
Maximising efficacy in HER2-positive breast cancer: immunoliposomal co-delivery of miR155 inhibitor and paclitaxel for targeted therapy†
Ramesh
Chaudhari
 a,
Vishva
Patel
a,
Bharti
Malvi
b,
Superb K.
Misra
b and
Ashutosh
Kumar
*a
a,
Vishva
Patel
a,
Bharti
Malvi
b,
Superb K.
Misra
b and
Ashutosh
Kumar
*a
aBiological & Life Sciences, School of Arts & Sciences, Ahmedabad University, Central Campus, Navrangpura, Ahmedabad 380009, Gujarat, India. E-mail: ashutosh.kumar@ahduni.edu.in
bMaterials Engineering, Indian Institute of Technology Gandhinagar, Gujarat, India
First published on 4th December 2024
Abstract
Breast cancer, particularly the HER2 positive subtype, presents a formidable challenge in clinical oncology, necessitating innovative therapeutic strategies. Here, we present a novel immunoliposome-based formulation designed for targeted delivery of paclitaxel and miRNA inhibitors to HER2-positive breast cancer cells. Through a rigorous preclinical evaluation encompassing in vitro cellular studies and an in vivo tumor xenograft model, we demonstrate the formulation's remarkable efficacy in inhibiting cell proliferation, inducing apoptosis, and suppressing tumor growth. Histopathological assessments reveal a favourable safety profile with minimal adverse effects on normal tissues. Furthermore, the study unveils the synergistic interaction between paclitaxel and miRNA inhibitor within the formulation, offering a potential avenue for combination therapy. The novelty of the study lies in the development of a precise and targeted therapeutic approach tailored to HER2-positive breast cancer, addressing critical gaps in current treatment modalities. Our findings underscore this innovative formulation's clinical relevance and translational potential, paving the way for personalised and effective therapies in HER2-positive breast cancer management.
1. Introduction
Breast cancer is a prevalent malignancy characterised by uncontrolled growth of abnormal cells in the breast tissue.1 Breast cancer ranks among the most prevalent cancers in women globally.2,3 In 2022, the world saw 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 295720 new cases and 15.4% of breast cancer-related deaths.4 Drug resistance and metastasis, particularly to vital organs like the lymph nodes, bones, lungs, and liver, are the primary causes of mortality.5 Breast cancer remains a formidable challenge in the field of oncology, with a significant subset of cases characterised by overexpression of the human epidermal growth factor receptor 2 (HER2).6 HER2 overexpression is associated with aggressive tumor behaviour and poor prognosis.7 Trastuzumab (Herceptin), a monoclonal antibody (mAb) targeting HER2, has emerged as a cornerstone therapy,8 substantially improving outcomes for patients with HER2-positive breast cancer.9
295720 new cases and 15.4% of breast cancer-related deaths.4 Drug resistance and metastasis, particularly to vital organs like the lymph nodes, bones, lungs, and liver, are the primary causes of mortality.5 Breast cancer remains a formidable challenge in the field of oncology, with a significant subset of cases characterised by overexpression of the human epidermal growth factor receptor 2 (HER2).6 HER2 overexpression is associated with aggressive tumor behaviour and poor prognosis.7 Trastuzumab (Herceptin), a monoclonal antibody (mAb) targeting HER2, has emerged as a cornerstone therapy,8 substantially improving outcomes for patients with HER2-positive breast cancer.9
However, despite the advancements in targeted therapies like trastuzumab, the development of drug resistance and tumor recurrence remains a major clinical challenge.10 One of the mechanisms implicated in chemotherapy resistance, particularly to agents like paclitaxel, is the dysregulation of microRNAs (miRNAs), among which miR-155 has gained significant attention.11–14 MicroRNA-155 (miR-155) has been identified as an oncogenic miRNA upregulated in breast cancer, contributing to tumor progression and resistance to chemotherapy.12,15 Notably, miR-155 overexpression has been associated with paclitaxel resistance by regulating TP53INP1, a critical mediator of apoptotic signalling pathways.14
In addition to microRNA dysregulation, drug resistance is significantly influenced by efflux pumps such as ATP-binding cassette (ABC) transporters, particularly P-glycoprotein (P-gp).16,17 These efflux pumps actively remove chemotherapeutic agents like paclitaxel from cells, leading to reduced intracellular drug concentrations and diminished therapeutic efficacy.18 Our immunoliposomal system is designed to address this challenge. By encapsulating paclitaxel within liposomes, the drug is shielded from immediate recognition and efflux by P-gp.19,20 Moreover, the targeting mechanism mediated by anti-HER2 antibodies ensures that liposomes bind specifically to HER2-overexpressing cells, facilitating endocytosis and enhancing drug retention within the target cells.21
In the context of overcoming drug resistance and improving therapeutic outcomes, immunoliposomes offer a promising strategy by combining the targeting capabilities of monoclonal antibodies with the drug delivery efficiency of liposomes.22 Originally developed in the 1980s, immunoliposomes have evolved to selectively deliver drugs to tumor cells by conjugating antibodies to the surface of liposomes, thereby enhancing the precision of drug delivery.22 Several immunoliposome formulations are currently in clinical trials, including trastuzumab-targeted liposomes encapsulating doxorubicin and paclitaxel, which aim to overcome drug resistance and enhance drug accumulation at the tumor site.22
To address the dual challenge of HER2-positive breast cancer and paclitaxel resistance, we developed a novel therapeutic approach utilising anti-HER2 immunoliposomes. These immunoliposomes are engineered by conjugating trastuzumab mAb onto liposomes via thiolation, enabling specific targeting of HER2-overexpressing cancer cells.
Additionally, it is important to consider the potential for allergic reactions to paclitaxel, often attributed to the use of excipients such as Cremophor EL in conventional formulations.23,24 These reactions can manifest as anaphylaxis or hypersensitivity, presenting significant challenges during treatment.25 Encapsulation of paclitaxel within the liposomal wall is anticipated to mitigate these allergic responses. By sequestering paclitaxel within liposomes, the formulation reduces the risk of direct contact between the drug and immune cells, thereby minimizing hypersensitivity reactions while maintaining therapeutic efficacy.24 Within the liposome, paclitaxel, a lipophilic chemotherapeutic agent, is encapsulated in the lipid bilayer, while a miR-155 inhibitor is entrapped within the liposome core.
The rationale for combining HER2-targeted therapy with a miR-155 inhibitor and paclitaxel chemotherapy in a dual payload delivery system is rooted in the quest for enhanced efficacy and reduced adverse effects in breast cancer management. This approach is geared explicitly towards minimising harm to healthy cells while maximising the therapeutic impact on cancerous cells. By targeting HER2-positive cancer cells with trastuzumab-conjugated liposomes, we can deliver therapeutic payloads directly to the tumor site, maximizing treatment effectiveness. Simultaneously, inhibiting miR-155 addresses a common mechanism of paclitaxel resistance, potentially overcoming this hurdle and allowing for lower doses of paclitaxel to induce cytotoxic effects. This combinatorial approach offers a multi-faceted attack on cancer cells, targeting multiple pathways involved in tumor growth and survival, thus minimising the likelihood of resistance development. Encapsulation of paclitaxel within liposomes not only reduces off-target effects and systemic toxicity but also exploits the enhanced permeability and retention effect for improved tumor penetration and accumulation. Overall, this strategy holds promise for optimising treatment outcomes and overcoming resistance mechanisms in HER2-positive breast cancer.
2. Materials and methods
2.1. Materials
Soy phosphatidylcholine (SPC), DSPE-PEG(2000) maleimide, cholesterol, paclitaxel, 2-iminothiolane hydrochloride (Traut's reagent), Sepharose™ CL-4B beads, chloroform, acetonitrile, negative control miRNA (CGGUACGAUCGCGGCGGGAUAUC), miR155 inhibitor miRNA (UUAAUGCUAAUCGUGAUAGGGGU), KBr, extruder filter support and polycarbonate membrane filters of 0.1 μM were purchased from Merck, Germany. USFDA-approved anti-Her2 antibodies were purchased from Biocon, India. PD-10 desalting columns were procured from GE Healthcare. Pierce™ BCA Protein Assay Kit, Milli Q water, loading dye, Roswell Park Memorial Institute medium (RPMI-1640), McCoy's 5A medium, sodium bicarbonate (NaHCO3), Gibco Antibiotic–Antimycotic solution, trypan blue, trypsin-EDTA, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethylsulfoxide (DMSO), FITC-annexin V, propidium iodide, PBS tablets, Maxima First Strand cDNA Synthesis Kit for RT-qPCR, PowerUp™ SYBR™ Green Master Mix for qPCR, TRIzol Reagent, and Triton-X100 were obtained from Thermo Fisher. Cyclosporine (Sandimmune) was obtained from Novartis, headquartered in Basel, Switzerland. Ketoconazole (Nizral) was sourced from Johnson & Johnson, based in New Brunswick, New Jersey, USA. Cyclophosphamide (Endoxan) was procured from Baxter, located in Halle, Germany. Ampoxin was purchased from Zydus, India.2.2. Formulation of paclitaxel-loaded miR155 inhibitor encapsulated liposomes
Liposomes were formulated using the conventional thin-film hydration method using SPC:Chol.:DSPE-PEG-melamide:drug in a molar ratio of 10:1:1:0.1. Initially, SPC, cholesterol, DSPE-PEG-melamide and paclitaxel drug were dissolved in chloroform and proceeded for solvent evaporation at 60 °C for 60 min using a rotary evaporator (evator). After forming a thin film, hydration was carried out with 10 mL sterile PBS (pH 7.4) containing 100 nM miR155 inhibitor, followed by 0.1 μM polycarbonate filter-based extrusion, and miR155 inhibitor was entrapped into the hydrophilic core. In contrast, the anti-neoplastic drug was entrapped in the hydrophobic bilayer of the liposomes.
2.3. Functionalisation of liposomes with trastuzumab antibody
The prepared liposome was functionalised with trastuzumab for target-specific delivery. Initially, trastuzumab antibodies were thiolated using Traut's reagent. For this, 1 mg trastuzumab was mixed in PBS buffer containing 5 mM EDTA (pH 8.0) and 18.8 μL of Traut's reagent was added (2 mg mL−1) (Traut's reagent:antibody 40:1, mol:mol) dropwise, under continuous stirring at 37 °C for 3 min. The Traut's reagent:antibody mixture was protected from light and kept at 37 °C for 1 hour. Thiolated antibodies were separated from unreacted Traut's reagent by the PD-10 desalting column. A total of 1 mL was passed through a 10 mL column, and 14 fractions of 1 mL were collected, each using PBS containing EDTA (pH 8.0). Protein estimation of each fraction was carried out using a BCA protein assay kit as per the manufacturer's instruction and combined with the fractions containing thiolated trastuzumab. Furthermore, thiolated trastuzumab was incubated with liposomes overnight at room temperature for immunoliposome formation. Formulated immune liposomes were purified using the Sepharose CL-4B column.
2.4. Characterisation of the formulated immunoliposome
The prepared formulation of immunoliposome was characterised for size and surface charge analysis by dynamic light scattering (DLS) Zetasizer Nano-ZS equipped with a 4.0 Mw, 633 nm laser (Model ZEN3600, Malvern Instruments). The samples were analysed three times at 25 °C. Infrared spectra of trastuzumab, thiolated trastuzumab, liposome and immunoliposome were obtained using a PerkinElmer FT-IR Spectrometer. The aqueous solution of the samples was dried and mixed with potassium bromide (KBr) to get a fine powder pressed onto the discs. All spectra were measured at a resolution of 1 cm−1 and over a wavelength range of 4000–400 cm−1. The morphology of the immunoliposomes was accessed using TEM (JEOL-1400 plus) at 120 kV, and samples were negatively stained using phosphotungstic acid on a 200 mesh carbon-coated copper grid. Bio-AFM (Bruker Nano wizard Sense) was used to analyse trastuzumab functionalisation on the liposomes.2.5. Encapsulation efficiency of paclitaxel and miR155 inhibitor
The loading efficiency of paclitaxel was determined using HPLC (Agilent 1220 Infinity II LC System) using acetonitrile:PBS (1:1) as a mobile phase after the overnight dialysis. Dialysis medium (PBS-pH 7.4) was mixed with acetonitrile in a 1:1 ratio and injected into the C18 HPLC column. Similarly, a standard plot was prepared for miRNA using Qubit (Invitrogen) and after overnight dialysis miRNA concentration in dialysis medium was estimated and % entrapment efficiency was calculated using the formula, drug/miRNA present in the formulation X 100/total drug/miRNA added into the system. For the release kinetics, free paclitaxel was removed via overnight dialysis and then transferred into a fresh dialysis medium. The sample was collected at multiple time points and analysed using HPLC.
2.6. Cell culture
The Her2 positive human breast cancer cell line, SKBR-3 and triple negative MDA-MB-231 cells were obtained from National Centre for Cell Sciences, Pune, India, cultured in McCoy's 5A and RPMI-1640 media respectively and supplemented with 10% FBS (heat inactivated), 0.2% sodium bicarbonate, and 10 mL L−1 antibiotic and antimycotic solution, at 37 °C under a humidified atmosphere of 5% CO2.2.7. Cellular internalisation of immunoliposome
The cellular uptake of immunoliposomes and targeting efficiency of immunoliposomes was evaluated by forming rhodamine-loaded immunoliposomes. SKBR-3 and MDA-MB-231 cells were cultured on Nunc Lab-Tek chamber slides (4 wells) and incubated with rhodamine-loaded immunoliposomes for 30 min. After incubation, the cells were washed with PBS, fixed using paraformaldehyde, and permeabilised using Triton X-100 and stained with fluorescein phalloidin and mounted in DAPI containing moviol. The prepared slides were analysed using a TCS SP8 (Leica) confocal microscope. Mean fluorescence intensity was measured using ImageJ software for relative quantification of uptake.2.8. Cell proliferation assay of the formulations
The cytotoxic effect of the formulation was determined by performing an MTT assay according to Mosmann et al.26 and with a slight modification with Chaudhari et al.27 In brief, the SKBR-3 cells (1 × 104 cells per well) in 100 μL of culture medium were seeded in 96-well plates and incubated for 24 h. The cells were then exposed to different concentrations of paclitaxel and miR155 inhibitor-loaded immunoliposome for 24, 48 and 72 h. Formulation interference with the assay reagents was also assessed using a cell-free system, where formulations alone were incubated with the assay reagents, including dye and buffers, and the absorption was monitored by spectroscopy. The results were assessed by measuring the absorbance of the end product at 595 nm wavelength. After accessing the concentration-dependent toxicity, we analysed the cytotoxic effect of all the formulation components in SKBR-3 and MDA-MB-231 cells.2.9. Apoptosis assay
Cells undergoing apoptosis after treatment were identified by staining with FITC-annexin V and PI according to the manufacturer's protocol (BD Biosciences, San Jose, CA, USA). Briefly, 3 × 105 cells per well were seeded in a 6 well culture plate for 24 h before treatment. The cells were treated with the formulation at different concentrations for 24 h. After removal of the treatment, the cells were harvested and washed with PBS, resuspended in 100 μL binding buffer containing 5 μL of FITC-annexin V and PI and kept at room temperature in the dark; after incubation, 400 μL of binding buffer was added to each sample and analysed using a flow cytometer (BD FACS Aria™ III Cell Sorter, BD Biosciences, CA).2.10. Wound healing (scratch) assay
A wound-healing assay was performed on SKBR-3 and MDA-MB-231 cells as described by Liang et al.28 Briefly, 3 × 105 cells per well were seeded in a 6-well culture and allowed to reach 100% confluency. Once the cells reached confluency, a straight wound was made with a 200 μL pipet tip and washed 3 times with PBS. In both the cell lines, wound healing potential was evaluated after treatment of 20 nM paclitaxel + 10.4 pM miR155 inhibitor loaded immunoliposomes with and without TGF-β stimulation. Images were captured at T-0 and T-48 hours, and the wound area was calculated using FIJI Image J software.2.11. Cell cycle analysis
The DNA content in the cell cycle was analysed with the help of flow cytometry as the method suggested by Patel et al.29,30 SKBR-3 cells at a density of 3 × 105 cells per well were seeded in a 6-well plate and treated with different formulation concentrations for 48 h. After the treatment, the cells were harvested and fixed in 70% ethanol (at −20 °C for 30 min). Cells were further lysed using 1 ml of 0.2% Triton X-100 in PBS at 4 °C for 30 min. The cells were then centrifuged and re-suspended in 500 μL of PBS comprising 20 μL RNase (10 mg mL−1) for 30 min at 37 °C. Finally, the cells were stained using 10 μL propidium iodide (1 mg mL−1) in 500 μL PBS for 10 to 15 min at 4 °C and analysed using FACS (BD FACS Aria™ III Cell Sorter, BD Biosciences, CA).2.12. Animals
Healthy C57 BL/6 mice were purchased from Zydus Research Centre, Ahmedabad, India. All the mice were kept in individually ventilated cages, with a relative humidity of 60 ± 5% and a temperature of 25 ± 2 °C. A 12:12 h light:dark cycle was also regulated for these animals. Balanced rodent food pellet and water was provided ad libitum. All experimental protocols were reviewed and accepted (LMCP/IAEC/22/0043) by the Institutional Animal Ethics Committee before the initiation of the experiment.
2.13. Tumor regression in an immunosuppressed tumor xenograft model
Immunosuppression was induced in 4–6 week old, healthy female mice (C57 BL/6) by administering 10 mg kg−1 ketoconazole by the oral route every day and 30 mg kg−1 cyclosporine by the intraperitoneal route every day for seven days. No treatment was given to the control group. All the animals were provided with autoclaved rodent food pellets and water ad libitum. Animals were given ampoxin (0.1 μg ml−1) by drinking water during the study. After completion of the study, haematology was carried out to determine the total white blood cells (WBC) and lymphocyte count to confirm immunosuppression. Cyclophosphamide was injected subcutaneously at a 60 mg kg−1 dose on day 3 and day 1 before cell injection. Blood samples were collected from all the animals from the retro-orbital sinus under isoflurane anaesthesia in a heparinised 1.5 mL microcentrifuge tube. Total WBC and lymphocyte counts were then performed in an automated haematology analyser. Hairs were removed by waxing from the abdomen region of each animal one day before the injection of 0.1 mL of cells (approximately 5 × 106 SKBR-3 cells) subcutaneously at the abdomen of the mice. Tumor growth was observed at the site of injection. Tumor volume was measured externally by Vernier calliper using the following formula: Tumor Volume (mm3) = (A) × (B2)/2, where A is the largest diameter (mm) and B the smallest (mm). Once the tumor volume reaches around 100 mm3 the mice were divided into different groups (n = 5) and treated with formulations (Table 1). At the end of the study, the animals’ tumours and major organs (i.e. liver, spleen, kidney, heart and lung) were excised from the animals and maintained in 10% neutral buffered formalin. The samples were cut into 5 μm sections and stained with haematoxylin and eosin for histopathological analysis.| Group | Mice/group | Formulation | Dose (IV) |
|---|---|---|---|
| 1 | 05 | Control | PBS (80 μL) |
| 2 | 05 | Immunosuppression | PBS (80 μL) |
| 3 | 05 | Tumor model control | PBS (80 μL) |
| 4 | 05 | Free neg. control miR | 11.22 μg kg−1 BW (80 μL) |
| 5 | 05 | Free PXl | 1.280 mg kg−1 BW (80 μL) |
| 6 | 05 | Free miR155 inhibitor | 11.22 μg kg−1 BW (80 μL) |
| 7 | 05 | Empty immunoliposome (IL) | IL- (80 μL) |
| 8 | 05 | Free neg. control miR IL | 11.22 μg kg−1 BW (80 μL) |
| 9 | 05 | miR155 inhibitor IL | 11.22 μg kg−1 BW (80 μL) |
| 10 | 05 | PXL IL | 1.280 mg kg−1 BW (80 μL) |
| 11 | 05 | PXL + miR155 inhibitor-IL (dose-1) | 0.426 mg kg−1 BW PXL + 3.74 μg kg−1 BW miR155 inhibitor (80 μL) |
| 12 | 05 | PXL + miR155 inhibitor-IL (dose-1) | 0.853 mg kg−1 BW PXL + 7.48 μg kg−1 BW miR155 inhibitor (80 μL) |
| 13 | 05 | PXL + miR155 inhibitor-IL (dose-1) | 1.280 mg kg−1 BW PXL + 11.22 μg kg−1 BW miR155 inhibitor (80 μL) |
2.14. Gene expression and protein analysis in harvested tumor tissues
Total RNA was extracted from these tissues using a FavorPrep™ Tissue total RNA Kit, with RNA yield and integrity assessed using the SYNERGY-HT multiwell plate reader (Bio-Tek, USA) and Gen5 software. One microgram of RNA from each sample was used for first-strand cDNA synthesis, performed with the Maxima First Strand cDNA Synthesis Kit (ThermoFisher Scientific, USA) as per the manufacturer's instructions. Real-time PCR was then conducted using gene-specific primers and the powerup™ SYBR® Green master mix (ThermoFisher Scientific, USA) on a QuantStudio 5 system (ThermoFisher Scientific, USA). GAPDH was utilised as an internal control, and gene expression levels were quantified by measuring the threshold cycle (Ct). Primer details are provided in the ESI,† Table S1.For protein analysis, the harvested tumor tissues were lysed in a buffer containing 1% NP-40, 0.1% SDS, 150 mM NaCl, 0.5% sodium deoxycholate, and 50 mM tris–HCl (pH 8) with a protease inhibitor cocktail (Sigma). Protein concentration was determined using the BCA assay kit (ThermoFisher Scientific, USA). Equal amounts of protein (30 μg) from control and treated groups were resolved on 8% SDS-PAGE gels and transferred to PVDF membranes by electroblotting. The membranes were dried, fixed, and blocked with skimmed milk, then incubated overnight at 4 °C with primary antibodies specific to TP53INP and GAPDH (Invitrogen). Following incubation with secondary antibodies, protein bands were visualised using chemiluminescence analysis in the ImageQuant LAS 500 software (GE Healthcare Biosciences, Sweden).
2.15. Statistical analysis
All the experiments were carried out in triplicate, and the results are presented as means ± standard error mean (SEM). ANOVA was used for statistical analysis followed by Dunnett's post hoc multiple comparison tests from Graph Prism-8.0. In all the cases, a p-value less than 0.05 was considered as statistically significant.3. Results and discussion
3.1. Synthesis of liposomes
The liposomes, which included paclitaxel-loaded and miR155 inhibitor-encapsulated variations, were prepared using the thin film hydration method. Their size, zeta potential, and PDI were evaluated using DLS. Different formulations were synthesised, such as empty liposomes, paclitaxel-loaded liposomes, miR155 inhibitor-loaded liposomes, empty immunoliposomes, paclitaxel-loaded immunoliposomes, miR155 inhibitor-loaded immunoliposomes, and paclitaxel combined with miR155 inhibitor-loaded immunoliposomes (Table 2). The size of the empty liposomes was measured at approximately 104 ± 0.5 nm, while paclitaxel and miR155 inhibitor-loaded liposomes exhibited 98 ± 1 nm and 119.5 ± 1.2 nm, respectively. The variation in size was attributed to differences in lipid bilayer compactness for paclitaxel and the presence of a hydrophilic core for the miR155 inhibitor. Similar size trends were observed in the immunoliposome formulations. Additionally, all formulations displayed favourable zeta potential values and low PDIs.| S. no. | Formulation | Hydrodynamic size (nm) | Zeta potential (mV) | PDI | |
|---|---|---|---|---|---|
| 1 | Empty liposome (EL) |

|
104 ± 0.5 | −44.66 ± 4.5 | 0.126 ± 0.003 |
| 2 | Paclitaxel-loaded liposome (PXL-L) |
|
98.4 ± 1.0 | −32.43 ± 6.6 | 0.111 ± 0.008 |
| 3 | miR155 inhibitor loaded liposome (miR155-L) |
|
119.5 ± 1.2 | −21.56 ± 0.4 | 0.082 ± 0.008 |
| 4 | Empty immunoliposome (E-IL) |
|
122 ± 2.3 | −32.23 ± 1.2 | 0.055 ± 0.016 |
| 5 | Paclitaxel loaded immunoliposome (PXL-IL) |
|
119.1 ± 0.6 | −20.43 ± 2.00 | 0.070 ± 0.006 |
| 6 | miR155 loaded immunoliposome (miR155-IL) |
|
143.4 ± 0.3 | −22.36 ± 1.8 | 0.056 ± 0.019 |
| 7 | Paclitaxel and miR155 loaded immunoliposome (PXL + miR155-IL) |
|
135.6 ± 1.5 | −17.6 ± 0.9 | 0.074 ± 0.005 |
3.2. Thiolation of trastuzumab
Thiolation of monoclonal antibodies trastuzumab was carried out via Traut's reagent, and thiolated antibodies were purified using a PD-10 desalting column. Traut's reagent present in excess was removed, fractions having thiolated antibodies were collected, and protein concentration was estimated using a BCA protein assay kit. Furthermore, FTIR analysis confirmed successful thiolation by comparing thiolated and un-thiolated antibodies. The presence of the –SH group in thiolated antibody fractions confirms the successful addition of a thiol group on the antibody (ESI,† Fig. S1).3.3. Conjugation of thiolated antibodies on the liposomes
In synthesising immunoliposomes, a targeted approach is achieved through noncovalent coupling, wherein biotinylated antibodies bind to avidin-derivatized liposomes.31 Alternatively, covalent attachment of antibodies to liposomes can be accomplished by directly coupling them to functional groups on the liposomal surface or utilising micelles of the antibody–lipid conjugate.32 Various functionalised PEG–lipids with reactive terminal groups such as hydrazine, maleimide, and cyanuric chloride are utilised for covalent methods.31 Sterically stabilised immunoliposomes are obtained by attaching PEG and antibodies to the liposomal surface, enhancing antibody–antigen binding by preventing steric hindrance and ensuring proper orientation for interaction with cell surface receptors.33 Thiolated antibodies are covalently attached to the liposomes via a thioether bond, confirmed by a C–S–C bond in immunoliposomes as indicated by FTIR analysis. Furthermore, the primary structural integrity of the antibodies is confirmed through SDS-PAGE analysis, showing heavy and light chain bands at 50 kDa and 25 kDa, respectively, for both thiolated antibodies and immunoliposomes. These findings validate the successful formation of immunoliposomes with a recovery rate of around 88% (ESI,† Fig. S2). These results confirm the successful formation of immunoliposomes.3.4. Characterisation of miRNA-encapsulated drug-loaded immunoliposomes
The physicochemical properties of miRNA-encapsulated drug-loaded immunoliposomes were comprehensively characterised to evaluate their suitability as drug-delivery vehicles (Fig. 1). Dynamic light scattering (DLS) analysis demonstrated consistent size distribution and zeta potential at both 4 °C and room temperature (RT), indicating stability over 30 days (Fig. 1(a)). Transmission electron microscopy (TEM) imaging revealed the uniform spherical morphology of the vesicles (Fig. 1(b)). Bio-atomic force microscopy (AFM) further revealed the morphology and topography of the liposomes (Fig. 1(c)), with immunoliposomes showing antibody aggregates on the surface, confirming successful antibody conjugation (Fig. 1(d)). This characterisation underscores the stability, structural integrity, and targeted functionality of miRNA-encapsulated drug-loaded immunoliposomes, offering promising potential for therapeutic applications.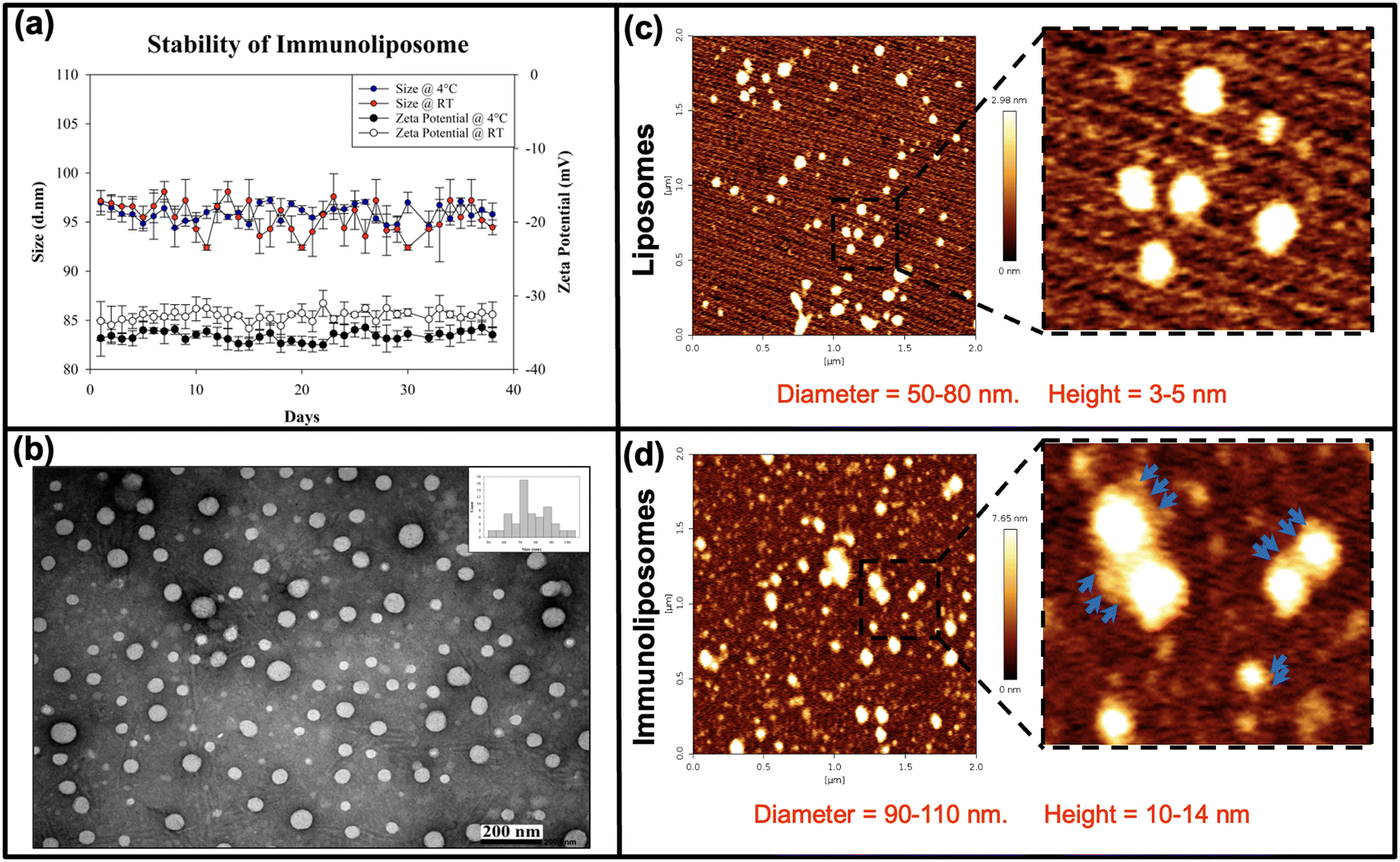 | ||
| Fig. 1 Characterisation of miRNA-encapsulated drug-loaded immunoliposomes. (a) Size and zeta potential analysis of immunoliposomes at 4 °C and room temperature (RT) using dynamic light scattering (DLS). (b) Transmission electron microscopy (TEM) image of miRNA-encapsulated drug-loaded immunoliposomes, demonstrating uniformity and spherical shape of the vesicles. (c) Bio-atomic force microscopy (AFM) image of liposomes, revealing the morphology and topography of the liposomal surface. (d) Bio-AFM image of immunoliposomes showcasing the presence of antibody aggregates on the surface, absent in the liposomes. | ||
3.5. Paclitaxel and miRNA inhibitor entrapment efficiency and release kinetics
The entrapment efficiency of paclitaxel and miRNA inhibitor within the immunoliposomal formulation was determined, with paclitaxel showing an entrapment efficiency of 82.8% and miRNA at 84.82%. Paclitaxel was detected through UV-Vis spectrophotometry, with absorbance peaks at 230 nm facilitating quantification. High-performance liquid chromatography (HPLC) confirmed paclitaxel's presence and retention time at 5 min, with a standard plot enabling quantification (ESI,† Fig. S3). The sustained release of paclitaxel over 72 h was observed, highlighting the formulation's controlled drug delivery potential (Fig. 2). Recent studies emphasise the benefits of immunoliposomal formulations in enhancing pharmacokinetics and therapeutic efficacy. Recent studies have highlighted the advantages of immunoliposomal formulations in improving pharmacokinetics and enhancing therapeutic efficacy.34,35 The high entrapment efficiencies of paclitaxel and miRNA and sustained release kinetics underscore the promising application of the developed immunoliposomal formulation for targeted drug delivery.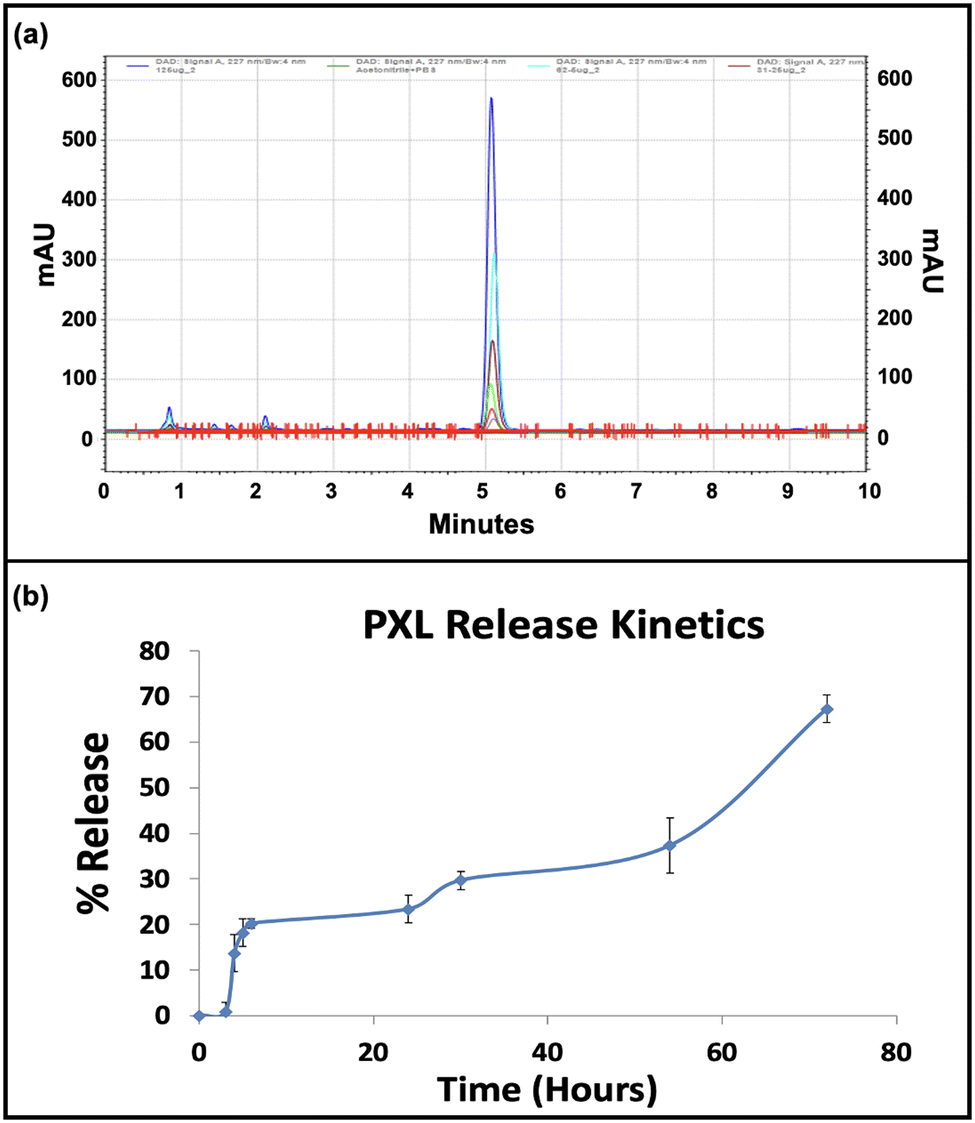 | ||
| Fig. 2 Characterization of paclitaxel (PXL) release. (a) Chromatogram depicting different concentrations of PXL for the standard plot. (b) Percentage release of paclitaxel (PXL) over time. The y-axis represents the percentage release, while the x-axis denotes the time in hours. | ||
3.6. Evaluation of targeting efficiency
To evaluate the targeting efficiency of our immunoliposomal formulation, uptake studies were conducted using HER2-positive SKBR-3 and triple-negative MDA-MB-231 breast cancer cell lines. Confocal microscopy images (Fig. 3) revealed distinct uptake patterns, with higher rhodamine immunoliposome uptake observed in SKBR-3 cells compared to MDA-MB-231 cells. Quantitative analysis confirmed significantly higher rhodamine intensity in SKBR-3 cells, indicating targeted delivery mediated by the formulation. This uptake pattern validates the specificity of our formulation towards HER2-positive cells, aligning with our targeted delivery strategy leveraging antibody-conjugated immunoliposomes. This finding is consistent with previous literature demonstrating the efficacy of antibody-conjugated nanoparticles in targeting receptor-overexpressing cancer cells.36,37 These findings underscore the clinical potential of our formulation in personalised cancer therapy for HER2-positive breast cancer patients.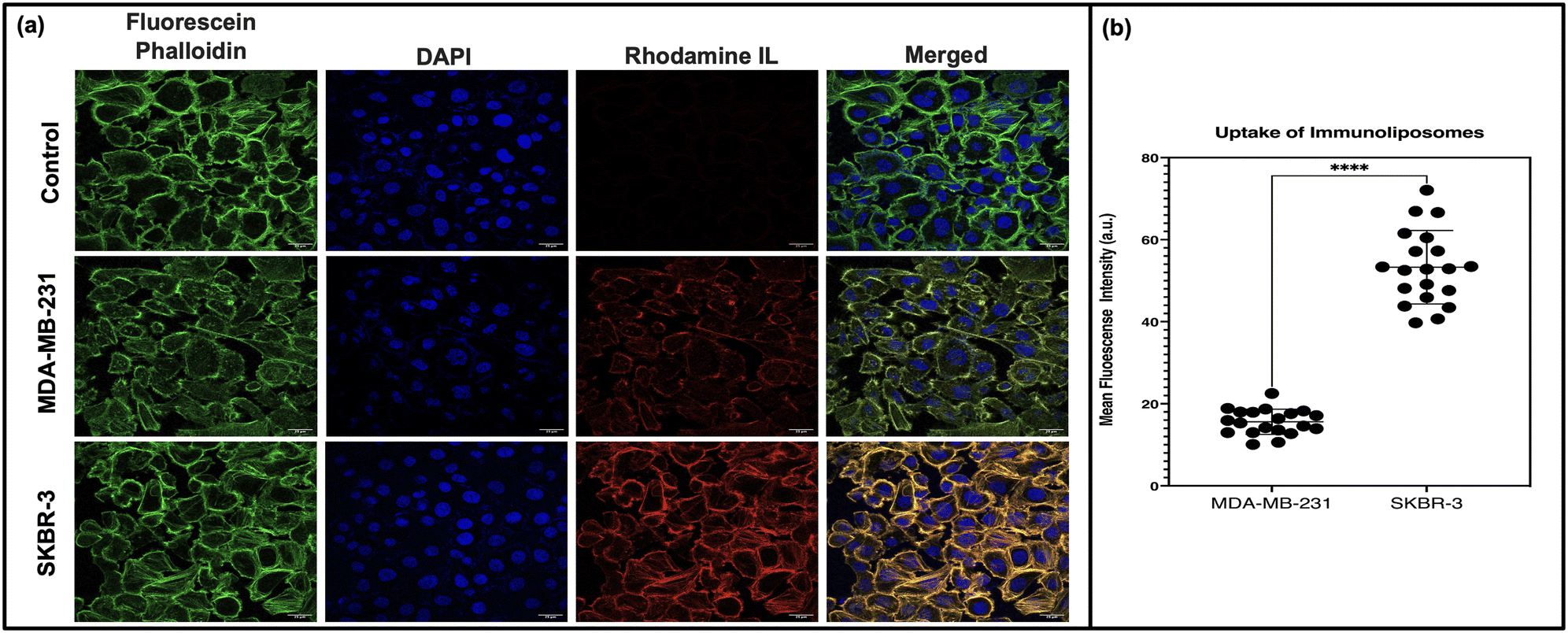 | ||
| Fig. 3 Uptake of the immunoliposome. (a) Illustrates the uptake of rhodamine-loaded immunoliposomes in SKBR-3 and MDA-MB-231 cells. SKBR-3 cells show significantly higher uptake compared to MDA-MB-231 cells. Fluorescein phalloidin staining reveals the actin cytoskeleton, DAPI staining highlights the cell nuclei, rhodamine staining demonstrates the presence of liposomes, and merged images provide an overall visualisation of the channels. The graph in (b) quantifies the relative uptake of rhodamine immunoliposomes, confirming the significantly higher uptake in SKBR-3 cells compared to MDA-MB-231 cells. | ||
3.7. Comparative analysis of antineoplastic activity
The combination of PXL and a miR155 inhibitor encapsulated in IL exhibited promising anticancer activity against HER2-positive breast cancer cells. SKBR-3 cells treated with concentrations ranging from 25 nM PXL + 13 pM miR155 inhibitor to 100 nM PXL + 53 pM miR155 inhibitor in IL showed dose-dependent reductions in cell viability over 48 h and 72 h (ESI,† Fig. S4). At the highest concentration, a 50% reduction in cell viability was observed at 48 h. Li et al. demonstrated a similar effect with a dendrimer-engineered nanocarrier, albeit at a much higher PXL concentration of 23.44 μM.38 Lee et al. reported a synergistic effect with a co-delivery system of PXL and Herceptin, resulting in a 60.2% reduction in cell viability in BT474 cells at a PXL concentration of 6.7 mM.39The concentration of 100 nM PXL + 53 pM miR155 inhibitor was chosen for further investigation due to its notable cytotoxic effect. Evaluation of individual formulation components revealed insignificant effects on cell viability compared to the control group. However, miR155 inhibitor-loaded IL, PXL-loaded IL, and PXL + miR155 inhibitor-loaded IL showed significant reductions in SKBR-3 cell viability after 48 h (Fig. 4), consistent with studies by Zhang et al. demonstrating the efficacy of miR155 inhibitors in sensitising HER2-positive breast cancer cells to chemotherapy.40 Extended treatment (72 h) with 100 nM PXL + 53 pM miR155 inhibitor led to a substantial reduction in SKBR-3 cell viability, reaching up to 80% for the combination treatment (Fig. 4(a)). In MDA-MB-231 cells, only 100 nM PXL + 53 pM miR155 inhibitor exhibited modest reductions in cell viability at both 48 h and 72 h, suggesting potential effectiveness against HER2-positive breast cancer cells (Fig. 4(b)). The formulation's efficacy at low doses highlights its clinical significance, with significant reductions achieved below typical clinical concentrations, indicating its potential as a targeted therapy for HER2-positive breast cancer.
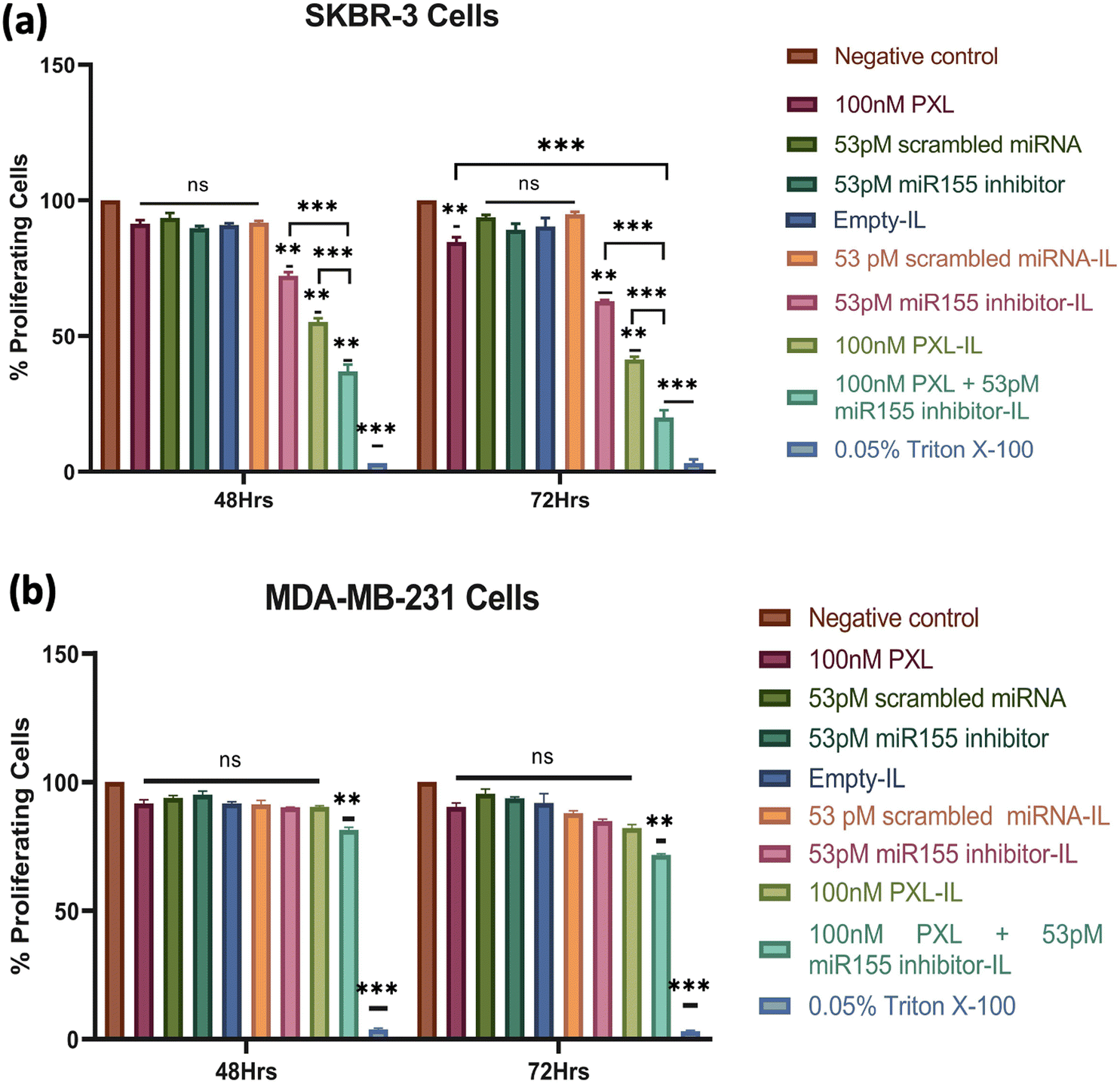 | ||
| Fig. 4 Effects of immunoliposome treatment on cell proliferation in SKBR-3 and MDA-MB-231 cells. (a) MTT assay results of SKBR-3 cells treated with different components of the immunoliposome. The graph demonstrates the impact of individual elements on cell proliferation over time, and (b) MTT assay results of MDA-MB-231 cells treated with the same components of the immunoliposome as in (a). Statistical significance was determined using a two-way ANOVA (*p < 0.05, **p < 0.01, ***p < 0.001). | ||
In the assessment of cell viability at 24 h, no significant reduction was observed in SKBR-3 cells. A subsequent annexin V-FITC apoptosis assay revealed cells in the early apoptotic phase at this time point (ESI,† Fig. S5). This observation provides critical insights into the cellular responses following treatment with our formulation. Although the reduction in cell viability may not be apparent at 24 hours, the induction of early apoptosis suggests an initial response to the treatment. Understanding the temporal dynamics of apoptosis induction is essential for elucidating the mechanisms underlying the antineoplastic activity of our formulation.
3.8. Cell cycle analysis: investigating the effects of combination treatment on cell proliferation
In light of paclitaxel's known mechanism of action, which involves blocking cells in the G2/M phase by inhibiting microtubules, we conducted a cell cycle assay using flow cytometry (Fig. 5). We aimed to elucidate the impact of our combination treatment on cell cycle progression in SKBR-3 cells. We employed the Watson Pragmatic model in FlowJo software to analyse the cell cycle data. This enabled us to accurately quantify and characterise cells' distribution across different cell cycle phases. The results of our analysis revealed a concentration-dependent effect on the cell cycle. Specifically, we observed an increase in the cell population in the G2 phase and a reduction in cells in the G0/G1 phase. This finding suggests that paclitaxel effectively inhibits cell proliferation in SKBR-3 cells even at lower concentrations in combination treatment.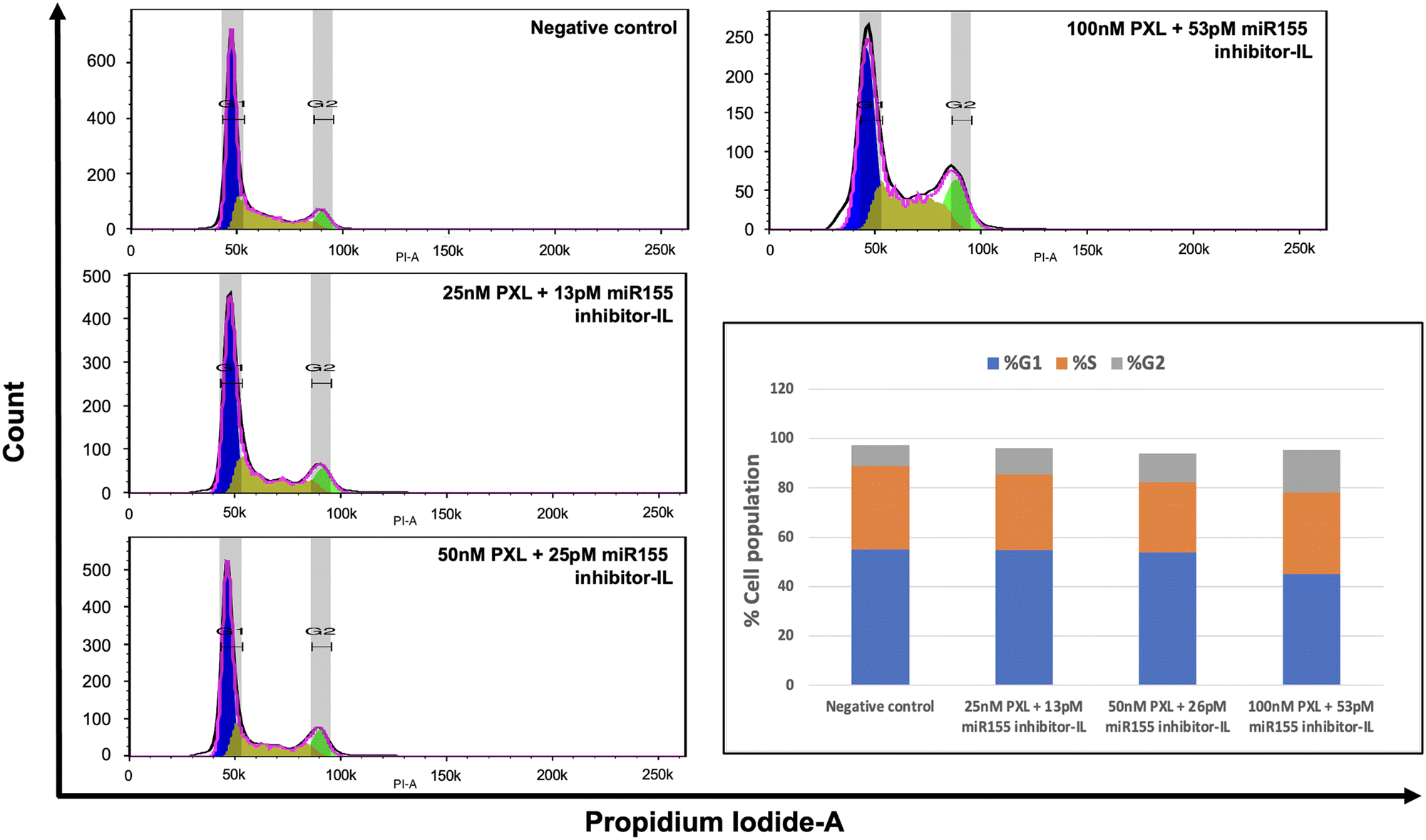 | ||
| Fig. 5 Investigating cell cycle modulation by combination therapy. The histogram illustrates DNA content distribution in SKBR-3 cells treated with varying concentrations of paclitaxel (PXL) and miR155 inhibitor-loaded IL. Concentration-dependent increases in the G2/M phase suggest the formulation's potential to induce cell cycle arrest. | ||
3.9. Assessment of the formulation's impact on cell migration: wound healing assay
miR-155 has been associated with promoting epithelial–mesenchymal transition (EMT) in breast cancer cells, a crucial step in metastasis.41,42 In this study, we evaluated the wound-healing capacity of our formulation and its ability to inhibit cell migration in SKBR-3 and MDA-MB-231 cells, with and without TGF-Beta stimulation (Fig. 6). Notably, treatment with our formulation significantly inhibited cell migration, leading to minimal reduction in wound size, particularly in SKBR-3 cells (Fig. 6(a)). A similar trend was observed in MDA-MB-231 cells, albeit to a lesser extent (Fig. 6(b)). The differential response between the two cell lines may be attributed to molecular profile variations and metastatic potential. SKBR-3 cells, being HER2-positive, may be more susceptible to the inhibitory effects of our formulation due to the targeted delivery approach. Conversely, MDA-MB-231 cells, representing a triple-negative subtype, may exhibit differential sensitivity to the formulation. Our findings underscore the potential therapeutic efficacy of our formulation in suppressing metastatic progression by impeding cell migration, particularly in HER2-positive breast cancer cells. By targeting miR-155, our formulation disrupts critical signalling pathways involved in cell migration, thereby hindering metastatic spread.42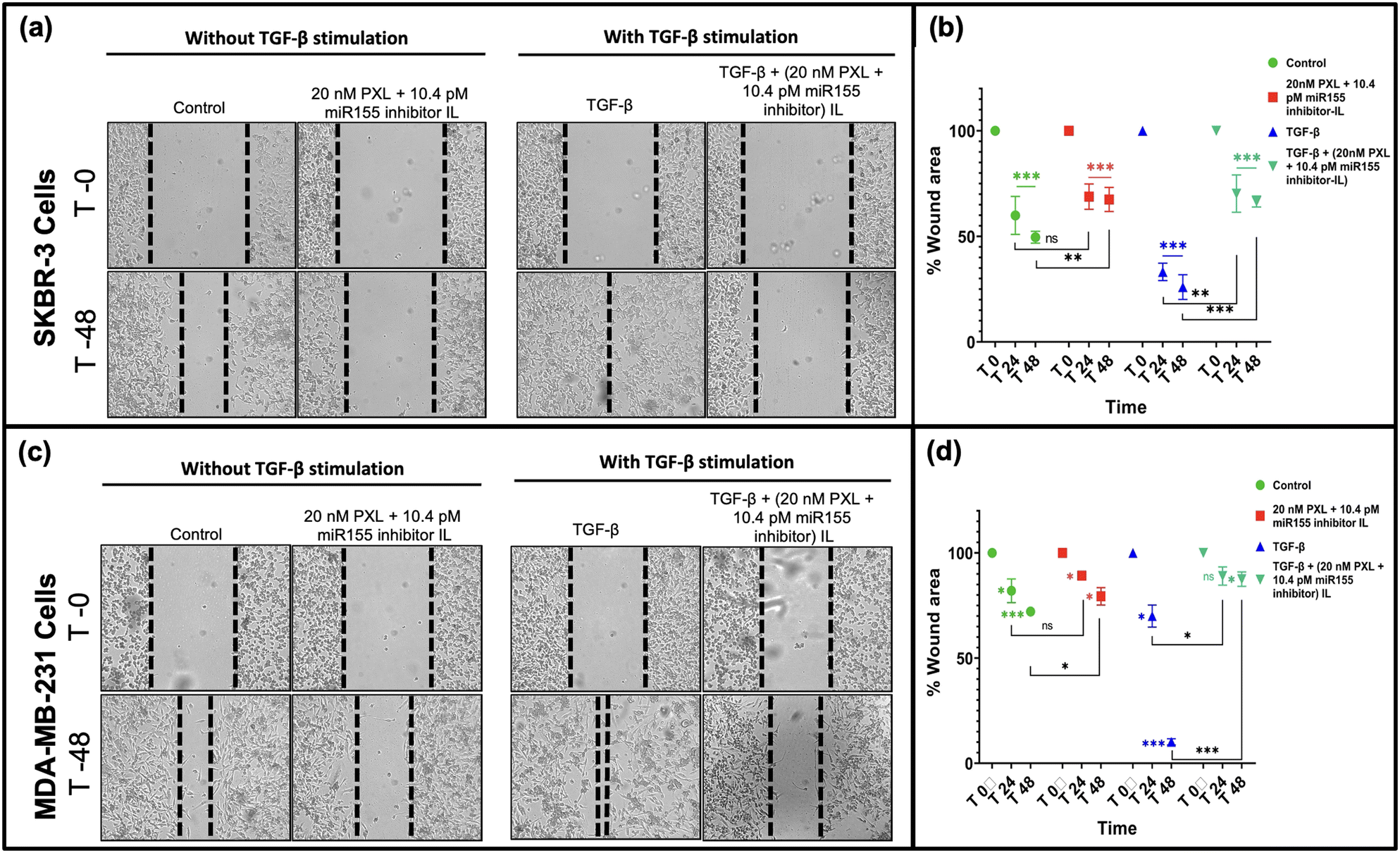 | ||
| Fig. 6 Wound healing assay comparing cell migration in SKBR-3 and MDA-MB-231 cell lines. Representative images of (a) SKBR-3 and (c) MDA-MB-231 cells at 0 and 48 hours post-wounding. Dashed lines indicate the scratch wounds. Graph showing the percentage of wound area that remained open in (b) SKBR-3 and (d) MDA-MB-231 cells over time. Statistical significance was determined using a two-way ANOVA (*p < 0.05, **p < 0.01, ***p < 0.001). | ||
3.10. Evaluation of tumor regression in C57BL/6 mice: SKBR-3 cell-based tumor xenograft study
In this study, we established a SKBR-3 cell-based tumor xenograft model in C57BL/6 mice by pharmacologically suppressing immunity and subsequently injecting the SKBR-3 cells subcutaneously to induce tumor growth. Once the tumor volume reached 100 mm3, mice were randomly allocated into different treatment groups, as outlined in the ESI,† Fig. S6, and administered the formulation via tail vein injection. Continual monitoring of tumor volume revealed a significant reduction in tumor size following treatment with immunoliposomes (IL) containing miR155 inhibitor, paclitaxel (PXL), and a combination of both (Fig. 7). Interestingly, there were no notable changes in the average body weight of animals across the various treatment groups, indicating the tolerability of the formulations. Moreover, compared to treatment with PXL-IL and miR155 inhibitor-IL alone, the combination of PXL and miR155 inhibitor-loaded IL exhibited a statistically significant reduction in tumor volume, suggesting a synergistic effect. These findings highlight the potential therapeutic efficacy of the formulation in tumor regression. Our formulation shows more promising results with previous studies, such as those by Eloy et al., who demonstrated the efficacy of paclitaxel-based nanocarriers in reducing tumor growth.43 Furthermore, the observed synergistic effect of combining paclitaxel with miR155 inhibition is supported by the work of Zhang et al., where the downregulation of miR155 enhanced the therapeutic effect of paclitaxel in breast cancer cells.40 Overall, our findings suggest that the combined delivery of paclitaxel and miR155 inhibitor via immunoliposomes holds promise as a therapeutic strategy for breast cancer treatment.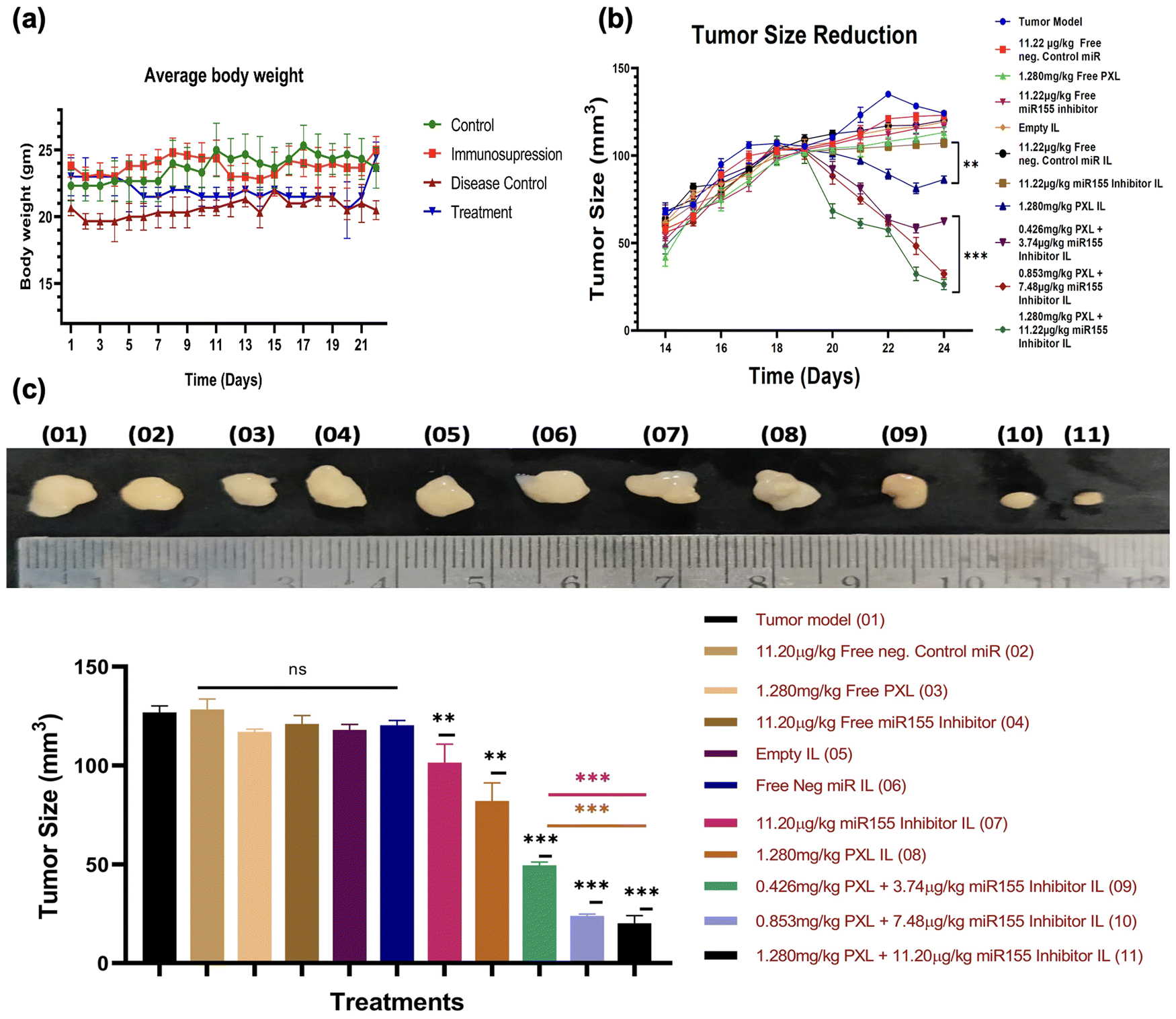 | ||
| Fig. 7 Comprehensive analysis of tumor regression and systemic effects of treatment. This figure encompasses multiple aspects of the experimental study, illustrating the impact of treatments on tumor regression and systemic responses. (a) Line graph depicting the average body weight of mice across different experimental groups, (b) the significant reduction in tumor size when treated with 1.280 mg kg−1 PXL + 11.22 g kg−1 miR155 inhibitor IL. A comparison is made against the tumor control group, emphasizing the efficacy of the combined treatment and (c) composite images displaying tumors post-treatment, including controls and various treatment conditions. (d) Bar graph presenting the tumor volume for each experimental group after 24 days. | ||
We conducted a comprehensive histopathological evaluation using haematoxylin and eosin (H&E) staining of major organs including the heart, lungs, liver, spleen, and kidneys to assess the safety and efficacy of our formulation (ESI,† Fig. S7). Remarkably, no significant alterations in tissue architecture were observed in any of the examined organs, indicating the safety profile of our formulation. This observation is consistent with the notion that our formulation does not induce overt toxicity in vital organs, which is a critical aspect of drug safety assessment. Additionally, we analysed tumor tissues from various treatment groups and compared them with the tumor model (disease model group). Our analysis revealed a higher number of apoptotic cells in the treatment groups compared to other groups, as observed through histopathological analysis (ESI,† Fig. S7 last panel), suggesting the potential of our formulation to target HER2-positive cells and induce apoptosis. This finding underscores the therapeutic efficacy of our formulation in inhibiting tumor growth and promoting cell death specifically in HER2 positive breast cancer cells. Our results align with previous studies demonstrating the safety and efficacy of liposome-based drug delivery systems in cancer treatment. For instance, Al et al. reported similar findings of minimal histological changes in major organs following treatment with nanoparticle formulations.44 Furthermore, the observed increase in apoptotic events in tumor tissues is consistent with the mechanism of action of paclitaxel and miRNA inhibitors in inducing apoptosis in cancer cells, as supported by multiple studies.45
Overall, our histopathological evaluation provides valuable insights into the safety and efficacy of our formulation, supporting its potential as a promising therapeutic strategy for HER2 positive breast cancer treatment.
3.11. Functional validation of miR155 inhibitor: gene expression and western blot analysis
miR155 is a well-characterized oncogenic microRNA that contributes to tumor progression by targeting and downregulating several tumor suppressor genes, including those involved in the p53 signalling pathway.40 The p53 protein is a critical regulator of cell cycle arrest, apoptosis, and genomic stability.46 Dysregulation of the p53 pathway, often through miR155 overexpression, leads to unchecked cell proliferation and survival, contributing to tumor growth.47Post-treatment, we observed a concentration-dependent increase in the expression levels of key apoptotic and cell cycle regulatory genes, including p53, p21, BAX, caspase-9, caspase-3, and APAF1. The upregulation of p53 and its downstream effectors p21 and BAX indicates the reactivation of p53-mediated apoptotic pathways. The increased expression of caspase-9 and caspase-3, along with APAF1, suggests the initiation of the intrinsic apoptotic pathway, culminating in programmed cell death. In contrast, BCL-2, a major anti-apoptotic gene, was significantly downregulated following treatment, reinforcing the pro-apoptotic impact of our formulation (Fig. 8).
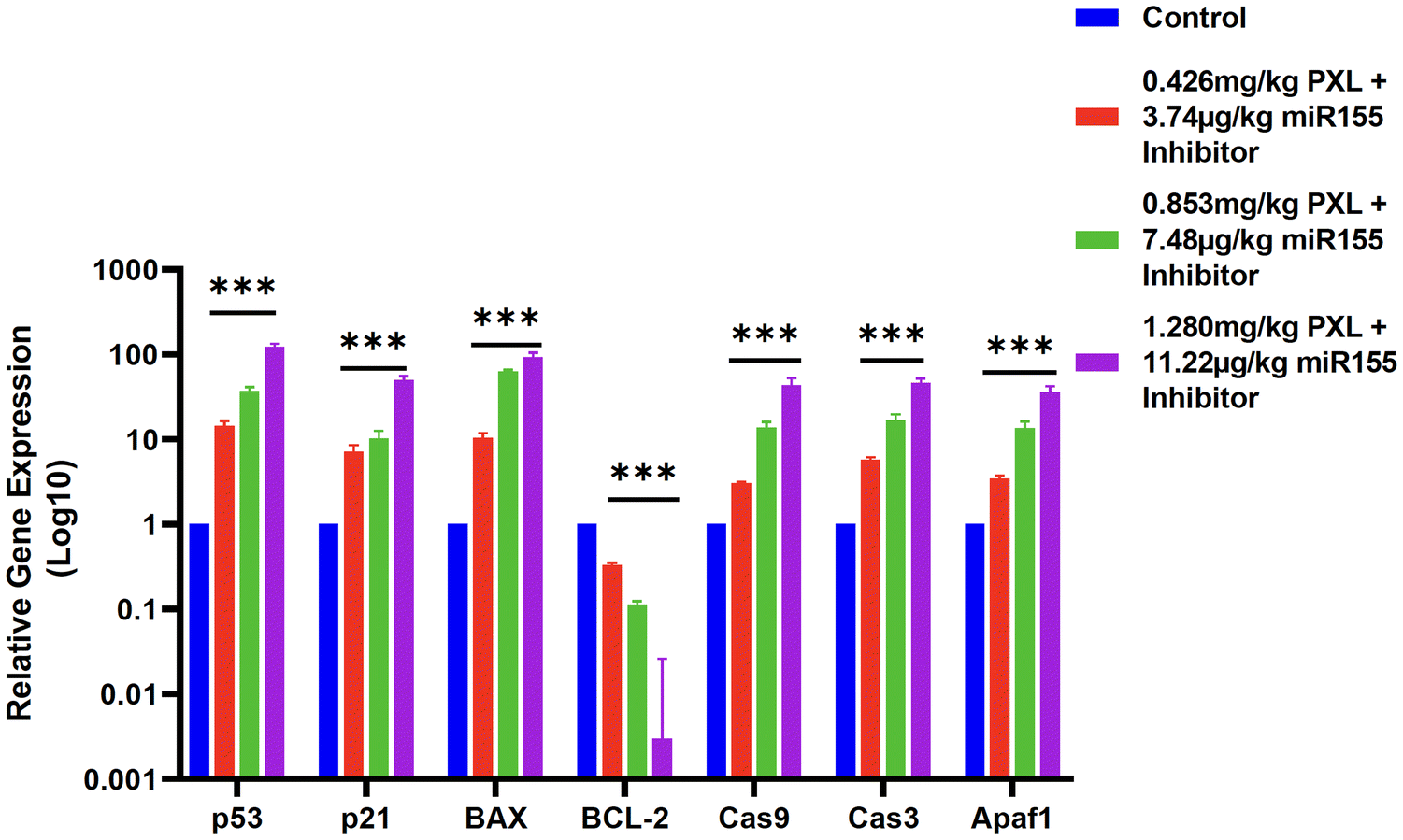 | ||
| Fig. 8 RT-qPCR analysis of the tumor tissue bar graph representing the relative fold change in gene expression levels for p53, p21, BAX, cas9, cas3, APAF1, and BCL-2 in tumor samples treated with three different concentrations of 0.426 mg kg−1 PXL + 3.74 g kg−1 miR155 inhibitor, 0.853 mg kg−1 PXL + 7.48 g kg−1 miR155 inhibitor, and 1.280 mg kg−1 PXL + 11.22 g kg−1 miR155 inhibitor. GAPDH was used as the housekeeping gene for normalization. The results show upregulation of p53, p21, BAX, cas9, cas3, and APAF1, whereas BCL-2 expression was downregulated. | ||
To confirm the functionality of the miR155 inhibitor within the tumor microenvironment, we conducted a western blot analysis for TP53INP, a direct target of miR155. The western blot results demonstrated a significant increase in TP53INP protein levels post-treatment, validating the inhibition of miR155 activity (ESI,† Fig. S8). This suggests that the miR155 inhibitor effectively counteracts miR155-mediated suppression of tumor suppressor genes, thereby facilitating tumor regression.
4. Conclusion
Our comprehensive investigation demonstrates the remarkable potential of the formulated immunoliposomes encapsulating paclitaxel and miRNA inhibitors as a targeted therapeutic approach for HER2 positive breast cancer. Through a meticulously designed series of in vitro and in vivo experiments, we have elucidated the formulation's capacity to effectively deliver therapeutic payloads to cancerous cells, resulting in profound inhibitory effects on cell proliferation, apoptotic induction, and tumor growth suppression. Notably, our formulation exhibits a favourable safety profile, as evidenced by histopathological assessments of major organs, indicating minimal adverse effects on normal tissues.Furthermore, our study unveils the synergistic interplay between paclitaxel and miRNA inhibitors within the formulation, highlighting the potential benefits of combination therapy. The observed ability of our formulation to selectively target HER2 positive cells and induce tumor regression underscores its clinical relevance in addressing the complexities of breast cancer heterogeneity. These findings contribute significantly to the burgeoning field of nanomedicine-based cancer therapeutics, offering a promising avenue for personalised and targeted treatment strategies.
Moving forward, future investigations should delve deeper into elucidating the underlying molecular mechanisms driving the therapeutic efficacy of our formulation. Optimisation of the formulation parameters to enhance effectiveness and minimise potential off-target effects remains a crucial avenue for further exploration. Additionally, clinical translation and rigorous evaluation through human trials are imperative steps towards validating the therapeutic potential of our formulation in a clinical setting. Ultimately, our study lays a solid foundation for advancing precision medicine approaches tailored to HER2 positive breast cancer, aiming to improve patient outcomes and quality of life.
Author contributions
Ramesh Chaudhari: conceptualization, methodology, investigation, formal analysis, writing – original draft. Vishva Patel: investigation, visualization, software. Bharti Malvi: software, data curation, writing – review & editing Superb K Misra: resources, validation, writing – review & editing, supervision. Ashutosh Kumar: conceptualization, methodology, writing – review & editing, visualization, supervision, project administration, funding acquisition.Data availability
The data supporting this study's findings are available on request from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Indian Council of Medical Research (ICMR), Government of India [grant number: EM/Dev/SG/217/1078/2023]. Financial assistance from The Gujarat Institute for Chemical Technology (GICT) for establishing a facility for environmental risk assessment of chemicals and nanomaterials is also acknowledged.Notes and references
- C. DeSantis, R. Siegel and A. Jemal, Breast cancer facts and figures 2013–2014, Am. Cancer Soc., 2013, 2013, 1–38 Search PubMed.
- W. Globocan, Estimated cancer incidence, mortality and prevalence worldwide in 2020, Int. Agency Res. Cancer, 2020, 209–249 Search PubMed.
- A. N. Giaquinto, et al., Breast cancer statistics, Ca-Cancer J. Clin., 2022, 72(6), 524–541 CrossRef.
- S. K. Ong, et al., Feasibility of monitoring Global Breast Cancer Initiative Framework key performance indicators in 21 Asian National Cancer Centers Alliance member countries, EClinicalMedicine, 2024, 67 DOI:10.1016/j.eclinm.2023.102365.
- S. K. Singh, et al., Drug delivery approaches for breast cancer, Int. J. Nanomed., 2017, 6205–6218 CrossRef CAS.
- N. R. Raghani, et al., Revolutionizing cancer treatment: comprehensive insights into immunotherapeutic strategies, Med. Oncol., 2024, 41(2), 51 CrossRef PubMed.
- M. H. Mohammed, et al., Evaluation of Immunohistochemical Expression of Human Epidermal Growth Factor Receptor 2 (HER2) and its Association with Clinicopathological Variables in Patients with Pancreatic Duct Adenocarcinoma in Upper Egypt, Res. Oncol., 2024, 1–8 Search PubMed.
- H. Muley, et al., Drug uptake-based chemoresistance in breast cancer treatment, Biochem. Pharmacol., 2020, 177, 113959 CrossRef CAS PubMed.
- K. Zhu, et al., HER2-targeted therapies in cancer: a systematic review, Biomarker Res., 2024, 12(1), 1–17 CrossRef PubMed.
- Y. Huang, P. Fu and W. Fan, Novel targeted therapies to overcome trastuzumab resistance in HER2-overexpressing metastatic breast cancer, Curr. Drug Targets, 2013, 14(8), 889–898 CrossRef CAS.
- W. Si, et al., The role and mechanisms of action of microRNAs in cancer drug resistance, Clin. Epigenet., 2019, 11(1), 1–24 CrossRef PubMed.
- D.-d Yu, et al., Role of miR-155 in drug resistance of breast cancer, Tumor Biol., 2015, 36, 1395–1401 CrossRef CAS.
- R. Bayraktar and K. Van Roosbroeck, miR-155 in cancer drug resistance and as target for miRNA-based therapeutics, Cancer Metastasis Rev., 2018, 37, 33–44 CrossRef.
- Y. Li, et al., MicroRNA-155-5p promotes tumor progression and contributes to paclitaxel resistance via TP53INP1 in human breast cancer, Pathol., Res. Pract., 2021, 220, 153405 CrossRef CAS.
- Z. Chen, et al., The pivotal role of microRNA-155 in the control of cancer, J. Cell. Physiol., 2014, 229(5), 545–550 CrossRef CAS PubMed.
- P. D. Eckford and F. J. Sharom, ABC efflux pump-based resistance to chemotherapy drugs, Chem. Rev., 2009, 109(7), 2989–3011 CrossRef CAS.
- F. Sharom, The P-glycoprotein efflux pump: how does it transport drugs?, J. Membr. Biol., 1997, 160, 161–175 CrossRef CAS PubMed.
- Y. Luqmani, Mechanisms of drug resistance in cancer chemotherapy, Med. Princ. Pract., 2005, 14(Suppl. 1), 35–48 CrossRef PubMed.
- J. Kopecka, et al., Insights in the chemical components of liposomes responsible for P-glycoprotein inhibition, Nanomedicine, 2014, 10(1), 77–87 CrossRef CAS PubMed.
- C. Mamot, et al., Liposome-based approaches to overcome anticancer drug resistance, Drug Resist. Updates, 2003, 6(5), 271–279 CrossRef CAS.
- X. Xue and X.-J. Liang, Overcoming drug efflux-based multidrug resistance in cancer with nanotechnology, Chin. J. Cancer, 2012, 31(2), 100 CrossRef CAS PubMed.
- P. Abasian, et al., Polymeric nanocarriers in targeted drug delivery systems: A review, Polym. Adv. Technol., 2020, 31(12), 2939–2954 CrossRef CAS.
- M. Picard and M. C. Castells, Re-visiting hypersensitivity reactions to taxanes: a comprehensive review, Clin. Rev. Allergy Immunol., 2015, 49, 177–191 CrossRef CAS.
- J. Szebeni, C. R. Alving and F. M. Muggia, Complement activation by Cremophor EL as a possible contributor to hypersensitivity to paclitaxel: an in vitro study, J. Natl. Cancer Inst., 1998, 90(4), 300–306 CrossRef CAS.
- F. Fitzpatrick and R. Wheeler, The immunopharmacology of paclitaxel (Taxol®), docetaxel (Taxotere®), and related agents, Int. Immunopharmacol., 2003, 3(13–14), 1699–1714 CrossRef CAS PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65(1–2), 55–63 CrossRef CAS.
- R. Chaudhari, et al., MiR-206 conjugated gold nanoparticle based targeted therapy in breast cancer cells, Sci. Rep., 2022, 12(1), 4713 CrossRef CAS.
- C.-C. Liang, A. Y. Park and J.-L. Guan, In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro, Nat. Protoc., 2007, 2(2), 329–333 CrossRef CAS.
- P. Patel, et al., Cell cycle dependent cellular uptake of zinc oxide nanoparticles in human epidermal cells, Mutagenesis, 2016, 31(4), 481–490 CrossRef CAS.
- R. Chaudhari, et al., Fabrication of methotrexate-loaded gold nanoconjugates and its enhanced anticancer activity in breast cancer, 3 Biotech, 2021, 11, 1–13 CrossRef.
- A. Schnyder, et al., Targeting of skeletal muscle in vitro using biotinylated immunoliposomes, Biochem. J., 2004, 377(1), 61–67 CrossRef CAS PubMed.
- X. Pan, et al., Synthesis of cetuximab-immunoliposomes via a cholesterol-based membrane anchor for targeting of EGFR, Bioconjugate Chem., 2007, 18(1), 101–108 CrossRef CAS.
- R. Petrilli, et al., Preparation of immunoliposomes by direct coupling of antibodies based on a thioether bond, Recombinant Glycoprotein Production: Methods and Protocols, 2018, pp. 229–237 Search PubMed.
- V. P. Torchilin, Multifunctional nanocarriers, Adv. Drug Delivery Rev., 2006, 58(14), 1532–1555 CrossRef CAS.
- M. Alavi and R. S. Varma, Overview of novel strategies for the delivery of anthracyclines to cancer cells by liposomal and polymeric nanoformulations, Int. J. Biol. Macromol., 2020, 164, 2197–2203 CrossRef CAS PubMed.
- R. Petrilli, et al., Immunoconjugates for cancer targeting: a review of antibody-drug conjugates and antibody-functionalized nanoparticles, Curr. Med. Chem., 2021, 28(13), 2485–2520 CrossRef CAS.
- A. Juan, et al., Antibody conjugation of nanoparticles as therapeutics for breast cancer treatment, Int. J. Mol. Sci., 2020, 21(17), 6018 CrossRef CAS.
- X. Li, et al., Amphiphilic dendrimer engineered nanocarrier systems for co-delivery of siRNA and paclitaxel to matrix metalloproteinase-rich tumors for synergistic therapy, NPG Asia Mater., 2018, 10(4), 238–254 CrossRef CAS.
- A. L. Lee, et al., The co-delivery of paclitaxel and Herceptin using cationic micellar nanoparticles, Biomaterials, 2009, 30(5), 919–927 CrossRef CAS PubMed.
- C.-M. Zhang, J. Zhao and H.-Y. Deng, MiR-155 promotes proliferation of human breast cancer MCF-7 cells through targeting tumor protein 53-induced nuclear protein 1, J. Biomed. Sci., 2013, 20, 1–10 CrossRef.
- S. Mattiske, et al., The oncogenic role of miR-155 in breast cancer, Cancer Epidemiol., Biomarkers Prev., 2012, 21(8), 1236–1243 CrossRef CAS PubMed.
- X. Liu, et al., miR-155 promotes proliferation and epithelial-mesenchymal transition of MCF-7 cells., Exp. Ther. Med., 2021, 21(3), 1 Search PubMed.
- J. O. Eloy, et al., Anti-HER2 immunoliposomes for co-delivery of paclitaxel and rapamycin for breast cancer therapy, Eur. J. Pharm. Biopharm., 2017, 115, 159–167 CrossRef CAS PubMed.
- A. K. Al Asmari, et al., Preparation, characterization, and in vivo evaluation of intranasally administered liposomal formulation of donepezil, Drug Des., Dev. Ther., 2016, 205–215 CAS.
- M. S. Franco, et al., Investigation of the antitumor activity and toxicity of long-circulating and fusogenic liposomes co-encapsulating paclitaxel and doxorubicin in a murine breast cancer animal model, Biomed. Pharmacother., 2019, 109, 1728–1739 CrossRef CAS PubMed.
- P. H. Shaw, The role of p53 in cell cycle regulation, Pathol., Res. Pract., 1996, 192(7), 669–675 CrossRef CAS PubMed.
- P. M. Neilsen, et al., Mutant p53 drives invasion in breast tumors through up-regulation of miR-155, Oncogene, 2013, 32(24), 2992–3000 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01090f |
| This journal is © The Royal Society of Chemistry 2025 |