions confirms that the strong interaction between NrGO and controlled electrolyte accessibility retains the structural integrity of the NPCo catalyst.
Fig. 4G shows the comparative cyclic voltammograms (CV) of PCo, NrGO, NPCo(phy), and NPCo catalysts recorded in N2-saturated 0.1 M KOH. An observable redox peak with a higher current density observed for the NPCo catalyst indicates the improved intrinsic conductivity of NPCo due to the effective interaction between NrGO and PCo. In contrast, PCo and NPCo(phy) have shown lower redox peak current densities corresponding to the semiconducting nature of pristine PCo and the least interaction between NrGO and PCo, respectively. The linear sweep voltammetry (LSV) analysis of the catalysts recorded at a scan rate of 5 mV s−1 shows the characteristic oxygen evolution peak at a higher current density (Fig. 4H). The prepared catalysts, NrGO, PCo, NPCo(phy), and NPCo, showed overpotentials of 560, 454, 410, and 353 mV, respectively, at 10 mA cm−2. The obtained overpotential for NPCo is significantly closer to the state-of-the-art (RuO2) catalyst, which has an overpotential of 346 mV@10 mA cm−2. However, the NPCo catalyst has outperformed the RuO2 catalyst at a higher current density with the overpotential of 517 mV@100 mA cm−2, whereas RuO2 could not achieve a higher current density. The lowest overpotential and higher OER current density were demonstrated by the NPCo catalyst, indicating the active role of the catalytic active sites and the effectively modified electronic structure of the NPCo catalyst due to the interaction between NrGO and pristine PCo.
Furthermore, the Tafel slope analysis of the obtained LSV curves showed the OER kinetics of the catalysts (Fig. 4I). The NPCo catalyst has displayed the lowest Tafel slope of ∼85 mV dec−1 as compared to the counterpart catalysts such as NrGO (∼187 mV dec−1), PCo (∼99 mV dec−1), and NPCo(phy) (∼91 mV dec−1). The observed Tafel slope for NPCo is distinctly lower than the other catalysts, suggesting the higher OER kinetics exhibited by the NPCo catalyst. Also, the NPCo catalyst has OER kinetics exhibited by the state-of-the-art (RuO2) catalyst, demonstrating effective electron transfer during the catalytic process. In addition, the faradaic efficiency (FE) of the NPCo catalyst was estimated (Fig. 4J) using an RRDE electrode (glassy carbon: disc; Pt: ring) rotating at 1600 rpm. The NPCo catalyst-coated disc electrode was subjected to a series of current steps (0–10 mA cm−2), and simultaneously a constant potential of 0.5 V vs. RHE was applied to the ring electrode. The obtained ring current is mainly ascribed to the reduction of disc electrode-generated O2 at the ring electrode. NPCo showed an FE of ∼95% at a lower applied current, which decreases to ∼63% at 10 mA cm−2 current density. The decreased FE at higher current density is ascribed to the escaped O2 molecules due to the increased concentration generated at a higher current density. The observed FE estimated by considering the ring electrode reaction current density due to the applied ORR potential suggests that the product generated at the disc electrode is O2 molecules because of the OER process. The comparative chronoamperometric stability analysis of RuO2, PCo, and NPCo (Fig. 4K and S19, ESI†) showed a better durability exhibited by the NPCo catalyst with the retention of ∼97% current density after 10 h. The pristine PCo catalyst displayed almost similar activity up to 4 h with a sudden decrease in activity to ∼19% current retention. The observed variation in current retention for the PCo catalyst is ascribed to the structural integrity possessed by PCo up to a certain period, owing to its structural designing advantages. However, the RuO2 catalyst has the lower durability compared to NPCo, with a current retention of ∼85% after 10 h. Fig. 4L shows the electrochemical potential required to drive the bifunctional catalytic activity (ORR and OER) of Pt/C + RuO2 and NPCo. The NPCo catalyst showed a 0.09 V potential difference needed for the overall oxygen reaction compared to the state-of-the-art catalyst, indicating the efficiency of the designed NPCo catalyst. The catalysts' comprehensive ORR and OER performances are summarized in Fig. S21, S22, ESI.†
Post-durability characterization of the NPCo catalyst was done using XRD, Raman, EPR, and SEM analyses (Fig. S23–S27, ESI†). XRD analysis showed new diffraction peaks at 19.8, 32.1, and 39.8, respectively, corresponding to the Co oxide. Similarly, Raman analysis also evidenced that the Co–O peaks arise after the long-term applied potential during the catalytic process. The comparative EPR analysis of the pristine NPCo and after the ORR and OER durability showed a significant reduction in the signal peak intensity. The peak intensity reduction signifies the destruction of NrGO and PCo interaction, which might be due to the exposure of the catalyst to the aq. KOH electrolyte under applied potential. In addition, the SEM and EDAX analyses of the NPCo catalyst showed the destruction of the wrapped structure and exposure of Co metal ions. So, the observed decrease in the current retention during the catalytic process is due to the Co oxide formation and the destruction of NrGO–perovskite interactions, as evidenced in the post-analysis of the NPCo catalyst.
Zinc–air battery performance of the catalyst electrodes
Owing to the higher structural stability in aqueous KOH electrolyte and the catalytic ORR/OER activity of the NPCo catalyst, a rechargeable zinc–air battery (ZAB) was fabricated using it as a cathode electrode. Fig. 5A shows the schematic illustration of a zinc–air battery where KOH + Zn(CH3COO)2 and zinc foil were employed as an electrolyte and anode, respectively. The ZABs with NPCo electrodes have shown an open-circuit voltage of 1.42 V (Fig. S28, ESI†). The comparative steady-state polarization of ZABs with NPCo and state-of-the-art ORR and OER catalyst (Pt/C + RuO2) cathode electrodes (Fig. 5B) showed peak power densities of 116 and 105 mW cm−2, respectively. The superior ZAB performance by NPCo, compared to the Pt/C + RuO2 electrode, correlates with the efficient O2 diffusion of the catalytic active sites. As the KOH concentration increased to 6 M KOH in ZAB, the O2 diffusion to the active sites was hindered due to the increased viscosity, leading to slower ORR rates despite improved reaction kinetics. Also, at higher KOH concentrations, due to increased ion–ion interactions, there may be a decrease in the ionic conductivity, causing the lowering of ORR kinetics. The ion conductivity and reactant mass transport are evidenced in the electrochemical impedance spectroscopy (EIS) analysis of the ZABs (Fig. 5C). The closer equivalent series resistance (∼1.7 Ω) of the NPCo and Pt/C + RuO2 based ZAB demonstrates similar conditions maintained for assembling the ZABs. However, there is a significant difference in the charge transfer resistance (RCT) of 0.1 and 2.1 Ω for NPCo and Pt/C + RuO2 ZABs, which verifies the higher ionic resistance exhibited by Pt/C + RuO2 compared to NPCo. In addition, the Warburg region of the comparative EIS spectra shows that the Pt/C + RuO2 electrode has experienced restricted O2 mass transport during the battery operation. These results demonstrate the active role of porosity and accessibility of the reactant molecules to the active sites. Fig. 5D shows the galvanostatic discharge curve of the Pt/C + RuO2 and NPCo electrode batteries at the discharge rate of 10 mA cm−2, displaying specific capacities of 736 and 760 mA hZn−1, respectively. The fabricated batteries showed a discharge time of ∼20 h at a constant discharge rate of 10 mA cm−2. A similar steady flat discharge profile has been obtained at higher discharge current densities (Fig. S29, ESI†). A smaller potential drop was observed during the discharge of ZABs, displaying that the fabricated batteries offer the least contact resistance from the electrodes.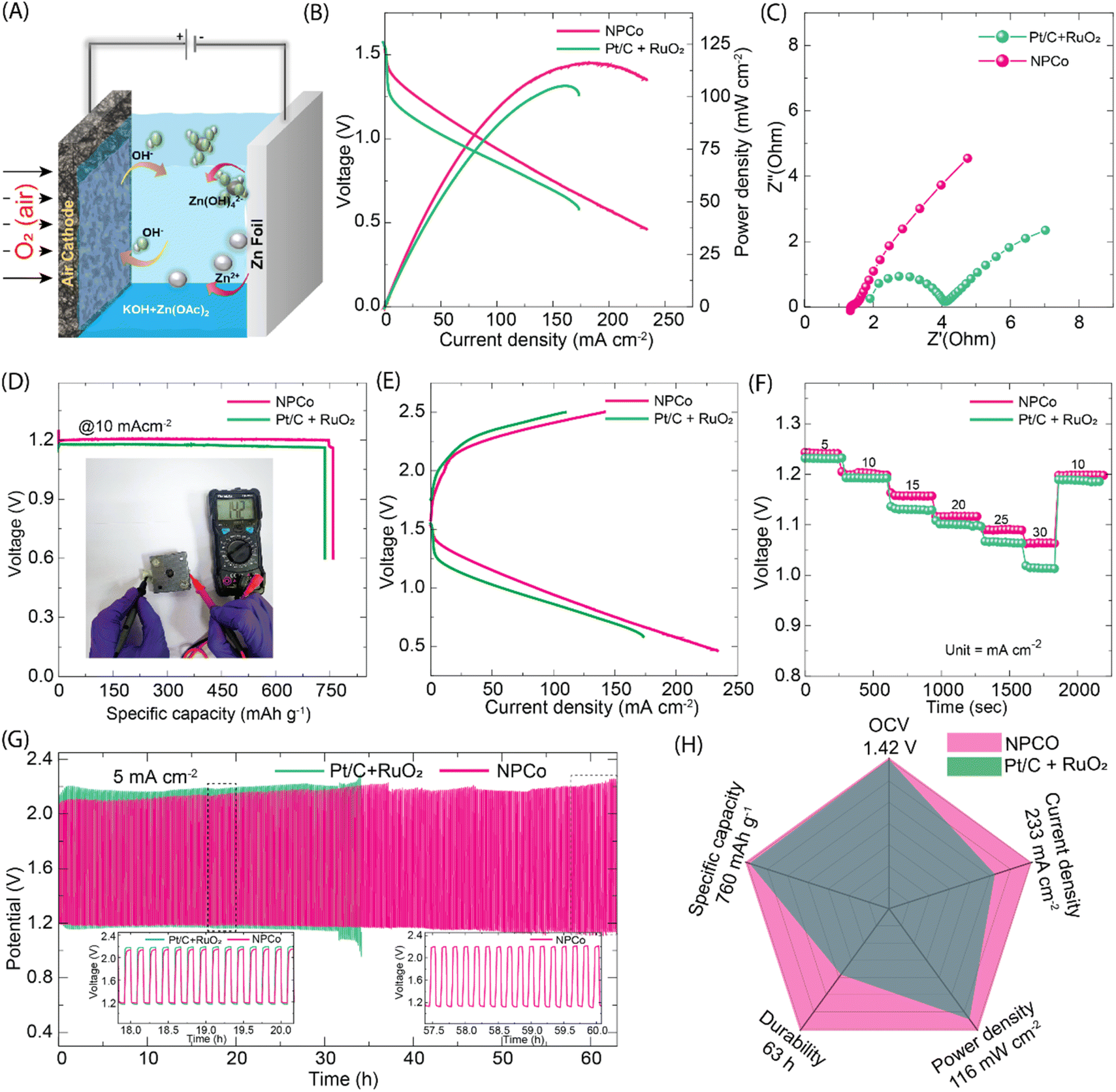 | ||
| Fig. 5 Primary and rechargeable zinc–air battery performance fabricated by using Pt/C + RuO2 and NPCo catalyst electrodes: (A) schematic illustration of the zinc–air battery system with Pt/C + RuO2 and NPCo as the air electrode catalyst; (B) polarization and power density curves of zinc–air batteries, showing maximum power densities of 116 mW cm−2 for NPCo and 105 mW cm−2 for Pt/C + RuO2 catalysts; (C) Nyquist plots from electrochemical impedance spectroscopy (EIS) measurements, with NPCo showing a lower charge-transfer resistance than Pt/C + RuO2; (D) discharge-specific capacity curves at a current density of 10 mA cm−2, with NPCo achieving a specific capacity of 760 mA h g−1 compared to 736 mA h g−1 for Pt/C + RuO2. Inset: the photographic image of the zinc–air battery setup under test fabricated by using the 3D