
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigning covalent organic frameworks for organic catalysis: bridging reticular and molecular chemistry
Mohammad Ali Shirania,
Mohaddeseh Afsharia,
Ignacio Romero-Muñiz *bc,
Ana E. Platero-Pratsde,
Mohammad Dinari*a and
Félix Zamora*de
*bc,
Ana E. Platero-Pratsde,
Mohammad Dinari*a and
Félix Zamora*de
aDepartment of Chemistry, Isfahan University of Technology, Isfahan, 84156-83111, Iran. E-mail: dinari@iut.ac.ir
bDepartment of Frontiers in Materials Chemistry, Instituto de Ciencia de Materiales de Madrid - CSIC, Madrid, 28049, Spain. E-mail: ignacio.romero@csic.es
cDepartamento de Química Orgánica, Facultad de Ciencias, Universidad Autónoma de Madrid, Madrid 28049, Spain
dDepartamento de Química Inorgánica, Facultad de Ciencias, Universidad Autónoma de Madrid, Madrid 28049, Spain. E-mail: felix.zamora@uam.es
eCondensed Matter Physics Center (IFIMAC), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 22nd May 2025
Abstract
Covalent organic frameworks (COFs) have gained significant attention over the past decade as heterogeneous catalysts for a wide range of applications. Among the most prominent is organic synthesis, a key area in disciplines such as medicinal chemistry, materials science, and designing molecules with optoelectronic properties. In this review, we provide an overview of the fundamental advances in the synthetic design of COFs, specifically focusing on strategies to enhance their catalytic activity for organic transformations. The discussion is organized around four key areas: catalysis driven by the material's backbone, ligand design for organocatalysis, support for metals, and the hosting of nanostructures such as nanoparticles or enzymes.
1. Introduction
Covalent organic frameworks (COFs) are purely organic crystalline materials with permanent porosity. They have a large variety of proven applications, from energy1 and environmental applications2,3 to catalytic transformations.4 Their high crystallinity and permanent porosity arise from the reversible assembly of topologically designed building blocks. The geometry and connectivity of the building blocks provide these materials with structural micro or mesoporosity. We can reach different network topologies depending on the length, shape, and connection of the selected starting materials.Since its discovery 20 years ago, the field of covalent organic frameworks has experienced continuous expansion, attracting chemists from diverse backgrounds, including organic chemistry, crystallography, and materials chemistry. This interdisciplinary collaboration underscores the dynamic and inclusive nature of COF research, fostering a rich exchange of ideas and insights across various scientific disciplines.
1.1. The chemistry of covalent organic frameworks
The development of COFs has progressed alongside advancements in other reticular materials, such as metal–organic frameworks (MOFs). However, despite sharing many similar qualities, some differences make these materials substrates of great interest in the field of catalytic materials. A very important point differentiating COFs from their inorganic counterpart5 is the pure organic nature of the framework. This feature confers enormous versatility and chemical stability to the systems and the possibility of using highly tailorable building blocks. On the other hand, the stronger bonds used for the network polymerisation make it more challenging to work in reversible conditions and reach the desired crystallinity. In this regard, many reversible or pseudo-reversible bonds have been used to form covalent organic frameworks.The design of COFs requires a careful balance between structural stability and crystallinity.6 A higher degree of bond reversibility enhances the material's self-healing capability (Fig. 1). However, excessive bond reversibility can lead to structural instability, making the material synthetically impractical. For this reason, COF chemistry over the past decade has focused on identifying bond types that provide sufficient strength for chemical stability while maintaining enough reversibility to enable self-healing.
 | ||
| Fig. 1 Schematic representation of the crystallisation process of COFs. | ||
To address this challenge, the synthetic community has turned to dynamic covalent chemistry – a strategy extensively explored in other areas of organic synthesis, such as the construction of mechanically interlocked molecules and supramolecular polymers.7,8 Building on this approach, researchers identified imines as promising bonding groups for COF synthesis.8 Imine-based COFs rapidly gained attention in the community9 because of their high thermal and chemical stability as well as their compatibility with a range of synthetic procedures, from high-temperature ones to room-temperature protocols.
In recent years, significant progress has been made in developing various types of bonds that enable the synthesis of crystalline organic materials. Fig. 2 highlights some of these advances, with nitrogen-based bonds constituting the largest group among them.
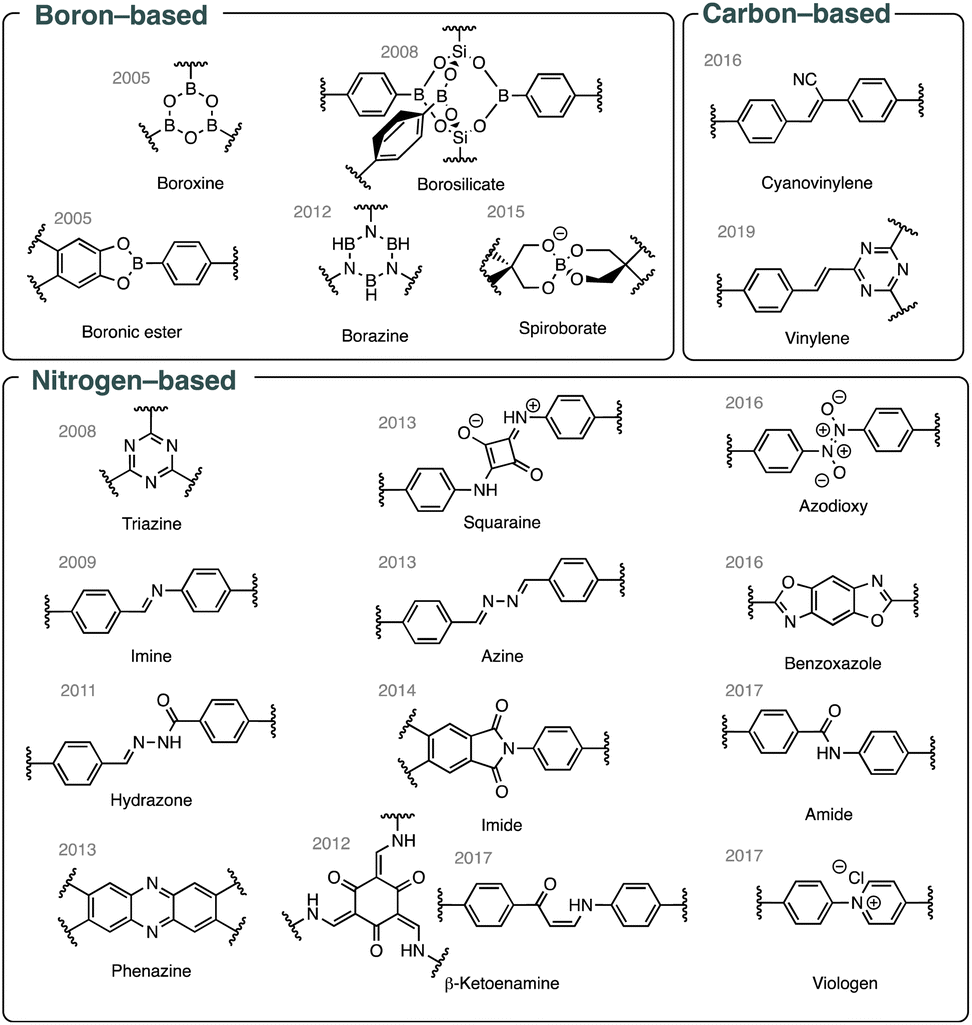 | ||
| Fig. 2 Selected bond types commonly used in COF synthesis. | ||
Most COFs have been synthesized using solvothermal methods, where reaction conditions require high temperatures (strictly temperatures above the boiling point of the solvent). This elevated temperature is essential to achieve the desired solubility and reactivity of building blocks and promote the reversibility of the reactions.13 Normally, this reaction occurs under an inert atmosphere (i.e., with the reaction mixture degassed via freeze–pump–thaw cycles). The most used solvent of choice typically involves a combination of mesitylene and dioxane in varying ratios, with acetic acid as the catalyst. Reactions can also be heated using microwave-assisted methods.14 To develop a greener alternative to solvothermal methods, mechanochemical methods stand out as a versatile methodology.15 In the mechanochemical synthesis, the monomers are placed in a mortar with a few drops of acid catalyst, or a solid one (tosylic acid), and ground using a pestle at room temperature to yield the COF.16 Sonochemical methods represent an emerging technique that is adaptable for use with conventional solvents like mesitylene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dioxane and for environmentally friendly synthesis in aqueous acetic acid, enabling the synthesis of COFs on a multigram scale within shorter reaction times, typically on the order of hours. This reduction in reaction times constitutes a major advancement in enabling high-throughput screening of materials.17 Microfluidics-based approaches are increasingly attracting attention as a viable option for producing large quantities of COF.18 In this sense, the reactor conditions remain steady, with the amount of COF generated being a function of time. The synthesis occurs in flow reactors with micron-sized channels, facilitating rapid mixing of building block solutions. This efficient approach allows the formation of crystalline COFs in remarkably short timeframes.
:dioxane and for environmentally friendly synthesis in aqueous acetic acid, enabling the synthesis of COFs on a multigram scale within shorter reaction times, typically on the order of hours. This reduction in reaction times constitutes a major advancement in enabling high-throughput screening of materials.17 Microfluidics-based approaches are increasingly attracting attention as a viable option for producing large quantities of COF.18 In this sense, the reactor conditions remain steady, with the amount of COF generated being a function of time. The synthesis occurs in flow reactors with micron-sized channels, facilitating rapid mixing of building block solutions. This efficient approach allows the formation of crystalline COFs in remarkably short timeframes.
De novo synthesis and post-synthetic modifications are two primary approaches for constructing and tailoring COFs. De novo synthesis refers to the bottom-up assembly of framework materials using pre-designed building blocks with specific geometries to achieve a target network under reversible conditions. This method relies heavily on the utilization of strong, directional covalent bonds that define the framework's stability and functionality. The selection of appropriate geometries for the building blocks involves applying topological abstractions to simplify complex networks into more manageable representations. These topological models describe the connections between structural components, providing a clear and systematic strategy for designing the framework (Fig. 3). Once the network has been topologically reduced, a process analogous to retrosynthetic analysis in organic chemistry is performed. This step involves dissecting the target structure into smaller, conceptual fragments, ensuring that the desired connectivity and symmetry are maintained. The next challenge is selecting molecular building blocks that possess the required geometry and functional groups to reconstruct the framework. These building blocks must align precisely with the topological blueprint, facilitating their assembly under reversible conditions into the intended structure (Fig. 4). The selection of molecular fragments is critical for preserving the symmetry of the desired topological abstraction. Most fragments are constructed using sp2 carbons, which inherently limit connectivity due to their planar geometry, resulting in predominantly low-connectivity di- tri- or tetratopic topologies. However, three-dimensional networks can still be designed without relying on sp3 carbon-based fragments. In these cases, steric hindrance becomes a pivotal factor in overcoming planarity constraints, enabling the formation of more complex, three-dimensional architectures.19–21
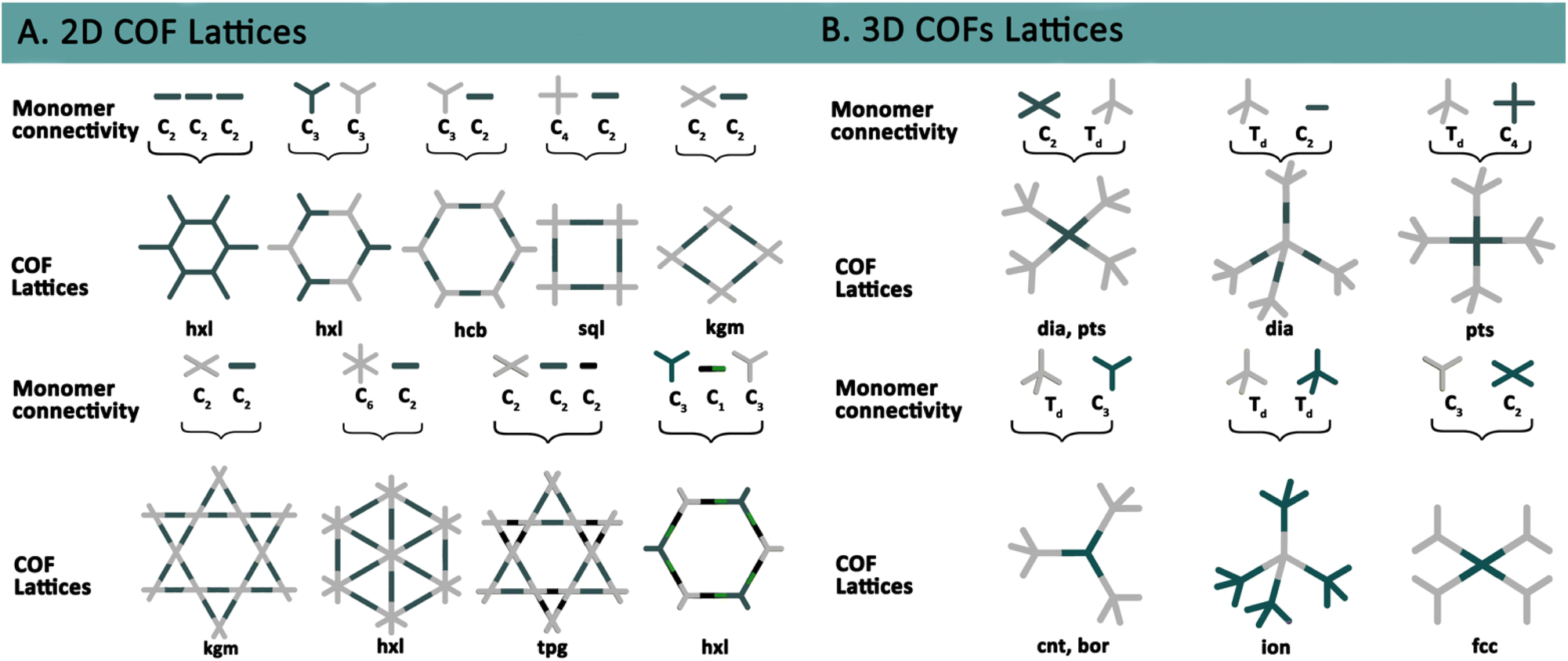 | ||
| Fig. 3 Topological diagrams illustrating representative designs of 2D and 3D COFs. | ||
 | ||
| Fig. 4 Building blocks used in the examples of this review. | ||
Through this rational design process, de novo synthesis enables the creation of COFs with predefined architectures and properties, serving as a foundational tool for materials development.
The post-synthetic strategy offers a versatile and alternative pathway for creating novel materials, diverging from the conventional de novo synthesis approach. This methodology has proven highly effective for fine-tuning the structures of COFs, leveraging the vast repertoire of organic chemistry to introduce modifications to pre-synthesized building blocks. Incorporating reactive groups into these building blocks opens numerous functionalization opportunities, enabling precise tailoring of material properties to suit specific applications. This strategy is particularly advantageous for introducing functionalities that might be incompatible with the harsh conditions of direct synthesis, thereby expanding the scope of possible COF structures.
However, the success of post-synthetic modifications relies on carefully selecting appropriate chemical reactions. The chosen reaction must exhibit quantitative efficiency to ensure complete conversion and avoid residual unreacted species that could interfere with the desired properties. It is also critical to select reactions that avoid generating insoluble by-products, as these can become trapped within the porous framework, compromising the material's structural integrity, surface area, and functionality. By addressing these considerations, post-synthetic strategies can serve as a powerful tool for the rational design and functionalization of advanced COF materials, enabling them to meet the demands of emerging technological applications.
1.2. Covalent organic frameworks in organic catalysis
Catalysis is a highly significant field in chemistry. Developing new catalysts with enhanced efficiency, selectivity, and stability is crucial in academia and industry to reduce costs, minimize waste, and save energy. Given the critical importance of discovering new catalysts, particularly solid ones, it is unsurprising that heterogeneous catalysis was among the first demonstrated applications of COFs.22COF-based catalysts uniquely combine the advantages of both homogeneous and heterogeneous catalysts, making them highly attractive for a wide range of catalytic applications. These materials possess isolated catalytic sites uniformly distributed within their ordered porous frameworks, ensuring consistent performance and enabling efficient substrate interaction and selective regioisomer product release.23 This structural uniformity enables precise control over the catalytic environment, mimicking the specificity and tunability often associated with homogeneous catalysts. At the same time, COFs exhibit the recyclability, stability, and robustness characteristic of heterogeneous catalysts, enabling their reuse in multiple reaction cycles without significant loss of activity or structural integrity.24 These dual advantages position COFs as versatile and durable catalysts for diverse chemical transformations. Simultaneously, they offer recyclability, stability, and robustness similar to heterogeneous catalysts.
Compared to traditional porous catalysts such as zeolites, silica, or activated carbon, COFs offer significantly enhanced chemical versatility and a broader range of pore sizes that are not only highly regular but also easily tunable. This level of control is particularly advantageous for the precise design of active sites tailored to specific reactions, allowing for unparalleled customization in catalytic applications. In this context, employing COFs as catalysts in organic transformations requires a comprehensive evaluation of several critical parameters, including their structural stability, surface area, pore accessibility, and functional group compatibility, to effectively characterize and optimize their catalytic performance.
This review focuses on COF-catalyzed chemical processes that have significantly impacted organic synthesis. Through the analysis of examples of both organo- and metal catalysis, we aim to present a range of material synthesis strategies focused on different applications, as well as the most important structural factors when evaluating a material as a potential catalyst for organic synthesis. In the present work, we summarise some of the most significant studies in the field of COFs used as catalysts for organic transformations.
The review is organized into four main sections. Firstly, we explore COFs where the intrinsic structure imparts catalytic properties to the material. This includes reactivity arising from network bonds, aromatic cores, and the extent of the p cloud in 2D layered structures. Secondly, we delve into materials specifically designed for targeted catalysis. These “synthetic enzymes” are crafted from building blocks already functionalized with desired functionalities, through de novo synthesis or post-synthetic modification, particularly in cases where achieving textural properties and crystallinity is particularly challenging. The third section discusses COFs incorporating metal centres into their structure, exploring various methodologies for effectively anchoring them within the crystalline network. The final topic discussed is the encapsulation, primarily of metallic nanoparticles within COF pores, is addressed alongside the main strategies for obtaining these composites.
In the conclusion, we provide a summary of the key points discussed and offer a brief outlook on the potential opportunities and advancements in COFs research that could enhance their application in organic catalysis.
2. COF backbone as organocatalyst
In this section, we explore the catalytic applications of non-specifically designed COFs, examining how their inherent structural features can be harnessed to catalyze organic transformations. The chemistry involved in the different parts of the linker, the core, the spacer, and the binding moiety, can be used to catalyse different organic processes. Upon analysis of the network structure, two intriguing aspects related to catalysis emerge the fully conjugated layers of 2D-COFs and the basicity of the nitrogen-based linkages.2.1. π-Extended networks and cores
The π-extension of the network plays a key role in the photoactivity of the material, as proven by Jiménez-Almarza et al.25 They explored how the morphology of two pristine COFs: 1,3,5-tri(4-aminophenyl)-benzene–1,3,5-benzenetricarbaldehyde (TAPB–BTCA) and tetra(p-aminophenyl)methane–benzene-1,4-dicarboxaldehyde (TAM–BDA) affects the activity towards the selective photooxidation of sulphur to sulfoxide. The more crystalline the layered COF, the greater the activity, presumably attributed to the enhanced conjugation of the network. This behaviour contrasts with the case of TAM–BDA, which exhibits a high yield regardless of the morphology presented due to the lack of conjugation along the network.More recently, Basak et al. reported three highly stable β-keto-enamine-based COFs which were investigated as photo-catalysts for metal-free C–B bond formation reactions.26 Three different COFs were evaluated for their photocatalytic performance TP-Azo, TpDpp and TpTab. The three of them were obtained by condensation of 1,3,5-triformylphloroglucinol (TP) with 4,4′-azodianiline, 2,8-diamino(6-phenylphenanthridine) and 1,3,5-tris(4-aminophenyl)benzene respectively. The materials were tested as C–H borylation reaction of quinolines, pyridines, and pyrimidines. The COFs exhibited moderate-to-high yields (from 20% up to 96%), depending on the substrate's molecular functionality (Fig. 5). Tp-Azo COF demonstrated the highest catalytic activity due to its high crystallinity, large surface area (2102 m2 g−1), low band gap (1.89 eV), and broader absorbance (400–600 nm). Mechanistic investigations highlighted the importance of light-harvesting capacity, charge separation efficiency, and current density during catalysis. The COFs' high photostability enabled reusability for several (>5) catalytic cycles. Other reactions have also been tested using β-keto-enamine-based COFs as catalysts, such as the synthesis of 2-arylbenzothiazoles is a cyclization reaction.27
 | ||
| Fig. 5 Material and catalytic reaction studied in ref. 26. | ||
In addition to the conjugation of the cores, the core itself serves as a versatile fragment for incorporating photoactive moieties. Unlike the linkages, constrained by those leading to crystalline structures or the spacers, restricted to those preserving the connectivity, the core has very high structural freedom. In this sense, we find COFs that possess simple aromatic structures like benzene or triazines and integrate more complex cycles such as pyrenes or perylenes, and even molecules macrocycles, as is the case with porphyrins.28
Using this approach, Y. Wu et al. prepared a pyrene–anthracene-based imine COF from 1,3,6,8-tetrakis(p-formylphenyl)pyrene (TFPPy), and 2,6-diaminoanthracene (DAAn), as a Diels–Alder reaction catalyst.29 This material was synthesized at mild conditions, resulting in high crystallinity. The structure presented columnar π-walls, which acted as catalytic beds, enhancing the reaction between N-substituted maleimide derivatives and 9-hydroxymethyl anthracene. The catalytic efficiency (99%) persisted even in the 4th cycle, showing the system's stability under reaction conditions.
Porphyrins stand out as prominent players in core-active COFs within the vast array of highly connected cores. A key attribute of these cores is their capacity to host a metal inside the pyrrolic cavity, resulting in metalloporphyrins, a topic included in Section 4.2. Moreover, pristine porphyrins exhibit strong absorption in the visible region and possess photoredox properties, making them ideal building blocks for the development of heterogeneous visible light catalysts. For instance, Hao et al. developed a metal-free porphyrin-based highly crystalline COF named A2B2-Por-COF through the self-polycondensation of an A2B2-Por monomer 4′,4′′′-(10,20-bis(4-aminophenyl)porphyrin-5,15-diyl)bis-(([1,1′-biphenyl]-4-carbal-dehyde)) with two-terminal amines and two terminal aldehydes attached to a porphyrin core.30 Compared to alternative co-condensation methods, the A2B2 monomer provided the two required functional groups, resulting in high crystallinity using the same monomer. The resulting material was employed for the selective oxidation of sulfides to sulphoxides with >99% selectivity and >96% conversion at room temperature. Additionally, owing to the basic groups of the cavity, A2B2-Por-COF exhibited excellent efficiency in the Knoevenagel reaction across a wide range of derivatives. Furthermore, it could be recovered over 10 consecutive cycles without structural changes, maintaining stability and experiencing only a minimal reduction in efficiency.
Following this approach, R. Chen et al. designed fully conjugated sp2-linked porphyrin COF (Por-sp2c-COF).31 The material was prepared by condensation of tetra(4-benzaldehyde)porphyrin and 1,4-phenylenediacetonitrile in basic media (5 M DBU in 1,2-dichlorobenzene). Incorporating the cyanovinylene linkage conferred high stability to the network, even in harsh acid or basic environments. Moreover, the fully conjugated porphyrin cores served as highly active catalysts (>85% yield) for the visible-light-induced aerobic oxidation of amines to imines, outperforming its imine-linked counterpart (no conversion) and the pyrene-based sp2c-COF analog (19% yield) (Fig. 6).
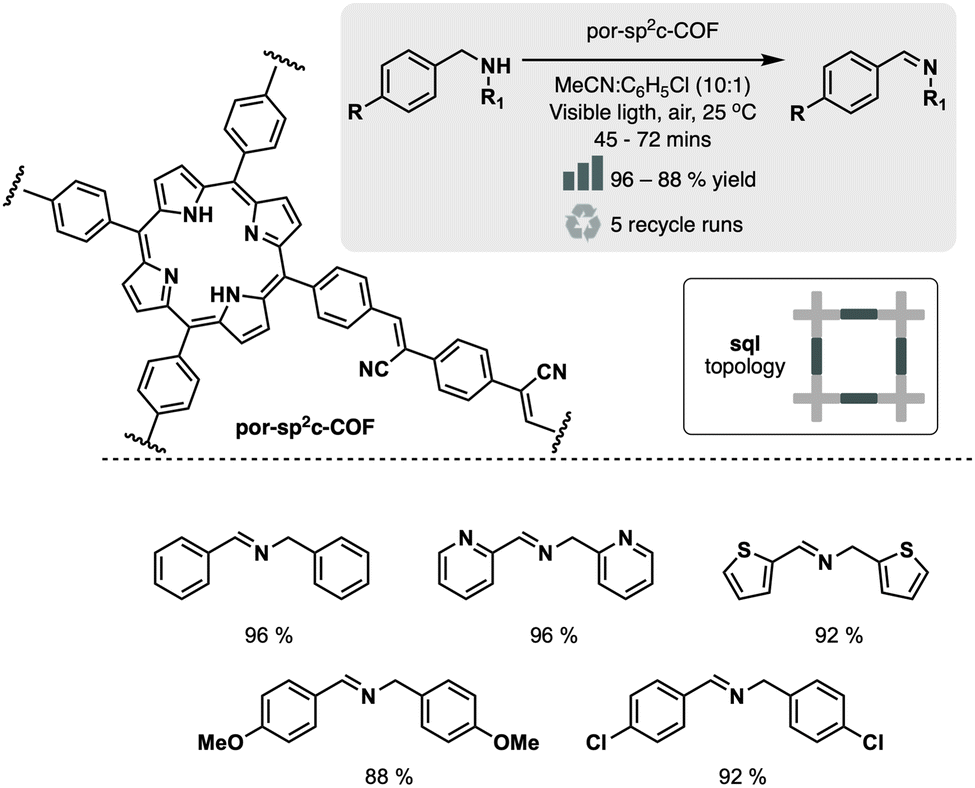 | ||
| Fig. 6 Material and catalytic reaction studied in ref. 31. | ||
Due to the predictable nature of COF structures and the conjugation between building blocks, intentional design allows for synergistic interactions at their cores. W. Qiu et al. synthesized a mixed anthracene–porphyrin COF by condensing tetra(4-aminophenyl)porphyrin and anthracene-9,10-dicarboxaldehyde.32 This COF displayed high activity in photocatalytic homocoupling of benzylamine derivatives, achieving 100% conversion rates for all reported substrates. DFT calculations supported the findings, demonstrating synergistic effects between anthracene and porphyrin moieties, forming a donor–acceptor system. The study offers valuable insights for constructing novel synergistic catalysts.
Beyond porphyrins, various other photoactive cores have been investigated as potential building blocks for COFs with unique photophysical properties. For instance, M. Roy et al.33 synthesized an imine COF by condensing TAPT with dithiophenedione functionalized with aldehydes at the 2 and 6 positions. This photoactive COF was evaluated for room-temperature amide synthesis under red-light irradiation. The catalyst demonstrated notable efficacy, promoting the formation of both primary and more challenging secondary amides under mild conditions, with catalytic activity retained over 10 cycles. The crystallinity of the COF enables precise integration of photochemical motifs into its backbone, facilitating efficient photoexcitation by low-energy photons. These excited photochromic units exhibit high reducibility, allowing radical-promoted dehydrogenation to occur under mild conditions.
A. Jati et al. demonstrated the construction of a series of β-ketoenamine COFs using TP and photoactive diamines, including DAAn, 2,6-diaminoanthraquinone, 2,7-diamino-9H-fluoren-9-one, and 2,7-diaminophenanthroquinone.34 These COFs were evaluated as photocatalysts (390 nm) for the decarboxylative fluorination of various aliphatic acids to yield alkyl fluorides using selectfluor in the presence of 2,6-lutidine. Among them, the anthraquinone derivative exhibited superior performance, achieving reaction rates 1.7 to 6 times faster than the other analogues and maintaining over 90% yield for up to seven cycles. The method was applied to late-stage functionalization of anti-inflammatory drugs like ibuprofen and ketoprofen and proved scalable in both batch and continuous flow modes. Furthermore, the protocol was extended to other halogenation reactions, enabling the production of alkyl chlorides, bromides, and iodides.
2.2. Linkages and spacers as active components
Numerous linkages contribute to achieving good crystallinity during COF synthesis. In this context, the linkages present in the structure can also serve as catalytic sites in organic reactions. This is the case for imine-based COFs, which could catalyse base-mediated processes such as the Knoevenagel condensation due to the intrinsic basicity of the imine linkage.Notably, Q. Fang et al. in 2014 explored the catalytic potential of two 3D microporous COFs, BF-COF-1 and BF-COF-2, based on imine and β-ketoenamine bonds, respectively.35 They demonstrated selective condensation between aldehydes of varying sizes by utilizing tetrahedral alkyl amine (TAA) and triangular trialdehyde. The COF architecture showcased size-dependent selectivity, providing yields comparable to those obtained with free ligands.
The basicity of the linkages can be enhanced by reducing the imine bonds to amine bonds.36 J. Hu et al. introduced H3PO3 as both a catalyst and a reducing agent for the conversion process.37 They synthesized the material by condensing BDA and 5,10,15,20-tetra(4-aminophenyl)porphyrin (TAPP) in the presence of H3PO3, resulting in the reduction of imine bonds and the formation of the amine-linked COF-366-R. In a Knoevenagel reaction between p-methyl benzaldehyde and malononitrile, COF-366-R exhibited higher yields (68%) compared to its imine counterpart (38%) due to its enhanced basicity (Fig. 7). Moreover, the irreversibility of the amine COF prevented the release of TAPP, avoiding crystalline loss and material deactivation after the reaction.
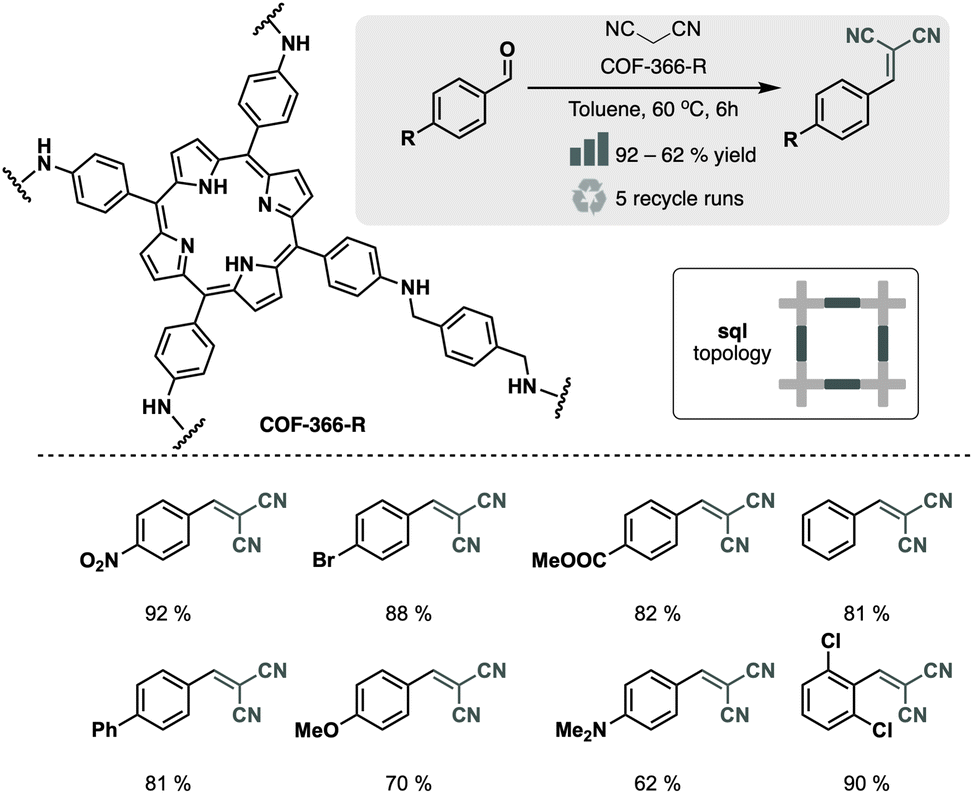 | ||
| Fig. 7 Material and catalytic reaction studied in ref. 37. | ||
Linkages play a critical role in controlling network conjugation, and thus, the optoelectronic properties of COFs, which is crucial for their photocatalytic performance. In this context, M. Traxler et al.38 examined the role of β-ketoenamine tautomerism in the performance of an acridine-containing COF. They synthesized three COFs based on 2,6-diaminoacridine (DAAcr) and various linkers: TP, 2,4-dihydroxybenzene-1,3,5-tricarbaldehyde, and 2-hydroxytribenzaldehyde (HDT). With the TP linker, tautomerization was irreversible to the keto form, while with the dihydroxy and monohydroxy linkers, it was reversible. The COFs were evaluated in Ni-mediated metallophotocatalytic C–N bond formation between 4-bromobenzotrifluoride and pyrrolidine. At 525 nm excitation, yields were low (3%) for the di- and mono-hydroxylated COFs, but moderate (38%) for the β-ketoenamine COF. However, using 440 nm irradiation, all COFs achieved quantitative yields after 16 hours. The superior activity of the β-ketoenamine COF likely relates to its enhanced charge carrier separation efficiency and larger surface area.
Y. Fan et al. provide another example of bond influence on network conjugation.39 In their work, they utilized fully conjugated cyanovinylene linkages to facilitate energy transfer between an photosensitizer unit based on pyrene (TFPPy) and a photoactive Ni-bipyridine building block (2,2′-BPyDCA) (see Section 4.2). This strategic linkage aims to optimize energy transfer within the COF framework, thereby enhancing the material's overall photoactivity. The same authors proposed using twisted cores based on spirobifluorenyl moieties to increase the charge transfer between this building block and the 2,2′-BPyDCA, combining both, core engineering and linkage engineering for the obtention of a highly photoactive material (see Section 4.2).40
In the development of catalytic reactions, spacers, alongside linkages, can play a crucial role. J. Hu et al. synthesized urea and thiourea-containing two-dimensional COFs acting as hydrogen bond donating catalysts in diverse reactions.41 Using amine monomers 1,3-bis(4-aminophenyl)urea (APU) and 1,3-bis(4-aminophenyl)thiourea (APTU), COF-TpU and COF-TpTU were created, showing high catalytic activity in epoxide ring opening, aldehyde acetalization, and Friedel–Craft reactions. With stability in mind, trialdehyde TpU and TpTU demonstrated impressive turnover numbers of 2 (30% yield) and 18 (99% yield), showcasing the site isolation effect. COF-TpTU maintained 70% enantiomeric excess for enantiopure styrene and 18% for racemic styrene. Superior to related MOFs, COF-TpTU exhibited consistently high efficiency over ten cycles. COFs outperformed monomers, APU and APTU, in Friedel–Crafts and aldehyde acetalization reactions, emphasizing the advantages of the site isolation effect in COFs.
Chemical customisation of the extension unit can also adjust the properties of the monomers. In this context, Chen et al. synthesised functionalised amide TABTA-COF-1 and TABTA-COF-2, via a reaction of N1,N3,N5-tris-(4-aminophenyl)benzene-1,3,5-tricarboxamide (TABTA) with BDA and 1,3,5-tri(4-formylphenyl)benzene (TFPB) respectively.42 Benzene-1,3,5-tricarboxamides are valuable building blocks for supramolecular assembly due to their intermolecular hydrogen bonds. Moreover, the arrangement of the amide group in the linker induces a donative effect on the aromatic ring bearing the imine bond nitrogen. This effect enhances the basicity of the imine bonds. Since the prepared TABTA-COFs possess intrinsic porosity and alkaline active sites, the Knoevenagel reaction between malononitrile and various benzaldehyde derivatives exhibited excellent efficiency and stability, enabling multiple recoveries. Control experiments involving similar COFs lacking amide functionality were conducted to confirm the enhanced basicity of the imine bonds. The amide-free COF displayed poor performance in this reaction, corroborating the catalytic role of amide groups in these structures.
3. Designing ligands for organocatalysis
Up to this point in the review, we have discussed the catalytic activity arising from the intrinsic components of COFs, such as linkages, cores, or spacers. However, one of the major advantages of COFs is the ability to tailor both monomers of the network. In this context, we can introduce specific fragments active in homogeneous catalysis into our material, facilitating the heterogenization of the catalyst.There are two main strategies to achieve this: the synthesis of already functionalized building blocks, or the functionalization of the building blocks after the network has been formed. The first strategy is referred as de novo synthesis of ligands, while the second is termed post-synthetic functionalization.
3.1. De novo synthesis of organo-functionalized COFs
The preparation via direct synthesis of organic decorated COFs assures the homogeneous dispersion and high loadings of a desired moiety relevant for catalytic transformations. However, several aspects must be considered before de novo synthesizing COFs with a functionality incorporated within (Fig. 9). The first one is the compatibility of the functional groups in the new fragment with those required to form the bond. Secondly, the incorporated moieties should be stable in the crystallization media, frequently requiring temperatures above 100 °C or very acidic conditions. Finally, the geometry of the core must allow the correct crystallization of the network. The two first issues can be predicted easily by the chemical properties of the selected groups. Regarding the third one, the reticular chemistry approach helps us to overcome this problem by designing the building block with a specific geometry. This could be achieved by incorporating the active moiety as a pendant group over the core, the spacer, or the linker of a molecule that yields a crystalline COF.The most straightforward strategy is the functionalization of the core. For instance, simple cores, such as the 1,3,5-trisubstituted TAPB, present unfunctionalized positions that can incorporate reactive groups, such as hydroxy, for anchoring active moieties. Following this strategy, Zhang et al. reported the synthesis of modified COFs based on 2,5-dimethoxyterephthalaldehyde (DMTA) and TAPB with a set of chiral pyrrolidine moieties anchored to the TABP molecule by an ether bond.43 These materials were active towards several asymmetric organic transformations including α-aminooxylation of aldehydes, aldol, and Diels–Alder reactions. This strategy can also be used to incorporate pendant moieties into the aldehyde building block44 or for enhancing stability of the network.45 This later is achieved by placing hydroxy moieties in α-position to aldehyde groups. This strategy is known to stabilize and contribute to the planarization of the layers of COF.46 In this regard, Y. Yusran et al. prepared two COFs based on hydrazone linkages between 1,3,5-tris(4-formyl-ethynylphenyl)benzene and 1,3,5-tris(3-hydroxy-4-formyl-ethynylphenyl)benzene with 1,3,5-benzenetricarbohydrazide.45 The later aldehyde yielded a COF with enhanced crystallinity and improved catalytic activity towards the amination of aryl bromides and chlorides.
A versatile molecule for incorporating side chains is 4,7-disubstituted benzimidazole. Its structural similarity with 1,4-disubstituted benzene, combined with the ability to introduce a pore-oriented pendant moiety in position 2, makes it an appealing scaffold for constructing de novo COFs. For instance, in 2016 Xu et al. reported the facile synthesis of two chiral COFs based on diamino building block 4,4′-(1H-benzo[d]imidazole-4,7-diyl)dianiline functionalized with L-proline and two trialdehydes, TAPB and TP.47 The chiral building block was constructed by direct cyclization of vicinal diamino derivative with the carboxylic moiety of L-proline. These two materials were active towards asymmetric aldol reaction in acetone at 30 °C, using trifluoroacetic acid as the proton donor. This strategy of using 4,7-disubstituted benzimidazoles can also be used to construct the aldehyde counterpart. Wang et al. reported a divergent strategy incorporating 8 different substituents in position 2 of the benzimidazole.48 In this case, incorporating a chloride group at position 2 allowed easy modification with a wide range of nucleophilic substituents. The fragments were active in the asymmetric amination of ethyl 2-oxocyclopentane-1-carboxylate with di-tert-butyl azodicarboxylate (Fig. 8).
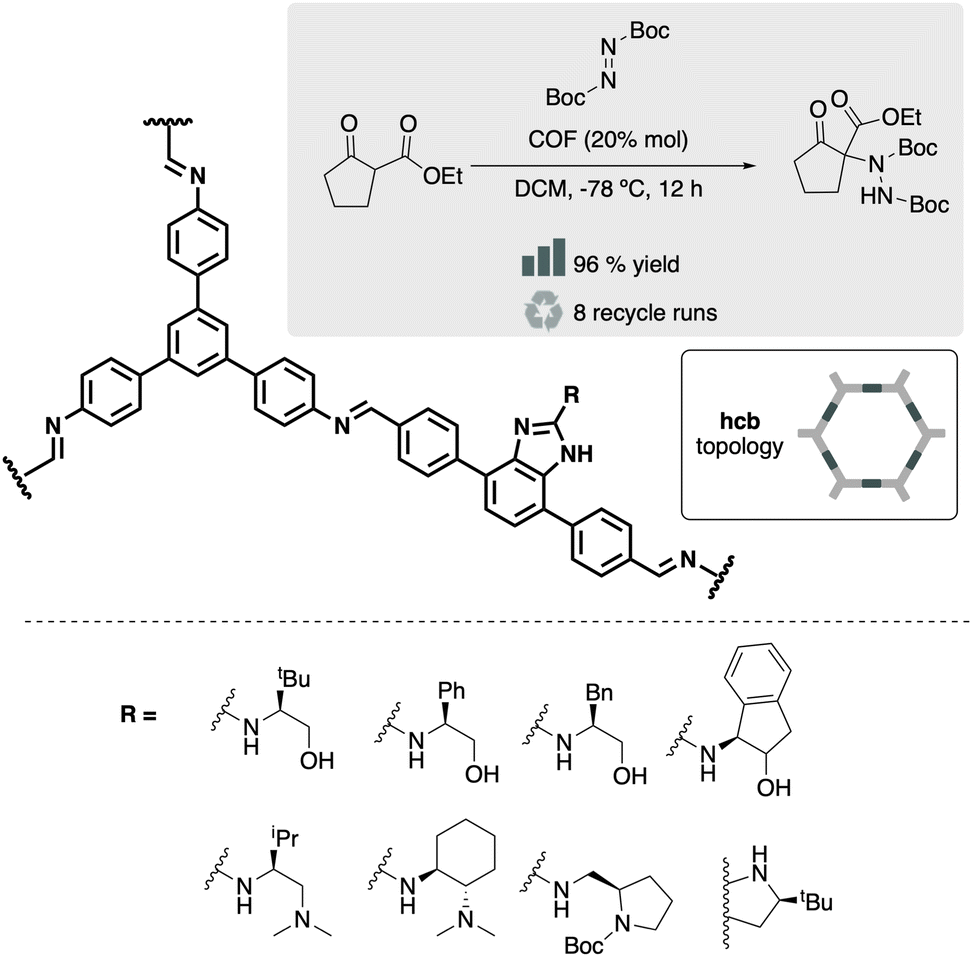 | ||
| Fig. 8 Material and catalytic reaction studied in ref. 48. | ||
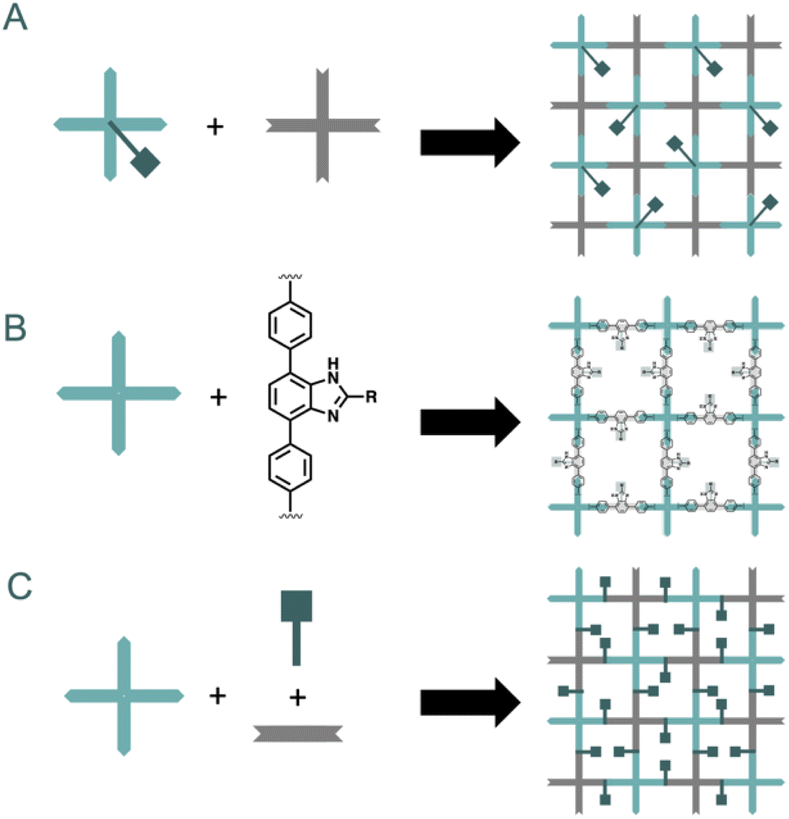 | ||
| Fig. 9 Different strategies for de novo construction of COFs: incorporation of a functionalizable moiety in a well-known core (A), incorporation of 4,7-disubstituted benzimidazole as building block core (B), assembly of the network using a multicomponent reaction (C). | ||
A more recent strategy for tuning the organic core of a COF is the use of multicomponent reactions in the construction of linkages. Wang et al. used a multicomponent reaction between DMTA, TAPB and phenylacetylene in the presence of a chiral catalyst, CuOTf–(S,S)-pybox (pyridine-2,6-bis(oxazolines)).49 This reaction mixture yielded a propargylamine-linked COF with an enantiopure phase due to the catalyst. Since the chirality comes from the Cu(I) catalyst, the enantiomeric material is easily obtained by replacing the pybox ligand CuOTf–(R,R)-pybox. The two materials were very active in Michael addition reactions with excellent enantiomeric excess in all the compounds of the proposed scope. This catalytic asymmetric polymerization could be further explored to prepare enantiopure materials without the requirement of chiral monomers or materials with enhanced stability of the bonds. This later was achieved by Yao et al. in 2021 by multicomponent polymerisation of TAPB, DMTA and 1-vinyl imidazole in the presence of Yb(OTf)3 and 2,3-dicyano-5,6-dichlorobenzoquinone (DDQ). This condensation yields a quinoline-linked COF, which can be further functionalized at the 3 position of the imidazole moiety (see Section 3.2).50
Using this strategy, H. Chen et al.51 synthesized a TAPP–BDA analogue functionalized with 4-vinylbenzenesulfonate, forming a quinoline-based bond. This dual-function, quinoline-containing COF photocatalyst integrates Brønsted acid sites into the photoactive COF framework, facilitating metal-free aerobic oxidative C–C bond cleavage in cycloalkanones. In this system, Brønsted acid sites activate the substrates, while the conjugated COF generates reactive species under visible light to cleave C–C bonds. The COF exhibited high catalytic activity in the oxidation of cyclobutyl, cyclopentyl, and cyclohexyl ketones to produce corresponding linear acids, as well as the corresponding esters in a one-step using a variety of alcohols. The catalyst preserved its performance, crystallinity, and surface area over five cycles.
3.2. Postsynthetic synthesis of organo-functionalized COFs
A post-synthetic strategy offers an alternative approach to the de novo synthesis of new frameworks. This strategy has been used to tune COF structures, taking advantage of the possibilities that organic chemistry offers to modify the building blocks (Fig. 10). A post-synthetic modification consists of a transformation done over a crystalline COF, retaining the long-range order of the system.52 The incorporation of reactive moieties in the building blocks opens new possibilities for the functionalization of the material in a post-synthetic fashion. However, some concerns must be considered before choosing a reaction. The reaction must be quantitative and must not generate insoluble by-products.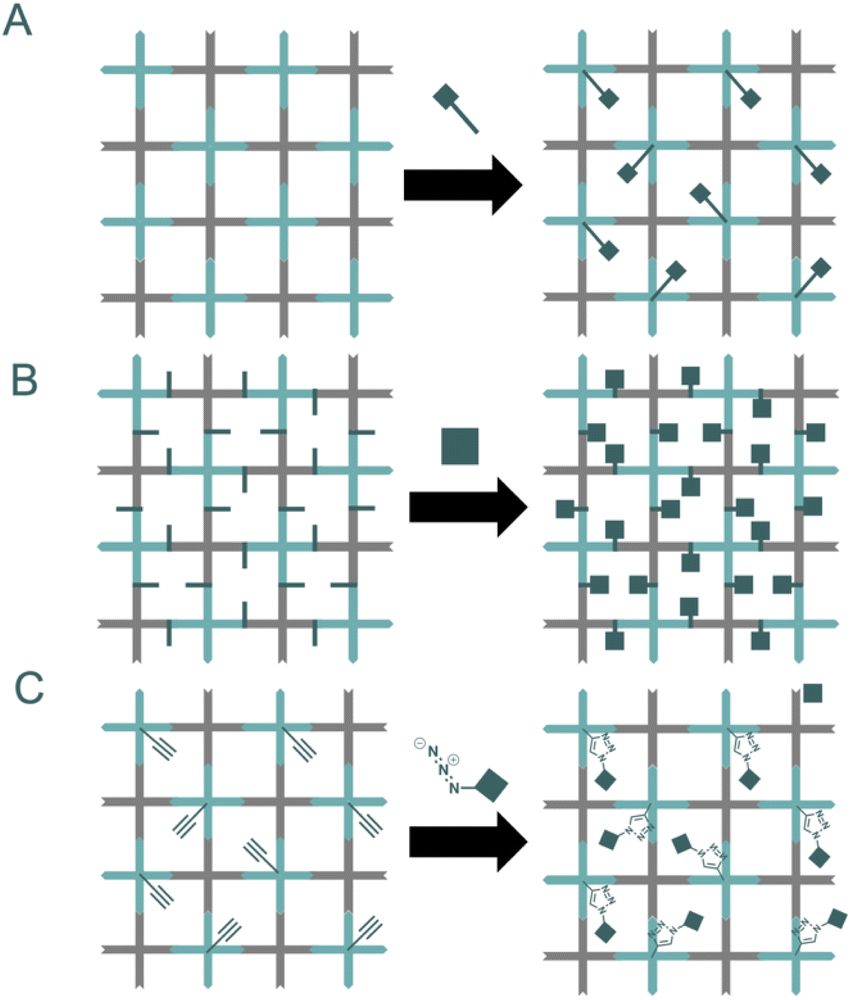 | ||
| Fig. 10 Post synthetic modification of COFs: incorporation of side chains via isosteric substitution of the core (A) or incorporation of pendant moieties (B). Finally, click chemistry approaches allow the rapid, quantitative functionalization of COFs (C). | ||
As in the case of de novo synthesis, the first and more direct way to functionalize a COF in this case, post-synthetically, is the transformation of a well-known structure. This is the case of the 2,5-dihydroxyterephthalaldehyde (DHTA) ligand, which presents free hydroxyl groups that can further react after the crystallization of the network. Following this strategy, Zhang et al. performed alkylation reactions of the hydroxyl groups of DHTA to incorporate ionic 1-alkyl-3-methylimidazolium moieties in DHTA–TAPP COF.53
The substitution of the benzene rings of the cores by isosteric pyridine analogues offers the possibility of extending the functionalities by alkylation reactions over the N atoms. This procedure generates cationic systems that can act as solid Lewis acids. Following this strategy, Zhang et al. prepared a COF based on a pyridinic derivative of 1,4-phenylenediamine, 1,4-diaminopyridine and trialdehyde TP. In a post-synthetic step, pyridinic N was alkylated with a 2-bromoacetic acid, leading to a zwitterionic species very active towards the cycloaddition between CO2 and an epoxide.54 In a similar way, Chen et al. prepared the pyridine derivative of TAPB, 2,4,6-tris(4-aminophenyl)pyridine (TAPPy), and after condensation with 2-hydroxobenzenetricarbaldehyde (HBTCA) a crystalline porous material was obtained. Alkylation of the N within the TAPP core with an (R)-proline derivative yielded a material very active in asymmetric Henry reaction due to the Lewis acidity of the pyridinium moiety.55
In addition to the isosteric substitution of the benzene ring to a triazine ring, nucleophilic sp2 N as pendant groups can be incorporated using multicomponent reactions such as the Povarov reaction (see Section 3.1).50 In this regard, the incorporation of a pendant imidazole group allows the post-synthetic incorporation of pore-oriented moieties with positive charges. Yao et al. took advantage of this sp2 N for the incorporation of sulfonated COF by the opening of 1,3-propanesultone. The Brønsted acidity of these sulfonic groups made the material active for the Biginelli reaction.
Finally, click chemistry is one of the most powerful strategies for the post-synthetic functionalization of COFs.56 Click chemistry consists of a set of high-yield, orthogonal reactions that allow the functionalization of an already crystalline material in a quantitative and chemoselective way. These two aspects are fundamental since the characterization of solid materials is more complex than molecular analogs.
In this regard, forming triazole moieties by cycloaddition of an alkyne and an azide is a powerful strategy to achieve the post-synthetic functionalization of a COF. In 2014, Xu et al. introduced a very versatile aldehyde building block that contains two alkyne moieties: 2,5-bis(2-propynyloxy)terephthalaldehyde (BPTA). This building block can replace DHTA in the synthesis of the TAPP–DHTA COF by controlling the ratio between both aldehydes. In this way, the authors prepared four COFs with different contents of alkynes 25%, 50%, 75%, or 100% of the aldehydes being BPTA, which can be post-synthetically modified with (S)-2-azidomethyl pyrrolidine. These modified materials were active towards the catalytic asymmetric Michael addition of β-nitrostyrene in aqueous media.57 The same strategy was tested for other COFS such as TAPB–DMTA, leading to very high yields and enantioselectivity even compared to molecular analogues in homogeneous phase.58
4. Metal single sites stabilization
Metallation of COFs is a commonly employed strategy to enhance their catalytic capabilities. This process involves the introduction of transition metal ions or atoms into the COF structure. There are various methods to achieve metallation, which can be broadly categorized into two main approaches. Firstly, by using a non-specific COF. This approach involves incorporating metal species into non-specifically designed COFs, typically through post-synthetic methods. This is possible in some COFs with intrinsic binding sites within their structure, such as the linkages. These inherent sites can be utilized for metallation. A second strategy consists of incorporating metal-binding functional groups in the building blocks. Specific functional groups or moieties can be incorporated into the COF structure to facilitate the binding of metal ions or atoms. The main groups consist of isosteric substitution of phenyl to pyridine, incorporation of hidroxy groups in a position to the amines, forming salen structures taking advantage of the imine present in the COF structure, or using porphyrinoid cores.Each of these approaches offers unique advantages and can be selected based on the specific requirements of the intended catalytic application.
4.1. COF linkages as binding site
N-rich COFs, as imine-COFs, present an intrinsic basicity, making them perfect scaffolds for post-synthetic metalation strategies. In 2011, Ding et al. pioneered the preparation of a Pd-loaded COF using this approach, showcasing the potential of metalation in pristine nitrogen-based COFs for catalysing C–C coupling reactions.59 The authors employed metalation over an imine COF based on 1,4-diaminobenzene (PDA) and (BTCA), capitalizing on the intrinsic basicity of the imine bonds involved in the crystallization process. To prepare the catalytic material, the imine COF PDA–BTCA was exposed to a Pd(OAc)2 solution, anchoring to the basic imine sites. After washing the material, the incorporation of the metal within the COF was confirmed through techniques such as XPS or ICP-MS. This binding to the framework was robust enough to facilitate the Pd-mediated catalytic reaction to form C–C bonds. This strategy for loading imine bonds was extended to 3D COFs, such as COF-300, as demonstrated by R. S. B. Gonçalves et al.60 The authors post-synthetically metalated COF-300 with Pd(OAc)2 and tested the material against a diverse range of molecules for Suzuki coupling (Fig. 11). Interestingly, they proposed the use of this material for continuous flow reactions by packing the COF inside a column, allowing reactants to flow through the packed-bed reactor, resulting in the desired product at the outlet of the setup. | ||
| Fig. 11 Material and catalytic reaction studied in ref. 60. | ||
This approach has been utilized in other related cases where imine bonds served as anchoring sites for Pd.61 In a more recent investigation, Romero-Muñiz et al. demonstrated how defects in the imine COF structure enhance the stability of Pd centres inserted within the framework.62 The study involved comparing two sets of post-synthetically metalated COFs (TAPB–BTCA) prepared at room temperature. In one approach, the metal salt (PdCl2(CN)2) was incorporated after the crystallization process. In contrast, in the other approach, the metal salt was introduced at an early stage when the material was still in an amorphous solid state. Both materials exhibited activity in Pd-catalyzed Suzuki couplings, but the yields in the case of the early metalation material were consistently higher. Advanced structural studies revealed that the late metalation process yielded a material that quickly formed Pd nanoparticles, which are less active than single-site catalysts. In contrast, the defective material stabilized the Pd single sites better, resulting in a more active catalyst.
Following this line, other metal salts were studied for their incorporation into the framework. This is the case of Cu2+ sites incorporated in β-ketoenamine COF TAPB–TP.63 The more basic amine groups present in this type of linkage are more suitable for binding hard metals, yielding catalytic active materials. Cu sites can be useful for the Huisgen reaction, one of the most important reactions in the field of click chemistry. The authors tested this material using the early metalation approach, which resulted in an increasing in the reversibility of the β-ketoenamine bond formation, yielding a high crystalline metallated material as demonstrated by PXRD and textural analysis. This Cu-loaded TAPB–TP catalysed effectively the regioselective synthesis of 1,4-disubstituted 1,3,5-triazoles.
X. Kan et al. introduced isolated Cu(II) centres into a β-ketoenamine COF using a tautomerised hydrazone linkage,64 which offered a more favourable coordination environment for Cu2+ ions compared to earlier systems. This was supported by single-crystal X-ray diffraction of molecular analogues and X-ray photoelectron spectroscopy. The COF was synthesized from 1,4-dibenzoylhydrazinyl-2-methylbenzene and TP, then metalated with copper(II) chloride to yield a highly crystalline material. The resulting COF showed excellent catalytic performance in alkyne–dihalomethane–amine coupling reactions, achieving up to 99% yield and sustaining activity over five catalytic cycles.
4.2. Specifically designed building blocks
While pristine COFs exhibit the potential to host metal species, the synthetic design of specific building blocks that facilitate the anchoring of metals within the COFs normally results in a more practical and efficient strategy. This approach is particularly advantageous when leveraging the possibilities afforded by the reticular synthesis methodology, which enables precise control over the integration of metal centres within the framework. By incorporating functional groups that promote metal coordination, the stability and activity of the resulting metal-loaded COFs can be significantly enhanced, thus expanding their applicability in various catalytic and adsorption processes.Following this approach, isosteric 4,4-biphenyl spacers could be substituted by bipyridine systems. Indeed, Ma et al. proposed the preparation of a COF containing bipyridine fragments analogous to biphenyl spacers.66 The authors used these N-containing moieties, 2,2′-bipyridine-5,5′-dicarbaldehyde or 2,2′-BPyDCA for anchoring Ir(I) ions within the 2D structure of the COF. Iridium bipyridine complexes are widely studied photocatalysts due to their excellent photophysical properties, including high stability, strong light absorption, and efficient intersystem crossing to generate long-lived excited states.67 The imine PyTTA–2,2′-BPyDCA COF was prepared by condensation between 2,2′-BPyDCA and 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetraaniline (PyTTA) monomers, followed by the postsynthetic immobilisation of Ir ions on the structure. They explored the efficacy of Ircod(I)@PyTTA–2,2′-BPyDCA COF in the C–H borylation reaction. The material exhibited high catalytic activity, especially compared to the control experiments with PyTTA–2,2′-BiPyDCA COF, [Ir(OMe)(cod)2], and the physical mixture of Py-2,2′-BiPyDCA COF and [Ir(OMe)(cod)2]. The material was also stable even after four catalytic cycles.
The highly ordered and periodic nature of COFs facilitates the construction of more intricate frameworks, such as bimetallic systems for tandem or dual catalysis. A powerful strategy to achieve this is sequential metallation. For instance, the BPyDCam–TP COF was first metallated with [Ir(ppy)2(CH3CN)2]PF6, followed by NiCl2, yielding the bimetallic Ni–Ir@COF (Fig. 12).68 The incorporation of the Ir centre significantly enhances the framework's photosensitizing capabilities, enabling efficient dual photocatalysis when complemented by the Ni decoration. The resulting Ni–Ir@TpBpy catalyst demonstrates exceptional catalytic performance, stability, and reusability, maintaining 80% yield after 10 catalytic cycles, thereby showing its potential for diverse applications.
 | ||
| Fig. 12 Material and catalytic reaction studied in ref. 68. | ||
The incorporation of imine bonds and bipyridine moieties within the structure offers an alternative pathway for the formation of bimetallic systems. These functional groups enable the coordination of different metal ions, allowing for the creation of highly efficient, cooperative catalytic sites. By leveraging the unique properties of both metal centres, bimetallic COFs can exhibit enhanced catalytic performance in a variety of reactions, offering advantages such as improved selectivity and reactivity compared to monometallic counterparts. The sequential incorporation of metals into the bipyridine moiety and the imine bond, respectively, allowed the preparation of Mn–Pd69 and Rh–Pd70 bimetallic materials. The Mn–Pd material, synthesized through the condensation of PyTTA and 2,2′-BPyDCA monomers via imines, displayed activity in a Heck-epoxidation cascade reaction. Imine bonds anchored Pd centres for the C–C coupling, while bipyridine moieties facilitated Mn incorporation for oxidation to epoxide. Rh(I) was loaded onto bipyridine moieties in the Rh–Pd heterometallic system. The authors varied bipyridine content in covalent organic frameworks (COFs) by condensing PyTTA with a mixture of 2,2′-BPyDCA and 4,4′-biphenyldicarboxaldehyde. Four materials with 25%, 59%, 75%, and 100% 2,2′-BPyDCA exhibited activity in adding phenylboronic acid to benzaldehyde (Rh(I) catalysis) and subsequent oxidation to benzophenone (Pd(II) catalysis). Rh(I)/Pd(II)@75% BPy COF achieved the highest yield (90%), showcasing the tunability of metal quantity and ratio through COF synthetic design.
P. Heintz et al.71 prepared an β-ketoenamine using the inversed building blocks: the 2,2′-bipyridyl-5,5-diamine (2,2′-BPyDA) and the aldehyde TP. The condensation of the COF was achieved via mechanochemical synthesis. The material exhibited activity in facilitating the conjugate additions of a broad spectrum of aryl boronic acids to β,β-disubstituted enones in aqueous media, forming quaternary carbon centres. The same material resulted in another study, also active in adding arylboronic acids to α,β-unsaturated carboxylic acids in an aqueous media.72
2,2′-bipyridine-based ligands and be used synergistically to achieve higher activities taking advantage of fully conjugated systems such as those provided by cyanovinylene linkage. In this line Y. Fan et al.39 reported a COF synthetised by the condensation of 2,2′-([2,2′-bipyridine]-5,5′-diyl)diacetonitrile and TFPPy. In the resulting system, photoexcited pyrenes in the COF efficiently transferred energy to Ni catalytic sites, enabling radical-based borylation and trifluoromethylation of aryl halides. The enhanced energy transfer within this π-conjugated COF significantly improved excited-state Ni catalysis, achieving a catalytic efficiency two orders of magnitude higher than that of its homogeneous counterpart. Notably, the COF maintained its catalytic activity and crystallinity, allowing for up to three cycles of reuse without significant degradation. The same material was also tested in a variety of bond forming reactions: C–O (esterification), C–S (thioetherification and sulfonation), C–N (amination and sulfonamidation), borylations, phosphonylation and allylation reactions.73 The charge transfer efficiency was further enhanced in a spirobifluorene-based analogue. Through the Knoevenagel condensation of 2,2′-([2,2′-bipyridine]-5,5′-diyl)diacetonitrile with the twisted building block spirobifluorenyl tetrabenzaldehyde, the authors successfully synthesized a cyanovinylene COF that exhibited 23 times higher activity than the homogeneous control experiment for C–N bond formation over aryl bromides.40 This study underscores the critical role of core geometries in tailoring COFs for specific applications.
Apart from 2,2′-BPyDCA, other cores such as porphyrins74 and phenanthroline derivative has also been employed for the metalation of COFs. In this regard, A. López-Magano et al. prepared an imine COF based on TAPB and 4,4′-(1,10-phenanthroline-3,8-diyl)dibenzaldehyde or (PhenDBA).75 The authors successfully anchored photoactive Ir(dF(CF3)ppy)2 and NiCl2 to the phenanthroline groups by exposing the pristine COF to a solution of both salts. The obtained Ir,Ni@TAPB–PhenDBA COF presented high catalytic activity toward photocatalytic cross-couplings between aryl bromides and four different radical precursors: N-protected proline, organic alkyl silicates, and potassium benzyl- and alkoxy-trifluoroborates. Additionally, comparative experiments with the pristine material, exclusively Ni-loaded and exclusively Ir-loaded materials, showed the synergistic effect between both metals, which was supported by mechanistic studies.
:1), attributed to the stabilising effect of MeOH on Cu+ centres.This strategy proved to be particularly effective for softer metals such as Pd, enhancing their catalytic performance. J. Han et al. inserted Pd2+ metals into an imine-based COF prepared by condensation of 3,3′-dihydroxybenzidine and BTCA (COF-BTDH).77 The hydroxy group in α into the imine provided the Pd2+ salt enhanced stability. The authors explored the catalyst performance using various olefins for the Heck coupling reaction. In the presence of Pd-COF-BTDH, even aliphatic olefins lacking heteroatoms or induction effects underwent high conversion to (E)-linear products with excellent selectivity. A performance comparison between crystalline and amorphous material showed a worse yield with poorer regioselectivity in the case of the amorphous solid. This demonstrated that structural order and regular channels significantly influenced selectivity.
This strategy is also applicable to other bonds, such as hydrazone bonds. Through the reaction of TP with 2,5-dihydroxyterephthalohydrazide and 2,3-dihydroxyterephthalohydrazide, Kundu et al. synthesized two mesoporous COFs (2.5 nm pore size) NUS-50 and NUS-51, respectively.16 The hydroxyl groups enriched building blocks provide proper sites to decorate Co2+ in the COF structures. Due to the uniform distribution of pore size, stability, and availability of active sites containing Co2+ with good Lewis acidic properties, they studied the catalytic activity of systems in aldehydes cyanosilylation reaction. Both catalysts gave almost the same efficiency and comparable performance to MOF-based catalysts. The conversion rate for both was similar to Co(OAc)2, indicating that the catalytic activity of Co(OAc)2 remained constant after the heterogeneity in the COF structures. The selectivity of NUS-50 and NUS-51 was studied by using larger aldehydes. The results presented that as the aldehyde gets larger, the efficiency decreases drastically, as far as, for 9-anthracenecarboxaldehyde only a trace yield was obtained, indicating that the catalytic process occurs inside cavities and channels. So, prepared COFs are highly selective against the size of the reactants. Both NUS-50 and NUS-51 displayed excellent stability after five recovery cycles without decreasing performance.
By incorporating a second hydroxy functional group into the building block, we can create a catechol moiety, providing an effective solution for chelating metal salts. For this purpose, DHTA can be used as a building block. Indeed, H. Vardhan et al. prepared two COFs by imine condensation of DHTA with PyTTA and 1,3,5-tris(4-aminophenyl)triazine (TAPT).78 These materials can host vanadyl ions through post-synthetic VO(acac)2 treatment. Both were tested as catalysts in the Mannich-type reaction, achieving high yields (70–99%) across a wide range of substrates. They exhibited excellent activity, and stability, retained crystalline structure, and could be recycled up to five times. Control experiments using metal-free ligands and metal salts demonstrated the superior activity of the COFs, attributed to their textural properties and the confinement effect of COF pores. Additionally, VO–DHTA–PyTTA COF was effective in the Prins condensation between β-pinene and formaldehyde, aiding the synthesis of NOPOL, a crucial primary alcohol. The material showed enhanced activity compared to the free salt and the pristine material.79
A prominent family of ligands frequently employed in catalysis is based on the condensation of salicylaldehyde and ethylenediamine, known as salen ligands. These ligands are tetradentate (2 imines and 2 hydroxy) and exhibit C2-symmetry. One of their notable characteristics is their ability to coordinate a diverse array of metal ions, often stabilizing them in various oxidation states. This versatility makes salen ligands highly valuable in catalytic applications.
Based on these ligands, Li et al. reported the synthesis of a COF based on the salen (Schiff base) framework.79 They achieved this by in situ formation of salen skeletons through the heating of ethylenediamine with tris(tert-butyl-salicylaldehyde)benzene (TTHB) under ambient air conditions. Subsequently, the salen moieties within the COF were metallized with Co, Mn, Cu, and Zn. The researchers then investigated the catalytic activity of these single-site COF catalysts in the cycloaddition reaction of CO2 with propylene oxide, in the presence of various co-catalysts. The study found that the catalytic performance increased by acidic strength (Cu2+ < Mn2+ < Zn2+ < Co2+) and coordination ability of the metal sites, which influence their binding to epoxides and the formation of oxyanion intermediates in the catalytic reaction.
This strategy could also yield chiral versions of the COF by using a chiral amino such as (R,R)-1,2-cyclohexane-diamine. Indeed, Han et al. reported the metal-directed synthesis of two chiral covalent organic frameworks (COFs) using Zn2+-directed imine condensation.80 They employed enantiopure (R,R)-1,2-cyclohexane-diamine in conjunction with TTHB or tris(salicylaldehyde)benzene derivatives to obtain CCOF-4-M and CCOF-3-M, respectively. CCOF-4 resulted in enhanced crystallinity due to the tert-butyl moieties. By post-synthetic metal exchange of Zn2+ ions, they reached Fe2+, Mn2+, Co2+, Cr2+ and V2+ loaded COFs. They used these COFs for various reactions, including aldehyde cyanation with TMSCN, asymmetric Diels–Alder reaction, oxidations to epoxides, and aminolysis of epoxides (Fig. 13).
 | ||
| Fig. 13 Material and catalytic reaction studied in ref. 80. | ||
N-Heterocyclic carbene (NHC) ligands represent a significant class of ligands in coordination chemistry. A classic example involves the derivation of NHC from N,N′-disubstituted imidazolium salts by deprotonating the acidic hydrogen at the C-2 position. The resulting free carbene is stable enough to be isolated, particularly when the R groups, such as adamantyl, are bulky. Indeed, NHC ligands are highly valuable in a wide range of catalytic applications because of their strong binding to metals and their ability to effectively promote various catalytic cycles, much like phosphine ligands. Yang et al. successfully incorporated NHCs as pendant moieties into an imine-based COF.81 This was achieved through the condensation of a disubstituted 1,4-di(4-formylbenzyl)benzene (NFB) aldehyde with 1,3,5-triaminobenzene under ionothermal conditions, yielding a COF-NHC material. Subsequently, this material was metallated with Pd(OAc)2, resulting in a catalyst that exhibited activity in the Suzuki–Miyaura reaction (Fig. 14) and C–C cross-coupling of triarylbismuths with aryl halide. This, together with the reusability of COF, demonstrates the possibilities that these materials offer in terms of heterogeneous catalysts, which can be reused up to 8 times without appreciable loss of activity.
 | ||
| Fig. 14 Material and catalytic reaction studied in ref. 81. | ||
Another strategy for incorporating NHCs into COFs involves leveraging the benzoimidazole moieties that have been previously discussed. Li et al. synthesized NHC–AuCl-COF, an imine COF based on TAPT and NHC–AuCl, exploring its catalytic activity in the direct carboxylation of terminal alkynes.82 Under optimized conditions, the catalyst demonstrated high efficiency with various alkynes, maintaining its structural integrity and efficacy during catalyst recovery cycles. Additionally, the catalyst's versatility was evaluated in the hydration of alkynes in an aqueous medium, which initially showed low efficiency. To enhance its performance, chloride groups were replaced by [SbF6]−1, resulting in NHC–AuSbF6-COF, which exhibited exceptional efficiency and reusability in the hydration reaction.
Finally, the reduction of imine bonds to amines offers new possibilities for pore-wall functionalization.83 In this regard, Yan et al. conducted a separate study where they synthesized a thermally stable Co2+@TFPPy–PyTTA-COF ionic catalyst based on pyrene moieties PyTTA and TFPPy.84 This catalyst was created by incorporating cobalt alkyl sulfonate groups onto amine linkages formed through the reduction of imine linkages using NaBH4. The main objective was to design a structure that combines acidic and nucleophilic sites within a single framework, eliminating the need for an additional catalyst and minimizing the potential for side reactions. The results revealed that the CO2 cycloaddition reaction with various epoxides, using the Co2+@TFPPy–PyTTA-COF catalyst, achieved an efficiency exceeding 90% under conditions of 100 °C, 3.0 MPa CO2 pressure and a 48 hour reaction time, highlighting the catalyst's exceptional catalytic activity.
Y. Li et al. utilized TAPP to host Co2+ centres, synthesizing a TFPPy–TAPP COF from a pre-metallated porphyrin.85 The cobalt within the ligand was retained in over 90% of the TAPP cavities. This metal species demonstrated activity in the cycloaddition of epichlorohydrin and CO2, with the presence of a TBAI co-catalyst, achieving an efficiency of 92% under 0.5 MPa of CO2.
In 2020, H. Ma et al. introduced a method for synthesizing (S)-CIK via the Strecker reaction using a COF with a unique sidewall design.86 The authors synthesised (R)-Cu-TAPBN-COF and (S)-CuTAPBN-COF, both with distinct pore sizes through imine condensation reaction between Cu2+–TAPP and optically active 6,6′-dichloro-2,2′-diethoxy-1,1′-binaphthyl-4,4′-dialdehyde ((R)-BINOL-DA or (S)-BINOL-DA). These COFs offered stability in harsh conditions, strong Lewis acidity, and light-to-heat conversion potential, making them promising for Strecker reaction catalysis (Fig. 15). (R)-Cu-TAPBN-COF displayed high catalytic efficiency (94–98% yield), enantiomeric excess (91–94% ee), and good reusability under visible light. Remarkably, (S)-CuTAPBN-COF effectively produced the mirror-image product, (R)-CIK, under the same conditions. (R)-Cu-TAPBN-COF could even drive the reaction using sunlight at 47 °C, showing potential for solar-powered asymmetric reactions. Control experiments revealed the COF's role in inducing chirality, while copper acted as a catalyst. The Strecker reaction primarily occurred at the catalyst's internal surface, yielding high yields and excellent enantiomeric excess with smaller-sized aldehydes.
 | ||
| Fig. 15 Material and catalytic reaction studied in ref. 86. | ||
5. Nanostructures encapsulation
Nanoparticles (NPs) are among the most widely used nanostructures in organic catalysis, often outperforming other common systems such as metal oxides, polyoxometalates (POMs) or enzymes. NPs are well-known catalysts in organic synthesis, especially in redox catalysis,87 and C–C/C–N cross-coupling reactions.88 Their catalytic activity strives for a high surface-to-mass ratio (specific surface area). Despite their high activity, nanoparticles tend to aggregate and stabilize in a sintering process. Homogeneous composites are usually prepared to preserve the nanoparticles that are isolated and, therefore, active, to avoid the nanoparticle deactivation process. In this context, COFs' intrinsic porosity makes them a perfect scaffold to host and grow inside metal nanoparticles.There are three main strategies for encapsulating nanoparticles within a COF: the “ship-in-bottle” approach, the “bottle-around-ship” approach, and the one-step synthesis approach (Fig. 16).
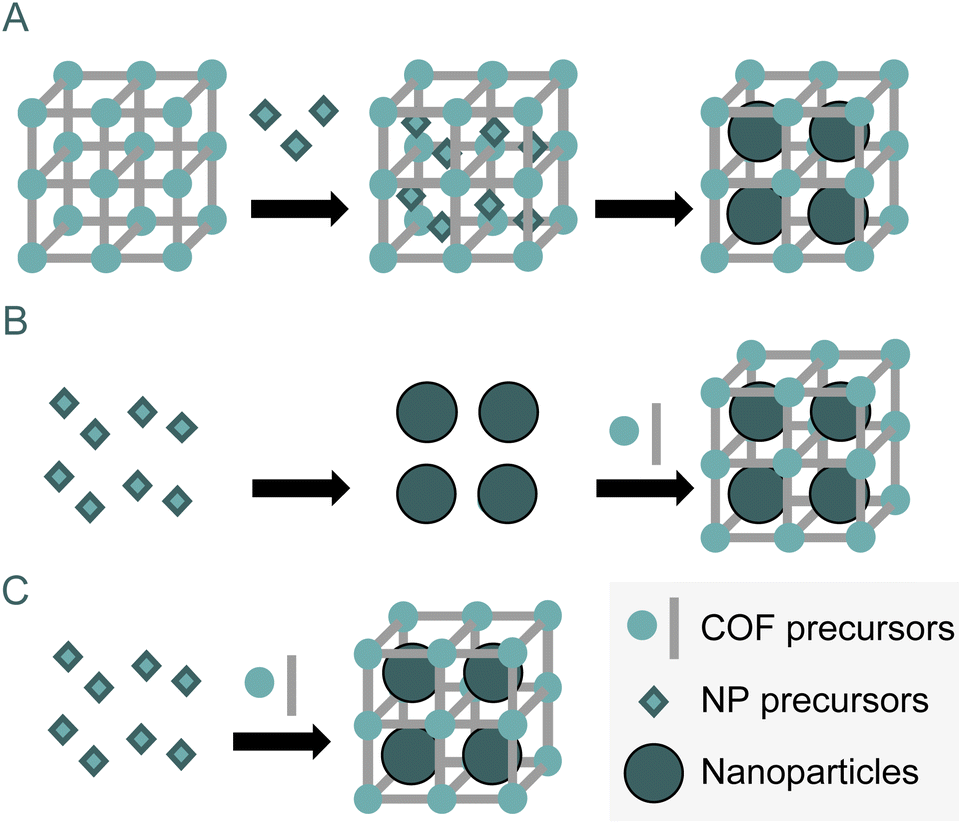 | ||
| Fig. 16 Different strategies for encapsulation of nanoparticles inside COFs. Ship-in-bottle approach (A), ship-around-bottle approach (B) and one-step synthesis approach (C). | ||
The ship-in-bottle strategy involves impregnating the COF with a metal precursor, which is further reduced (or oxidised in the case of metal oxides) inside the pores. This process is the most used for loading nanoparticles inside COFs. It presents an additional advantage to the stabilization of nanoparticles avoiding the sintering process. This method allows controlling the size of the nanoparticle since the cavity limits the nanoparticle growth, yielding a homogeneous size distribution of NP. The second method consists of growing the COF around well-formed and dispersed nanoparticles. This method is known as bottle-around-ship. Finally, the formation of both the COF and the nanoparticles can be done in a single step, a one-step strategy to achieve the composite. Of the three existing strategies, the first is by far the most widely used.
The stabilization of NPs within COFs can be achieved by incorporating heteroatoms like N or P into the structure of the building blocks. In this context, Ghosh et al. synthesized two distinct COFs using TAPT or PDA condensed with TP.89 The presence of a triazine core in TAPT allowed for slightly higher Ag loadings, then resulting in enhanced performance in the solvent-free synthesis of iodomethyl group-substituted 5- and 6-membered cyclic carbamates from unsaturated amines in the presence of N-iodosuccinimide under ambient conditions and involved the conversion of CO2 to cyclic carbamates. The improved performance was attributed to the larger surface areas of these COFs, which were also related to the planarity of the triazine cores.
Phosphorus can also be used for the stabilization of nanoparticles as demonstrated by Tao et al.90 They prepared an imine COF Phos-COF-1 by condensation of tris-(4-formylphenyl)phosphine (TFPP) and PDA. The authors prepared both monometallic (Pd) and bimetallic (Pd–Au) versions of Phos-COF-1 by ship-in-bottle strategy. The well-structured Phos-COF-1 allowed for uniform dispersion of sub-2 nm metal nanoparticles, enhancing catalytic activity. Pd-loaded COF acted as catalysts for Suzuki coupling reactions. They also developed a tandem catalytic system, combining Pd-catalyzed coupling and Au-catalyzed reduction using PdAuNPs@Phos-COF-1. With 0.5 mol% of PdAuNPs@Phos-COF-1, they reached over 90% efficiency within 2 hours, reduced to 60% overall efficiency at 25 °C with NaBH4.
Continuing along this line of NP stabilization, the presence of nitrogen within the linkages of the COFs proves to be beneficial for stabilizing NPs. In this context, Chen et al. successfully stabilized Ru0 NPs within a hydrazone-linked COF.91 They initially prepared the COF by condensing benzene-1,3,5-tricarbohydrazide and terephthaldehyde, resulting in COF-ASB. In a subsequent post-synthetic step, they incorporated Ru nanoparticles with a loading of 5%. This material demonstrated catalytic activity in the synthesis of imine compounds from benzyl alcohols and aniline derivatives, all without the need for a solvent. It is important to note that the optimal conditions for the two reaction steps involved in this cascade process were quite divergent. The high temperature required for the alcohol oxidation step was unfavourable for the subsequent reaction. However, the introduction of the nanoparticle-encapsulated catalyst effectively addressed this challenge, resulting in an overall efficiency of up to 93%.
Introducing heteroatoms into linkages and post-synthetically incorporating side chains onto COF building blocks can foster metal interactions within the pores. M. Guo et al.92 utilized this approach for Pt nanoparticle formation within a COF constructed from TAPB with varying ratios of DHTA and 2,5-divinyl-terephthalaldehyde (DVTA). Through a thiol–ene reaction, 1-propanethiol was introduced post-synthetically. The resulting Pt@COF demonstrated selective activity in reducing 1,4-chloronitrobenzene, with the Pt/S ratio significantly influencing both conversion and selectivity. Optimal results were achieved with lower Pt/S ratios, underscoring its importance in catalytic performance.
The Povarov reaction with styrene derivatives offers an effective strategy for pore-wall functionalization in COFs.93 Introducing a series of 4-substituted styrenes (–CH3, –H, –CF3, and –CN) enabled controlled growth of bimetallic Pd–Cu nanoparticles within the channels of a TAPB–DMTA COF. This modified COF exhibited selective catalytic activity, facilitating the reduction of alkynes to alkenes with high efficiency.
An alternative to the ship-in-bottle strategy is the one-step synthesis, in which COF crystallization and NP formation occur simultaneously, avoiding the use of a strong external reducing agent or a capping agent. Bhadra et al. proposed using Pd-loaded building block 2,2′-bipyridine-5,5′-diamine palladium chloride (Bpy-PdCl2) for the construction of β-ketoenamine COF by condensation with TP.94 This material was active in the synthesis of 2-substituted benzofurans from 2-bromophenols and terminal alkynes.
Alternatively, mechanochemical methods enable the encapsulation of nanoparticles within COFs, as shown by Brown et al.95 They synthesized TAPT–DMTA loaded with Pd nanoparticles using Pd(AcO)2 as a precursor. After one hour of reaction at 20 Hz, a crystalline COF was obtained that retained catalytic activity in C–C coupling reactions across five cycles, with minimal Pd leaching. This approach can also be applied to other systems, such as TAPT condensed with 2,4,6-tris(4-formylphenyl)-1,3,5-triazine, broadening its applicability for COF-based catalysis, and scaled up to gram scale while using less solvent than traditional syntheses.
In addition to incorporating low-valence metal nanoparticles, researchers have also successfully integrated metal oxides and POMs into COFs. For example, Das et al. carried out a post-synthetic incorporation of CuO into a β-ketoenamine COF known as TAPT–TP. This modified material exhibited catalytic activity in the C–C homocoupling reaction of arylboronic acids.96 Zhang et al. prepared an imine COF based on TAPB–DHTA. The free hydroxy moiety allowed the incorporation of a cationic moiety for stabilizing anionic POM [H3PW12O40]3−.97 Despite losing almost all the pore volume, the material resulted in activity towards the cycloaddition of CO2 with epoxides.
Finally, COFs can also serve as a platform for encapsulating enzymes,98 similar to the encapsulation of metallic or metal oxide nanoparticles previously covered. A good example is found in the study by Paul et al., where they utilized Tp-Azo COF to encapsulate the enzymes β-glucosidase (BGL), endoglucanase (EG), and cellobiohydrolase (CBH).99 The loading weights were 2.8%, 23%, and 7% respectively. This loading capacity could be enhanced by forming a COF foam using CO2.100
These materials were tested as catalysts for the hydrolysis of carboxymethylcellulose into glucose under conditions of pH 6 and 55 °C. The materials remained active for up to 10 cycles, with the composite remaining stable for up to 120 days and retaining half of its initial activity.
In addition to immobilizing proteins, the COF itself can serve as an active, non-innocent scaffold in catalytic processes. L. Ran et al.101 proposed a COF as a photosensitizer for the kinetic resolution of amines into amides, employing the enzyme Candida antarctica lipase B. The COF scaffold includes PhenDBA units coordinated with Ir, which facilitate proton transfer, leading to two possible isomers in the secondary amine. This secondary amine is then kinetically selected by the enzyme for amide coupling, achieving stereoselective coupling with high enantiomeric excess. Remarkably, the catalyst maintains its activity even after five cycles.
6. Conclusions and outlook
In this review, we have explored the fundamental aspects of COFs as catalysts in organic reactions, emphasizing their unique properties and the exciting opportunities they present for the future of catalysis. In this regard, four key points were proposed as the foundation on which to develop the text. The review starts by introducing COFs: polymers composed of purely organic monomers that, due to the reversibility of the bonds connecting them, can arrange into an orderly network in space (i.e., crystalline) with cavities or pores within their structure. The review discusses the common characteristics of COFs that enhance their performance as catalysts, such as their extended π cloud and high connectivity, which enable the incorporation of chemically complex fragments like porphyrins and polycyclic aromatic hydrocarbons. It has been demonstrated that COFs based on imines and those based on β-ketoenamines can function as photocatalysts for reactions relevant to organic synthesis.24,26 Additionally, the high connectivity (c > 2) required in at least one of the two building blocks of these materials facilitates the incorporation of more chemically complex heterocyclic fragments, such as porphyrins,29,30 or polycyclic aromatic hydrocarbons like anthracenes.28 The redox properties of these cores inherently provide the material with utility in organic transformations. Additionally, the links, moieties that might initially seem inconsequential in catalyzing reactions, can play critical roles as basic catalytic centres in the case of imines,35 β-ketoenamines,35 and amines,37 while still maintaining their central role to be sufficiently reversible to achieve long-range order in the network.This crystallinity is a crucial feature of these materials. Unlike other organic polymers, their long-range order allows their periodic structure to be defined through repeating units of hundreds of atoms, known as unit cells. This makes their synthetic design predictable, enabling the preparation of ad hoc materials for specific applications due to the foreseeability of the network assembly. This synthetic approach is known as reticular chemistry. New COFs can be designed based on known structures by substituting structural ligands with others possessing similar geometric characteristics, such as connectivity and spatial dimensions, and chemicals, such as functional groups, using reticular chemistry. In Section 3.1, “De novo synthesis of organo-functionalized COFs”, we discussed how the chemical structure of our ligands can be modified while maintaining the structural/geometric properties that are known to generate topologies leading to COF formation. Incorporating side chains outside the building block's structural unit has been crucial. This approach is exemplified in the functionalization of 1,3,5-trisubstituted cores43,44 and the incorporation of 4,7-disubstituted benzimidazole moieties as replacements for 1,4-disubstituted benzene cores.47,48 Lastly, we explored the potential of multicomponent reactions as a promising strategy for incorporating side chains into the pores while preserving the geometry of dynamic covalent bonds.49,50
Apart from the design of structures relevant to organocatalysis, we have seen how the incorporation of functional groups, or even certain cores, within the structure of COFs, while always respecting the principles of reticular chemistry, can be of vital importance for hosting metal salts within the structure of our material. For instance, the inclusion of nitrogen atoms by substituting benzenes with nitrogenated heterocycles is one of the most direct strategies for stabilising metal centres. This approach leads to commonly used ligands such as 2,2′-BPyDCA66–72 or its phenanthroline-based analogue, PhenDBA.75 The porous nature of COFs allows for the hosting of coordinating groups through pore functionalization, enabling chemical complexity to flourish, limited only by imagination. As discussed, the incorporation ranges from simple hydroxyls,16,76–78 to NHCs,81,82 and includes examples where the geometry of the ligands and the bonds formed act synergistically, giving rise to classic coordinating groups such as salen.80 These transformations are possible thanks to reticular design, which guides the synthesis by filtering out the most suitable geometries and providing boundary conditions, thereby allowing a focused approach to ligand synthesis.
COF networks can also serve as substrates for chemical transformations. In this regard, a single COF with a functionalizable group can give rise to a family of materials that possess similar physical-structural properties but with different chemical characteristics tailored “à la carte” for specific applications. These types of transformations, which occur on the already formed framework, are known as post-synthetic modifications. Post-synthetic modifications enable rapid chemical diversification of materials, facilitating detailed studies of the structure–activity relationship. We have seen in numerous examples how COFs with different side chains have been compared for organocatalysis,50,53–58 or the incorporation of different metals explored to carry out various chemical transformations.16,59–80,84
Throughout this review, we have explored how the meticulous selection of building blocks in constructing COFs enhances their performance as heterogeneous catalysts in organic reactions. The unique structure of COFs, which clusters discrete active centres throughout the network, imparts characteristics akin to both heterogeneous catalysts (recyclability, robustness, ease of handling) and homogeneous catalysts (high specificity, selectivity, and activity). This underscores the critical role of the structure–activity relationship in COFs. Their crystalline structure allows for the predictable generation of networks, making COFs highly adaptable heterogeneous catalysts with extensive chemical versatility. Although this review has focused on chemical modifications, it is essential to emphasize that the microstructural ordering of the material is crucial for its application. Controlling the morphology resulting from the synthesis of COFs is one of the most critical parameters for their future applications. Typically, these materials can form aggregates in various dimensionalities, including 0D, 1D, 2D, and 3D crystals, as detailed by Sasmal et al.102 These aggregates can be synthesized using top-down strategies, such as exfoliation,103 which involves starting with a larger crystal and reducing its size. Alternatively, bottom-up approaches can be employed, where molecular monomers are assembled directly into the desired nanostructure, modulating the morphology through the selection of the monomers.104 In addition to modulating the microstructure through aggregation control, macrostructure control can also be achieved. This approach presents a significant opportunity for the application of COFs in reactor design, enabling the selection and shaping of the macroscopic structure of the resulting solids to meet specific requirements. For example, 3D printing of pure COFs can be used to create aerogels with specific shapes tailored for particular applications.105 The so-called material processing emerges as a crucial factor in obtaining practical materials for industrial applications. Transforming COFs into pellets,106 gels,107 aerogels,108,109 or membranes110 enhances their value in terms of recyclability, ease of handling, and textural properties, including the generation of microporous structures. This advancement could significantly promote the widespread adoption of COFs as leading catalysts for organic synthesis.
Despite all the advancements covered in this review, numerous improvements still lie ahead. In the field of catalysis, general advances with COFs can be specifically applied to organic catalysis. Due to their structural features, particularly the precise atomic-level design of COFs, they exhibit remarkable selectivity for specific chemical reactions despite being heterogeneous catalysts. This makes them ideal for creating reactor beds in columns for continuous reactions. While this approach has been used with MOFs,111 it remains relatively unexplored with COFs, presenting a promising avenue for future research and application in terms of recyclability, sustainability and efficiency.
Another important aspect we have covered in the review is the structural characterization at an atomic level of the network, which is one of the key points that differentiates most COFs from other organic polymers. The design of COFs becomes increasingly intricate, and advancements in characterization techniques must keep pace. Electron single-crystal diffraction emerges as a promising tool for structural analysis in our field.112 Not only does it enable the examination of material morphology, but it also offers atomic-level resolution of the network. Though still evolving, electron diffraction has already resolved some structures. For COFs used in organic synthesis, investigating active sites would facilitate a comprehensive analysis of the structure–activity relationship in these systems.
The discovery of novel linkages for COF crystallization enhances the stability of these networks against solvents and thermal conditions, expanding the range of reactions they can catalyze. Linkages like dioxine or cyanovinylene-based networks hold potential to revolutionize COF applications in organic catalysis. Particularly, networks built on sp2 carbon could mark a significant leap forward in producing fully conjugated materials for catalysing photochemical processes. In this context, the development of single crystals with highly stable bonds could truly revolutionize the field.113
While significant progress has been made, numerous opportunities for advancing COFs in catalysis still lie ahead, particularly in areas like continuous flow reactions, structural characterization, and the development of highly stable linkages for photochemical catalysis. The future of COFs in organic synthesis looks promising, with challenges to be met by the collective effort of the research community.
Abbreviations
| 2,2′-BPyDA | 2,2′-Bipyridyl-5,5-diamine |
| 2,2′-BPyDCA | 2,2′-Bipyridyl-5,5-dialdehyde |
| A2B2-Por | 4′,4′′′-(10,20-Bis(4-aminophenyl)porphyrin-5,15-diyl)bis-(([1,1′-biphenyl]-4-carbaldehyde)) |
| APTU | 1,3-Bis(4-aminophenyl)thiourea |
| APU | 1,3-Bis(4-aminophenyl)urea |
| BDA | Benzene-1,4-dicarboxaldehyde |
| BGL | β-Glucosidase |
| BPTA | 2,5-Bis(2-propynyloxy)terephthalaldehyde |
| Bpy-PdCl2 | (2,2′-Bipyridine)dichloropalladium(II) |
| BTCA | 1,3,5-Benzenetricarbaldehyde |
| CBH | Cellobiohydrolase |
| COFs | Covalent organic framework |
| DAAn | 2,6-Diaminoanthracene |
| DFP | Pyridine-2,6-dicarbaldehyde |
| DHIP | 6,7-Dihydro-5H-pyrrolo[1,2-a]imidazole |
| DHNDA | 2,6-Dihydroxynaphthalene-1,5-dicarbaldehyde |
| DHTA | 2,5-Dihydroxyterephthalaldehyde |
| DMTA | 2,5-Dimethoxyterephthalaldehyde |
| EG | Endoglucanase |
| HBTCA | 2-Hydroxobenzenetricarbaldeheyde |
| H2TAPP | 5,10,15,20-Tetra(p-amino-phenyl)porphyrin |
| HTD | 2-Hydroxyl tribenzaldehyde |
| MA | Melamine |
| MOF | Metal organic framework |
| NaTFA | Sodium trifluoroacetate |
| NHC–M | N-Heterocyclic carbene metal complex |
| NIS | N-Iodosuccinimide |
| NP | Nanoparticle |
| PA | Phenylacetylene |
| PDA | 1,4-Diaminobenzene |
| PhenDBA | 4,4′-(1,10-Phenanthroline-3,8-diyl)dibenzaldehyde |
| POM | Polyoxometalates |
| PyTTA | 4,4′,4′′,4′′′-(Pyrene-1,3,6,8-tetrayl)tetraaniline |
| BINOL-DA | 6,6′-Dichloro-2,2′-diethoxy-1,1′-binaphthyl-4,4′-dialdehyde |
| (S)-CIK | (S)-2-(2-Chlorophenyl)-2-(6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)acetonitrile |
| (S,S)-Pybox | (S,S)-2,6-Bis(4-phenyl-2-oxazolinyl)pyridine |
| SC-CO2 | Supercritical CO2 |
| ss-NMR | Solid-state nuclear magnetic resonance |
| TAA | 1,3,5,7-Tetraaminoadamantane |
| TABTA | N1,N3,N5-Tris-(4-aminophenyl)benzene-1,3,5-tricarboxamide |
| TAM | Tetra(p-aminophenyl)methane |
| TAPB | 1,3,5-Tri(4-aminophenyl)-benzene |
| TAPP | 5,10,15,20-Tetra(4-aminophenyl)porphyrin |
| TAPPy | 2,4,6-Tris(4-aminophenyl)pyridine |
| TAPT | 1,3,5-Tris(4-aminophenyl)triazine |
| TFPB | 1,3,5-Tri(4-formylphenyl)benzene |
| TFPP | Tris-(4-formylphenyl)phosphine |
| TFPPy | 1,3,6,8-Tetrakis(p-formylphenyl)pyrene |
| TP | 1,3,5-Triformylphloroglucinol |
| TTHB | Tris(salicylaldehyde)benzene |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Writing – original draft: I. R. M., M. A. S. and M. A. Writing – review & editing: I. R. M, M. A. S., M.A., A. E. P. P., M. D. and F. Z. Conceptualization: I. R. M., M. D. and F. Z. Project administration: I. R. M., M. D. and F. Z. Funding acquisition: M. D. and F. Z.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
M. A. S., M. A. and M. D., gratefully acknowledge the Iranian National Science Foundation (INSF) for their financial (Grant No. 4026090) support and the Research Affairs Division of the Isfahan University of Technology (IUT), for partial financial support. I. R.-M. and F. Z., acknowledge financial support from the Comunidad de Madrid and the Spanish State through the Recovery, Transformation and Resilience Plan [“Materiales Disruptivos Bidimensionales (2D)” (MAD2D-CM) (UAM2)-MRR Materiales Avanzados], and the European Union through the Next Generation EU funds. F. Z. acknowledges the grants PDC2022-133498-I00, TED2021-129886B-C42, and PID2022-138908NB-C31. F. Z. also acknowledges support from the European Innovation Council under grant agreement 101047081 (EVA). A. E. P.-P. acknowledges the grants PID2021-123839OB-I00, RYC2018-024328-I, and CNS2022-135261 funded by MICIU/AEI/10.13039/501100011033. F. Z. and A. E. P.-P. acknowledge support from the Spanish Ministry of Science, Innovation and Universities through the “María de Maeztu” Programme for Units of Excellence in R&D (CEX2023-001316 M). I. R.-M. also acknowledges Juan de la Cierva programme JDC2023-051645-I, funded by MCIU/AEI/10.13039/501100011033 and by the FSE+.Notes and references
- L. Yao, A. Rodríguez-Camargo, M. Xia, D. Mücke, R. Guntermann, Y. Liu, L. Grunenberg, A. Jiménez-Solano, S. T. Emmerling, V. Duppel, K. Sivula, T. Bein, H. Qi, U. Kaiser, M. Grätzel and B. V. Lotsch, J. Am. Chem. Soc., 2022, 144, 10291–10300 CrossRef CAS PubMed.
- Z. Meng and K. A. Mirica, Chem. Soc. Rev., 2021, 50, 13498–13558 RSC.
- P. Albacete, A. López-Moreno, S. Mena-Hernando, A. E. Platero-Prats, E. M. Pérez and F. Zamora, Chem. Commun., 2019, 55, 1382–1385 RSC.
- J. Guo and D. Jiang, ACS Cent. Sci., 2020, 6, 869–879 CrossRef CAS PubMed.
- H. Vardhan, G. Verma, S. Ramani, A. Nafady, A. M. Al-Enizi, Y. Pan, Z. Yang, H. Yang and S. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 3070–3079 CrossRef CAS PubMed.
- F. Haase and B. V. Lotsch, Chem. Soc. Rev., 2020, 49, 8469–8500 RSC.
- J. Yu, M. Gaedke and F. Schaufelberger, Eur. J. Org Chem., 2023, 26, e202201130 CrossRef CAS.
- T. Jiao, G. Wu, Y. Zhang, L. Shen, Y. Lei, C.-Y. Wang, A. C. Fahrenbach and H. Li, Angew. Chem., Int. Ed., 2020, 59, 18350–18367 CrossRef CAS PubMed.
- Y. Chen, H. Tang, H. Chen and H. Li, Acc. Chem. Res., 2023, 56, 2838–2850 CrossRef CAS PubMed.
- J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- Y. Li, W. Chen, G. Xing, D. Jiang and L. Chen, Chem. Soc. Rev., 2020, 49, 2852–2868 RSC.
- F. Haase and B. V. Lotsch, Chem. Soc. Rev., 2020, 49, 8469–8500 RSC.
- J. Martín-Illán, D. Rodríguez-San-Miguel, C. Franco, I. Imaz, D. Maspoch, J. Puigmartí-Luis and F. Zamora, Chem. Commun., 2020, 56, 6704–6707 RSC.
- S. Karak, S. Kandambeth, B. P. Biswal, H. S. Sasmal, S. Kumar, P. Pachfule and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 1856–1862 CrossRef CAS PubMed.
- T. Kundu, J. Wang, Y. Cheng, Y. Du, Y. Qian, G. Liu and D. Zhao, Dalton Trans., 2018, 47, 13824–13829 RSC.
- W. Zhao, P. Yan, B. Li, M. Bahri, L. Liu, X. Zhou, R. Clowes, N. D. Browning, Y. Wu, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9902–9909 CrossRef CAS PubMed.
- P. Martinez-Bulit, A. Sorrenti, D. R. S. Miguel, M. Mattera, Y. Belce, Y. Xia, S. Ma, M. H. Huang, S. Pané and J. Puigmartí-Luis, Chem. Eng. J., 2022, 435, 135117 CrossRef CAS.
- Z. Shan, M. Wu, D. Zhu, X. Wu, K. Zhang, R. Verduzco and G. Zhang, J. Am. Chem. Soc., 2022, 144(13), 5728–5733 CrossRef CAS PubMed.
- W. Liu, L. Gong, Z. Liu, Y. Jin, H. Pan, X. Yang, B. Yu, N. Li, D. Qi, K. Wang, H. Wang and J. Jiang, J. Am. Chem. Soc., 2022, 144, 17209–17218 CrossRef CAS PubMed.
- M. Lu, S.-B. Zhang, R.-H. Li, L.-Z. Dong, M.-Y. Yang, P. Huang, Y.-F. Liu, Z.-H. Li, H. Zhang, M. Zhang, S.-L. Li and Y.-Q. Lan, J. Am. Chem. Soc., 2024, 146, 25832–25840 CrossRef CAS PubMed.
- R. K. Sharma, P. Yadav, M. Yadav, R. Gupta, P. Rana, A. Srivastava, R. Zbořil, R. S. Varma, M. Antonietti and M. B. Gawande, Mater. Horiz., 2020, 7, 411–454 RSC.
- T.-Y. Yu, Q. Niu, Y. Chen, M. Lu, M. Zhang, J.-W. Shi, J. Liu, Y. Yan, S.-L. Li and Y.-Q. Lan, J. Am. Chem. Soc., 2023, 145, 8860–8870 CrossRef CAS PubMed.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- A. Jiménez-Almarza, A. López-Magano, L. Marzo, S. Cabrera, R. Mas-Ballesté and J. Alemán, ChemCatChem, 2019, 11, 4916–4922 CrossRef.
- A. Basak, S. Karak and R. Banerjee, J. Am. Chem. Soc., 2023, 145, 7592–7599 CrossRef CAS PubMed.
- Z. Liu, Z. Chen, H. Tong, M. Ji and W. Chu, Green Chem., 2023, 25, 5195 RSC.
- S. Dalapati, M. Addicoat, S. Jin, T. Sakurai, J. Gao, H. Xu, S. Irle, S. Seki and D. Jiang, Nat. Commun., 2015, 6, 7786 CrossRef CAS PubMed.
- Y. Wu, H. Xu, X. Chen, J. Gao and D. Jiang, Chem. Commun., 2015, 51, 10096–10098 RSC.
- W. Hao, D. Chen, Y. Li, Z. Yang, G. Xing, J. Li and L. Chen, Chem. Mater., 2019, 31, 8100–8105 CrossRef CAS.
- R. Chen, J. L. Shi, Y. Ma, G. Lin, X. Lang and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 6430–6434 CrossRef CAS PubMed.
- W. Qiu, Y. He, L. Li, Z. Liu, S. Zhong and Y. Yu, Langmuir, 2021, 37, 11535–11543 CrossRef CAS PubMed.
- M. Roy, B. Mishra, S. Maji, A. Sinha, S. Dutta, S. Mondal, A. Banerjee, P. Pachfule and D. Adhikari, Angew. Chem., Int. Ed., 2024, 63, e202410300 CrossRef CAS PubMed.
- A. Jati, A. K. Mahato, D. Chanda, P. Kumar, R. Banerjee and B. Maji, J. Am. Chem. Soc., 2024, 146, 23923–23932 CrossRef CAS PubMed.
- Q. Fang, S. Gu, J. Zheng, Z. Zhuang, S. Qiu and Y. Yan, Angew. Chem., Int. Ed., 2014, 126, 2922–2926 CrossRef.
- H. Liu, J. Chu, Z. Yin, X. Cai, L. Zhuang and H. Deng, Chem, 2018, 4, 1696–1709 CAS.
- J. Hu, F. Zanca, G. J. McManus, I. A. Riha, H. G. T. Nguyen, W. Shirley, C. G. Borcik, B. J. Wylie, M. Benamara, R. D. Van Zee, P. Z. Moghadam and H. Beyzavi, ACS Appl. Mater. Interfaces, 2021, 13, 21740–21747 CrossRef CAS PubMed.
- M. Traxler, S. Gisbertz, P. Pachfule, J. Schmidt, J. Roeser, S. Reischauer, J. Rabeah, B. Pieber and A. Thomas, Angew. Chem., Int. Ed., 2022, 61, e202117738 CrossRef CAS PubMed.
- Y. Fan, D. W. Kang, S. Labalme, J. Li and W. Lin, Angew. Chem., Int. Ed., 2023, 62, e202218908 CrossRef CAS PubMed.
- Y. Fan, D. W. Kang, S. Labalme, J. Li and W. Lin, J. Am. Chem. Soc., 2023, 145, 25074–25079 CrossRef CAS PubMed.
- J. Hu, F. Zanca, P. Lambe, M. Tsuji, S. Wijeweera, S. Todisco, P. Mastrorilli, W. Shirley, M. Benamara, P. Z. Moghadam and M. H. Beyzavi, ACS Appl. Mater. Interfaces, 2020, 12, 29212–29217 CrossRef CAS PubMed.
- Y. Li, W. Chen, R. Gao, Z. Zhao, T. Zhang, G. Xing and L. Chen, Chem. Commun., 2019, 55, 14538–14541 RSC.
- J. Zhang, X. Han, X. Wu, Y. Liu and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8277–8285 CrossRef CAS.
- J. Zhang, X. Han, X. Wu, Y. Liu and Y. Cui, ACS Sustain. Chem. Eng., 2019, 7, 5065–5071 CrossRef CAS.
- Y. Yusran, J. Xing, Q. Lin, G. Wu, W.-C. Peng, Y. Wu, T. Su, L. Yang, L. Zhang, Q. Li, H. Wang, Z.-T. Li and D.-W. Zhang, Small, 2023, 19, 2303069 CrossRef CAS PubMed.
- S. T. Emmerling, R. Schuldt, S. Bette, L. Yao, R. E. Dinnebier, J. Kästner and B. V. Lotsch, J. Am. Chem. Soc., 2021, 143, 15711–15722 CrossRef CAS PubMed.
- H. S. Xu, S. Y. Ding, W. K. An, H. Wu and W. Wang, J. Am. Chem. Soc., 2016, 138, 11489–11492 CrossRef CAS.
- L. K. Wang, J. J. Zhou, Y. B. Lan, S. Y. Ding, W. Yu and W. Wang, Angew. Chem., Int. Ed., 2019, 58, 9543–9547 CrossRef.
- Y. B. Dong, J. C. Wang, X. Kan, J. Y. Shang and H. Qiao, J. Am. Chem. Soc., 2020, 142, 16915–16920 CrossRef PubMed.
- B. J. Yao, W. X. Wu, L. G. Ding and Y. B. Dong, J. Org. Chem., 2021, 86, 3024–3032 CrossRef CAS PubMed.
- H. Chen, W. Liu, C. Liu, J. Sun, L. Bourda, R. Morent, N. De Geytere, R. Van Deun, K. Van Hecke, K. Leus and P. Van Der Voort, Appl. Catal., B, 2022, 319, 121920 CrossRef CAS.
- J. L. Segura, S. Royuela and M. Mar Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- Y. Zhang, H. Hu, J. Ju, Q. Yan, V. Arumugam, X. Jing, H. Cai and Y. Gao, Chin. J. Catal., 2020, 41, 485–493 CrossRef CAS.
- H. He, Q. Q. Zhu, W. W. Zhang, H. W. Zhang, J. Chen, C. P. Li and M. Du, ChemCatChem, 2020, 12, 5192–5199 CrossRef CAS.
- M. Chen, J. Zhang, C. Liu, H. Li, H. Yang, Y. Feng and B. Zhang, Org. Lett., 2021, 23, 1748–1752 CrossRef CAS PubMed.
- W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572–2590 CrossRef CAS.
- H. Xu, X. Chen, J. Gao, J. Lin, M. Addicoat, S. Irle and D. Jiang, Chem. Commun., 2014, 50, 1292–1294 RSC.
- H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905–912 CrossRef CAS PubMed.
- S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- R. S. B. Gonçalves, A. B. V. Deoliveira, H. C. Sindra, B. S. Archanjo, M. E. Mendoza, L. S. A. Carneiro, C. D. Buarque and P. M. Esteves, ChemCatChem, 2016, 8, 743–750 CrossRef.
- Y. Hou, X. Zhang, J. Sun, S. Lin, D. Qi, R. Hong, D. Li, X. Xiao and J. Jiang, Microporous Mesoporous Mater., 2015, 214, 108–114 CrossRef CAS.
- I. Romero-Muñiz, A. Mavrandonakis, P. Albacete, A. Vega, V. Briois, F. Zamora and A. E. Platero-Prats, Angew. Chem., Int. Ed., 2020, 59, 13013–13020 Search PubMed.
- I. Romero-Muñiz, P. Albacete, A. E. Platero-Prats and F. Zamora, ACS Appl. Mater. Interfaces, 2021, 13, 54106–54112 CrossRef PubMed.
- X. Kan, J.-C. Wang, J.-L. Kan, J.-Y. Shang, H. Qiao and Y.-B. Dong, Inorg. Chem., 2021, 60, 3393–3400 CrossRef CAS PubMed.
- Q. Sun, B. Aguila and S. Ma, Mater. Chem. Front., 2017, 1, 1310–1316 RSC.
- H. Vardhan, Y. Pan, Z. Yang, G. Verma, A. Nafady, A. M. Al-Enizi, T. M. Alotaibi, O. A. Almaghrabi and S. Ma, APL Mater., 2019, 7, 101111 CrossRef.
- F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS PubMed.
- A. Jati, K. Dey, M. Nurhuda, M. A. Addicoat, R. Banerjee and B. Maji, J. Am. Chem. Soc., 2022, 144, 7822–7833 CrossRef CAS PubMed.
- W. Leng, R. Ge, B. Dong, C. Wang and Y. Gao, RSC Adv., 2016, 6, 37403–37406 RSC.
- W. Leng, Y. Peng, J. Zhang, H. Lu, X. Feng, R. Ge, B. Dong, B. Wang, X. Hu and Y. Gao, Chem.–Eur. J., 2016, 22, 9087–9091 CrossRef CAS PubMed.
- P. M. Heintz, B. P. Schumacher, M. Chen, W. Huang and L. M. Stanley, ChemCatChem, 2019, 11, 4286–4290 CrossRef CAS.
- M. Wen, S. Lu, C. Fan, K. Shen, S. Lin and Q. Pan, Appl. Organomet. Chem., 2021, 35, e6263 CrossRef CAS.
- A. Jati, S. Dam, S. Kumar, K. Kumar and B. Maji, Chem. Sci., 2023, 14, 8624–8634 RSC.
- Y. Li, K. Zuo, T. Gao, J. Wu, X. Su, C. Zeng, H. Xu, H. Hu, X. Zhang and Y. Gao, RSC Adv., 2022, 12, 4874–4882 RSC.
- A. López-Magano, B. Ortín-Rubio, I. Imaz, D. Maspoch, J. Alemán and R. Mas-Ballesté, ACS Catal., 2021, 11, 12344–12354 CrossRef PubMed.
- T. Gao, X. Su, H. Xu, H. Hu, C. Zeng and Y. Gao, ChemistrySelect, 2020, 5, 15010–15014 CrossRef CAS.
- J. Han, X. Sun, X. Wang, Q. Wang, S. Hou, X. Song, Y. Wei, R. Wang and W. Ji, Org. Lett., 2020, 22, 1480–1484 CrossRef CAS PubMed.
- H. Vardhan, L. Hou, E. Yee, A. Nafady, M. A. Al-Abdrabalnabi, A. M. Al-Enizi, Y. Pan, Z. Yang and S. Ma, ACS Sustain. Chem. Eng., 2019, 7, 4878–4888 CrossRef CAS.
- H. Li, X. Feng, P. Shao, J. Chen, C. Li, S. Jayakumar and Q. Yang, J. Mater. Chem. A, 2019, 7, 5482–5492 RSC.
- X. Han, Q. Xia, J. Huang, Y. Liu, C. Tan and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8693–8697 CrossRef CAS PubMed.
- J. Yang, Y. Wu, X. Wu, W. Liu, Y. Wang and J. Wang, Green Chem., 2019, 21, 5267–5273 RSC.
- Y. Li, Y. Dong, J. L. Kan, X. Wu and Y. B. Dong, Org. Lett., 2020, 22, 7363–7368 CrossRef CAS PubMed.
- L. Grunenberg, G. Savasci, M. W. Terban, V. Duppel, I. Moudrakovski, M. Etter, R. E. Dinnebier, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2021, 143, 3430–3438 CrossRef CAS PubMed.
- Q. Yan, H. Xu, H. Hu, S. Wang, C. Zeng, Y. Gao, H. Xu and X. Jing, RSC Adv., 2020, 10, 17396–17403 RSC.
- Y. Li, J. Zhang, K. Zuo, Z. Li, Y. Wang, H. Hu, C. Zeng, H. Xu, B. Wang and Y. Gao, Catalysts, 2021, 11, 1133 CrossRef CAS.
- H. C. Ma, G. J. Chen, F. Huang and Y. Bin Dong, J. Am. Chem. Soc., 2020, 142, 12574–12578 CrossRef CAS PubMed.
- A. Corma and H. Garci, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- N. Nouruzi, M. Dinari, B. Gholipour, M. Afshari and S. Rostamnia, ACS Appl. Nano Mater., 2022, 5, 6241–6248 CrossRef CAS.
- S. Ghosh, T. S. Khan, A. Ghosh, A. H. Chowdhury, M. A. Haider, A. Khan and S. M. Islam, ACS Sustain. Chem. Eng., 2020, 8, 5495–5513 CrossRef CAS.
- R. Tao, X. Shen, Y. Hu, K. Kang, Y. Zheng, S. Luo, S. Yang, W. Li, S. Lu, Y. Jin, L. Qiu and W. Zhang, Small, 2020, 16, 1906005 CrossRef CAS PubMed.
- G. J. Chen, X. B. Li, C. C. Zhao, H. C. Ma, J. L. Kan, Y. Bin Xin, C. X. Chen and Y. Bin Dong, Inorg. Chem., 2018, 57, 2678–2685 CrossRef CAS PubMed.
- M. Guo, X. Guan, Q. Meng, M.-L. Gao, Q. Li and H.-L. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202410097 CrossRef CAS PubMed.
- M. Guo, Q. Meng, W. Chen, Z. Meng, M.-L. Gao, Q. Li, X. Duan and H.-L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202305212 CrossRef CAS PubMed.
- M. Bhadra, H. S. Sasmal, A. Basu, S. P. Midya, S. Kandambeth, P. Pachfule, E. Balaraman and R. Banerjee, ACS Appl. Mater. Interfaces, 2017, 9, 13785–13792 CrossRef CAS PubMed.
- N. Brown, Q. Zhang, Z. Alsudairy, C. Dun, Y. Nailwal, A. Campbell, C. Harrod, L. Chen, S. Williams, J. J. Urban, Y. Liu and X. Li, ACS Sustainable Chem. Eng., 2024, 12, 13535–13543 CrossRef CAS.
- S. K. Das, B. K. Chandra, R. A. Molla, M. Sengupta, S. M. Islam, A. Majee and A. Bhaumik, Mol. Catal., 2020, 480, 110650 CrossRef CAS.
- Y. Zhang, D. H. Yang, S. Qiao and B. H. Han, Langmuir, 2021, 37, 10330–10339 CrossRef CAS PubMed.
- C. Wang and K. Liao, ACS Appl. Mater. Interfaces, 2021, 13, 56752–56776 CrossRef CAS PubMed.
- S. Paul, M. Gupta, K. Dey, A. K. Mahato, S. Bag, A. Torris, E. B. Gowd, H. Sajid, M. A. Addicoat, S. Datta and R. Banerjee, Chem. Sci., 2023, 14, 6643–6653 RSC.
- S. Karak, K. Dey, A. Torris, A. Halder, S. Bera, F. Kanheerampockil and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 7572–7581 CrossRef CAS PubMed.
- L. Ran, Y. Chen, Y. Zhu, H. Cai, H. Pang, D. Yan, Y. Xiang and H. Teng, Angew. Chem., Int. Ed., 2024, 63, e202319732 CrossRef CAS PubMed.
- H. S. Sasmal, A. K. Mahato, P. Majumder and R. Banerjee, J. Am. Chem. Soc., 2022, 144(26), 11482–11498 CrossRef CAS PubMed.
- D. W. Burke, C. Sun, I. Castano, N. C. Flanders, A. M. Evans, E. Vitaku, D. C. McLeod, R. H. Lambeth, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Angew. Chem., Int. Ed., 2020, 59, 5165–5171 CrossRef CAS PubMed.
- K. Koner, A. Sadhukhan, S. Karak, H. S. Sasmal, Y. Ogaeri, Y. Nishiyama, S. Zhao, M. Položij, A. Kuc, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2023, 145, 14475–14483 CrossRef CAS PubMed.
- S. Royuela, S. Sevim, G. Hernanz, D. Rodríguez-San-Miguel, P. Fischer, C. Franco, S. Pané, J. Puigmartí-Luis and F. Zamora, Adv. Funct. Mater., 2024, 34, 2314634 CrossRef CAS.
- S. Karak, S. Kandambeth, B. P. Biswal, H. S. Sasmal, S. Kumar, P. Pachfule and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 1856–1862 CrossRef CAS PubMed.
- C. Arqueros, L. Welte, C. Montoro and F. Zamora, J. Mater. Chem. A, 2024, 12, 20121–20128 RSC.
- Y. Li, J.-M. Wang, J.-L. Kan, F. Li, Y. Dong and Y.-B. Dong, Inorg. Chem., 2022, 61, 2455–2462 CrossRef CAS PubMed.
- J. Martín-Illán, L. Sierra, A. Guillem-Navajas, J. A. Suárez, S. Royuela, D. Rodríguez-San-Miguel, D. Maspoch, P. Ocón and F. Zamora, Adv. Funct. Mater., 2024, 34, 2403567 CrossRef.
- H. S. Sasmal, A. Halder, S. Kunjattu H, K. Dey, A. Nadol, T. G. Ajithkumar, P. R. Bedadur and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 20371–20379 CrossRef CAS PubMed.
- P. Ji, X. Feng, P. Oliveres, Z. Li, A. Murakami, C. Wang and W. Lin, J. Am. Chem. Soc., 2019, 141, 14878–14888 CrossRef CAS PubMed.
- Z. Huang, E. S. Grape, J. Li, A. K. Inge and X. Zou, Coord. Chem. Rev., 2021, 427, 213583 CrossRef CAS.
- S. Li, S. Xu, E. Lin, T. Wang, H. Yang, J. Han, Y. Zhao, Q. Xue, P. Samorì, Z. Zhang and T. Zhang, Nat. Chem., 2025, 17, 226–232 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |