
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cohesive Co and Mo2C heterostructure catalysts strongly confined to hollow carbon support for enhanced kinetics and durability in the alkaline hydrogen evolution reaction†
Hyein Lee‡
 a,
Miyeon Kim‡b,
Jeonghan Roha,
KwangHo Leea,
Jooyoung Shinc,
Jeong Woo Han*b,
MinJoong Kim*d and
EunAe Cho*a
a,
Miyeon Kim‡b,
Jeonghan Roha,
KwangHo Leea,
Jooyoung Shinc,
Jeong Woo Han*b,
MinJoong Kim*d and
EunAe Cho*a
aDepartment of Materials Science and Engineering, KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: eacho@kaist.ac.kr
bDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: jwhan98@snu.ac.kr
cHydrogen Research Department, Korea Institute of Energy Research, 152 Gajeong-ro, Yuseong-gu, Daejeon, 34129, Republic of Korea
dDepartment of Materials Science and Engineering, Kyung Hee University, 1732 Deogyeong-daero, Yongin, Gyeonggi-do 17104, Republic of Korea. E-mail: mj.kim@khu.ac.kr
First published on 23rd May 2025
Abstract
Water electrolysis technologies in alkaline environments have drawn considerable interest for their potential in low-cost hydrogen production without noble metals. However, it remains challenging to achieve high performance and durability in the alkaline hydrogen evolution reaction (HER) using non-noble metal catalysts because of their intrinsically poor water dissociation kinetic properties. Here, we address this challenge by introducing a cohesive Co and Mo2C heterostructure catalyst strongly confined to N-doped carbon hollow polyhedron (NCHP) supports. Computational analysis and X-ray spectroscopic analysis results reveal that the charge redistribution between metallic Co and Mo2C not only promotes water dissociation on Mo2C sites but also accelerates hydrogen gas evolution kinetics at their interfaces, leading to enhanced HER performance. The Co–Mo2C/NCHP catalyst reduces the HER overpotential by 66% at 10 mA cm−2 compared to the single-active-site catalyst (Co/NCHP). Moreover, the Co–Mo2C/NCHP catalyst demonstrates enhanced durability, exhibiting a 38% lower HER overpotential than commercial Pt/C at 10 mA cm−2 after 1000 cycles. By forming a cohesive structure of Co and Mo2C within the hollow carbon framework, the formation of interfaces is maximized, and physical and chemical degradation is prevented. This study presents key strategies for designing interfaces to overcome the limitations of alkaline HER kinetics and durability.
Introduction
Hydrogen energy is one of the most important alternatives to fossil fuels because of its abundance and lack of harmful emissions.1,2 Among hydrogen production methods, water electrolysis, particularly anion exchange membrane water electrolysis (AEMWE), has attracted significant interest in recent years.3–7 AEMWE offers a promising solution by addressing the high material costs of polymer electrolyte membrane water electrolysis (PEMWE) and overcoming the low operating current density and limited dynamic response of liquid alkaline water electrolysis.8,9A key challenge in advancing AEMWE is achieving efficient hydrogen evolution reaction (HER) with low-cost materials, comparable to Pt.10 In alkaline environments, water dissociation significantly impacts HER kinetics.11–13 Thus, water dissociation energy, along with the hydrogen adsorption energy, has been identified as a key activity descriptor for the alkaline HER.14 While some 3d transition metals, like Co and Ni, have shown potential as cost-effective catalysts for alkaline HER,15–18 their intrinsic activity remains low19–22 primarily due to unfavorable water dissociation energetics.23,24 Consequently, designing catalysts that can effectively modulate both water dissociation and hydrogen adsorption energies is crucial for enhancing HER activity in alkaline environments.
A feasible solution to overcome this limitation is the construction of synergistic dual active sites for water dissociation and hydrogen evolution,14 particularly to improve the water dissociation activity on the 3d transition metal catalyst surfaces. Furthermore, heterogeneous structures incorporating these dual active species can modulate their electronic structure at the interface, thereby further enhancing the intrinsic catalytic performance for each reaction.25–27 Among various candidates, Mo-based compounds, particularly Mo2C, have been reported to achieve remarkable improvements in HER activity due to their ability to alter the Volmer reaction kinetics relating to water dissociation.28–31 This enhanced water dissociation capability is attributed to the unique Pt-like electronic structure of Mo2C, which optimizes the interaction between water molecules and active sites, thereby lowering the energy barrier for hydrogen evolution. For instance, S. Sun et al. demonstrated that protons generated by the Mo2C sites via water dissociation can be immediately reduced by adjacent MoOx sites to promote overall HER activity.28 Y. Zhan et al. also demonstrated the synergistic effect between highly conductive metallic Ni and Mo2C–MoN for improving HER activity.29
Despite these advantages, the primary limitation of Mo compounds is the poor stability of Mo2C in alkaline environments, which leads to its transformation into less active phases, such as MoOx, thereby degrading long-term performance.32–34 To achieve an effective dual active site for alkaline HER with Mo2C, structural strategies are needed to address the instability and to foster the formation of a synergistic interface.
Metal–organic frameworks (MOFs) offer a versatile platform for catalyst design by forming well-defined structures through the coordination of metal ions with organic ligands.35 Moreover, the catalytic properties of MOFs can be further tailored through a carbonization process, which enables the formation of unique carbon structures with embedded metal components. The strong confinement of metallic particles within MOF-derived carbon can create heterogeneous structures with synergistic properties,36 while also effectively protecting them from the harsh dissolution conditions often encountered in alkaline environments.37,38 Additionally, MOF-derived carbon provides the numerous nanopores adjacent to metallic particles, which can enhance gas bubble detachment and improve catalyst performance during HER.39,40 Evolved gas bubbles can block active sites and hinder ion transport, reducing catalyst performance.41–43 Therefore, the unique ability of carbonized MOF to create heterogeneous structures, provide strong confinement of active sites, and facilitate gas bubble detachment makes it a promising strategy for developing high-performance and durable catalysts for alkaline HER.
In this study, we propose a catalyst design strategy that employs two distinct MOFs to construct a unique hollow catalyst structure with cohesive and integrated dual active sites of Co and Mo2C. The strategy utilizing MOFs as a framework introduces and effectively confines Co and Mo2C nanoparticles within an N-doped carbon hollow polyhedron (NCHP) after the carbonization process. This cohesive structure generates abundant Co–Mo2C dual active sites with strong interfacial interactions, enhancing both water dissociation and hydrogen evolution kinetics. Additionally, the carbon-embedded catalyst structure has beneficial effects in suppressing chemical degradation of active phases, such as Mo leaching.29 Furthermore, the NCHP, with sub-nanometer-sized pores, can facilitate early and efficient bubble desorption, mitigating the physical degradation of active phases.44 Consequently, the Co–Mo2C/NCHP catalyst exhibited enhanced alkaline HER performance and durability, compared to the single-site catalysts and commercial Pt/C. These strategies hold immense promise for advancing AEMWE research by addressing the key challenges to achieving efficient HER with non-noble metal catalysts, and paving the way for the development of next-generation high-performance and durable catalysts.
Experimental section
Chemicals and materials
All chemicals were purchased from commercial sources and directly used without further pretreatment; zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 96%) and cobalt nitrate hexahydrate (Co(NO3)2·6H2O, 97%) were obtained from Junsei Chemical. 2-Methylimidazole (C4H6N2, 99%), ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O, 99%), polyvinylpyrrolidone (PVP, (C6H9NO)n, Mw = ∼55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), and N,N-dimethylformamide (DMF, 99.8%) were obtained from Sigma-Aldrich. Commercial Pt/C catalyst (TEC10V20E, 20 wt% Pt) was purchased from Tanaka Kikinzoku Kogyo. Pt/C (TEC10E50E, 47 wt% Pt) and IrO2 (Premion®, Ir 84.5%) catalysts for single-cell electrode fabrication were purchased from Tanaka Kikinzoku Kogyo and Alfa Aesar, respectively. Ethanol (C2H5OH, 95%) was purchased from Samchun Chemicals. Deionized water with a specific resistance of 18.2 MΩ cm was obtained using a Millipore-DQ5 purification system.
000), and N,N-dimethylformamide (DMF, 99.8%) were obtained from Sigma-Aldrich. Commercial Pt/C catalyst (TEC10V20E, 20 wt% Pt) was purchased from Tanaka Kikinzoku Kogyo. Pt/C (TEC10E50E, 47 wt% Pt) and IrO2 (Premion®, Ir 84.5%) catalysts for single-cell electrode fabrication were purchased from Tanaka Kikinzoku Kogyo and Alfa Aesar, respectively. Ethanol (C2H5OH, 95%) was purchased from Samchun Chemicals. Deionized water with a specific resistance of 18.2 MΩ cm was obtained using a Millipore-DQ5 purification system.
 | ||
| Fig. 1 (a) Schematic illustration of the preparation, (b) TEM image, (c) HRTEM image, and (d) HAADF-STEM elemental mapping results of Co–Mo2C/NCHP. (e) XRD patterns and (f) Raman spectra of Co/NCHP, Mo2C/NCHP, Mo2C/Co/NCHP, and Co–Mo2C/NCHP. (g) BJH desorption pore size distribution and N2 adsorption/desorption isotherms (inset) of Pt/C, Co/NCHP, and Co–Mo2C/NCHP. | ||
In this model, Co(111) p(3 × 2), which had lattice parameters of 14.93 Å × 9.95 Å × 26.10 Å with α = 90°, β = 90°, and γ = 120°, was combined with Mo2C (101) p(2 × 1), which had lattice parameters of 15.28 Å × 6.03 Å × 25.83 Å with α = 90°, β = 90°, and γ = 113.3°, through the fully relaxed optimization. The optimized Co–Mo2C model had lattice parameters of 14.93 Å × 16.69 Å × 26.10 Å with α = 90°, β = 90°, and γ = 120°. Mo2C experienced a 2.33% compressed strain along with the a-axis and a 5.22% tensile strain in the gamma angle, which was the interaxial angle between a-axis and b-axis. The model where C was directly bonded with Co showed the most stable surface energy of 0.26 eV Å−2. As shown in Fig. S1c,† the Co–Mo2C heterostructure was divided into two regions based on the interface: the Co/C interface, where C atoms of Mo2C bonded with Co, and the Co/Mo interface, where Mo atoms of Mo2C bonded with Co. The models were isolated with a vacuum of 20 Å, and the bottom half of the layers were fixed to simulate bulk properties.
The DFT models are shown in Fig. S1a–c,† and the d-band center of metals in Co–Mo2C exhibited similar values to the individual Co and Mo2C models in Fig. S1d.† This similarity in the d-band centers indicated that the heterostructure did not undergo excessive distortion during the modeling process. For density of states (DOS) calculations, a Monkhorst–Pack k-point grid of 6 × 6 × 1 was used for the Co and Mo2C, and 2 × 2 × 1 was used for Co–Mo2C. The Methfessel–Paxton smearing with a width of 0.2 eV and a cutoff energy of 450 eV were applied. Ionic relaxations were converged until the energy change was less than 10−5 eV, and electronic optimizations were converged until the forces were smaller than 0.03 eV Å−1. VESTA was utilized to visualize the results of calculations and models.48 Activation energies for water dissociation were calculated using the climbing image-nudged elastic band (CI-NEB) method.49,50 The transition states (TS) were confirmed by a single imaginary mode. The Gibbs free energy was calculated with ΔG = ΔE0 + ΔZPE − TΔS at 298.15 K and 1 atm.
The charge of each model (ρ) was calculated by Bader charge analysis.51 The charge density difference (Δρ) was calculated using the following equation.
| Δρdiff = ρCo–Mo2C − ρCo − ρMo2C |
A positive sign indicates electron depletion, while a negative sign indicates electron accumulation.
Results and discussion
Fig. 1a schematically illustrates the Co–Mo2C/NCHP catalyst prepared via a two-step approach. The synthesis begins with the formation of a core–shell metal–organic framework (MOF) structure, where a cobalt–organic framework (ZIF-67) is grown on a zinc–organic framework (ZIF-8) (Fig. S2†).52 This core–shell ZIF-8-ZIF-67 (ZIF-8@ZIF-67) structure is then subjected to Mo ion incorporation. The XRD analysis (Fig. S3†) indicates that the Mo incorporation process into the ZIF-8@ZIF-67 structure does not alter the overall crystal structure, as there are no significant changes in peak positions compared to the ZIF-8@ZIF-67 structure. This observation confirms that the crystal structure remains intact during the Mo incorporation process, but it does not directly confirm the presence of Mo ions.Subsequently, the Mo incorporated ZIF-8@ZIF-67 (Mo-ZIF-8@ZIF-67) structure undergoes pyrolysis under an inert atmosphere at 900 °C. This pyrolysis process leads to the decomposition of the thermally less stable ZIF-67 shell and the collapse of the ZIF-8 core (Fig. S4a–c†). As the shell decomposes, it forms graphitized carbon that contributes to the creation of N-doped carbon polyhedron (NCHP). During this process, cobalt nanoparticles and Mo2C are embedded within the NCHP structure, and they form interfaces that play a key role in enhancing the catalytic activity. These dual active sites—Co and Mo2C—supported on NCHP are essential for the observed catalytic performance. The utilization of Zn as the sacrificial metal in the core–shell Mo-ZIF-8@ZIF-67 structure offers several advantages. During pyrolysis and subsequent acid leaching, Zn is effectively removed due to its volatility and solubility, which suppresses metal sintering and contributes to the formation of porous NCHP. This is further supported by X-ray photoelectron spectroscopy (XPS) of Co–Mo2C/NCHP (Fig. S5†), which confirms negligible Zn content after thermal treatment.
To elucidate the synergetic effects of the Co and Mo2C, catalysts with different metal compositions and configurations were synthesized for comparison (Fig. S6†). The Mo2C supported on NCHP (Mo2C/NCHP) catalyst was prepared through the selective acid leaching of Co from Co–Mo2C/NCHP, resulting primarily in Mo2C within the NCHP. The Co supported on NCHP (Co/NCHP) catalyst was synthesized by the pyrolysis of the core–shell ZIF-8@ZIF-67 without Mo addition. The Mo2C supported on Co/NCHP (Mo2C/Co/NCHP) catalyst was synthesized using an additional heat treatment of the Co/NCHP catalyst mixed with Mo precursor. This additional Mo introduction method may lead to a different Mo distribution pattern within the NCHP compared to the Co–Mo2C/NCHP catalyst prepared with the one-step heat treatment. Furthermore, the XPS Zn 2p spectra (Fig. S5†) indicate effective removal of Zn from Co/NCHP, Mo2C/NCHP, and Mo2C/Co/NCHP during the synthetic process.
Fig. 1b and c depict the characterization of the Co–Mo2C/NCHP catalyst after thermal treatment. The transmission electron microscope (TEM) images reveal a uniform distribution of both Co and Mo throughout the NCHP structure, with their interfaces formed between them. The Co and Mo2C particles are embedded in the graphitized carbon structure, as evidenced by TEM images that show the layering of graphite (Fig. S7a†). This uniform distribution can be attributed to the inherent properties of the MOF precursor, where metal ions are arranged regularly alongside organic ligands.53 Notably, the high-resolution TEM (HRTEM) image further showcases crystalline domains within the catalysts with d-spacings of 0.21 nm and 0.23 nm, corresponding to the (111) planes of metallic Co and the (101) planes of Mo2C, respectively. As illustrated in Fig. 1d, S7b and c,† the sizes of the Co nanoparticles range from single atoms to ∼5 nm, while the Mo2C particles also exist as small nanoparticles, several nanometers in size. The EDX spectrum (Fig. S7d†) confirms the presence of both Co and Mo, and the inset of Fig. S7d† displays the elemental compositions determined by inductively coupled plasma optical emission spectrometry (ICP-OES), which shows that Mo is present at 5.5 wt% and Co at 4.5 wt%. The cohesive Co–Mo2C heterostructure embedded within the NCHP framework can lead to the formation of numerous interfaces.
Fig. 1e presents the X-ray diffraction (XRD) patterns of the Co/NCHP, Mo2C/NCHP, Co–Mo2C/NCHP, and Mo2C/Co/NCHP catalysts. Peaks observed at 42.5°, 44.3°, and 51.5° correspond to the diffraction angles of C (101) and Co (PDF#15-0806), confirming the successful reduction of Co ions to metallic Co nanoparticles within the NCHP support after pyrolysis. Additionally, peaks at 34.0°, 38.0°, 39.5°, 52.0°, 62.1°, and 69.8° correspond to the diffraction angles of β-Mo2C (PDF#35-0787), a molybdenum carbide known for its exceptional hydrogen evolution reaction (HER) activity.54 This observation confirms the successful incorporation of Mo into the catalyst structure as Mo2C.
For comparison purposes, TEM images (Fig. S8†) and high-angle annular dark-field scanning TEM (HAADF-STEM) elemental mapping and line profiles (Fig. S9†) were obtained for the Co/NCHP, Mo2C/NCHP, and Mo2C/Co/NCHP catalysts. In the case of the Co/NCHP catalyst, Co nanoparticles are observed in the NCHP structure. The Mo2C/NCHP catalyst exhibits a lower Co content and a relatively larger amount of Mo in the form of larger nanoparticles compared to the Co–Mo2C/NCHP catalyst. The Mo2C/Co/NCHP catalyst displays a noticeable depletion of Mo in specific regions, as evidenced by the elemental line profile. This observation suggests there is a non-uniform Mo distribution pattern compared to the Co–Mo2C/NCHP catalyst.
The Raman spectrum presented in Fig. 1f provides insights into the structural disorder and graphitic nature of the carbon support. The spectra display two characteristic bands at 1345 cm−1 and 1582 cm−1, corresponding to the D and G bands, respectively.55 Notably, the intensity ratio (ID/IG) of Co–Mo2C/NCHP catalyst (0.67) is lower compared to the Co/NCHP (0.73), Mo2C/NCHP (0.80), and Mo2C/Co/NCHP (0.73) catalysts. This suggests a higher degree of graphitization in the Co–Mo2C/NCHP catalyst, potentially catalyzed by the carbonization of Mo.56 Enhanced graphitization translates to improved electrical conductivity, potentially benefiting HER catalytic activity.57
In contrast, the ID/IG for the Mo2C/Co/NCHP catalyst remained unchanged compared to Co/NCHP. This indicates that Mo introduction after the carbonization process of the MOFs does not influence the further graphitization of carbon. The highest ID/IG observed for the Mo2C/NCHP catalyst can be attributed to the introduction of surface defects caused by an additional acid-leaching step, which consequently reduces the degree of graphitization.58 Fourier transform infrared spectra (FT-IR) (Fig. S10†) show peaks corresponding to activated carbon59 Co–O,60 and Mo–O61 bonds, indicating the presence of these components in the catalyst.
Fig. 1g displays the Barrett–Joyner–Halenda (BJH) desorption pore size distribution, revealing that the NCHP support possesses a network of fine nanoscale pores. The inset graph shows the N2 adsorption/desorption isotherms of the measured catalysts. The Co/NCHP (390 m2 g−1) and Co–Mo2C/NCHP (351 m2 g−1) catalysts exhibited a significantly larger specific surface compared to the commercial Pt/C (186 m2 g−1), highlighting the enhanced surface area provided by the NCHP support.
X-ray absorption fine structure (XAFS) analysis was conducted to investigate the coordination environment of the Co and Mo ion centers in the Co/NCHP, Mo2C/NCHP, Mo2C/Co/NCHP, and Co–Mo2C/NCHP catalysts. Fig. 2a presents the X-ray absorption near-edge structure (XANES) spectra at the Co K-edges of the investigated catalysts. Reference spectra of commercial Co foil, CoO, and Co3O4 are included for comparison. The absorption edge energy is known to be sensitive to the oxidation state of Co. It can be obtained by performing a second-derivative analysis of the XANES spectral edge.62 The inset in Fig. 2a depicts the correlation between the threshold energy (E0) and the Co oxidation state in the reference compounds. A monotonic increase in E0 is observed with increasing Co oxidation states. The E0 values for the Co/NCHP, Mo2C/Co/NCHP, and Co–Mo2C/NCHP catalysts are 7721.69 eV, 7721.39 eV, and 7708.78 eV, respectively. These values suggest an effective oxidation state of Co atoms between 0 and +2 (the E0 values for Co and CoO are 7709.09 eV and 7721.1 eV, respectively). Notably, the XANES spectrum of the Co–Mo2C/NCHP catalyst exhibits a pre-edge absorption feature similar to that of the Co foil, indicating that Mo introduction during the ZIF-8@ZIF-67 stage effectively facilitates the reduction of Co to metallic form. In contrast, the Co K-edge in the Co/NCHP and Mo2C/Co/NCHP catalysts is shifted to a higher energy compared to the Co–Mo2C/NCHP catalyst, implying a partially oxidized state for Co in these catalysts.
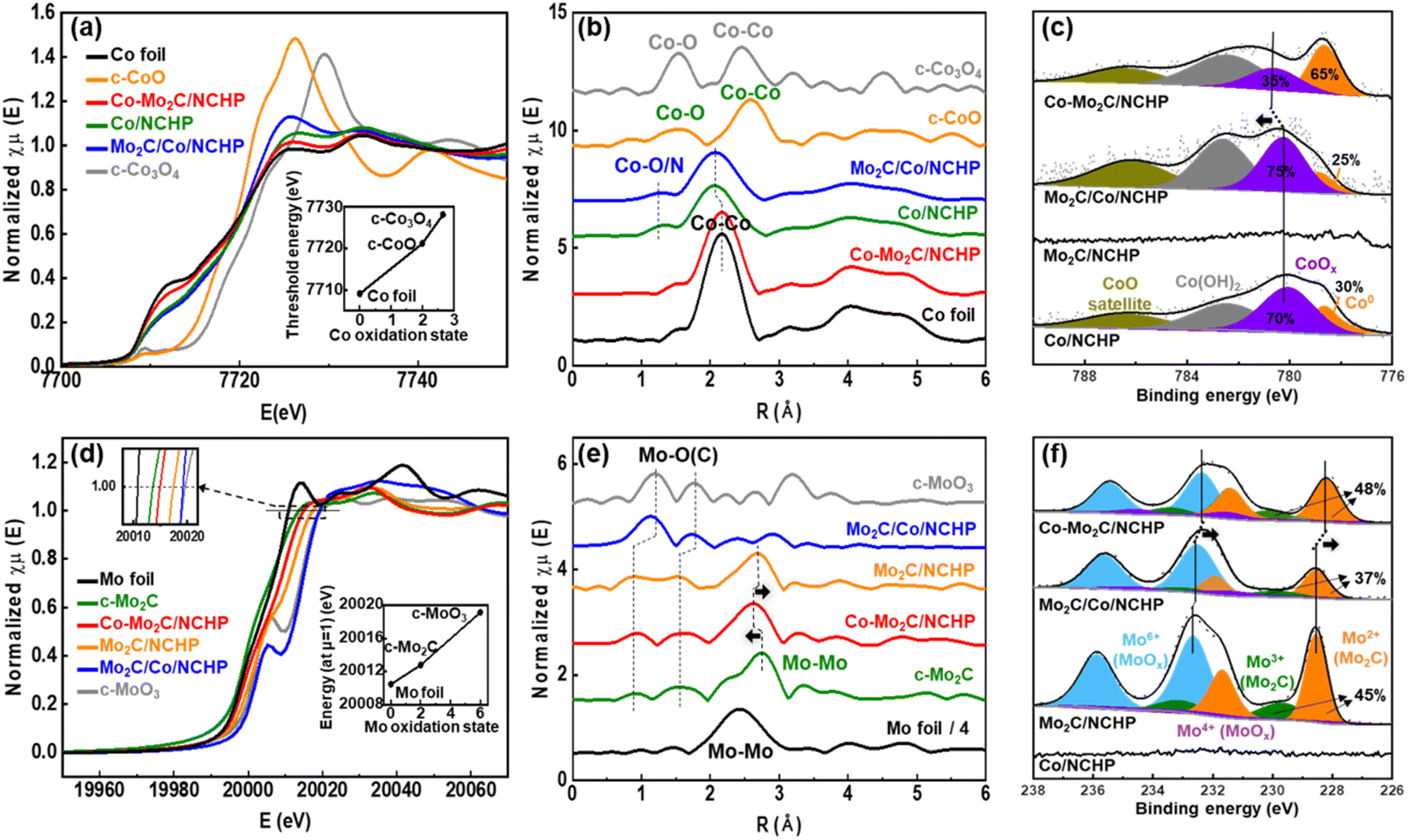 | ||
| Fig. 2 Normalized XANES and FT-EXAFS spectra (a and b) at the Co K-edge and (d and e) Mo K-edge of different catalysts and reference samples (Co foil, CoO, Co3O4, Mo foil, Mo2C, and MoO3). XPS patterns of (c) Co 2p and (f) Mo 3d of Co/NCHP, Mo2C/NCHP, Mo2C/Co/NCHP, and Co–Mo2C/NCHP. | ||
Extended X-ray absorption fine structure (EXAFS) analysis at the Co K-edge was performed to investigate the configuration and local coordination environment of the Co ion center using Fourier transform (FT) analysis (Fig. 2b).63 The Co foil spectrum exhibits a strong peak at 2.2 Å corresponding to Co–Co bonds, while the peak at 1.5 Å is associated with Co–O bonds in the CoOx clusters. Interestingly, the peak at 1.4 Å, indicative of the Co–O(N) bonds, is absent in the Co–Mo2C/NCHP catalyst but is present in the Co/NCHP and Mo2C/Co/NCHP catalysts. Additionally, the Co–Co bond peak appears at 2.0 Å in Co/NCHP and Mo2C/Co/NCHP, while it is observed at 2.2 Å in Co–Mo2C/NCHP. These differences likely stem from variations in the oxygen content within the catalysts. This observation suggests that Co in the Co–Mo2C/NCHP catalyst exists primarily in the metallic state, whereas Co in the Co/NCHP and Mo2C/Co/NCHP catalysts is partially oxidized.64
To further elucidate the distinct coordination environments of the Co atoms, EXAFS fitting based on the experimental data is presented in Fig. S11 and Table S1.† As expected for a metallic Co phase, the Co foil exhibits a Co–Co bond length of 2.5 Å. Similarly, the Co/NCHP and Mo2C/Co/NCHP catalysts show a Co–Co bond length of 2.5 Å. In contrast, the Co–Mo2C/NCHP catalyst displays a unique coordination environment with a slightly shorter Co–Co bond length compared to 2.5 Å. This reduction in the Co–Co bond length can be attributed to the smaller ionic radius of Co induced by electron donation from Co to Mo.65 The presence of a Co–Mo scattering path with a coordination number (CN) of 0.5 further supports the existence of an interface facilitating these interactions.
Unlike the Co–Mo2C/NCHP catalyst, the Mo2C/Co/NCHP catalyst does not exhibit a decrease in bond length. This is likely because the additional introduction of Mo2C to Co/NCHP, does not influence the pre-existing Co–Co bonds. Fig. 2c displays the Co 2p XPS spectra of the Co/NCHP, Mo2C/Co/NCHP, and Co–Mo2C/NCHP catalysts. In the Co 2p3/2 region of the Co/NCHP spectrum, four peaks at 778.8 eV, 781.2 eV, 782.5 eV, and 786.4 eV correspond to the Co metal, CoOx, Co(OH)2, and satellite peaks, respectively.66 Consistent with the XANES data, a prominent metallic Co peak is evident in the spectrum of the Co–Mo2C/NCHP catalyst. Additionally, the Co2+ peak of the Co–Mo2C/NCHP catalyst is observed at 781.9 eV, which is shifted positively compared to those of the Co/NCHP and Mo2C/Co/NCHP catalysts. These results reveal that the Mo plays a crucial role in facilitating the formation of the Co–Mo2C interface, which reduces the Co oxidation state to its metallic state. This metallic Co–Mo2C interface promotes charge redistribution between Co and Mo2C, which alters their local electronic environment and strengthens interfacial interactions. As revealed by XAFS and XPS analyses, the formation of this interface supports the design strategy of creating dual active sites within the NCHP framework, underscoring the critical role of Co–Mo2C interactions.
Fig. 2d displays the XANES spectra at the Mo K-edges of the Mo2C/NCHP, Mo2C/Co/NCHP, and Co–Mo2C/NCHP catalysts. Reference spectra of commercial Mo foil, Mo2C, and MoO3 are included for comparison. The inset in Fig. 2d depicts the dependence of the K-edge position at μ = 1 on the Mo oxidation states in the reference compounds.67 These compounds exhibit a monotonic increase in the K-edge position at μ = 1 with increasing Mo oxidation states. The K-edge positions at μ = 1 for the Mo2C/NCHP, Mo2C/Co/NCHP, and Co–Mo2C/NCHP catalysts are 20016.5 eV, 20019 eV, and 20015.8 eV respectively. These values suggest an effective oxidation state of Mo atoms between +2 and +6 (the K-edge position at μ = 1 for Mo2C and MoO3 are 20012.8 eV and 20019.1 eV, respectively).
For the Mo2C/Co/NCHP catalyst, Mo2C is primarily exposed on the surface, since it was introduced after the construction of the NCHP. Consequently, it undergoes oxidation in air, exhibiting an oxidation state similar to MoO3. Conversely, in the Co–Mo2C/NCHP catalyst, where Mo2C resides in close proximity to metallic Co within the NCHP structure, the Mo is significantly protected from oxidation. Mo2C in the Mo2C/NCHP catalyst, after Co leaching, shows a relatively increased oxidation. Therefore, the following trend can be observed for the Mo oxidation state: Mo2C/Co/NCHP > Mo2C/NCHP > Co–Mo2C/NCHP.
EXAFS analysis at the Mo K-edge (Fig. 2e) reveals that the peaks at 1.50 Å and 2.74 Å in commercial Mo2C correspond to Mo–O(C), Mo–Mo bonds, respectively.68 In the Co–Mo2C/NCHP catalyst, a Mo–Mo scattering peak is observed at a shorter bond distance (around 2.60 Å) compared to commercial Mo2C (around 2.74 Å). Conversely, in the Mo2C/NCHP catalyst, where Co was removed by harsh acid leaching, the Mo–Mo scattering peak shifts back at a longer bond distance (around 2.70 Å) similar to that of commercial Mo2C. This peak shift suggests the presence of charge redistributions between metallic Co and Mo2C. Additionally, the Mo2C/Co/NCHP catalyst exhibits predominantly Mo–O scattering peaks. This indicates that molybdenum oxide was probably in an amorphous state.
The EXAFS fitting based on the experimental data is presented in Fig. S12 and Table S2.† The Co–Mo2C/NCHP catalyst shows a CN of 0.5 for the Mo–Co scattering path, 5.3 for the Mo–Mo scattering path, and 1.8 for the Mo–C scattering path, confirming the presence of an interface for Co–Mo charge redistributions. The presence of both Mo–Mo1 and Mo–Mo2 bonds in the Mo based structure complicates the interpretation of the EXAFS fitting results. In the Co–Mo2C/NCHP catalyst, due to the electron donation from Co, a large number of electrons are involved in Mo bonding, leading to an elongation of the Mo–Mo1 bond length (from 2.89 Å to 2.98 Å).65
Furthermore, Fig. 2f displays the Mo 3d XPS spectra of Mo2C/NCHP, Mo2C/Co/NCHP, and Co–Mo2C/NCHP catalysts. In the Mo 3d5/2 region, four peaks are observed at 228.7 eV, 229.5 eV, 231.4 eV, and 232.6 eV, corresponding to Mo2+, Mo3+, Mo4+, and Mo6+ from Mo2C and MoOx, respectively.69 The MoOx peak likely originates from the oxidation of Mo2C upon exposure to air. This suggests a reduced state for Mo in the Co–Mo2C/NCHP catalyst, as evidenced by the higher Mo2C to MoOx ratio. In the Co–Mo2C/NCHP catalyst, consistent with the results from the Co2+ peak shift observed in Fig. 2c, charge redistributions between the metallic Co and Mo2C lead to a negative shift in the Mo peaks. These findings are consistent with the observations from XANES and EXAFS analyses.
Fig. 3a presents polarization curves of the HER performance of electrodes utilizing various catalysts: Co/NCHP, Mo2C/NCHP, Co–Mo2C/NCHP, Mo2C/Co/NCHP, and commercial Pt/C. At the current density of 10 mA cm−2, the overpotentials for HER follow this trend: Mo2C/NCHP (highest, 397 mV) > Mo2C/Co/NCHP (266 mV) > Co/NCHP (182 mV) > Co–Mo2C/NCHP (125 mV) > Pt/C (63 mV). The Co–Mo2C/NCHP catalyst exhibits significantly enhanced HER activity due to the synergistic presence of metallic Co for the hydrogen evolution sites, Mo2C for the water dissociation sites, and abundant interfaces facilitating charge redistribution between metallic Co and Mo2C.70,71 Notably, the Co–Mo2C/NCHP catalyst outperforms other reported powder-based HER catalysts at a current density of 10 mA cm−2 (refer to Fig. S13† for overpotential values). To facilitate a direct comparison of both intrinsic and practical HER activities, LSV curves with and without iR correction are presented in Fig. S14.†72
 | ||
| Fig. 3 (a) IR-corrected HER polarization curves, (b) Tafel slopes, (c) Nyquist plots at −300 mV. (d) The Gibbs free energy of H adsorption on Co, Mo2C, Co/C interface in Co–Mo2C, and Co/Mo interface in Co–Mo2C. (e) The d-band center of Co and Mo in Co–Mo2C, the Co/C interface, and the Co/Mo interface. (f) The activation energy for water dissociation on Co, Mo2C, and Co–Mo2C. (g) HER polarization curves of Co–Mo2C/NCHP before (black) and after (blue) 1000 potential cycles and after bubble removal (red) from 0.05 to −0.55 VRHE at a scan rate of 0.05 V s−1 in 1.0 M KOH. (h and i) Overpotential at 10 and 100 mA cm−2. | ||
However, the Mo2C/Co/NCHP catalyst exhibits higher overpotentials compared to the Co–Mo2C/NCHP catalyst. This can be attributed to Co oxidation and MoOx formation. Oxidation of Co weakens its interaction with Mo2C, while surface exposure of the Mo leads to the formation of MoOx, which has lower HER activity (as discussed in Fig. 2f).
Fig. 3b illustrates the Tafel slopes of the catalysts. Pt/C exhibits a slope of 45 mV dec−1, indicating that the rate-determining step (RDS) is the Tafel reaction step (H* + H* ↔ H2).73 In contrast, the Mo2C/NCHP, Co/NCHP, Mo2C/Co/NCHP, and Co–Mo2C/NCHP catalysts display a slope of approximately 120 mV dec−1. This similarity suggests that the Volmer reaction step (H2O + e− ↔H* + OH−) is the RDS for these non-platinum group metal catalysts. Interestingly, introducing Mo, which is known to lower water dissociation energy compared to Co,14,74 improves the Volmer reaction kinetics and decreases Tafel slopes. Notably, the Co–Mo2C/NCHP catalyst exhibits the lowest slope (110 mV dec−1).
The Nyquist plots (Fig. 3c) show the electrochemical impedance characteristics of the catalysts. In the high frequency region, the system resistance is solely the solution resistance (Rs). Conversely, the low frequency range reflects the sum of Rs and charge transfer resistance (Rct), represented by the diameter of the semicircle. A smaller diameter signifies a faster reaction rate. Among the catalysts, Co–Mo2C/NCHP exhibits the lowest Rct, indicating the fastest electron transfer and most favorable HER kinetics at the electrolyte interface. This aligns with the observations from the polarization curves (Fig. 3a).
As an estimate of the electrochemical active surface area (ECSA) of the catalysts, the double-layer capacitance (Cdl) was determined from cyclic voltammetry (CV) measurements, performed in the non-faradaic region (1.0–1.1 VRHE) at various scan rates ranging from 10 mV s−1 to 50 mV s−1 in H2-saturated 1 M KOH solution (Fig. S15†). At 1.05 VRHE, the capacitive current density was calculated as the sum of the anodic and cathodic current densities and plotted as a function of scan rate (Fig. S16†). The Cdl was calculated based on the following relationship between the capacitive current (i) and the scan rate (ν):
| i = Cdl·ν |
The calculated Cdl values followed the order: Mo2C/NCHP (0.32 mF cm−2) < Mo2C/Co/NCHP (0.99 mF cm−2) < Co/NCHP (1.16 mF cm−2) < Co–Mo2C/NCHP (1.67 mF cm−2), suggesting that the Co–Mo2C/NCHP catalyst possesses the largest accessible electrochemical surface area among the samples. These results further support the superior HER activity of Co–Mo2C/NCHP, as a larger surface area offers more active sites for catalytic reactions.
Fig. S17† shows the single-cell performance of the Co–Mo2C/NCHP catalyst-coated cathode assembled with commercial AEM and iridium(IV) oxide-coated anode (catalyst loading: 0.85 ± 0.30 mg cm−2), compared with the commercial Pt/C-coated cathode. A 1 M KOH solution at 60 °C was supplied to both the anode and cathode inlets. Although only a small amount of active material (Co + Mo2C loading: 0.14 ± 0.03 mg cm−2) was loaded onto the electrode, a voltage of 2.02 V was observed at a current density of 1 A cm−2. Interestingly, as the current density increases, the single-cell performance difference compared to the Pt/C electrode decreases, showing only a 30 mV voltage difference at 3 A cm−2. This result indicates that the Co–Mo2C/NCHP material possesses potential characteristics suitable for high-current-density hydrogen production. To achieve better performance, it will be necessary to increase the amount of metallic active materials within the NCHP support and further optimize the membrane electrode assembly (MEA). However, the present results demonstrate the potential applicability of this approach to practical cells. A 50-hour stability test at 0.5 A cm−2 showed a voltage increase of ∼0.09 V, indicating the need for further optimization to improve durability under practical AEMWE conditions (Fig. S18†).
The origin of the improved HER activity of Co–Mo2C/NCHP was elucidated through DFT calculations. A theoretical model of Co–Mo2C was simulated to identify the active sites for hydrogen desorption and water dissociation. To compare hydrogen desorption capability, the Gibbs free energy of hydrogen adsorption was calculated.74 Specifically, the Co–Mo2C heterostructure presented unique electronic structures at the interfaces between the Co and Mo2C. The division of the Co/C and Co/Mo interfaces, as shown in Fig. S1,† helps to identify the favorable electronic properties of the specific interfaces in the Co–Mo2C. Fig. 3d and S19† illustrate that Mo2C possesses the strongest H binding energy (−0.79 eV), followed by the Co/Mo interface (−0.65 eV), Co (−0.42 eV), and the Co/C interface (−0.18 eV).
From these results, the Co/C interface in the Co–Mo2C is the most thermoneutral result, facilitating hydrogen gas production. Meanwhile, the Co/Mo interface exhibits an intermediate adsorption strength between individual Co and Mo2C.
To explain why the Co–Mo2C/NCHP catalyst has the highest HER activity, the charge density difference was calculated in the Co–Mo2C model, where sufficient charge transfer was observed at the interfaces, as shown in Fig. S20.† At the Co/C interface, charges are transferred from Co to Mo2C, enabling moderate adsorption of hydrogen atoms. This charge transfer direction at the Co/C interface is consistent with a previous study of Co/Mo2C@C.75 Conversely, at the Co/Mo interface, charges are transferred from Mo2C to Co. The detailed charge density difference values are summarized in Table S3.†
Further, the d-band centers of metals at different interfaces within Co–Mo2C were analyzed, as shown in Fig. 3e. While Co presents similar d-band centers across interfaces, Mo2C displays notable differences. The average d-band center of Mo in the Co–Mo2C is −0.71 eV. Compared to this value, at the Co/C interface, the Mo d-band center shifts downward to −0.85 eV. In contrast, at the Co/Mo interface, the Mo d-band center shifts upward to −0.58 eV. The lower and more distant Mo d-band center from the Fermi level at the Co/C interface supports the thermoneutral Gibbs free energy of hydrogen adsorption.
Additionally, Fig. S21† illustrates that the Co and Mo2C sites within Co–Mo2C have hydrogen adsorption energies similar to that of individual Co and Mo2C, as depicted in Fig. 3d. Consequently, these results highlight that the interfaces in Co–Mo2C are key active sites of the improved HER activity, based on the synergistic effects between Co and Mo2C. Especially, the accumulation of charge on Mo2C and the lower d-band center of Mo promote moderate hydrogen adsorption and thus accelerate the hydrogen evolution.
The water dissociation ability is also crucial for generating proton sources for hydrogen gas in alkaline HER.14 The Gibbs free energy profile of water dissociation indicates the activation energies of 0.70 eV for Co, 0.36 eV for Mo2C, and 0.16 eV for Co–Mo2C in Fig. 3f and S22.† The lowest energy barrier of Co–Mo2C highlights its excellent water splitting capability, which surpasses that of individual Co and Mo2C. While Co is advantageous for hydrogen desorption, as indicated in Fig. 3d, it has poor water dissociation ability compared to Mo2C and Co–Mo2C.19 On the other hand, Mo2C exhibits strong hydrogen adsorption strength but demonstrates a lower activation barrier for water dissociation than Co.28 This combination of properties in Co–Mo2C, with efficient hydrogen desorption by Co and enhanced water dissociation by Mo2C, results in the improved HER performance.
To further evaluate the pH-dependent catalytic behavior, we measured the HER performance of all catalysts in neutral electrolyte (1.0 M PBS), and the iR-corrected polarization curves are presented in Fig. S23.† It is well established that HER kinetics in neutral media are significantly influenced by both the Gibbs free energy of hydrogen adsorption and the efficiency of water dissociation, particularly due to the sluggish Volmer step under near-neutral pH conditions. Consistent with this, all non-precious metal catalysts showed a notable decrease in activity in PBS compared to 1.0 M KOH, while the commercial Pt/C catalyst retained high activity with only a slight decline, owing to its intrinsic ability to efficiently adsorb protons and dissociate water even at low proton concentrations.
Among the catalysts, Mo2C/NCHP exhibited the lowest HER performance in neutral media, followed by Mo2C/Co/NCHP, Co/NCHP, and Co–Mo2C/NCHP, which demonstrated the highest activity. This catalytic activity trend strongly correlates with the hydrogen adsorption and water dissociation, as shown in Fig. 3d–f. These results highlight the synergistic effect of the Co–Mo2C interface, which facilitates both hydrogen adsorption and water dissociation, thereby maintaining favorable HER kinetics even under proton-limited neutral conditions.
In addition, the charge redistribution between Co and Mo2C further improves the water splitting performance beyond that of Mo2C. This can contribute to the lower Tafel slopes experimentally observed in both Co–Mo2C/NCHP and Mo2C/Co/NCHP. As both catalysts exhibit similar Tafel slopes in Fig. 3b, it can be interpreted that the charge redistribution between Co and Mo2C promotes water dissociation through the heterostructure strategy. Furthermore, the order of activation energies for water dissociation correlates well with that of the Tafel slopes in Fig. 3b. These results suggest that both thermoneutral hydrogen adsorption at the Co/C interface and the lowest water dissociation barrier contribute to the noteworthy HER activity of Co–Mo2C/NCHP, driven by the synergistic effects between Co and Mo2C. In line with this, the theoretical results further support that the Co–Mo2C interface facilitates optimal hydrogen adsorption and efficient water dissociation. The synergistic effects between Co and Mo2C, combined with the structural confinement within the NCHP support, enhance HER performance by overcoming kinetic limitations at each single active site.
Following the accelerated durability test (ADT) experiments, an increase in overpotential for the HER was observed. This can be attributed to two factors: catalytic degradation and the adverse effect of bubbles on the catalysts.32,76 To distinguish between these factors, durability testing was conducted. Linear sweep voltammetry (LSV) polarization curves were recorded at the three stages: pre-ADT (before the test), post-ADT with surface microbubbles, and post-drying (after removing surface microbubbles). The post-drying process was performed by separating the RDE and vacuum drying it at room temperature for 20 minutes. After this drying step, LSV measurements were recorded again to evaluate the changes in the catalytic performance after the removal of surface microbubbles. As a representative example, Fig. S24† illustrates the polarization curves for commercial Pt/C during the ADT experiments. The initial overpotential at 10 mA cm−2 is 63 mV. After ADT, the overpotential increased significantly to 222 mV. However, drying to remove surface microbubbles led to a slight reduction in overpotential (to 184 mV).
This observation suggests that microbubbles blocking active sites is a major contributor to the overpotential increase after ADT for Pt/C. The detachment of Pt nanoparticles from the carbon support during ADT can also contribute to the remaining overpotential increase after drying. Therefore, the change in overpotential between post-ADT and post-drying stages is attributed to bubble blocking, while the difference between pre-ADT and post-drying stages reflects the true catalytic degradation.
This phenomenon can be accelerated in commercial carbon supported Pt catalysts by the destruction of Pt anchoring sites by the harsh ADT conditions.77 Additionally, delayed bubble desorption during HER can lead to the formation of larger bubbles, which can cause extensive peeling of Pt nanoparticles from the surface.
Similar trends in overpotential changes are observed for the Co/NCHP, Mo2C/NCHP, Mo2C/Co/NCHP, and Co–Mo2C/NCHP catalysts (Fig. 3g and S25†). To quantify the contributions of catalytic degradation and bubble blocking, the overpotentials of these catalysts and Pt/C at current densities of 10 mA cm−2 and 100 mA cm−2 were investigated and are presented in Fig. 3h and i, respectively. The red region represents the overpotential increase attributed to catalytic degradation, while the blue region corresponds to the overpotential increase resulting from bubble blocking. In the red region (catalytic degradation), the overpotential increase for NCHP-supported catalysts is relatively small, with a maximum increase of 44 mV compared to Pt/C (121 mV). Similarly, in the blue region (bubble blocking), the overpotential increase for NCHP-supported catalysts is minimal, with a maximum increase of 16 mV compared to Pt/C (38 mV). The Co–Mo2C/NCHP catalyst reduces the HER overpotential by 66% at 10 mA cm−2 compared to the single active site catalyst (Co/NCHP). Moreover, the Co–Mo2C/NCHP catalyst demonstrates enhanced durability, exhibiting a 38% lower HER overpotential than commercial Pt/C at 10 mA cm−2 even after 1000 cycles. These findings are consistent with the ADT results obtained at a current density of 100 mA cm−2. Overall, both catalytic degradation and bubble blocking are significantly lower for all catalysts supported on NCHP, compared to Pt/C. These durability results highlight the effectiveness of the NCHP support in mitigating performance degradation during prolonged HER cycles through efficient bubble desorption.
Particle agglomeration and dissolution are known to be the main mechanisms leading to catalytic degradation and performance loss during HER. TEM is a valuable tool for investigating the morphological changes of catalysts after undergoing ADT.78 To understand the factors contributing to the high durability of NCHP-supported catalysts, TEM images of the Co–Mo2C/NCHP and Pt/C catalysts were obtained. Fig. 4 presents TEM images and corresponding particle size distributions for Co–Mo2C/NCHP (Fig. 4a and b) and Pt/C (Fig. 4c and d) before and after ADT.
 | ||
| Fig. 4 TEM images of (a and b) Co–Mo2C/NCHP and (c and d) Pt/C, taken before and after 1000 potential cycles. Insets show the corresponding particle size distributions, along with the average particle sizes. | ||
Interestingly, the Co–Mo2C/NCHP catalyst exhibits minimal change in particle size, with an increase from 4.08 ± 1.10 nm to 4.16 ± 0.92 nm. In contrast, Pt/C particles show a more significant increase, from 2.57 ± 0.53 nm to 3.01 ± 0.61 nm. This slight increase in particle size for the Co–Mo2C/NCHP catalyst correlates with the observed 8 mV increase in catalytic degradation after the durability test at 10 mA cm−2. However, it is important to note that this increase is not attributed to bubble blocking. This exceptional durability of the Co–Mo2C/NCHP catalyst can be attributed to the presence of micro-pores (Fig. 1g) and its three-dimensional structure. This unique structure facilitates easy access of the electrolyte to active particles and allows for the early desorption of generated hydrogen gas. Consequently, it reduces both catalytic degradation and the negative impact of bubbles on HER performance.79,80
Fig. S26† shows the TEM images and the amounts of Mo measured in the electrolyte using ICP analysis, which helps to quantify the Mo dissolution from the Co–Mo2C/NCHP and Mo2C/Co/NCHP catalysts. Both catalysts exhibit minimal structural changes before and after ADT. However, the Co–Mo2C/NCHP catalyst displays a lower concentration of Mo in the electrolyte compared to the Mo2C/Co/NCHP catalyst. This indicates that the Mo loss is associated with the surface exposure of Mo2C. As a result, Co–Mo2C/NCHP exhibits the lowest catalytic degradation due to the protected Mo2C, in conjunction with metallic Co within the NCHP structure.
Even after 1000 cycles of ADT testing, the overpotential of the Co–Mo2C/NCHP catalyst increased by only 16 mV at 100 mA cm−2, indicating minimal bubble blocking and catalytic degradation. In contrast, the Mo2C/NCHP (+44 mV @ 10 mA cm−2, +27 mV @ 100 mA cm−2) and Mo2C/Co/NCHP (+31 mV @ 10 mA cm−2, +35 mV @ 100 mA cm−2) catalysts exhibit considerable catalytic degradation compared to the Co–Mo2C/NCHP. This can be attributed to the presence of surface exposed Mo2C (as showed in Fig. 2f), which is susceptible to leaching. The slight increase in particle size and the low Mo dissolution observed after ADT suggest that the NCHP structure effectively prevents the common issues of particle agglomeration and Mo leaching. These findings emphasize the importance of structural design in preventing the loss of active material, thereby enhancing catalyst durability. These results demonstrate the critical role of the NCHP support in enhancing the operational durability of Co–Mo2C/NCHP catalysts in an alkaline HER environment.
The actual catalytic substances in the catalytic process are of great significance for the design of efficient and practical electrodes. Fig. S27a and b† shows the XRD patterns and Raman spectra of the Co–Mo2C/NCHP before and after the ADT testing. The results indicate that the crystalline Co and Mo2C nanoparticles, as well as the ID/IG ratio of the sample, remained unchanged, demonstrating the robust nature of the nanoparticles and the porous carbon framework during the stability test.
After the HER ADT testing in alkaline solution, the XPS spectra of Co–Mo2C/NCHP were measured to investigate changes in surface properties (Fig. S27c and d†). After ADT testing, the metallic Co signal was weakened and the peaks corresponding to cobalt hydroxides became increased as exposed to alkaline solution, coinciding with previous reports.81,82 Meanwhile, the peaks corresponding to MoOx decreased, indicating the dissolution of air-formed MoOx on the Mo2C surface. Even after prolonged HER operation, a significant portion of metallic Co and Mo remains, and the Co–Mo2C/NCHP catalyst maintains strong interfacial interactions between Co and Mo2C, contributing to high HER durability.
Overall, the cohesive integration of Co and Mo2C dual active sites within the NCHP addresses key limitations in alkaline HER kinetics, offering improvements in both catalytic efficiency and long-term stability. This strategy dual-active sites and provides a clear path forward for advancing non-noble metal catalysts in hydrogen evolution systems, with potential for further optimization in future studies.
Conclusions
To overcome the challenges of sluggish water dissociation kinetics associated with non-noble metal catalysts, we have introduced a catalyst design featuring cohesive Co and Mo2C dual active sites, which are strongly confined within an N-doped carbon hollow polyhedron (NCHP). Carbonizing a core–shell ZIF structure containing Co and Mo facilitates the formation of abundant metallic Co–Mo2C interfaces. Charge redistribution between the metallic Co and Mo2C at active Co–Mo2C interfaces was corroborated by several observations: a shorter peak in the Mo K-edge EXAFS spectra, distinct coordination environments of Co–Co and Mo–Mo revealed by EXAFS fitting, and the peak shifts in the Co 2p and Mo 3d XPS spectra. These findings demonstrate that strategic incorporation of Mo effectively maximizes the formation of metallic Co–Mo2C interfaces, promoting efficient HER performance.DFT calculations reveal that the Co–Mo2C interface can promote the optimal hydrogen binding strength and facilitate water dissociation. The Co–Mo2C/NCHP catalyst also demonstrated superior durability, compared with the commercial Pt/C catalyst. This was attributed to the strongly confined Co–Mo2C structure within the microporous NCHP, which not only enhanced bubble desorption but also mitigated Mo leaching. Consequently, the Co–Mo2C/NCHP catalyst exhibits a low overpotential of 125 mV at a current density of 10 mA cm−2, outperforming many other reported non-noble metal catalysts, along with improved durability. This catalyst design strategy paves the way for the development of next-generation non-noble metal catalysts with enhanced efficiency, stability, and durability for large-scale hydrogen production by addressing key challenges such as sluggish water dissociation kinetics, Mo leaching, and bubble desorption.
Data availability
All data supporting this article, including raw electrochemical measurements, TEM images, and XRD patterns, are provided in the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
H. L. and M. K. contributed equally to this work. This work was partly supported by Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government (MOTIE) (20214000000650, Energy Innovation Research Center for Fuel Cell Technology) and the National Research Foundation of Korea (NRF) Grant funded by the Ministry of Science and ICT, Republic of Korea (RS-2024-00466554).References
- A. Z. Arsad, M. A. Hannan, A. Q. Al-Shetwi, M. Mansur, K. M. Muttaqi, Z. Y. Dong and F. Blaabjerg, Hydrogen energy storage integrated hybrid renewable energy systems: A review analysis for future research directions, Int. J. Hydrogen Energy, 2022, 47, 17285–17312, DOI:10.1016/j.ijhydene.2022.03.208.
- H. Ishaq, I. Dincer and C. Crawford, A review on hydrogen production and utilization: Challenges and opportunities, Int. J. Hydrogen Energy, 2022, 47, 26238–26264, DOI:10.1016/j.ijhydene.2021.11.149.
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Anion-exchange membrane water electrolyzers, Chem. Rev., 2022, 122, 11830–11895, DOI:10.1021/acs.chemrev.1c00854.
- S. A. Lee, J. Kim, K. C. Kwon, S. H. Park and H. W. Jang, Anion exchange membrane water electrolysis for sustainable large-scale hydrogen production, Carbon Neutralization, 2022, 1, 26–48, DOI:10.1002/cnl2.9.
- D. Hua, J. Huang, E. Fabbri, M. Rafique and B. Song, Development of anion exchange membrane water electrolysis and the associated challenges: a review, Chemelectrochem, 2023, 10, e202200999, DOI:10.1002/celc.202200999.
- Y. Sugawara, S. Sankar, S. Miyanishi, R. Illathvalappil, P. K. Gangadharan, H. Kuroki, G. M. Anilkumar and T. Yamaguchi, Anion exchange membrane water electrolyzers: An overview, J. Chem. Eng. Jpn., 2023, 56, 2210195, DOI:10.1080/00219592.2023.2210195.
- K. Chand and O. Paladino, Recent developments of membranes and electrocatalysts for the hydrogen production by anion exchange membrane water electrolysers: A review, Arabian J. Chem., 2023, 16, 104451, DOI:10.1080/00219592.2023.2210195.
- B. Yang and Z. Cunman, Progress in constructing high-performance anion exchange membrane: Molecular design, microphase controllability and in-device property, Chem. Eng. J., 2023, 457, 141094, DOI:10.1016/j.cej.2022.141094.
- W. K. Ng, W. Y. Wong, N. A. H. Rosli and K. S. Loh, Commercial anion exchange membranes (AEMs) for fuel cell and water electrolyzer applications: Performance, durability, and materials advancement, Separations, 2023, 10, 424, DOI:10.3390/separations10080424.
- M. Chen, J. Ma, Y. Feng, Y. Wu, G. Hu and X. Liu, Advanced characterization enables a new era of efficient carbon dots electrocatalytic reduction, Coord. Chem. Rev., 2025, 535, 216612, DOI:10.1016/j.ccr.2025.216612.
- R. P. M, N. B. G, B. Neppolian and A. Sengeni, Co-W (Hydr)oxide with ultralow Ru promotes water dissociation coupled H+ abstraction in alkaline HER, ChemCatChem, 2024, 16, e202401163, DOI:10.1002/cctc.202401163.
- S. Anantharaj, S. Noda, V. R. Jothi, S. Yi, M. Driess and P. W. Menezes, Strategies and perspectives to catch the missing pieces in energy-efficient hydrogen evolution reaction in alkaline media, Angew. Chem., Int. Ed., 2021, 60, 18981, DOI:10.1002/anie.202015738.
- S. Anantharaj, Hydrogen evolution reaction on Pt and Ru in alkali with volmer-step promotors and electronic structure modulators, Curr. Opin. Electrochem., 2022, 33, 100961, DOI:10.1016/j.coelec.2022.100961.
- W. Gou, H. Sun and F. Cheng, Dual active sites engineering of electrocatalysts for alkaline hydrogen evolution, Nano Res. Energy, 2024, 3, e9120121, DOI:10.26599/NRE.2024.9120121.
- M. Miles and M. Thomason, Periodic variations of overvoltages for water electrolysis in acid solutions from cyclic voltammetric studies, J. Electrochem. Soc., 1976, 123, 1459, DOI:10.1149/1.2132619.
- Y. Zhao, J. Li, K. Li, L. Liang, J. Zhu, M. Xiao, C. Liu and W. Xing, The synergistic effect of Ni-NiMo4N5 heterointerface construction and Fe-doping enables active and durable alkaline water splitting at industrial current density, J. Mater. Chem. A, 2025, 13, 9184, 10.1039/d5ta00038f.
- M. Yang, J. Ding, Z. Wang, J. Zhang, Z. Peng and X. Liu, NiMo-based alloy and its sulfides for energy-saving hydrogen production via sulfion oxidation assisted alkaline seawater splitting, Chin. Chem. Lett., 2025, 110861, DOI:10.1016/j.cclet.2025.110861.
- S. Anantharaj and S. Noda, Amorphous catalysts and electrochemical water splitting: An untold story of harmony, Small, 2020, 16, 1905779, DOI:10.1002/smll.201905779.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices, J. Am. Chem. Soc., 2015, 137, 4347–4357, DOI:10.1021/ja510442p.
- Y. Cheng, H. Guo, X. Li, X. Wu, X. Xu, L. Zheng and R. Song, Rational design of ultrahigh loading metal single-atoms (Co, Ni, Mo) anchored on in-situ pre-crosslinked guar gum derived N-doped carbon aerogel for efficient overall water splitting, Chem. Eng. J., 2021, 410, 128359, DOI:10.1016/j.cej.2020.128359.
- H. J. Kim, H. Y. Kim, J. Joo, S. H. Joo, J. S. Lim, J. Lee, H. Huang, M. Shao, J. Hu and J. Y. Kim, Recent advances in non-precious group metal-based catalysts for water electrolysis and beyond, J. Mater. Chem. A, 2022, 10, 50–88, 10.1039/D1TA06548C.
- D. T. Dung, E. Roh, S. Ji, J. M. Yuk, J.-H. Kim, H. Kim and S.-M. Lee, Ni/Co/Co3O4@C nanorods derived from a MOF@MOF hybrid for efficient overall water splitting, Nanoscale, 2023, 15, 1794–1805, 10.1039/D2NR05686K.
- V. Bagotzky and N. Osetrova, Investigations of hydrogen ionization on platinum with the help of micro-electrodes, J. Electroanal. Chem., 1973, 43, 233–249, DOI:10.1016/S0022-0728(73)80494-2.
- H. Kim, Y. Lee, D. Song, Y. Kwon, E.-J. Kim and E. Cho, Cu3P/PAN derived N-doped carbon catalyst with non-toxic synthesis for alkaline hydrogen evolution reaction, Sustain. Energy Fuels, 2020, 4, 5247–5253, 10.1039/D0SE00923G.
- F. Yu, Y. Gao, Z. Lang, Y. Ma, L. Yin, J. Du, H. Tan, Y. Wang and Y. Li, Electrocatalytic performance of ultrasmall Mo2C affected by different transition metal dopants in hydrogen evolution reaction, Nanoscale, 2018, 10, 6080–6087, 10.1039/C8NR00908B.
- T. Wang, P. Wang, W. Zang, X. Li, D. Chen, Z. Kou, S. Mu and J. Wang, Nanoframes of Co3O4–Mo2N heterointerfaces enable high-performance bifunctionality toward both electrocatalytic HER and OER, Adv. Funct. Mater., 2022, 32, 2107382, DOI:10.1002/adfm.202107382.
- W. Liu, M. Zhou, J. Zhang, W. Liu, D. Qin, Q. Liu, G. Hu and X. Liu, Construction of a CoP/MnP/Cu3P heterojunction for efficient methanol oxidation-assisted seawater splitting, Mater. Chem. Front., 2025, 9, 953–964, 10.1039/D4QM01067A.
- S. Sun, Y. Liu, G. Xu, C. Jiang, J. Li, L. Fan and W. Cai, Controllably constructed carbide/oxide heterointerfaces of molybdenum for efficient hydrogen evolution, Fuel, 2023, 335, 127084, DOI:10.1016/j.fuel.2022.127084.
- Y. Zhan, F. Xie, H. Zhang, Z. Lin, J. Huang, W. Zhang, X. Sun, H. Meng, Y. Zhang and J. Chen, Metallic Ni promoted Mo2C-MoN particles supported on N-doped graphitic carbon as bifunctional catalyst for oxygen and hydrogen evolution reaction in alkaline media, J. Electrochem. Soc., 2018, 165, F75, DOI:10.1149/2.0891802jes.
- G. Liu, H. Bai, Y. Ji, L. Wang, Y. Wen, H. Lin, L. Zheng, Y. Li, B. Zhang and H. Peng, A highly efficient alkaline HER Co–Mo bimetallic carbide catalyst with an optimized Mo d-orbital electronic state, J. Mater. Chem. A, 2019, 20, 12434–12439, 10.1039/c9ta02886b.
- M. Hou, L. Zheng, D. Zhao, X. Tan, W. Feng, J. Fu, T. Wei, M. Cao, J. Zhang and C. Chen, Microenvironment reconstitution of highly active Ni single atoms on oxygen-incorporated Mo2C for water splitting, Nat. Commun., 2024, 15, 1342, DOI:10.1038/s41467-024-45533-3.
- M. Schalenbach, F. D. Speck, M. Ledendecker, O. Kasian, D. Goehl, A. M. Mingers, B. Breitbach, H. Springer, S. Cherevko and K. J. Mayrhofer, Nickel-molybdenum alloy catalysts for the hydrogen evolution reaction: Activity and stability revised, Electrochim. Acta, 2018, 259, 1154–1161, DOI:10.1016/j.electacta.2017.11.069.
- W. Du, Y. Shi, W. Zhou, Y. Yu and B. Zhang, Unveiling the in situ dissolution and polymerization of Mo in Ni4Mo alloy for promoting the hydrogen evolution reaction, Angew. Chem., Int. Ed., 2021, 60, 7051–7055, DOI:10.1002/anie.202015723.
- X. Yang, J. Cheng, X. Yang, Y. Xu, W. Sun and J. Zhou, Facet-tunable coral-like Mo2C catalyst for electrocatalytic hydrogen evolution reaction, Chem. Eng. J., 2023, 451, 138977, DOI:10.1016/j.cej.2022.138977.
- H. Kang, J. Shin, T.-H. Kim, Y. Lee, D. Lee, J. Lee, G. Kim and E. Cho, Metal–organic framework-derived magnesium oxide@carbon interlayer for stable lithium–sulfur batteries, ACS Sustainable Chem. Eng., 2023, 11, 1344–1354, DOI:10.1021/acssuschemeng.2c05064.
- M. Subramaniam, S. Ramakrishnan, S. Sidra, S. C. Karthikeyan, S. Vijayapradeep, J. Huang, M. Mamlouk, D. H. Kim and D. J. Yoo, Carbon core-shell Pt nanoparticle embedded porphyrin Co-MOF derived N-doped porous carbon for the alkaline AEM water electrolyzer application, J. Mater. Chem. A, 2024, 12, 5967–5979, 10.1039/d3ta06745a.
- X. Zhang, D. Xue, S. Jiang, H. Xia, Y. Yang, W. Yan, J. Hu and J. Zhang, Rational confinement engineering of MOF-derived carbon-based electrocatalysts toward CO2 reduction and O2 reduction reactions, InfoMat, 2022, 4, e12257, DOI:10.1002/inf2.12257.
- Y. Tang, H. Kang, J. Zheng, H. Li, R. Wang, L. Zhang, Q. Ma, X. Xiong, T. Zhou and C. Zhang, Metal-organic framework derived Bi2S3 hybrid nanofibers for enhanced lithium-ion storage, J. Power Sources, 2022, 520, 230895, DOI:10.1016/j.jpowsour.2021.230895.
- T. Groß, J. Bauer, G. Ludwig, D. Fernandez Rivas and P. Pelz, Bubble nucleation from micro-crevices in a shear flow: Experimental determination of nucleation rates and surface nuclei growth, Exp. Fluid, 2018, 59, 1–10, DOI:10.1007/s00348-017-2459-y.
- Z. Duan, Y. Liu, Y. Wang, M.-K. Kim, Y. Fang, Q. Yuan, Y. Zhang, P. Xiong and J. Suhr, Laser-Induced controllable porosity in additive manufacturing boosts efficiency of electrocatalytic water splitting, Nano Lett., 2024, 24, 8558–8566, DOI:10.1021/acs.nanolett.4c01450.
- D. Zhang and K. Zeng, Evaluating the behavior of electrolytic gas bubbles and their effect on the cell voltage in alkaline water electrolysis, Ind. Eng. Chem. Res., 2012, 51, 13825–13832, DOI:10.1021/ie301029e.
- C. A. Sequeira, D. M. Santos, B. Šljukić and L. Amaral, Physics of electrolytic gas evolution, Braz. J. Phys., 2013, 43, 199–208, DOI:10.1007/s13538-013-0131-4.
- A. Taqieddin, R. Nazari, L. Rajic and A. Alshawabkeh, Review—Physicochemical hydrodynamics of gas bubbles in two phase electrochemical systems, J. Electrochem. Soc., 2017, 164, E448, DOI:10.1149/2.1161713jes.
- A. Angulo, P. van der Linde, H. Gardeniers, M. Modestino and D. F. Rivas, Influence of bubbles on the energy conversion efficiency of electrochemical reactors, Joule, 2020, 4, 555–579, DOI:10.1016/j.joule.2020.01.005.
- J. Miao, F.-X. Xiao, H. B. Yang, S. Y. Khoo, J. Chen, Z. Fan, Y.-Y. Hsu, H. M. Chen, H. Zhang and B. Liu, Hierarchical Ni-Mo-S nanosheets on carbon fiber cloth: A flexible electrode for efficient hydrogen generation in neutral electrolyte, Sci. Adv., 2015, 1, 14, DOI:10.1126/sciadv.1500259.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758, DOI:10.1103/PhysRevB.59.1758.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865, DOI:10.1103/PhysRevLett.77.3865.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276, DOI:10.1107/S0021889811038970.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904, DOI:10.1063/1.1329672.
- G. Henkelman and H. Jónsson, Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points, J. Chem. Phys., 2000, 113, 9978–9985, DOI:10.1063/1.1323224.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360, DOI:10.1016/j.cej.2021.132697.
- Y. Pan, K. Sun, S. Liu, X. Cao, K. Wu, W.-C. Cheong, Z. Chen, Y. Wang, Y. Li and Y. Liu, Core–shell ZIF-8@ZIF-67-derived CoP nanoparticle-embedded N-doped carbon nanotube hollow polyhedron for efficient overall water splitting, J. Am. Chem. Soc., 2018, 140, 2610–2618, DOI:10.1021/jacs.7b12420.
- P. K. Chattopadhyay and N. R. Singha, MOF and derived materials as aerogels: Structure, property, and performance relations, Coord. Chem. Rev., 2021, 446, 214125, DOI:10.1016/j.ccr.2021.214125.
- C. Wan, Y. N. Regmi and B. M. Leonard, Multiple phases of molybdenum carbide as electrocatalysts for the hydrogen evolution reaction, Angew. Chem., 2014, 126, 6525–6528, DOI:10.1002/ange.201402998.
- G. S. Bajad, S. K. Tiwari and R. Vijayakumar, Synthesis and characterization of CNTs using polypropylene waste as precursor, J. Mater. Sci. Eng. B, 2015, 194, 68–77, DOI:10.1016/j.mseb.2015.01.004.
- S. Sharma, B. P. Jaisi, M. I. Araby, S. Elnobi, M. E. Ayhan, G. Kalita and M. Tanemura, The Mo catalyzed graphitization of amorphous carbon: an in situ TEM study, RSC Adv., 2019, 9, 34377–34381, 10.1039/C9RA05936A.
- Z. Wan, L. Wang, Y. Zhou, S. Xu, J. Zhang, X. Chen, S. Li, C. Ou and X. Kong, A frogspawn inspired twin Mo2C/Ni composite with a conductive fibrous network as a robust bifunctional catalyst for advanced anion exchange membrane electrolyzers, Nanoscale, 2024, 16, 5845–5854, 10.1039/D3NR06242B.
- J. L. Qi, X. Wang, J. H. Lin, F. Zhang, J. C. Feng and W.-D. Fei, A high-performance supercapacitor of vertically-oriented few-layered graphene with high-density defects, Nanoscale, 2015, 7, 3675–3682, 10.1039/C4NR07284G.
- M. Jahan and F. Feni, Environmentally friendly bifunctional catalyst for ORR and OER from coconut shell particles, Adv. Mater. Phys. Chem., 2022, 12, 106–123, DOI:10.4236/ampc.2022.125008.
- H. Cai, Q. Wei, H. Xiao, H. Liu and J. Wang, Preparation and microwave absorption properties of petal CoO/CNFs composites, J. Mater. Sci. Mater. Electron., 2020, 31, 7606–7615, DOI:10.1007/s10854-020-03231-y.
- N. Maheswari and G. Muralidharan, Controlled synthesis of nanostructured molybdenum oxide electrodes for high performance supercapacitor devices, Appl. Surf. Sci., 2017, 416, 461–469, DOI:10.1016/j.apsusc.2017.04.094.
- Z. Lin, W. Lai, Z. Wu, J. Liu and Y. An, Investigations of the valence states, cobalt ion distribution, and defect structures in Co-doped ITO films, J. Mater. Res., 2018, 33, 2336–2341, DOI:10.1557/jmr.2018.184.
- L. van Haandel, G. Smolentsev, J. A. van Bokhoven, E. J. Hensen and T. Weber, Evidence of octahedral Co–Mo–S sites in hydrodesulfurization catalysts as determined by resonant inelastic X-ray scattering and X-ray absorption spectroscopy, ACS Catal., 2020, 10, 10978–10988, DOI:10.1021/acscatal.0c03062.
- Y. Zhu, J. Sokolowski, X. Song, Y. He, Y. Mei and G. Wu, Engineering local coordination environments of atomically dispersed and heteroatom-coordinated single metal site electrocatalysts for clean energy-conversion, Adv. Energy Mater., 2020, 10, 1902844, DOI:10.1002/aenm.201902844.
- O. Breuer, A. Chakraborty, J. Liu, T. Kravchuk, L. Burstein, J. Grinblat, Y. Kauffman, A. Gladkih, P. Nayak and M. Tsubery, Understanding the role of minor molybdenum doping in LiNi0.5Co0.2Mn0.3O2 electrodes: from structural and surface analyses and theoretical modeling to practical electrochemical cells, ACS Appl. Mater. Interfaces, 2018, 10, 29608–29621, DOI:10.1021/acsami.8b09795.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, Synthesis and characterization of cobalt hydroxide, cobalt oxyhydroxide, and cobalt oxide nanodiscs, J. Phys. Chem. C, 2010, 114, 111–119, DOI:10.1021/jp908548f.
- F. Farges, R. Siewert, G. E. Brown, A. Guesdon and G. Morin, Structural environments around molybdenum in silicate glasses and melts. I. Influence of composition and oxygen fugacity on the local structure of molybdenum, Can. Mineral., 2006, 44, 731–753, DOI:10.2113/gscanmin.44.3.731.
- H. Zhang, P. Zhang, M. Qiu, J. Dong, Y. Zhang and X. W. Lou, Ultrasmall MoOx clusters as a novel cocatalyst for photocatalytic hydrogen evolution, Adv. Mater., 2019, 31, 1804883, DOI:10.1002/adma.201804883.
- Y. Yang, Y. Qian, Z. Luo, H. Li, L. Chen, X. Cao, S. Wei, B. Zhou, Z. Zhang and S. Chen, Water induced ultrathin Mo2C nanosheets with high-density grain boundaries for enhanced hydrogen evolution, Nat. Commun., 2022, 13, 7225, DOI:10.1038/s41467-022-34976-1.
- M. Kim, S. Kim, D. Song, S. Oh, K. J. Chang and E. Cho, Promotion of electrochemical oxygen evolution reaction by chemical coupling of cobalt to molybdenum carbide, Appl. Catal., B, 2018, 227, 340–348, DOI:10.1016/j.apcatb.2018.01.051.
- L. Zhang, Y. Zhu, Z. Nie, Z. Li, Y. Ye, L. Li, J. Hong, Z. Bi, Y. Zhou and G. Hu, Co/MoC nanoparticles embedded in carbon nanoboxes as robust trifunctional electrocatalysts for a Zn–air battery and water electrocatalysis, ACS Nano, 2021, 15, 13399–13414, DOI:10.1021/acsnano.1c03766.
- S. Anantharaj and S. Noda, Dos and don’ts in screening water splitting electrocatalysts, Energy Adv., 2022, 1, 511–523, 10.1039/D2YA00076H.
- K. J. Vetter, Electrochemical Kinetics: Theoretical and Experimental Aspects, Academic Press, 1967 Search PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc., 2005, 152, J23, DOI:10.1149/1.1856988.
- S. Yuan, M. Xia, Z. Liu, K. Wang, L. Xiang, G. Huang, J. Zhang and N. Li, Dual synergistic effects between Co and Mo2C in Co/Mo2C heterostructure for electrocatalytic overall water splitting, Chem. Eng. J., 2022, 430, 132697, DOI:10.1016/j.cej.2021.132697.
- H. A. El-Sayed, A. Weiß, L. F. Olbrich, G. P. Putro and H. A. Gasteiger, OER catalyst stability investigation using RDE technique: a stability measure or an artifact?, J. Electrochem. Soc., 2019, 166, F458–F464, DOI:10.1149/2.0301908jes.
- A. Zadick, L. Dubau, N. Sergent, G. Berthomé and M. Chatenet, Huge instability of Pt/C catalysts in alkaline medium, ACS Catal., 2015, 5, 4819–4824, DOI:10.1021/acscatal.5b01037.
- M. Li, S. Saedy, S. Fu, T. Stellema, R. Kortlever and J. R. van Ommen, Enhancing the durability of Pt nanoparticles for water electrolysis using ultrathin SiO2 layers, Catal. Sci. Technol., 2024, 14, 1328–1335, 10.1039/D3CY00996C.
- Q. Song, Z. Xue, C. Liu, X. Qiao, L. Liu, C. Huang, K. Liu, X. Li, Z. Lu and T. Wang, General strategy to optimize gas evolution reaction via assembled striped-pattern superlattices, J. Am. Chem. Soc., 2019, 142, 1857–1863, DOI:10.1021/jacs.9b10388.
- T. Fujimura, M. Kunimoto, Y. Fukunaka and T. Homma, Analysis of the hydrogen evolution reaction at Ni micro-patterned electrodes, Electrochim. Acta, 2021, 368, 137678, DOI:10.1016/j.electacta.2020.137678.
- D. Xu, J. Yao, X. Ma, Y. Xiao, C. Zhang, W. Lin and H. Gao, F, N neutralizing effect induced Co-P-O cleaving endows CoP nanosheets with superior HER and OER performances, J. Colloid Interface Sci., 2022, 619, 298–306, DOI:10.1016/j.ijhydene.2018.01.161.
- Z. Chen, Q. Kang, G. Cao, N. Xu, H. Dai and P. Wang, Study of cobalt boride-derived electrocatalysts for overall water splitting, Int. J. Hydrogen Energy, 2018, 43, 6076–6087, DOI:10.1016/j.ijhydene.2018.01.161.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ta02607e |
| ‡ H. L. and M. K. contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2025 |