printing technique; (E) charge–discharge polarization curves of the batteries, highlighting a smaller overpotential for NPCo across the current density range of 0–250 mA cm−2; (F) voltage profiles during stepwise charge–discharge cycling at varying current densities (5–30 mA cm−2), with NPCo showing a stable operation and higher voltage efficiency; (G) durability test at a constant current density of 5 mA cm−2, showing voltage stability over 62 hours for NPCo and 35 hours for Pt/C + RuO2; insets: zoomed-in voltage profiles highlighting stable cycling behaviour; (H) radar chart summarizing performance metrics: NPCo shows superior specific capacity (760 mA h g−1), power density (116 mW cm−2), current density (233 mA cm−2), durability (63 h), and voltage efficiency compared to Pt/C + RuO2. | ||
A rechargeable zinc–air battery (rZABs) analysis was carried out owing to the bifunctional (ORR/OER) catalytic activity of the NPCo catalyst; Fig. 5E shows the charge–discharge profile of the rZABs recorded at the current density of 10 mA cm−2. The NPCo-based rZABs have achieved a higher discharge current density with a lower charging current than the Pt/C + RuO2 battery. The reversibility analysis of the batteries measured at variable charge–discharge current densities (5–30 mA cm−2 and reversing back at 10 mA cm−2) (Fig. 5F and S30, ESI†) shows a highly reversible nature, reflecting in its tendency of attending back to the initial voltage at 10 mA cm−2 in the reverse cycle. The obtained reversible battery performance at various current densities demonstrates the effective catalytic OER activity of the developed NPCo, as observed in the half-cell studies (Fig. 4). For the durability test of the catalyst electrode under rZABs’ operating conditions, the rZABs fabricated using Pt/C + RuO2 and NPCo were cycled at 5 mA cm−2 for ∼62 h. The poor durability of the Pt/C + RuO2 catalyst electrode as compared to NPCo is reflected in Fig. 5G, where the Pt/C + RuO2 battery showed an increased charge–discharge potential difference after 32 h and later stopped working. Fig. 5G (inset) shows the comparative cycling durability test of the batteries, displaying a nominal change in the battery voltage composed of NPCo. In contrast, the Pt/C + RuO2 battery has displayed a remarkable change in the charge–discharge voltage, reflecting its lower stability. Fig. 5H shows the comparative analysis of the overall performance of the Pt/C + RuO2 and NPCo electrodes in ZABs.
Fig. 6 demonstrates the fabricated solid-state and flexible zinc–air battery devices. Two solid-state ZABs connected in series, giving a maximum voltage of 2.84 V, illuminated the 54 LEDs connected in parallel (Fig. 6A). The LED light illumination demonstration video is provided in video VS4, ESI.† It reveals the high performance of the developed NPCo catalyst in solid-state devices. Furthermore, the NPCo catalyst-coated gas diffusion layer was used as the cathode, employing the KOH-soaked PVA polymer and zinc foil as the membrane electrolyte and anode, respectively, to fabricate the flexible zinc–air batteries (fZABs) (Fig. 6B). Fig. 6C shows the charge–discharge cycle of fZABs at 3 mA cm−2, which resembles the solid-state ZAB activity, demonstrating the fabrication technique's effectiveness. The fZAB presents stable galvanostatic discharge–charge curves at various bending angles for >100 cycles. This confirms the good mechanical stability of the constructed fZAB, validating the robustness of the NPCo catalyst and fabricated fZAB (Fig. 6D and E).
 | ||
| Fig. 6 Demonstration of the fabricated solid-state ZAB by illuminating LEDs and flexible zinc–air battery performance at various bending angles: (A) demonstration of illumination of 54 LED lights by using two solid-state ZABs assembled by using NPCo as an air cathode and batteries connected in series; (B) schematic diagram of the fabrication strategy used for the flexible ZAB using the NPCo catalyst as the air electrode; (C) charge–discharge analysis of the fabricated flexible ZAB at 3 mA cm−2 current density; (D) and (E) charge–discharge profiles of the flexible ZAB at 90° and 180° bending angles of the device for >100 cycles. | ||
Conclusions
This study presents a significant advancement in developing aqueous alkaline pH-stable inorganic–organic hybrid perovskite materials for energy storage and conversion applications. Incorporation of N-doped reduced graphene oxide (NrGO) into the (PEA)2CoCl4 perovskite structure enhances its stability under aqueous alkaline conditions and improves its electronic conductivity and catalytic activity. Extensive characterization and ab initio MD simulations reveal that the NrGO coating provides a hydrophobic protective layer, effectively preventing water-induced degradation while maintaining accessibility to catalytic active sites. The resulting composite material, NPCo, exhibits excellent bifunctional catalytic performance for oxygen evolution and reduction reactions (OER/ORR), with significantly lower overpotentials, higher current densities, and superior Tafel slopes compared to conventional catalysts like Pt/C and RuO2. The superior stability and catalytic performance of NPCo enable its successful application in rechargeable zinc–air batteries (ZABs) and flexible ZABs. The NPCo-based ZABs demonstrate outstanding specific capacity, peak power density, and prolonged durability, outperforming state-of-the-art catalysts. This study highlights the importance of material design strategies, such as surface passivation and electronic interaction modulation, and sets a new benchmark for perovskite-based materials in energy storage technologies. By addressing critical challenges related to water stability and conductivity, this work paves the way for the broader utilization of halide perovskites in sustainable and flexible energy systems, offering a promising pathway for future advancements in renewable energy technologies.Data availability
The data that support this study are available in the corresponding ESI.†Author contributions
The manuscript was written with contributions from all authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors confirm that they have no known financial or personal conflicts of interest that could have influenced the work presented in this paper.Acknowledgements
P. B. gratefully acknowledges the financial support from the Shiv Nadar Institution of Eminence (SNIoE), India. P. B. and S. K. S. acknowledge the central instrumentation facilities (XRD, Raman, AFM, EPR, FESEM) available at the SNIoE. The calculations have been performed on the high-performance computing facility Magus and Magus02 of SNIoE. S. K. S. acknowledges the financial grant (SRG/2022/000223) provided by DST-SERB, India. The computational work was done using the high-performance computing facility Magus at SNIoE.References
- L. Mao, C. C. Stoumpos and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 1171–1190 CrossRef CAS PubMed.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS.
- D. A. Egger, A. M. Rappe and L. Kronik, Acc. Chem. Res., 2016, 49, 573–581 CrossRef CAS PubMed.
- Z. Xiao, Z. Song and Y. Yan, Adv. Mater., 2019, 31, 1803792 CrossRef CAS PubMed.
- S. Yang, S. Chen, E. Mosconi, Y. Fang, X. Xiao, C. Wang, Y. Zhou, Z. Yu, J. Zhao, Y. Gao, F. De Angelis and J. Huang, Science, 2019, 365, 473–478 CrossRef CAS PubMed.
- D. Bi, W. Tress, M. I. Dar, P. Gao, J. Luo, C. Renevier, K. Schenk, A. Abate, F. Giordano, J.-P. Correa Baena, J.-D. Decoppet, S. M. Zakeeruddin, M. K. Nazeeruddin, M. Grätzel and A. Hagfeldt, Sci. Adv., 2016, 2, e1501170 CrossRef PubMed.
- Y. Han, Z. Guo, S. Liu, Y. Wu, X. Li, G. Cui, S. Zhou and H. Zhou, Adv. Mater., 2024, 2413895 CrossRef.
- L. Lei, Q. Dong, K. Gundogdu and F. So, Adv. Funct. Mater., 2021, 31, 2010144 CrossRef CAS.
- J. S. Manser, J. A. Christians and P. V. Kamat, Chem. Rev., 2016, 116, 12956–13008 CrossRef CAS PubMed.
- L. Polavarapu, B. Nickel, J. Feldmann and A. S. Urban, Adv. Energy Mater., 2017, 7, 1700267 CrossRef.
- S.-T. Ha, R. Su, J. Xing, Q. Zhang and Q. Xiong, Chem. Sci., 2017, 8, 2522–2536 RSC.
- E. L. Unger, L. Kegelmann, K. Suchan, D. Sörell, L. Korte and S. Albrecht, J. Mater. Chem. A, 2017, 5, 11401–11409 RSC.
- D. B. Straus and R. J. Cava, ACS Appl. Mater. Interfaces, 2022, 14, 34884–34890 CrossRef CAS PubMed.
- S. Cheng and H. Zhong, J. Phys. Chem. Lett., 2022, 13, 2281–2290 CrossRef CAS.
- J. Hidalgo, W. Kaiser, Y. An, R. Li, Z. Oh, A.-F. Castro-Méndez, D. K. LaFollette, S. Kim, B. Lai, J. Breternitz, S. Schorr, C. A. R. Perini, E. Mosconi, F. De Angelis and J.-P. Correa-Baena, J. Am. Chem. Soc., 2023, 145, 24549–24557 CAS.
- H.-C. Wang, S.-Y. Lin, A.-C. Tang, B. P. Singh, H.-C. Tong, C.-Y. Chen, Y.-C. Lee, T.-L. Tsai and R.-S. Liu, Angew. Chem., Int. Ed., 2016, 55, 7924–7929 CrossRef CAS PubMed.
- A. Loiudice, S. Saris, E. Oveisi, D. T. L. Alexander and R. Buonsanti, Angew. Chem., Int. Ed., 2017, 56, 10696–10701 CrossRef CAS.
- T. Leijtens, B. Lauber, G. E. Eperon, S. D. Stranks and H. J. Snaith, J. Phys. Chem. Lett., 2014, 5, 1096–1102 CrossRef CAS PubMed.
- Y. Wei, X. Deng, Z. Xie, X. Cai, S. Liang, P. a. Ma, Z. Hou, Z. Cheng and J. Lin, Adv. Funct. Mater., 2017, 27, 1703535 CrossRef.
- H. Yu, X. Cheng, Y. Wang, Y. Liu, K. Rong, Z. Li, Y. Wan, W. Gong, K. Watanabe, T. Taniguchi, S. Wang, J. Chen, Y. Ye and L. Dai, ACS Photonics, 2018, 5, 4520–4528 CrossRef CAS.
- Y. Wu, P. Wang, X. Zhu, Q. Zhang, Z. Wang, Y. Liu, G. Zou, Y. Dai, M.-H. Whangbo and B. Huang, Adv. Mater., 2018, 30, 1704342 CrossRef.
- B. A. Rosales, M. P. Hanrahan, B. W. Boote, A. J. Rossini, E. A. Smith and J. Vela, ACS Energy Lett., 2017, 2, 906–914 CrossRef CAS.
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS.
- A. Babayigit, A. Ethirajan, M. Muller and B. Conings, Nat. Mater., 2016, 15, 247–251 CrossRef CAS PubMed.
- A. G. Gilman, L. S. Goodman and A. Gilman, Goodman & Gilman's The Pharmacological Basis of Therapeutics, Macmillan, New York, 1980, pp. 380–390 Search PubMed.
- A. Ara and J. A. Usmani, Interdiscip. Toxicol., 2015, 8, 55–64 CrossRef PubMed.
- Q. Fan, G. V. Biesold-McGee, J. Ma, Q. Xu, S. Pan, J. Peng and Z. Lin, Angew. Chem., Int. Ed., 2020, 59, 1030–1046 CrossRef CAS.
- I. López-Fernández, D. Valli, C. Y. Wang, S. Samanta, T. Okamoto, Y. T. Huang, K. Sun, Y. Liu, V. S. Chirvony and A. Patra, Adv. Funct. Mater., 2024, 34, 2307896 CrossRef.
- J. Wu, N. Akhtar, R. Y. N. Gengler, T. T. M. Palstra and P. Rudolf, J. Mater. Chem. C, 2017, 5, 1782–1788 RSC.
- Y. Asensio, S. Marras, D. Spirito, M. Gobbi, M. Ipatov, F. Casanova, A. Mateo-Alonso, L. E. Hueso and B. Martín-García, Adv. Funct. Mater., 2022, 32, 2207988 CrossRef CAS.
- R. Willett, H. Place and M. Middleton, J. Am. Chem. Soc., 1988, 110, 8639–8650 CrossRef CAS.
- H. Peng, L. Xu, Y. Sheng, W. Sun, Y. Yang, H. Deng, W. Chen and J. Liu, Small, 2021, 17, 2102149 CrossRef CAS PubMed.
- G. Lin, Z. Zhang, Q. Ju, T. Wu, C. U. Segre, W. Chen, H. Peng, H. Zhang, Q. Liu, Z. Liu, Y. Zhang, S. Kong, Y. Mao, W. Zhao, K. Suenaga, F. Huang and J. Wang, Nat. Commun., 2023, 14, 280 CrossRef CAS PubMed.
- M. Hamdan and A. K. Chandiran, Angew. Chem., Int. Ed., 2020, 59, 16033–16038 CrossRef CAS.
- D. Wu, C. Wang, B. Huo, K. Hu, X. Mao, Z. Geng, Q. Huang, W. Zhang, J. Zeng and X. Tang, EEM, 2023, 6, e12411 CAS.
- Y. Nakayama, S. Nishihara, K. Inoue, T. Suzuki and M. Kurmoo, Angew. Chem., Int. Ed., 2017, 56, 9367–9370 CrossRef CAS.
- Y. Wang, C. Wang, Y. Wei, F. Wei, L. Kong, J. Feng, J.-Q. Lu, X. Zhou and F. Yang, Chem.–Eur. J., 2022, 28, e202201832 CrossRef CAS PubMed.
- R. Babu, A. K. Vardhaman, V. M. Dhavale, L. Giribabu and S. P. Singh, Chem. Commun., 2019, 55, 6779–6782 RSC.
- N. Navarro, C. Núñez, D. Espinoza, K. Gallardo, I. Brito and R. Castillo, Inorg. Chem., 2023, 62, 17046–17051 CrossRef CAS.
- X. Ren, Y. Zhai, P. Wang, Z. Xu, S. Gao, X. Chen, Q. Gu, B. Wang, J. Li and S. Liu, Adv. Mater., 2023, 35, 2301166 CrossRef CAS PubMed.
- K. P. Lindquist, T. Lee, X. Xu and R. J. Cava, Chem. Mater., 2024, 36, 7610–7618 CrossRef CAS.
- P. Jiang, D. Acharya, G. Volonakis, M. Zacharias, M. Kepenekian, L. Pedesseau, C. Katan and J. Even, APL Mater., 2022, 10, 060902 CrossRef CAS.
- I. H. Oh, D. Kim, Y. D. Huh, Y. Park, J. M. Park and S. H. Park, Acta Crystallogr., Sect. E:Crystallogr. Commun., 2011, 67, m522–m523 CrossRef CAS.
- A. Ghoudi, I. Chaabane, R. Naouari, A. Aydi, A. Oueslati, E. Dhahri, B. F. O. Costa, T. Nikitin, J. A. Paixão and R. Fausto, Inorg. Chem. Commun., 2024, 168, 112925 CrossRef CAS.
- H. Ben Attia, F. Bentahar, M. S. M. Abdelbaky, A. Elferjani, S. García-Granda and M. Dammak, J. Alloys Compd., 2023, 969, 172317 CrossRef CAS.
- F. Garci, H. Chebbi, N. Rouzbeh, L. Rochels, S. Disch, A. Klein and M. F. Zid, Inorg. Chim. Acta, 2022, 539, 121003 CrossRef CAS.
- L. Tan, Z. Luo, X. Chang, Y. Wei, M. Tang, W. Chen, Q. Li, P. Shen and Z. Quan, Inorg. Chem., 2021, 60, 6600–6606 CrossRef CAS PubMed.
- A. M. Anthony, M. K. Pandian, P. Pandurangan and M. Bhagavathiachari, ACS Appl. Mater. Interfaces, 2022, 14, 29735–29743 CrossRef CAS.
- A. O. Polyakov, A. H. Arkenbout, J. Baas, G. R. Blake, A. Meetsma, A. Caretta, P. H. M. van Loosdrecht and T. T. M. Palstra, Chem. Mater., 2012, 24, 133–139 CrossRef CAS.
- Q. Ba, A. Jana, L. Wang and K. S. Kim, Adv. Funct. Mater., 2019, 29, 1904768 CrossRef CAS.
- L. Romani, A. Bala, V. Kumar, A. Speltini, A. Milella, F. Fracassi, A. Listorti, A. Profumo and L. Malavasi, J. Mater. Chem. C, 2020, 8, 9189–9194 RSC.
- L. N. Quan, M. Yuan, R. Comin, O. Voznyy, E. M. Beauregard, S. Hoogland, A. Buin, A. R. Kirmani, K. Zhao, A. Amassian, D. H. Kim and E. H. Sargent, J. Am. Chem. Soc., 2016, 138, 2649–2655 CrossRef CAS PubMed.
- S. K. Singh, V. M. Dhavale and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 21138–21149 CrossRef CAS.
- M. Daub, R. Stroh and H. Hillebrecht, ZAAC, 2016, 642, 268–274 CrossRef CAS.
- S. K. Singh, V. M. Dhavale and S. Kurungot, ACS Appl. Mater. Interfaces, 2015, 7, 442–451 CrossRef CAS.
- B. K. Mahapatra, P. Barman, D. R. Panigrahi, S. Kochrekar, B. Paul, A. Panghal, U. Anil Kumar, V. M. Dhavale, M. Gupta, D. Kumar, V. Kumar and S. K. Singh, Small, 2025, 21, 2405530 CrossRef CAS PubMed.
- N. Zhang, C. He, Y. Jing, Y. Qian, M. Obuchi, R. Toyoshima, H. Kondoh, K. Oka, B. Wu, L. Li, A. Anzai, T. Toyao and K.-i. Shimizu, Environ. Sci. Technol., 2025, 59, 1598–1607 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta02493e |
| This journal is © The Royal Society of Chemistry 2025 |