
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect cytosol delivery of mRNA by micron-sized co-assembly with designer oligopeptides†
Ruilu
Feng‡
 ,
Mehrnoosh
Rafiei‡
,
Kalindu S.
Fernando‡
and
Ying
Chau
*
,
Mehrnoosh
Rafiei‡
,
Kalindu S.
Fernando‡
and
Ying
Chau
*
Chemical and Biological Engineering Department, The Hong Kong University of Science and Technology, Hong Kong SAR, China. E-mail: keychau@ust.hk
First published on 13th January 2025
Abstract
Inefficient endosomal escape has been regarded as the main bottleneck for intracellular nucleic acid delivery. While most research efforts have been spent on designing various nano-sized particles, we took a different path here, investigating micron-sized carriers for direct cytosol entry. Using the spontaneous co-assembly of mRNA and the designer 27 amino acid oligopeptide named pepMAX2, micron-sized co-assemblies were obtained with various sizes by altering the concentration of NaCl salt and time for pre-incubation. Surprisingly, transfection was much more effective using micron-sized than nano-sized co-assemblies, and the efficiency surpasses that of a widely used lipid-based commercial reagent. The study was complemented by computational simulations, inhibitor studies and live-cell confocal imaging to shed light on the role of electrostatic interaction on assembly and the mechanism of uptake and intracellular trafficking. These micron-sized co-assemblies directly enter the cytosol and then release mRNA, bypassing conventional pathways and thus avoiding the lysosomal degradation. This simple approach involving short oligopeptides and salt addition to create optimal micron-sized co-assembly with mRNA should open new avenues to overcome endosomal barriers for intracellular delivery of nucleic acid therapeutics.
Introduction
Messenger RNA (mRNA) serves as a template for protein synthesis. This feature promotes a variety of biomedical applications, such as the vaccines by Pfizer/BioNTech1 and Moderna2 to combat the COVID-19 pandemic.3 Other applications, such as protein replacement or augmentation, would expand the scope of pharmacy beyond conventional treatments. One key technical challenge lies in delivering intact and functional mRNA into the cytosol of target cells. For non-viral vehicles, numerous studies suggested that endosomal escape is the limiting step of delivery.4 Although endosomal escape has been chemically encoded in these delivery systems to enhance cytosol delivery, a previous study on lipid-based carriers showed that only 1–2% of administered nucleic acid was detected in the cytosol5,6 and the endosomal escape efficiency of a pH-responsive polymer, poly(2-diisopropylamino ethyl methacrylate) (PDPAEMA), was less than 2%.7 Besides endocytosis, approaches taking advantage of the non-endocytic entry mechanisms could be an alternative option to avoid the endosomal barriers.8 Recent studies on a phase-separated peptide assembly demonstrated its ability to bypass the classical endocytic pathways to achieve efficient cytosolic delivery of diverse macromolecules.9 Unlike the conventional nanoscale carriers typically below 200 nm,8 this peptide interacted with the macromolecules to form micron-sized structures.10 For the delivery of Cas9 protein and sgRNA delivery, gold particles with a diameter of 500 nm (by TEM) showed successful cytosol delivery in 90% of treated cells, in contrast to less than 10% of cells when treated with ∼100 nm particles.11 These investigations inspired us to explore micron-sized structures which might be able to circumvent the classical endocytic pathways for cell entry.Compared with the traditional delivery systems, peptides supported automated synthesis, allowed fast single-step formulation, and exhibited good biocompatibility.12 Inspired by virus structures, we have developed various peptides capable of delivering RNA13 or DNA.14–17 Although variation and optimization existed in these peptide series, general design principles were followed – a positively changed N-terminal for nucleic acid binding, a hydrophobic segment to promote self-assembly, and a flexible linker to connect these two segments. We discovered that a subtle change in amino acid resulted in dramatic changes in the peptide self-assembly morphologies and also its transfection efficiency when co-assembled with nucleic acids.16 After several rounds of design evolution, we reported a 27-amino acid peptide sequence, named pepMAX, capable of co-assembling with mRNA into 100–150 nm nanostructures for efficient transfection of multiple cell lines.13 The percentage of cells transfected (>80%) was comparable to commercially available mRNA-specific vectors Lipofectamine™ MessengerMAX™ (LipoMMAX).
Our previous sequence modifications only focused on nano-scaled changes. In this study, we explore whether the performance of pepMAX could be further improved, not only by sequence design but also by manipulating the co-assemblies of peptide/mRNA, specifically, by changing the size from nano to micron-scale. The sequence used in this study is pepMAX2 (PKKKKKVGHHHH-C12-C′HHC′C′HHC′GSPHHD-amide) (Fig. 1). PepMAX2 differs from pepMAX only in its N-terminal with the removal of an Fmoc group. Since peptide assembly was coordinated by various interactions, we hypothesized that pepMAX2 tended to be more susceptible to altering charge–charge interactions, as removal of Fmoc resulted in a reduced proportion of hydrophobic interactions and other short-distance interactions (such as π–π). While the assembly was dependent on various physiochemical factors, such as peptide concentration, pH, ionic strength, temperature, etc.,18 the charge–charge interaction between positively charged peptides and mRNA would play a crucial role in regulating the co-assembling process by electrically screening ionic interactions.19 As the incorporation of salt ions triggered changes in the assemblies’ structure19–21 and had the capacity to induce size changes in polymer-based delivery systems,22,23 we hypothesized that the size of our pepMAX2/mRNA co-assemblies would be adjusted by the addition of salt during particle formation. By optimizing the salt concentration and pre-incubation time, micron-sized co-assemblies could be obtained.
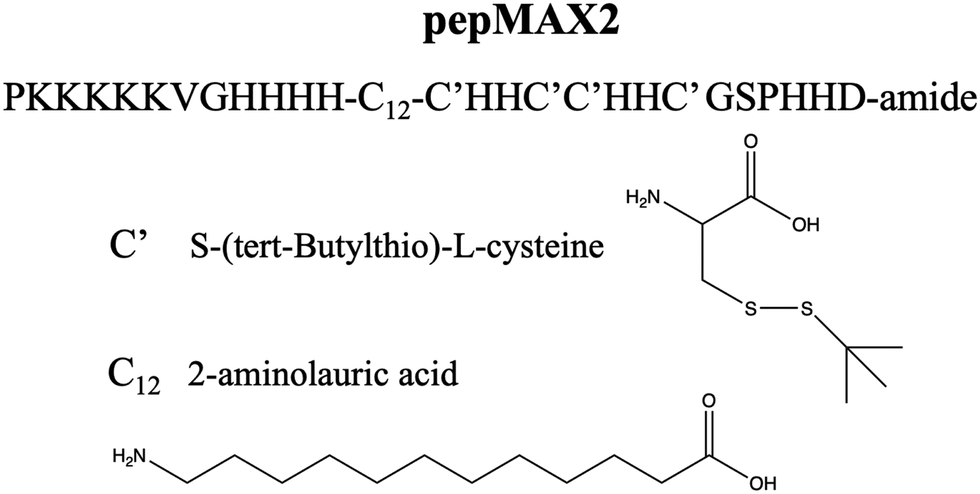 | ||
| Fig. 1 The chemical structure of pepMAX2. | ||
Experimental
Materials
All the Fmoc-protected amino acids and rink amide resins were purchased from GL Biochem (Shanghai, China) or Chemenu (Shanghai, China). LysoTracker™ Red DND-99 and Lipofectamine™ MessengerMAX™ were purchased from ThermoFisher. CleanCap EGFP mRNA (5moU, L-7201) and CleanCap Cy5-EGFP mRNA (5moU, L-7701) were purchased from TriLink™ BioTechnologies Inc. The rest of the chemicals were purchased from Sigma-Aldrich unless otherwise stated. HeLa cells and SKNMC cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. The cells were grown at 37 °C in a 5% CO2 atmosphere. Cells were passaged at ∼80% confluence.Peptide synthesis
The peptide was synthesized on a microwave-assisted peptide synthesizer (biotage, Sweden) in our laboratory based on Fmoc-Solid-Phase Peptide Synthesis (SPPS) as reported previously.16,24 The synthesis was on a 0.1 mmol scale, with 4 equivalents of the amino acids for each coupling. The resin used here was rink amide resin for amide formation at the C-terminal. The amino acids were protected as recommended conventionally, including K (lysine) with Boc, H (histidine) with Trt, S (serine) with tBu and D (asparagine) with OtBu. A coupling system using 1-hydroxybenzotriazole (HOBt)/2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU)/N,N-diisopropylethylamine (DIEA) was employed. Once the synthesis was completed, the peptide was deprotected and simultaneously cleaved from dry resins with TFA for 3 h. The filtrate was collected and precipitated with cold ether. The precipitate was washed twice with cold ether and air-dried for HPLC purification.Peptide purification
The dried crude peptide was dissolved in 50% water/acetonitrile with a 0.1% TFA mixture by sonication. The clear supernatant was collected after filtration with a 220 nm filter and purified on the reverse-phase HPLC (Agilent 1260 Infinity II) through the VYDAC protein and peptide C-18 preparative column (19 × 250 mm) with a linear gradient increase at 1% per min from 30% at 6 mL min−1. The collected fractions were evaporated on the rotavapor to remove acetonitrile, and the rest of the peptide aqueous solution was frozen in liquid nitrogen and freeze-dried on a freeze-dryer (FreeZone Benchtop Freeze Dryer, Labconco) to obtain the white peptide powder, which was stored at 4 °C until use. Peptide purity, 98.4%, was confirmed by analytical HPLC (Agilent 1260 Infinity II) through the Poroshell 120 EC-C18 4 μm column (4.6 × 150 mm) with a linear gradient increase at 3% per min from 10% at 1 mL min−1. Peptide identity was confirmed by MALDI-TOF (BRUKER) analysis: observed, 3600.7 [calculated for (M + H)+, 3601.4] (Fig. S0, ESI†).Formation of peptide/mRNA co-assemblies
The peptide powder was dissolved in DMSO at 10 mM as stock and stored at 4 °C. The stock HEPES (100 mM, pH 9) was used as a buffer to make the final pH = 7.4 after peptide addition. Generally, the stock mRNA (Trilink™, 1 mg mL−1) was diluted in the HEPES buffer (10 mM) to reach a final concentration at 0.1 mg mL−1. To obtain the optimal NaCl concentration for pre-incubation, NaCl solution in HEPES buffer (10 mM) ranging from 0 mM to 5000 mM was mixed with the mRNA solution (0.1 mg mL−1) at a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Then the peptide stock solution was added into the buffered mRNA salt solution to make a final peptide concentration of 0.0867 mM. The mixture was vortexed for 5 seconds and incubate at room temperature for 30 min. The optimal transfection conditions were obtained from the one with an NaCl concentration of 250 mM, with 15–40 min pre-incubation time intervals (HEPES, 10 mM; mRNA, 0.033 mg mL−1; NaCl, 166.7 mM; pepMAX2, 0.0867 mM).
:1 ratio. Then the peptide stock solution was added into the buffered mRNA salt solution to make a final peptide concentration of 0.0867 mM. The mixture was vortexed for 5 seconds and incubate at room temperature for 30 min. The optimal transfection conditions were obtained from the one with an NaCl concentration of 250 mM, with 15–40 min pre-incubation time intervals (HEPES, 10 mM; mRNA, 0.033 mg mL−1; NaCl, 166.7 mM; pepMAX2, 0.0867 mM).
Encapsulation efficiency of mRNA by RiboGreen assay
To assess the encapsulation efficiency of mRNA, the RiboGreen assay (RiboGreen Assay Kit, Invitrogen, USA) was utilized to quantify the amount of unloaded mRNA. The RiboGreen dye specifically binds to free mRNA, allowing for the determination of unencapsulated RNA in the sample. Three different salt concentrations (0 mM, 250 mM, and 5000 mM) were utilized for pepMAX2–mRNA co-assemblies, with a pre-incubation time of 30 minutes and a peptide concentration of 2 μM for loading 80 ng of mRNA. The encapsulation efficiency (EE%) was calculated using the following formula: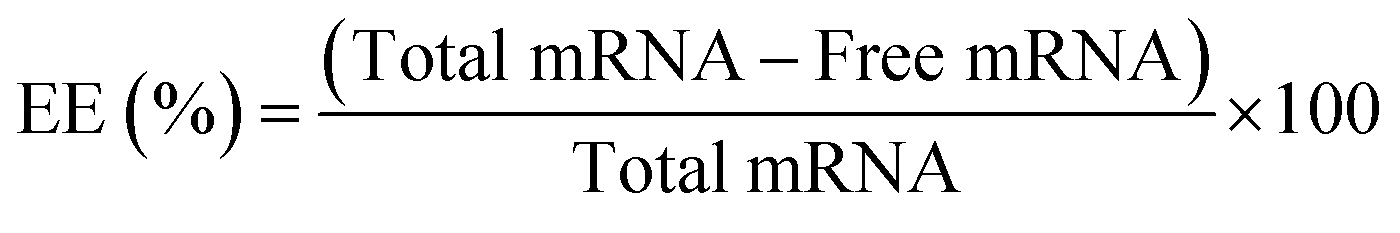
Hydrodynamic sizes by dynamic light scattering (DLS)
The hydrodynamic sizes and polydispersity of co-assemblies were measured by dynamic light scattering (DLS) with a particle size analyzer (90Plus, Brookhaven Instruments Corporation, NY, USA). The freshly prepared sample solution (1.8 μL) was diluted 40 times with DMEM medium (70 μL) and measured immediately, with a dilution ratio similar to the subsequent assessment of transfection efficiency.Determination of relative turbidity with a UV-Vis spectrometer
The relative turbidity of the peptide/mRNA co-assemblies was determined with a UV-Vis spectrometer (Varioskan LUX, ThermoFisher) in order to confirm the formation of micrometer-sized particles. The absorbance at 600 nm was used to calculate the relative turbidity as9| 100 − 100 × (10−A600) |
The freshly prepared sample solution (30 μL) was added into one of the 384-well assay plates and the change in its absorbance with increasing incubation time was immediately measured. To examine the turbidity and changes at different pH values, the sample (pH 7.4, pre-incubated for 30 min) was mixed at a 1:1 ratio with 10 mM phosphate buffers at various pH (ionic strength of 100 mM). Absorbance was measured immediately and the pH after mixing was recorded as the indicated pH value.
Structural characterization with transmission electron microscopy (TEM)
Carbon supported TEM copper grids (400-mesh, TED PELLA, Inc., USA) were pre-treated with a plasma cleaner for 1 min to clean the grid surface and increase the sample attachment. The sample solution (2.5 μL) was loaded onto a parafilm, followed by covering the copper grid face-down on the sample drops. After 2 min incubation, the excess solution on the copper grid was removed with filter paper, followed by covering it with 2% uranyl acetate staining solution (2.5 μL, Sigma-Aldrich). After another 2 min incubation, the excess staining solution was dipped away with filter paper, and the sample grids were placed in a grid holder and air-dried overnight before imaging. A JOEL 2010 transmission electron microscope was used to image the samples.Structural characterization with Cryo-EM (Cryo-EM)
For cryo-grid preparation, aliquots (3 μL) of samples were applied to glow-discharged Quantifoil Au grids (R2/2, 400 mesh) and blotted using an FEI Vitrobot IV. Cryo grids were loaded and inspected using Thermo Fisher Scientific Titan Krios operated at 300 kV. Images were taken using a Gatan Bio-quantum K3 camera (Gatan) in the counted mode, at a nominal magnification of 81000× and defocus range of −1.5 to −2.5 μm. A total exposure of 50 electrons per Å2 over 40 frames, with a total exposure time of 4.5 s.
Agarose gel electrophoresis for the release study of pepMAX2–mRNA co-assemblies
1% agarose gel electrophoresis was conducted using optimized pepMAX2–mRNA complexes (incubated with 250 mM NaCl for 30 minutes) in release media containing either HEPES (10 mM) or heparin (0.4 mg mL−1) at a volume ratio of 1:1. Samples were analyzed at various time points (30 minutes, 1 hour, and 4 hours) at 37 °C. Each sample was mixed with 6× loading dye (ThermoFisher), and a total of 10 μL containing 100 ng of mRNA was loaded into each well. The gel was run at 150 V for 20 minutes, followed by staining with SYBR Gold Nucleic Acid Gel Stain for 30 minutes in the dark. Images were captured using a GelDoc Go Gel Imaging System (Bio-Rad, Hercules, CA, USA).
Molecular dynamics (MD) simulations and molecular mechanics Poisson–Boltzmann surface area (MMPBSA) estimations
The structure of pepMAX2 was constructed in the fully extended configuration using Pymol (v1.7.4 (for academics)) software. A model mRNA sequence with 20 nucleotides (UAAAGGAGAAGAACUUUUCA) was selected, and energy minimized configuration was created and determined using the RNA composer25,26 online server. The topology parameters of pepMax2 and model mRNA for MD simulations were generated using the CHARMM-GUI.27,28 Then all atomistic MD simulations were performed using GROMACS29 (v2019.5.1) with explicitly presented water as the solvent30–32 for explicit water modeling. For pepMAX2 simulations, two pepMAX2 were inserted into a dodecahedron box with 1400 nm3 volume approximately. Then the system was dissolved with water, and NaCl was added up to the desired concentration (0 mM, 250 mM, 2500 mM). The total number of molecules in the simulation box was approximately 47000. The same procedure was used for pepMAX2 and model mRNA simulations by inserting a pepMAX2 molecule and a model mRNA.33
Periodic boundary conditions were applied in the x, y, and z directions. Long-range electrostatic interactions were evaluated using the particle mesh Ewald algorithm33,34 with a cutoff of 1.0 nm for the real space interactions. van der Waals interactions were computed using a cutoff distance of 1.0 nm. LINCS35 constrained all bond lengths, and a 2.0 fs time step was used to integrate the equations of motion.
The energy minimization of the system was performed using the steepest descent algorithm followed by 50000 equilibrium steps (100 ps) of an NVT ensemble (T = 300 K) and 50000 equilibrium steps (100 ps) of an NPT ensemble (T = 300 K, P = 1 atm). Temperature and pressure were controlled using a modified Berendsen thermostat and Parrinello–Rahman barostat schemes, respectively. Finally, a 400 ns production run trajectory was produced as an NPT ensemble for the analysis of structural properties and pairwise interaction energies of the pepMAX2. Pairwise binding energy was estimated using the MMPBSA36 method utilizing the g_mmpbsa open-source package utilizing all default parameters for peptides and RNAs to estimate interaction energies and their distribution.37,38 The AP parameter was estimated using the following formula:
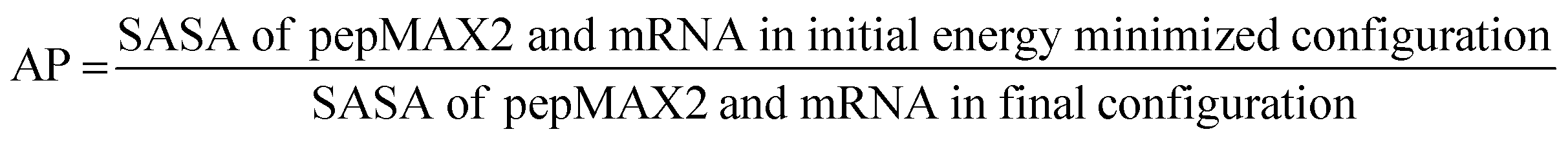
Three different MD simulations, MMPBSA calculations and AP parameter estimations were performed at three different salt conditions (0 mM, 250 mM, and 2500 mM) for both the pepMAX2 system and the pepMAX2 and model mRNA system.
Transfection by pepMAX2/mRNA co-assemblies
The day before the transfection, HeLa cells or SKNMC or PC12 cells were seeded with a density of 1.5 × 104 cells per well in a 96-well plate. 24 hours later, the cell confluence reached 80–90%. The cell medium (0.1 mL) was replaced with Opti-MEM (0.1 mL) with reduced serum (75% serum reduction), followed by the addition of 2.4 μL of the sample solution (the final mRNA concentration in the medium is 80 ng per well, the peptide concentration is 2 μM). As the control, LipoMMAX with mRNA was prepared according to the manufacturer's protocol with a LipoMMAX:mRNA ratio of 3 (80 ng mRNA, 0.24 μL LipoMMAX/well). After 24 hours of incubation at 37 °C in the presence of 5% CO2, the cells of GFP-expression were imaged under a fluorescence microscope (Nikon, Eclipse Ti Motorized Inverted Microscope) with ×10 objective at 200 ms with the light shutter at 8. The transfection efficiency was quantified with flow cytometry on a FACS Aria III flow cytometer (BD Biosciences, USA). The mean fluorescence intensity was calculated by averaging cells with positive signals. The above cells transfected with co-assemblies for 24 hours were trypsinized and resuspended in complete DMEM for FACS analysis with a 488 nm laser channel. For each sample, a minimum of 5000 events were analyzed, and three repeats were averaged.Cell viability by MTT
To assess cell viability, HeLa cells were seeded at a density of 1.5 × 104 cells per well in a 96-well plate one day before the MTT assay to achieve approximately 80% confluency. On the second day, the complete DMEM medium was replaced with Opti-MEM with reduced serum (75% serum reduction), followed by the addition of pepMAX2–mRNA under optimal conditions (30 min pre-incubation with 250 mM NaCl, with peptide C = 2 μM for 80 ng mRNA loading). The MTT assay was performed at 24, 48, and 72 hours post-transfection following the manufacturer's protocol. In brief, 10 μL of MTT stock solution (12 mM) was added to each well and incubated for 4 hours. Subsequently, all media were replaced with 100 μL of DMSO to dissolve the formazan crystals, and after a 10-minute incubation at room temperature, the absorbance was measured at 540 nm using a microplate reader to calculate cell viability relative to controls.Intracellular trafficking by confocal microscopy
Live cell imaging with an SP8 confocal microscope (Leica Microsystems, Germany) was used to assess the intracellular distribution of the delivered mRNA. HeLa cells at a density of 3.75 × 104 cells per well were seeded in a confocal dish (0.25 mL DMEM) with a central glass disc on the bottom. The following day, the complete DMEM medium (0.25 mL) was replaced with Opti-MEM (0.25 mL), then 6 μL (80 ng mRNA/100 μL) of freshly prepared Cy5-labeled co-assemblies was added and gently mixed into the cell culture medium. The late endosomes and lysosomes were stained by Lysotracker Red (DND99, 50 nM) immediately after the co-assemblies’ addition. The cells were observed under an SP8 confocal microscope equipped with ×63 objective and cell incubator for live cell imaging (Tokahit temperature & CO2 module). Cell images with the expressed EGFP proteins, red late-endo/lysosomes and blue mRNA with laser wavelengths of 488 nm, 574 nm and 633 nm were collected in layers under the sequential model in SP8. 10 locations (10–20 cells per location) were randomly selected as the observation points, and videos were recorded for 3 hours at 1 min time intervals, 20 min after sample addition. The sample-containing medium was removed after 6-hour incubation and washed twice and changed with complete DMEM. After medium change, the cells were directly observed under an SP8 confocal microscope equipped with ×63 objective. All the laser intensity and gain values were set to be the same under different time points with 488 nm at 1.0 intensity 100% gain, 574 nm at 0.3 intensity 100% gain and 633 nm at 1.0 intensity 100% gain, where the blank control showed no signals for all the channels. LipoMMAX was used as the control with the same mRNA concentration at a LipoMMAX:mRNA ratio of 3 (80 ng mRNA, 0.24 μL LipoMMAX/100 μL).
Internalization mechanism study
The same as for the transfection efficiency measurements, HeLa cells were seeded with a density of 1.5 × 104 cells per well in a 96-well plate, the day before the measurement. 24 hours later, the cell medium (0.1 mL) was replaced with Opti-MEM (0.1 mL) containing various inhibitors, including methyl-β-cyclodextrin (MβCD) (2.5 mM, 3.2 mg mL−1), sodium azide (NaN3) (125 mM, 8.13 mg mL−1), chlorpromazine (CPM) (17 μM, 6 μg mL−1), wortmannin (525 nM, 225 ng mL−1) or amiloride (AM) (30 μM, 7.983 μg mL−1). The concentration of inhibitors was based on literature reports,9,11,39 while adjusting for cell viability obtained in our own experiments to achieve 80–90% cell viability. After 1 hour treatment, 2.4 μL of the sample solutions (the final mRNA concentration in the medium is 80 ng per well, the peptide concentration is 2 μM) were added. After another 4 h of incubation, the cells were washed three times with PBS buffer containing 0.5 mg mL−1 heparin sodium salt to ensure the removal of particles attached to the cell surface. The above cells were trypsinized and resuspended in complete DMEM for FACS analysis with a 633–647 nm laser channel. The mean fluorescence intensity was calculated by averaging all the live cells. For each sample, a minimum of 5000 events were analysed, and three repeats were averaged. The inhibition rate (%) was calculated using the following formula:where Ip is the mean fluorescence intensity of cells incubated with co-assemblies only (particles), Ih is the fluorescence intensity of cells incubated with co-assemblies and inhibitors, and Ib is the fluorescent intensity of the control group where cells were untreated (blank).
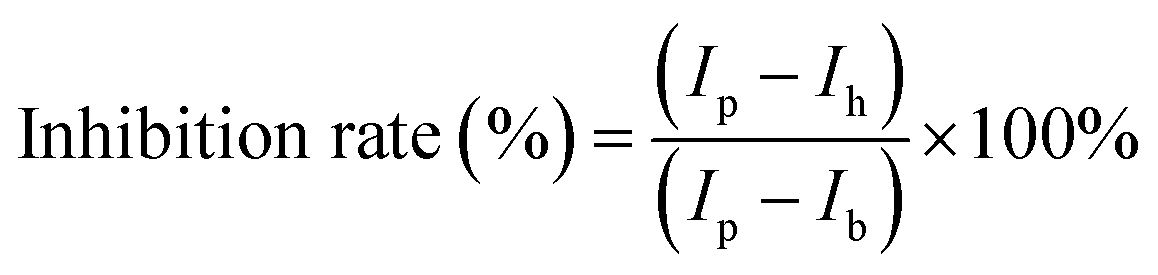
Statistical analysis
Statistical analyses were conducted using two-way analysis of variance (ANOVA) accompanied by Tukey's multiple comparison test within GraphPad Prism 10 software. This approach enabled comparisons between each treatment group and their respective control groups. Statistical significance was determined at p < 0.05, with the following thresholds for significance: **** for p < 0.0001, *** for p < 0.0002, ** for p < 0.0021, and * for p < 0.0332.Results
Obtain and characterize micron-sized peptide/mRNA co-assemblies
The size of co-assemblies was influenced by the salt concentration and the duration of pre-incubation. We first investigated the effect of salt concentration on the co-assemblies by keeping the incubation time as 30 minutes.As shown in Fig. 2(A), the hydrodynamic size and polydispersity index (PDI) of the co-assemblies were monitored by DLS. Without salt, pepMAX2 co-assembled with mRNA into ∼300 nm nano-sized particles with a polydispersity of 0.13. The particle size and polydispersity increase with increasing salt concentration initially, with a peak size of 3.11 μm and polydispersity of 0.35 at 750 mM NaCl. Further increase of the salt concentration resulted in a decrease in particle size and polydispersity, and the size decreased to ∼300 nm at a polydispersity of 0.08 when pre-incubated with 5000 mM NaCl. As shown, the co-assemblies started to form micron-sized particles when the salt concentration reached 200 mM. Therefore, there was a salt concentration window in which to obtain the micron-sized particles. We then investigated the effect of incubation time on particle sizes. A NaCl salt concentration of 250 mM was chosen as a representative for the salt window to monitor the size changes of micron-sized co-assemblies after different incubation time intervals. As shown in Fig. 2(B), the hydrodynamic size of the co-assembles increased with increasing incubation time. After 30 minutes, the size of the co-assemblies increased from 0.38 μm to 1.21 μm, and all the polydispersity values were below 0.30, indicating the formation of a narrowly distributed population of particles.40 Further incubation caused the size to increase to 1.3–1.5 μm, and the polydispersity increased to above 0.30, indicating that large aggregates (3–4 μm according to DLS) were formed after pre-incubation for more than 40 min. Therefore, the optimal time window for the formation of micron-sized particles at 250 mM NaCl was 20–40 min. Formation of the micron-sized particles was also confirmed by relative turbidity measurement (Fig. S1A, ESI†). The relative turbidity of the co-assemblies increased after incubation and reached the maximum at 40 min and dropped afterwards. The increased turbidity up to 40 min was due to the size enlargement of the co-assemblies, while the subsequent drop in turbidity (from 40–90 min) was likely a result of particle aggregates falling out of solution. Since the particles would be diluted into the medium before entering the cells, we also investigated the particle size change after dilution by 40 times with medium (DMEM, 110 mM NaCl, ionic strength 169 mM41). Here, the dilution factor was the same as in the subsequent transfection efficiency measurement and the size of the co-assemblies was monitored for 60 minutes. Fig. S1B (ESI†) showed that the nano-sized co-assemblies increased from 0.30 μm to 0.65 μm, while the size of micron-sized co-assemblies only changed negligibly (e.g., from 1.10 μm to 1.05 μm), indicating that the micron-sized co-assemblies remained stable after dilution.
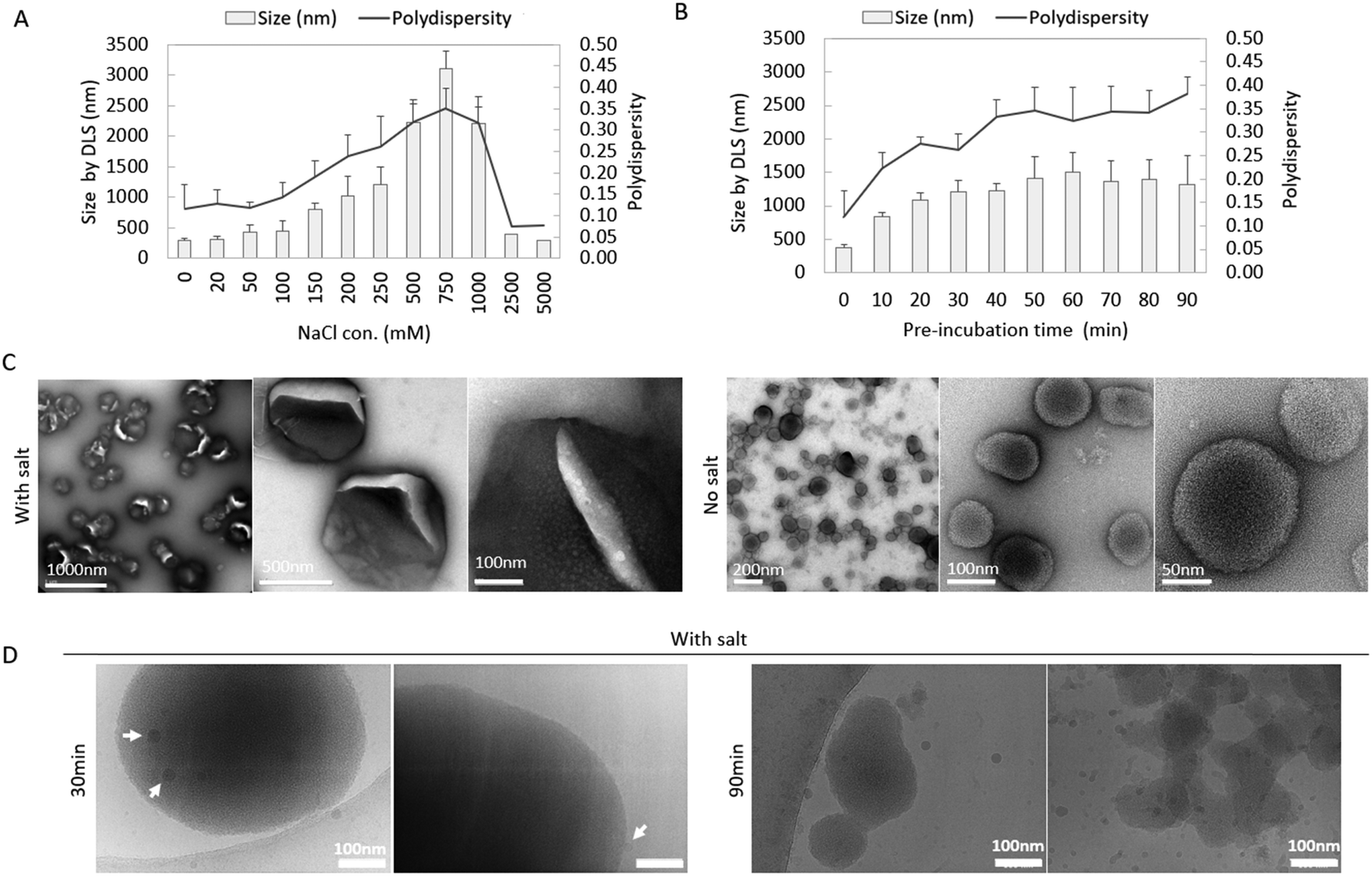 | ||
| Fig. 2 Characterization of peptide/mRNA co-assemblies. Size (bar) and polydispersity (line) of the co-assemblies pre-incubated (A) without/with salt solution for 30 min, and (B) with salt (250 mM NaCl) for different time intervals by DLS (data shown are mean ± SD, n ≥ 3). (C) TEM images of micron-sized and nano-sized co-assemblies formed in the presence/absence of salt. (D) Cryo-EM images of the co-assemblies pre-incubated in salt for 30 min and 90 min. White arrows indicate small particles likely formed from the peptide themselves. | ||
Morphologies of the co-assemblies (30 min pre-incubation with/without 250 mM NaCl) were observed by TEM (Fig. 2(C)), Cryo-EM (Fig. 2(D)) and confocal microscopy (Fig. S1C, ESI†). With Cy5-labled mRNA, micron-sized fluorescence round particles could be observed in the confocal images (Fig. S1C, ESI†). A RiboGreen assay was performed to quantify both the unencapsulated mRNA remaining in the solution and the amount successfully encapsulated. As illustrated in Fig. S2 (ESI†), the encapsulation efficiency across different salt concentrations (0 mM, 250 mM, and 5000 mM) ranged from 70% to 80%, with no significant differences detected (with two-way ANOVA statistics). The pepMAX2/mRNA co-assemblies with/without salt were observed in TEM images. As in Fig. 2(C), co-assemblies formed in salt had a diameter around 500 nm and there were cracks on the particles’ surface, where the cracks might be the drying effect from TEM sample preparation. Note that the size by TEM was much smaller than that of DLS, which might be due to not only the hydrodynamic layer, but also the shrinkage of the particle after drying. In comparison to the co-assemblies formed in salt, the co-assemblies formed without salt for 30 min showed a much smaller size at ∼100 nm, indicating the importance of the salt during micron-sized particles formation. For peptides co-assembled with mRNA in salt solution, the charges on these two components were partially shielded, which might lead into reduced charge repulsion and higher chance of hydrophobic interactions. To avoid any artefacts due to drying effects, we also observed the micron-sized co-assemblies by Cryo-EM (30 min pre-incubation with 250 mM NaCl) (Fig. 2(D)). At 30 min, large co-assemblies with sizes of 0.5–1.0 μm could be observed; at 90 min, amorphous aggregates formed, consistent with the polydispersity (PDI) increase observed by DLS (Fig. 2(B)). Cryo-EM also revealed an interesting phenomenon that small particles with a size of about 20 nm (indicated by white arrows in Fig. 2(D)) could be observed on the surface of the large particles (1.0 μm). We speculated that these small particles might be formed by the peptide themselves. According to a recently proposed peptide assembly process,42 peptides first self-assemble into solute-rich liquid droplets which act as nucleation sites for further assemblies’ formation. PepMAX2 might have also first assembled into metastable solute-rich droplets at 30 min and continued into aggregate formation. This solute-rich droplets hypothesis could explain the severe drying effect as observed from the TEM images. These findings suggested that our co-assemblies might have some liquid nature.
Release study of pepMAX2–mRNA co-assemblies
We hypothesize that a higher concentration of anionic macromolecules in the cytosol may trigger the release of mRNA from co-assemblies by competing for binding sites and disrupting its interaction with peptides. Gel electrophoresis was performed using optimized pepMAX2–mRNA (250 mM NaCl, incubated for 30 minutes) in release media of HEPES (10 mM) or heparin (0.4 mg mL−1) at various time points (30 minutes, 1 hour, and 4 hours) at 37 °C. Here, the negatively charged heparin is used as a model anionic macromolecule.17 The results demonstrated that the mRNA bands in the heparin release medium closely resembled those of the free mRNA control at all time points, indicating effective mRNA release (Fig. S3, ESI†). Conversely, no bands were detected in the HEPES condition, suggesting that HEPES does not facilitate mRNA release under these circumstances. The findings imply that heparin competes with the peptide for binding to the mRNA, thereby promoting its release from the complexes. This underscores the significance of ionic interactions in enhancing mRNA disassociation from these micron-sized co-assemblies. We have observed a similar phenomenon in other self-assembling peptide/nucleic acid complexes that were previously investigated by our research group.13,17Computational simulation of interactions in co-assemblies affected by salt
Computational simulation was carried out to understand the change in particle size with salt concentration and to reveal the possible liquid nature of the co-assemblies. We performed all atomistic molecular dynamics (MD) simulations and binding energy estimations to understand pepMAX2–pepMAX2 and pepMAX2–mRNA interactions at different salt conditions. PepMAX2–pepMAX2 showed positive binding energy at all three salt concentrations, which was mainly due to electrostatic repulsion. The result is consistent with our understanding of pepMAX2 because the peptide is positively charged at pH 7.4. As for pepMAX2–mRNA, negative binding energy was observed at all three salt concentrations, indicating the attractive force between pepMAX2 and mRNA. Interestingly, the absolute value of the overall binding energy increased from ∼8600 kJ mol−1 to ∼10500 kJ mol−1 when the salt concentration increased from 0 mM to 250 mM, while it decreased to ∼7200 kJ mol−1 when the salt concentration increased further to 2500 mM. The trend was attributed to the change in electrostatic interactions between pepMAX2 and mRNA, where the electrostatic energy changed from −10000 kJ mol−1, −11900 kJ mol−1 to −7900 kJ mol−1 at these three salt concentrations (Table S1, ESI†). This non-monotonic dependence of binding energy on salt concentration corresponded to the observed trend of the particle size.
We observed in MD simulations that pepMAX2 molecules are structurally stretched and repulse each other in the absence of salt, and supporting video S1 (ESI†) shows the dynamic repulsive interactions between two pepMAX2 molecules. When the NaCl concentration is at 250 mM, we observed partial electric screening of charges on pepMAX2 molecules by salt molecules and formation of co-assemblies (supporting video S2, ESI†). Further addition of salt to 2500 mM results in complete binding and formation of a counterion layer on peptides, completely shielding the charge interactions between peptide and mRNA, thus lowering the co-assembling propensity. Supporting video S3 (ESI†) shows a layer of salt ion clusters formed on pepMAX2 molecules, which appear to hinder the peptide from interacting with mRNAs. Consistent with the results in this study, Li et al. previously reported that forming ion clusters in the medium at very high salt concentrations reduces the assembly formation.43,44 These observations help to comprehend that the medium salt concentration provides optimum ion shielding for co-assembly formation.
The possible liquid nature of the co-assemblies was probed by calculating the aggregation propensity (AP) of pepMAX2 and model mRNA molecules. AP is defined as the ratio of solvent-accessible surface area (SASA) of pepMAX2–mRNA in the initial energy-minimized configuration to the final configuration.44–46 The AP values of pepMAX2–mRNA estimated using 400 ns trajectories of all atomistic MD simulations vary from 1.6 to 2.0 (Table S2 and Fig. S4 in the ESI†). With AP > 1.0 at all conditions, it is evident that pepMAX2 and mRNA form co-assemblies. When AP values fall within the range of 1.6 to 2.0, the assemblies are fluidic. As reported previously, dipeptides and tripeptides with AP values in the range of 1.5 to 2.0 exhibited liquid-like assemblies, as confirmed by microscopic observations,45 DOSY spectra (diffusion ordered spectroscopy) measured diffusion rates and secondary structures estimated using FTIR absorption spectra.44–46 The computational calculations corresponded with our experimental observations and provided indirect evidence of the liquid-like assembly behavior of co-assemblies at all three salt concentrations considered.
Transfection efficiency of co-assemblies
The transfection efficiency of co-assemblies was monitored under different formation conditions (varying salt concentration and incubation time). We first investigated the transfection efficiency of co-assemblies formed in different salt concentrations while keeping the incubation time identical. As shown in Fig. 3, the co-assemblies pre-incubated without salt (with sizes ∼0.30 μm) transfected 27% of the cell population. When the salt concentration was increased from 0 to 50 mM, the size of the co-assemblies barely increased, while the transfection efficiency dropped to 16%. This might be due to changes in interactions after salt addition, which altered other features of the co-assemblies, such as peptide/mRNA interaction, liquid/solid nature, or the stability. A further increase in salt concentration to 250 mM increased the transfection efficiency to 90%. Further increase in salt concentration from 250 mM to 750 mM resulted in an increase in the size of co-assemblies from 1.28 μm to 3.11 μm, and a progressive decrease in transfection efficiency, probably due to the upper bound for cellular uptake.47 While further increase of NaCl from 750 mM up to 5000 mM decreased the particle size to 0.30 μm with a drop in transfection efficiency. Although concentrations of 2500–5000 mM showed the same low transfection efficiency as the 0–150 mM condition, this similarity could be due to entirely different factors, such as variations in ionic strength and osmolarity, rather than nano size effects. However, this study primarily focuses on the relationship between size and transfection efficiency, which has been achieved within mild salt concentrations (0 mM and 250 mM). After settling at an optimal salt concentration (250 mM), we then investigated the effect of incubation time keeping the same salt concentration. As shown in Fig. 4, co-assemblies pre-incubated for 15 min to 40 min showed the highest transfection efficiency, while longer incubation time reduced the transfection efficiency. This trend mirrored the size measurements in Fig. 1(B) and reflected the colloidal stability – the co-assemblies formed micron-sized particles (∼1.0 μm) after 15 min and large aggregates after 40 min (∼1.4 μm, with PDI > 0.3). LipoMMAX with the same mRNA loading was compared with the co-assemblies. The co-assemblies had lower cytotoxicity, transfected a higher percentage of cells (90%), and attained a higher protein expression (2-fold of LipoMMAX). Therefore, the optimal transfection condition for pepMAX2/mRNA was pre-incubation in 250 mM salt for 15–40 min, where micron-sized particles of about ∼1–1.3 μm with PDI < 0.3 were formed.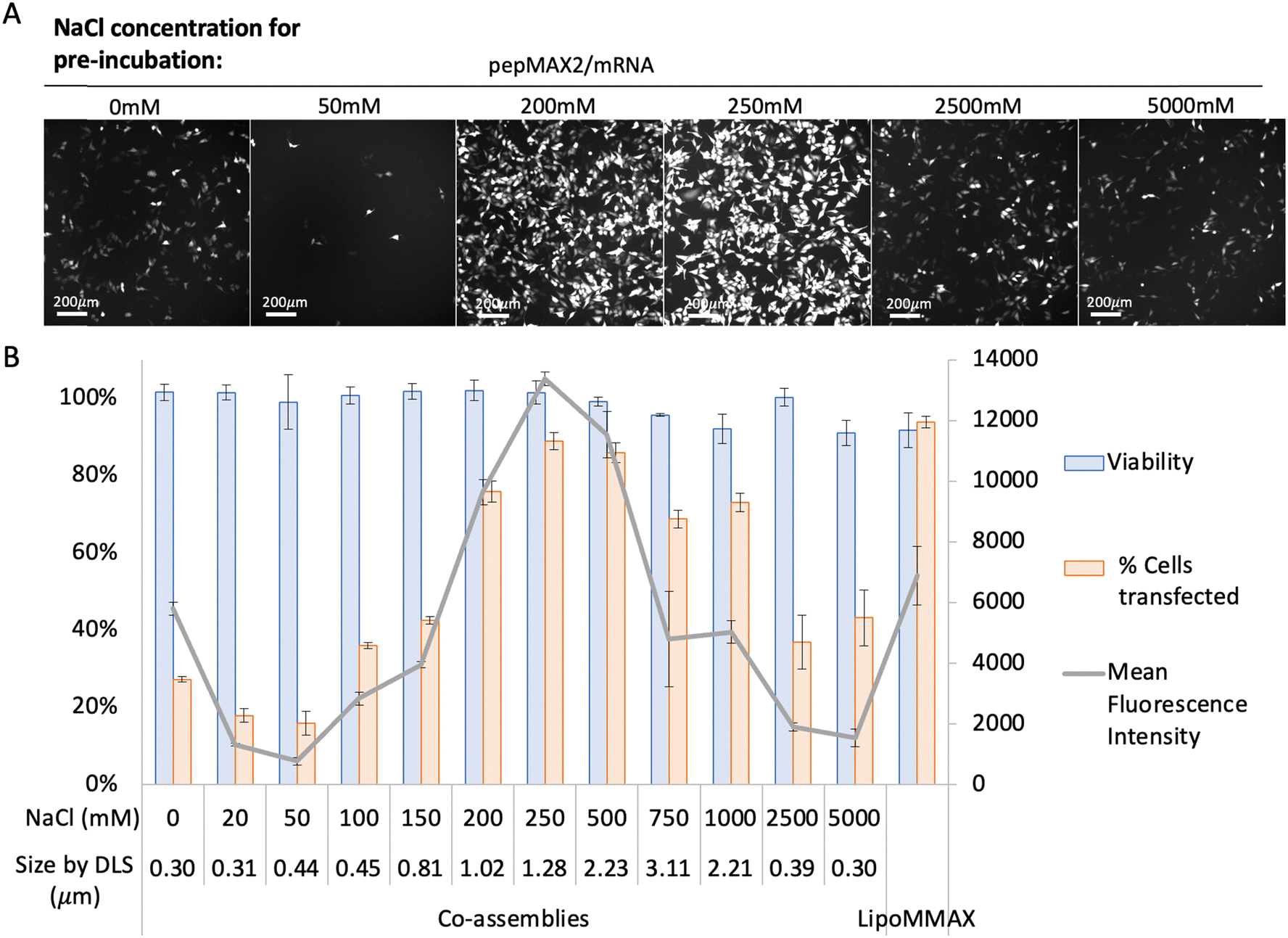 | ||
| Fig. 3 Transfection efficacy of pepMAX2/mRNA pre-incubated for 30 min in different concentrated NaCl solutions. The N/P ratio was kept the same at 4, with peptide concentration at 2 μM for 80 ng loading. The transfection efficiency was observed by (A) fluorescence microscopy and quantified by (B) flow cytometry, with cell viability, percentage of cells transfected and mean fluorescence intensity (MFI) of the positive cells. LipoMMAX served as a control with the same mRNA loading as in the co-assemblies (data shown are mean ± SD, n ≥ 3). | ||
 | ||
| Fig. 4 Transfection efficacy of pepMAX2/mRNA pre-incubated in 250 mM NaCl solution for different time intervals. The N/P ratio was kept the same at 4, with the peptide concentration at 2 μM for 80 ng loading. The transfection efficiency was observed by (A) fluorescence microscope and quantified by (B) flow cytometry, with cell viability, percentage of cells transfected and mean fluorescence intensity (MFI) of the positive cells. LipoMMAX served as a control with the same mRNA loading as 80 ng/100 μL in the co-assemblies (data shown are mean ± SD, n ≥ 3). | ||
Cell viability by MTT
In our study, we performed an MTT assay using the optimized PepMAX2 formulation (250 mM NaCl with a 30-minute incubation time) to evaluate the long-term effects of these micron-sized carriers at 24, 48, and 72 hours post-transfection. The results, presented in Fig. S5 (ESI†), demonstrated that cell viability remained high, exceeding 80% even at the 72-hour mark following transfection with pepMAX2. Statistical analysis conducted via two-way ANOVA indicated no significant differences in viability between the positive control and our treatment across triplicate data sets. These findings suggest that our optimized transfection reagent is safe and exhibits minimal toxicity in HeLa cells.Intracellular trafficking of co-assemblies
To study the cellular entry and intracellular release dynamics of micron-sized co-assemblies, time-lapse confocal imaging was recorded. We tracked the Cy5-labeled mRNA in HeLa cells and labelled the late endosome/lysosome with Lysotracker Red. Sustained cellular uptake was observed from 20 min after sample addition (supporting video 4, ESI†). One delivery event was shown as a representative to reveal that a fast cellular entry of co-assemblies could occur within 10 min (Fig. 5(A)). No colocalization between co-assemblies and Lysotracker signal was observed at this time point. Approximately 2 hours post sample addition, release of mRNA into the cytosol was evident. As shown from 120 min to 160 min, the peripheral area of the delivered mRNA (blue dot pointed by arrow) gradually brightened, indicating the slow release of mRNA into the cytosol (supporting video 5, ESI†). Therefore, we considered these co-assemblies to have successfully avoided the lysosomal digestion. At 6 hours, cells transfected by micron-sized co-assemblies started producing EGFP (Fig. 5(B)). Within these high-expressing cells, bright blue dots and evenly distributed blue colour could be observed. These bright dots were not colocalized with lysotracker and might serve as depots of mRNA in the cytosol, continuing to release mRNA and prolong EGFP production, resulting in a bright signal 24 hours after transfection. In comparison, cells treated with nano-sized co-assemblies showed many small blue dots with no observable spread of the dye-labelled mRNA in the cytosol. The lack of apparent diffusion might be due to the limitation of resolution under such low cytosol concentration or a result of different trafficking causing the nano-sized assemblies to be confined within some vesicles such as recycling endosomes or arrested endosomes, where they were trapped but cannot be revealed by lysotracker.48 For LipoMMAX with the same amount of mRNA loaded, no bright dots were observed, while the mRNA was evenly distributed inside the cytosol. We reasoned that the higher level of protein expression of the micron-sized co-assemblies was caused by the higher cellular uptake, more efficient endosomal escape and/or the sustained release of mRNA in the cytosol.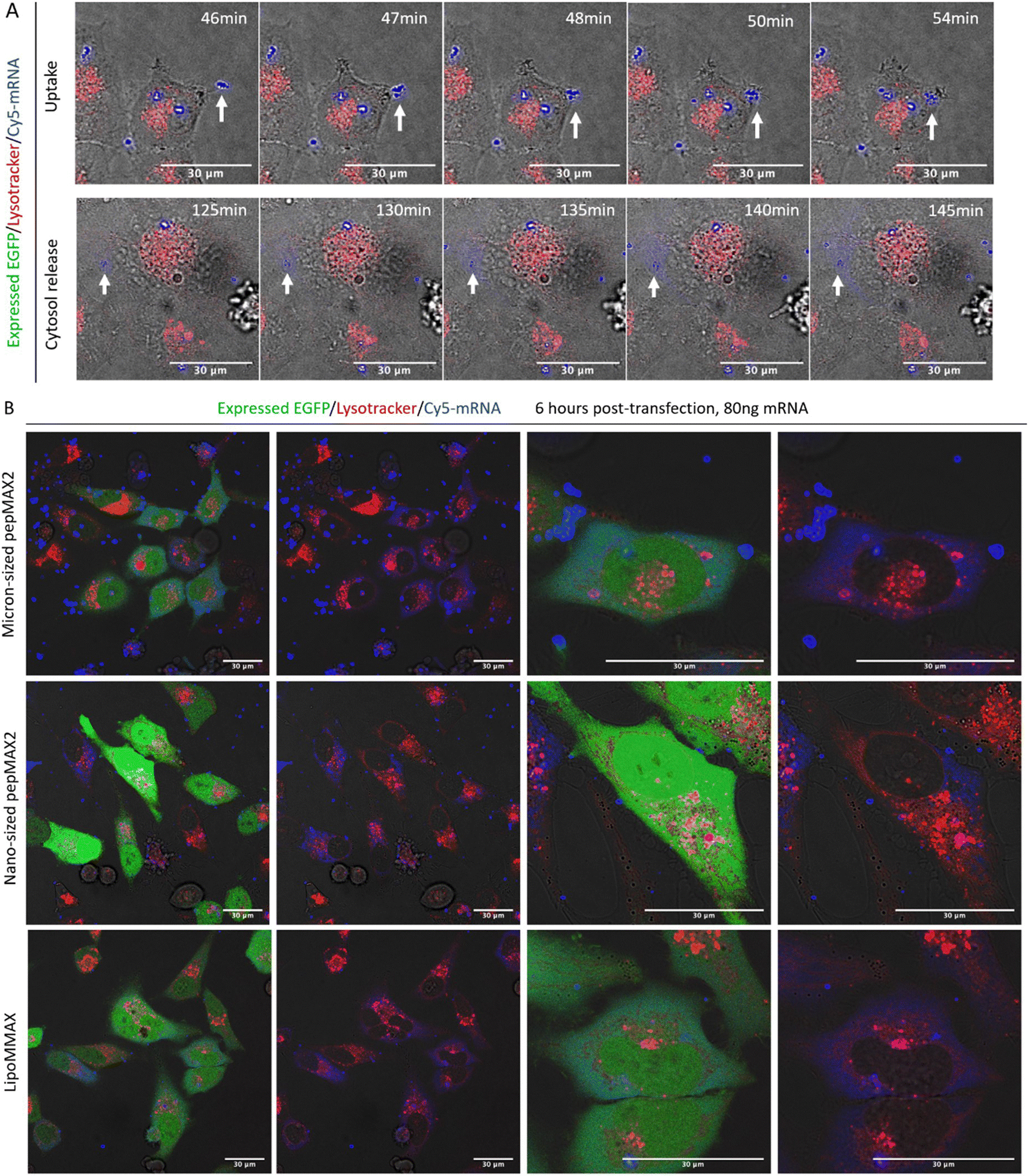 | ||
| Fig. 5 Confocal microscope images revealing subcellular location of co-assemblies. (A) Real-time tracking of a delivery event by micron-sized co-assemblies. Time-lapse imaging of mRNA delivery including cellular uptake 20–120 min post-transfection and mRNA release 120 min post-transfection. Direct cytosol delivery of mRNA without endosomal entrapment was observed (also see the supporting videos in the ESI†) (B) mRNA distribution inside cells at 6 h post transfection by micron-sized and nano-sized co-assemblies. LipoMMAX served as a control with the same mRNA loading. Color code: red for Lysotracker Red, green for EGPF, and blue for Cy5 labeled-mRNA. Scale bar: 30 μm. | ||
Endocytic pathways of co-assemblies
The internalization mechanism of the co-assemblies was explored by pre-treating the cells with a panel of endocytosis inhibitors. Methyl-β-cyclodextrin (MβCD) was used to inhibit cholesterol-dependent lipid rafting, sodium azide (NaN3) to inhibit the energy-dependent endocytosis, chlorpromazine (CPM) against the clathrin-mediated endocytosis, and wortmannin or amiloride (AM) against the macropinocytosis. As shown in Fig. 6, MβCD and NaN3 inhibited approximately 90% of the uptake, suggesting that the mechanism of micron-sized co-assemblies’ uptake was cholesterol-dependent lipid rafting and energy dependent. Other inhibitors were negligible, suppressing the uptake by less than 30%. Since the inhibition was dose-dependent and varied in different cells, the concentration of inhibitors was based on literature reports9,11,39 and adjusted for cell viability obtained in our own experiments. The concentration of inhibitors was able to maintain cell viability at 80–90%. We also investigated the endocytosis pathway for nano-sized co-assemblies formed in the absence of salt. The results indicated these nano-sized co-assemblies also entered by a lipid-raft energy-dependent pathway, similar to the micron-sized co-assemblies. Internalization via cholesterol-dependent lipid raft was employed by other systems with high cytosolic delivery, such as phase-separated peptides for protein and mRNA delivery9 and arginine nanoparticles (ArgNPs) carrying Cas9 protein complexed with a guide RNA (sgRNA).11 For non-phagocytic cells, the conventional endocytosis pathways include clathrin-coated pit-mediated endocytosis (CME), fast endophilin-mediated endocytosis (FEME), caveolae endocytosis (Cav) and macropinocytosis.49 The classical sizes for these endocytic vesicles (diameter) are ∼100 nm spherical for CME, ∼60–80 nm tubular for FEME, ∼60 nm for Cav and 200 nm–5 μm for macropinocytosis.49,50 However, studies on the relationship between particle sizes and internalization pathways suggested that particles of 500 nm in size predominantly entered the cells via caveolae endocytosis (Cav) and the delivery to the lysosomes was subtle.47 For our micron-sized particles, the major pathway was not macropinocytosis, where neither wortmannin nor amiloride (AM) inhibited the uptake, while amiloride (AM) even increased uptake. Therefore, among conventional endocytosis pathways, we considered that these co-assemblies were most likely to be endocytosed by Cav, as this trafficking was influenced by cholesterol and allowed entry of large particles. Novel pathways that are not yet fully understood could be involved. Another possible explanation is that the micron-sized particles mainly entered by direct fusion with the cell membrane. As we observed that mRNA was released into the cytosol (Fig. 5) before being transported to lysosomes, we considered that this cytosol delivery occurred prior to vesicle maturation and that efficient release might impede maturation of endosomal vesicles. Besides successful cytosol delivery by micron-sized co-assemblies, nano-sized co-assemblies entering cells also via a lipid-raft energy-dependent pathway failed to attain efficient cytosol delivery, suggesting that micron size is a critical prerequisite for direct cytosol entry. | ||
| Fig. 6 Cell internalization mechanism study of co-assemblies (pre-incubated with/without salt). Flow cytometry histogram of HeLa cells treated with various inhibitors before incubation with co-assemblies with Cy5-mRNA for 4 h. Two groups were included as the control: totally untreated cells (blank) and cells treated by co-assemblies without any inhibitors (particles). Both cholesterol-depletion compound MβCD and energy-depletion compound NaN3 inhibit cell uptake. | ||
Caveolae endocytosis occurs in many cell types, including endothelial cells, smooth muscle cells, adipocytes, and fibroblasts.51 To determine whether the observation could be generalized to other cell types, transfection experiments were repeated on SKNMC and PC12 cells, which also allow a caveolae endocytosis pathway.52,53 As shown in Fig. S6 (ESI†), 90% of cells were transfected using micron-sized particles (200 mM NaCl, 30 min pre-incubation), while only 50% were transfected by nano-sized co-assemblies. Transfection efficiency reached a maximum and started to plateau using co-assemblies pre-incubated for 15 min (Fig. S7, ESI†). Although the optimal co-assembly size of SKNMC (∼1.02 μm) was slightly different, a similar trend in transfection efficiency compared to HeLa cells supports our assumption that micron size is a prerequisite. For the PC12 cells, which are known to be difficult to transfect, we evaluated the efficacy of our pepMAX2–mRNA co-assembly system at an optimal NaCl concentration of 250 mM, utilizing pre-incubation times of 20 and 40 minutes. The results revealed that transfection efficiency reached a maximum of only 50% for cells treated with our system, irrespective of the incubation time. Additionally, the findings indicated no significant differences when compared to the results obtained with LipoMMAX, as assessed by two-way ANOVA statistics (Fig. S8, ESI†). Excellent transfection efficiency across various cell types supports our hypothesis regarding the relationship between micron-sized particles and caveolae-mediated endocytosis. Additionally, these findings highlight the potential of using this approach for mRNA delivery, particularly in cells that utilize caveolae pathways.
Discussion
Endosomal barriers are regarded as the bottleneck for nucleic acid delivery. Aside from classical approaches of engineering on nanoparticles, we demonstrated how micron-sized carriers circumvent the classical endocytic pathways and succeed in direct cytosol entry. By engineering peptide chemistry and controlling the assembling process with mRNA, we transformed a nano-sized delivery system into a micron-sized delivery system. These micron-sized co-assemblies successfully bypassed lysosomal digestion and delivered mRNA into the cytosol, transfecting approximately 90% of cells and doubling the protein expression levels compared to that achieved by a commercially available reagent, LipoMMAX. The size of the co-assemblies could be adjusted by salt during peptide/mRNA pre-incubation. From the experimental observation and computational results, we found that the charge–charge interaction between peptide and mRNA played a crucial role in regulating the co-assembling. The computational study showed that the optimal shielding of peptide by salt counterions at medium salt concentration facilitated the formation of micron-sized co-assemblies with high transfection efficiency. The non-monotonic change in binding energy with salt concentration verified the experimental trend of particle size. Moreover, the AP values indicated the liquid-like nature of the co-assemblies, which could be advantageous for efficient cytosolic delivery.9 Mechanisms of uptake and intracellular trafficking of these micron-sized co-assemblies were studied to understand the high transfection efficiency. Live cell confocal microscopy revealed fast entry of micron-sized particles, followed by subsequent mRNA release and diffusion in the cytosol for a prolonged period. This direct entry into the cytosol, bypassing the conventional endocytic pathways and avoiding lysosomal degradation, is attributed to the high transfection efficiency. Although the trafficking mechanism is not fully understood yet, the intracellular pathway for these micron-sized co-assemblies involves cholesterol-dependent lipid rafting. While nano-sized co-assemblies also entered cells via the lipid-raft energy-dependent pathway, they failed to attain efficient cytosol delivery, suggesting that micron size is likely a critical prerequisite for direct cytosol delivery. A similar trend in the correlation between transfection efficiency and particle sizes in other caveolae-associated cell lines (SKNMC and PC12) suggests the potential utility of this direct entry in other caveolae cell types. This study demonstrates the potential of peptide-based micron-sized carriers for non-viral delivery of mRNA.Conclusion
This study presents a novel approach to improve the delivery of messenger RNA (mRNA) into cells, a critical step in developing various biomedical applications, such as vaccines and protein replacement therapies. We designed a unique 27-amino acid peptide called pepMAX2, which can spontaneously co-assemble with mRNA to form micron-sized particles. These particles can efficiently deliver mRNA directly into the cell's cytosol, bypassing the endosomal barriers that often limit the effectiveness of traditional nano-sized carriers. By optimizing the size and formation conditions of these peptide/mRNA co-assemblies, we could achieve a transfection efficiency that surpassed commercially available reagents, with minimal cytotoxicity and high protein expression levels. The findings of this study have broad implications for the design of synthetic carriers for mRNA delivery, offering a promising alternative to conventional nano-sized carriers. Furthermore, the study provides valuable insights into the cellular entry mechanisms of these micron-sized particles, which involve cholesterol-dependent lipid rafting and energy-dependent pathways. This knowledge can help guide the development of more efficient and targeted delivery systems for various therapeutic applications. Overall, the innovative approach presented in this study has the potential to advance the field of mRNA delivery significantly, opening new possibilities for treating a wide range of diseases.Author contributions
Y. Chau, R. Feng, and K.S. Fernando initiated and conceptualized the research project. R. Feng conducted experiments including (1) peptide synthesis and purification, (2) designed experiments to obtain nano- and micron-sized co-assemblies, (3) conducted co-assembly characterization including DLS, turbidity, TEM and Cryo-EM, (4) transfection efficiency optimization, (5) intracellular trafficking by confocal microscopy, (6) cellular uptake mechanism study, and (7) transfection efficiency study in SKNMC cells. M. Rafiei conducted experiments including (1) co-assemblies’ encapsulation, (2) release study, (3) cell viability by MTT, and (4) transfection efficiency study in PC12 cells. K.S. Fernando performed all the computer simulations, including (1) determination of pepMAX2 structure, (2) determination of necessary mRNA fragments, (3) perform MD simulations, and (4) calculate binding energy by MMPBSA and check fluidity with AP parameters. R. Feng and K. S. Fernando wrote the original draft. M. Rafiei supplemented experiments based on reviewer's suggestions and wrote the relevant sections. Y. Chau, R. Feng, K. S. Fernando and M. Rafiei collectively contributed to the writing, review, and editing process. Y. Chau provided supervision and guidance throughout the project.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Hong Kong Research Grants Council (grant no. GRF16309818) and the Chinese National Engineering Research Centre for Tissue Restoration and Reconstruction (grant no. ITC-CNERC14SC01). The EM dataset was collected at the Biological Cryo-EM Center, generously supported by a donation from the Lo Kwee Seong Foundation to HKUST. K.S. Fernando received financial support from the Hong Kong PhD Fellowship Scheme (HKPFS).References
- F. P. Polack, S. J. Thomas, N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, J. L. Perez, G. P. Marc, E. D. Moreira and C. Zerbini, N. Engl. J. Med., 2020, 2603–2615 CrossRef CAS.
- S. A. Meo, I. A. Bukhari, J. Akram, A. S. Meo and D. C. Klonoff, Eur. Rev. Med. Pharmacol. Sci., 2021, 25, 1663–1669 CAS.
- H. Ledford, Nature, 2020 DOI:10.1038/d41586-020-03593-7.
- T. Bus, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2018, 6, 6904–6918 RSC.
- S. C. Semple, A. Akinc, J. Chen, A. P. Sandhu, B. L. Mui, C. K. Cho, D. W. Y. Sah, D. Stebbing, E. J. Crosley, E. Yaworski, I. M. Hafez, J. R. Dorkin, J. Qin, K. Lam, K. G. Rajeev, K. F. Wong, L. B. Jeffs, L. Nechev, M. L. Eisenhardt, M. Jayaraman, M. Kazem, M. A. Maier, M. Srinivasulu, M. J. Weinstein, Q. Chen, R. Alvarez, S. A. Barros, S. De, S. K. Klimuk, T. Borland, V. Kosovrasti, W. L. Cantley, Y. K. Tam, M. Manoharan, M. A. Ciufolini, M. A. Tracy, A. De Fougerolles, I. MacLachlan, P. R. Cullis, T. D. Madden and M. J. Hope, Nat. Biotechnol., 2010, 28, 172–176 CrossRef CAS PubMed.
- M. Maugeri, M. Nawaz, A. Papadimitriou, A. Angerfors, A. Camponeschi, M. Na, M. Hölttä, P. Skantze, S. Johansson, M. Sundqvist, J. Lindquist, T. Kjellman, I. L. Mårtensson, T. Jin, P. Sunnerhagen, S. Östman, L. Lindfors and H. Valadi, Nat. Commun., 2019, 10, 4333 CrossRef CAS PubMed.
- M. A. Beach, S. L. Y. Teo, M. Z. Chen, S. A. Smith, C. W. Pouton, A. P. R. Johnston and G. K. Such, ACS Appl. Mater. Interfaces, 2022, 14, 3653–3661 CrossRef CAS.
- R. Goswami, T. Jeon, H. Nagaraj, S. Zhai and V. M. Rotello, Trends Pharmacol. Sci., 2020, 41, 743–754 CrossRef CAS PubMed.
- Y. Sun, S. Y. Lau, Z. W. Lim, S. C. Chang, F. Ghadessy, A. Partridge and A. Miserez, Nat. Chem., 2022, 14, 274–283 CrossRef CAS PubMed.
- J. Liu, R. Feng and Y. Chau, Matter, 2022, 5, 1637–1639 CrossRef CAS.
- R. Mout, M. Ray, G. Yesilbag Tonga, Y. W. Lee, T. Tay, K. Sasaki and V. M. Rotello, ACS Nano, 2017, 11, 2452–2458 CrossRef CAS PubMed.
- P. Boisguérin, K. Konate, E. Josse, E. Vivès and S. Deshayes, Biomedicines, 2021, 9, 583 CrossRef.
- R. Feng, A. C. Y. Chang, R. Ni, J. C. Y. Li and Y. Chau, ACS Appl. Bio Mater., 2022, 7, 3476–3486 CrossRef PubMed.
- R. Ni and Y. Chau, Angew. Chem., Int. Ed., 2017, 56, 9356–9360 CrossRef CAS PubMed.
- R. Ni and Y. Chau, Angew. Chem., Int. Ed., 2020, 132, 3606–3612 CrossRef.
- R. Feng, R. Ni and Y. Chau, ChemMedChem, 2021, 16, 3559–3564 CrossRef CAS PubMed.
- R. Ni, R. Feng and Y. Chau, ACS Appl. Nano Mater., 2023, 6, 3191–3201 CrossRef CAS.
- J. Kopeček and J. Yang, Acta Biomater., 2009, 5, 805–816 CrossRef PubMed.
- Y. Hong, M. D. Pritzker, R. L. Legge and P. Chen, Colloids Surf., B, 2005, 46, 152–161 CrossRef CAS.
- A. Iscen and G. C. Schatz, J. Phys. Chem. B, 2019, 123, 7006–7013 CrossRef CAS.
- Q. Zhang, Y. Liu, T. Xie, Y. Shang-guan, M. Tian, Q. Zhang and M. Cao, Colloids Surf., A, 2022, 637, 128252 CrossRef CAS.
- K. A. Curtis, D. Miller, P. Millard, S. Basu, F. Horkay and P. L. Chandran, PLoS One, 2016, 11, 1–20 Search PubMed.
- Y. Sang, K. Xie, Y. Mu, Y. Lei, B. Zhang, S. Xiong, Y. Chen and N. Qi, Cytotechnology, 2015, 67, 67–74 CrossRef CAS PubMed.
- M. Akishiba, T. Takeuchi, Y. Kawaguchi, K. Sakamoto, H. H. Yu, I. Nakase, T. Takatani-Nakase, F. Madani, A. Gräslund and S. Futaki, Nat. Chem., 2017, 9, 751–761 CrossRef CAS PubMed.
- M. Popenda, M. Szachniuk, M. Antczak, K. J. Purzycka, P. Lukasiak, N. Bartol, J. Blazewicz and R. W. Adamiak, Nucleic Acids Res., 2012, 40(14), e112 CrossRef CAS.
- M. Antczak, M. Popenda, T. Zok, J. Sarzynska, T. Ratajczak, K. Tomczyk, R. W. Adamiak and M. Szachniuk, Acta Biochim. Pol., 2016, 63(4), 737–744 CAS.
- B. R. Brooks, C. L. Brooks, A. D. Mackerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30(10), 1545–1614 CrossRef CAS.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29(11), 1859–1865 CrossRef CAS PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- K. S. Fernando and Y. Chau, A multi-scale computational model for liquid-liquid and liquid-solid phase separation of intrinsically disordered protein mimicking polymer peptide hybrids, ACS annual meeting Fall 2023, American Chemical Society, US, 2023.
- K. S. Fernando and Y. Chau, Modeling of Intrinsically Discorded Protein Mimicking Hybrids for Membraneless Organelles formation, MRS Annual Meeting 2023 spring, Material Research Society, US, 2023.
- K. S. Fernando, G. Jahanmir, J. Liu, I. C. Unarta and Y. Chau, Associate polymer theory-inspired modelling of intrinsically disordered protein-mimicking polymer-oligopeptide hybrids for the formation of artificial membranelles organelles, MRS spring meeting and exhibit 2021, Material Research Society, US, 2021.
- K. S. Fernando, F. Ruilu and Y. Chau, A molecular dynamics study for salt-mediated size-tunable designer peptide/mRNA co-assembly for efficient cytosol delivery, Controlled Release Society Annual meeting & Expo 2023, Control Release Society, US, 2023 DOI:10.13140/RG.2.2.33542.75845.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS.
- R. Kumari, R. Kumar and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef CAS PubMed.
- K. S. Fernando, G. Jahanmir, I. C. Unarta and Y. Chau, Langmuir, 2024, 40, 7607–7619 CrossRef CAS PubMed.
- K. S. Fernando and Y. Chau, J. Mater. Chem. B, 2024, 12(48), 12608–12617 RSC.
- J. Zhou and Y. Chau, Biomater. Sci., 2016, 4, 1462–1472 RSC.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. R. Mozafari, Pharmaceutics, 2018, 10, 1–17 CrossRef.
- D. Kwon, S. H. Lee, J. Kim and T. H. Yoon, J. Toxicol. Environ. Health Sci., 2010, 2, 78–85 CrossRef.
- C. Yuan, A. Levin, W. Chen, R. Xing, Q. Zou, T. W. Herling, P. K. Challa, T. P. J. Knowles and X. Yan, Angew. Chem., Int. Ed., 2019, 58, 18116–18123 CrossRef CAS PubMed.
- Y. Li, M. Girard, M. Shen, J. A. Millan and M. O. De La Cruz, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11838–11843 CrossRef CAS PubMed.
- P. W. J. M. Frederix, R. V. Ulijn, N. T. Hunt and T. Tuttle, J. Phys. Chem. Lett., 2011, 2, 2380–2384 CrossRef CAS PubMed.
- Y. Tang, S. Bera, Y. Yao, J. Zeng, Z. Lao, X. Dong, E. Gazit and G. Wei, Cell Rep. Phys. Sci., 2021, 2, 100579 CrossRef CAS.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- J. Rejman, V. Oberle, I. S. Zuhorn and D. Hoekstra, Biochem. J., 2004, 377, 159–169 CrossRef CAS PubMed.
- P. Paramasivam, C. Franke, M. Stöter, A. Höijer, S. Bartesaghi, A. Sabirsh, L. Lindfors, M. Yanez Arteta, A. Dahlén, A. Bak, S. Andersson, Y. Kalaidzidis, M. Bickle, M. Zerial, M. Y. Arteta, A. Dahlén, A. Bak, S. Andersson, Y. Kalaidzidis, M. Bickle and M. Zerial, J. Cell Biol., 2022, 221, 1–20 CrossRef PubMed.
- J. J. Rennick, A. P. R. Johnston and R. G. Parton, Nat. Nanotechnol., 2021, 16, 266–276 CrossRef CAS PubMed.
- H. S. Kruth, N. L. Jones, W. Huang, B. Zhao, I. Ishii, J. Chang, C. A. Combs, D. Malide and W.-Y. Zhang, J. Biol. Chem., 2005, 280, 2352–2360 CrossRef CAS PubMed.
- A. Echarri and M. A. Del Pozo, Curr. Biol., 2012, 22, 114–116 CrossRef.
- S. Il Choi, Y. S. Maeng, T. I. Kim, Y. Lee, Y. S. Kim and E. K. Kim, PLoS One, 2015, 10, 1–23 Search PubMed.
- S. Peiró, J. X. Comella, C. Enrich, D. Martín-Zanca and N. Rocamora, J. Biol. Chem., 2000, 275, 37846–37852 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01098a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |