
Regulation of macrophage uptake through the bio-nano interaction using surface functionalized mesoporous silica nanoparticles with large radial pores†
Juan
Wen
ab,
Chang
Lei
 *cdf,
Shu
Hua
ab,
Larry
Cai
c,
Huan
Dai
ab,
Siyuan
Liu
abef,
Yiwei
Li
df,
Saso
Ivanovski
ab and
Chun
Xu
*abef
*cdf,
Shu
Hua
ab,
Larry
Cai
c,
Huan
Dai
ab,
Siyuan
Liu
abef,
Yiwei
Li
df,
Saso
Ivanovski
ab and
Chun
Xu
*abef
aSchool of Dentistry, The University of Queensland, Brisbane, Queensland 4006, Australia. E-mail: chun.xu@sydney.edu.au
bCentre for Orofacial Regeneration, Reconstruction and Rehabilitation (COR3), School of Dentistry, The University of Queensland, Brisbane, Queensland 4006, Australia
cAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, St Lucia, QLD 4072, Australia
dSchool of Medical Sciences, Faculty of Medicine and Health, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: chang.lei@sydney.edu.au
eSydney Dental School, Faculty of Medicine and Health, The University of Sydney, Camperdown, NSW 2006, Australia
fCharles Perkins Centre, The University of Sydney, Camperdown, NSW 2006, Australia
First published on 29th October 2024
Abstract
Porous nanoparticles, such as mesoporous silica nanoparticles (MSNs), have garnered significant interest for biomedical applications. Recently, MSNs with large radial pores have attracted increased attention because their unique pore structure and large pore size are suitable for delivering large molecules such as proteins and genes. Upon entry into biological systems like the bloodstream, nanoparticles quickly form a ‘protein corona,’ leading to alterations in their interactions with immune cells. In this study, we investigated the formation of protein corona on MSNs with large radial pores and various surface modifications using mass spectrometry. We also examined the effects of protein corona on the interaction between MSNs and macrophages. We prepared MSNs with large, cone-shaped radial pores (>30 nm) and six different functional groups, resulting in nanoparticles with neutral, negative, and positive surface charges. Our findings indicate that surface functional groups significantly alter the composition of the protein corona, affecting the bio-nano interaction of these surface-modified MSNs with macrophages. Notably, nanoparticles with similar surface charges exhibited distinct corona characteristics and were internalized differently by macrophages. This underscores the crucial role of the protein corona in determining the fate, behavior, and biological responses of nanoparticles. Our research sheds light on the significance of understanding and controlling protein corona formation to optimize the design and functionality of nanoparticle-based biomedical applications.
 Chun Xu | Dr Chun Xu obtained his bachelor's and master's degrees from Wuhan University (China) in 2010 and 2012, respectively. He received his PhD in Chemistry and Bioengineering from The University of Queensland (Australia) under the supervision of Prof. Chengzhong Yu. Following his PhD, he undertook postdoctoral training at the University of California, Los Angeles (UCLA), and Stanford University. He is currently a senior lecturer and Sydney Horizon Fellow at the University of Sydney, Australia. Before that, he was a senior lecturer and group leader at the University of Queensland. His research focuses on the synthesis of novel nanomaterials and their biomedical applications. |
Introduction
Nanoparticles are increasingly recognized as promising platforms for biomedical application, particularly in drug delivery, due to their ability to enable controlled release, enhance safety profiles, and achieve targeted delivery.1,2 A wide range of therapeutic agents have been encapsulated within nanoparticles to protect them from degradation, prolong their circulation time, and improve their targeting efficiency.3–5 However, following administration, the mononuclear phagocyte system (MPS) often degrades and removes a significant portion of these nanoparticles from the bloodstream.6 Macrophages, as key components of the MPS, play a central role in the detection, uptake, and clearance of nanoparticles. Therefore, modulating macrophage recognition and phagocytosis is important for the effective design and application of nanomedicines.Upon interaction with biological fluids, such as blood, nanoparticles rapidly develop a protein corona within seconds due to the absorption of proteins and other biomolecules on their surfaces.7,8 This protein corona plays a crucial role in modulating the biomedical and physiological properties of nanoparticles, affecting key aspects such as circulation time, cytotoxicity, and targeting efficiency.9 Certain components of the corona, such as immunoglobulins and complement proteins (referred to as opsonins), promote cellular binding and uptake by macrophages, leading to the rapid clearance of nanoparticles. In contrast, dysopsonins, including histidine-rich glycoprotein, albumin, and clusterin, inhibit macrophage uptake, thereby prolonging the nanoparticles' circulation time.10–12 Surface functional groups are recognized as key determinants in protein binding, significantly affecting biological behaviors such as cellular uptake, biodistribution, and tissue diffusion.13,14 Understanding how surface engineering influences the composition of the protein corona and its recognition by macrophages is critical for optimizing nanoparticle blood residency time.
Among various nanoparticles, mesoporous silica nanoparticles (MSNs) are widely used for drug delivery due to their favorable properties, such as excellent biocompatibility, controllable particle sizes and morphologies, ease of surface modification, and tunable pore structures.15,16 Recently, MSNs with large radial pores (>10 nm) have garnered significant interest for their enhanced drug delivery capacity.2,17 Several studies have shown that the surface group,18 shape,19 and pores20 of MSNs affect protein corona formation. Tsang-Pai Liu et al.21 synthesized PEGylated MSNs with varying zeta potentials and demonstrated that culture conditions, specifically serum-free and serum-containing media, impact their uptake by Raw 264.7 macrophages. They identified the level of p-p38 on the MSN surface as a key factor, though detailed protein corona composition and macrophage endocytosis were not thoroughly investigated. A deeper understanding of the specific protein corona components on MSNs, particularly those with large radial pores, and their role in macrophage endocytosis would provide valuable insights for optimizing MSN-based drug delivery systems.
In this study, we systematically investigated the formation of specific protein coronas on mesoporous silica nanoparticles with a large radial pore (MSN-CC) featuring diverse surface chemistries, and evaluated their impact on macrophage uptake. We successfully modified MSN-CC with six different functional groups, resulting in nanoparticles possessing neutral, negative, and positive charges. Through comprehensive analysis, we characterized the protein corona formed on each surface-functionalized MSN-CC, demonstrating that its composition was dictated by the specific surface functional groups. Notably, even MSN-CC with similar surface charges exhibited distinct protein corona profiles and macrophage uptake behaviors depending on their functional groups. These findings highlight the critical role of surface chemistry in shaping protein corona formation, which in turn influences the fate, behavior, and biological responses of nanoparticles (Fig. 1b).
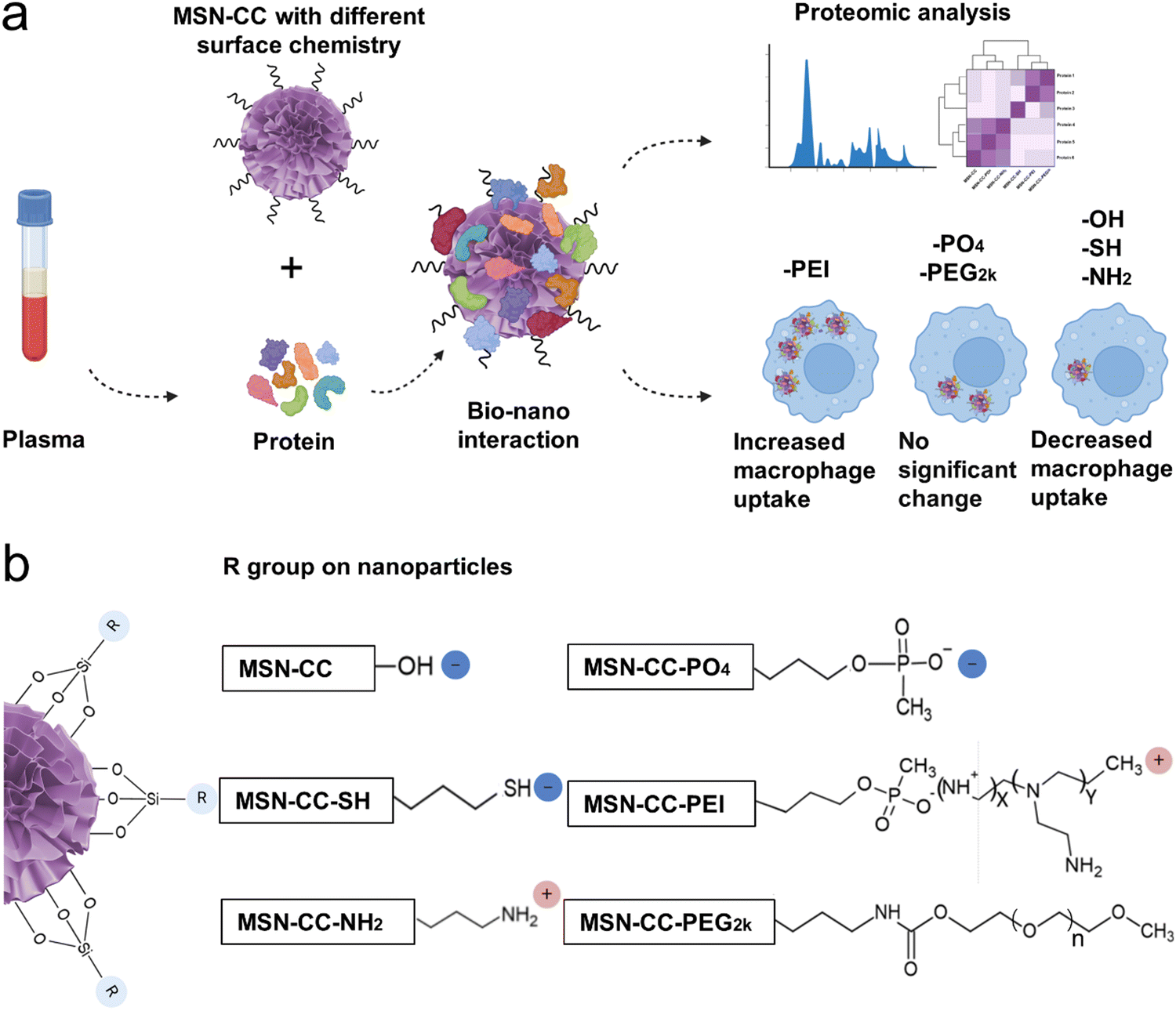 | ||
| Fig. 1 Schematic illustration on macrophage uptake through the bio-nano interaction using surface functionalized MSN-CC. (a) Overall design of this study. (b) Diagram illustrating the surface functionalization of core-cone structured mesoporous silica nanoparticles (MSN-CC). | ||
Material and methods
Chemicals
Cetyltrimethylammonium chloride (CTAC, 25 wt% in H2O), (3-aminopropyl)triethoxysilane (APTES, ≥98.0%), chlorobenzene (anhydrous, 99.8%), triethanolamine (TEA, ≥99.0%), tetraethyl orthosilicate (TEOS, 98%), ammonium hydroxide(28.0–30.0% NH3 basis), 3-(trihydroxysilyl)propyl methylphosphonate, monosodium salt solution, (THPMP, 50 wt% in H2O), 3-mercaptopropyl trimethoxysilane (MPTS, ≥80%), three-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT), phorbol 12-myristate 13-acetate (PMA) and fluorescein isothiocyanate isomer I (FITC) were purchased from Sigma-Aldrich, Australia without further purification. Dulbecco's modified Eagle's medium (DMEM, Gibco), fetal bovine serum (FBS, Gibco), βME, penicillin/streptomycin glutamine (Gibco), BCA protein assay, dimethyl sulfoxide (DMSO), 4′,6-diamidino-2-phenylindole (DAPI), and Phalloidin (Alexa Fluor® 555) were obtained from Thermo Fisher Scientific (Australia). 2× laemmli buffer, urea, CHAPS, 4–20% polyacrylamide gel, and precision protein standards were purchased from Bio Rad (Australia). Ethanol (absolute for analysis) was purchased from Supelco (Australia). Polyethylenimine (PEI, branched, M.W. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 99%) was purchased from Alfa Aesar (Australia). mPEG-silane, 2 K was purchased from Biopharma PEG scientific (Watertown, MA, USA). Thiourea was obtained from FUJIFILM Wako Pure Chemical Corporation (Richmond, VA, USA). All chemicals were used directly without further purification.
000, 99%) was purchased from Alfa Aesar (Australia). mPEG-silane, 2 K was purchased from Biopharma PEG scientific (Watertown, MA, USA). Thiourea was obtained from FUJIFILM Wako Pure Chemical Corporation (Richmond, VA, USA). All chemicals were used directly without further purification.
Preparation of different surface-functionalized MSN-CC
The synthesis of MSN-CC followed a previously protocol.17 In brief, 4.8 mL of CTAC (25% water solution), 0.08 g of TEA, and 7.2 mL of Milli-Q ultrapure water were mixed and stirred at 60 °C for 30 min. Subsequently, 3.5 mL of chlorobenzene and 0.5 mL of TEOS were added to the mixture and stirred at 60 °C for 12 h. The nanoparticles were centrifuged at 15000 rpm for 15 min and washed three times with ethanol. The CTAC templates were removed by reflux in acetone/HCl solution.
For MSN-CC-PO4 synthesis, 30 mg of MSN-CC were dispersed in 10 mL of water, and the pH was adjusted to 10 with ammonium hydroxide. Then, 10 mL 3(trihydroxysilyl) propylmethylphosphonate solution (56 mM) was added, and the mixture was stirred for 2 h at 40 °C. The nanoparticles were collected by centrifugation and washed three times with water.
For MSN-CC-NH2 synthesis, MSN-CC (100 mg) was added into 10 mL of ethanol, sonicated and mixed with 500 μL of ammonia, followed by the addition of 270 μL of 3-aminopropyltriethoxysilane (APTES). The mixture was stirred at room temperature overnight. The nanoparticles were collected by centrifugation and washed three times with ethanol.
To synthesize MSN-CC-SH, 20 mg of MSN-CC was added to 5 mL of ethanol and sonicated. Subsequently, 100 μL of 30 wt% ammonia was added, followed by the addition of 200 μL of 3-mercaptopropyl trimethoxysilane. After stirring overnight at room temperature, the products were collected by centrifugation and washed with ethanol.
For MSN-CC-PEI, MSN-CC was first modified with a -PO4 group using the above method. Then MSN-CC-PO4 (30 mg) was dispersed in 100 mM carbonate buffer (pH 9.6, 15 mL) containing 150 mg PEI (molecular weight of 10 kD). The mixture was stirred for 4 h at room temperature, and the PEI-modified nanoparticles were obtained by centrifugation, washed with water/ethanol, and dried at room temperature.
For modification of MSN-CC with mPEG-silane with PEG (molecular weight of 2000), the w/w ratio of PEG-to-MSN-CC was 20:1. MSN-CC (100 mg) were suspended in 30 mL of anhydrous toluene, followed by sonication for 2–3 min. The resulting MSN-CC suspension was heated to 110 °C, and mPEG-silane (2 g) in 20 mL of anhydrous toluene was added dropwise to the stirred suspension. The nanoparticles were stirred for 12 h and isolated by centrifugation, followed by washing with water/ethanol to remove unreacted chemicals. The obtained nanoparticles were then dispersed in ethanol and stored at 4 °C.
Fourier transform infrared (FTIR) spectra
FTIR spectra (ThermoNicolet Nexus iS20, ThermoFisher Scientific, Madison, USA) was employed to assess the surface functionalization on nanoparticles. The spectrum was obtained with 128 scans, at a resolution of 16 cm−1, covering the range from 500 to 4000 cm−1.Nitrogen sorption measurements
The nitrogen adsorption/desorption measurements were conducted at 77 K using a Micromeritics Tristar II system (Micromeritics Instrument Corp, USA). Prior to testing, the dried samples were degassed at 453 K overnight under vacuum. The total pore volume was determined from the amount adsorbed at a maximum relative pressure (P/P0) of 0.99. Specific surface areas were calculated using the Brunauer–Emmett–Teller (BET) method, while pore sizes were derived from the adsorption branches of the isotherms using the Barrett–Joyner–Halenda (BJH) method.X-ray photoelectron spectroscopy (XPS)
Surface-functionalized nanoparticles were subjected to XPS analysis using a Kratos Axis ULTRA X-ray Photoelectron Spectrometer (Kratos Analytical Ltd, Manchester, United Kingdom) equipped with a 165 mm hemispherical electron energy analyzer. Monochromatic Al Kα X-rays (1486.7 eV) were utilized at 150 W (15 kV, 10 mA). The scanning of vacuum-dried samples was performed with an analyzer pass energy of 160 eV over a binding energy range of 0–1200 eV, employing 1.0 eV steps and a dwell time of 100 ms for broad surveys. Additionally, scans were conducted at 20 eV with 0.05 eV steps and 250 ms dwell time for detailed high-resolution surveys. CasaXPS version 2.3.14 software was utilized for calculating the atomic concentrations.Thermogravimetric analyses (TGA)
TGA of silica samples was performed with an air atmosphere, employing a Mettler Toledo TGA/DSC1 STAR system (Mettler Toledo, Oregon, United States). Air was supplied at a flow rate of 20 mL min−1, with the temperature ranging from 25 to 900 °C.Dynamic light scattering (DLS)
For DLS, zeta potential and PDI measurement for both naked and corona-coated nanoparticles in PBS (pH = 7.4), the detections were carried out by using Malvern Zetasizer Nano (Malvern Panalytical, Worcestershire, United Kingdom). The measurement was conducted at room temperature (25 °C) with a scattering angle of 173°.Transmission electron microscopy (TEM)
The TEM images were acquired using a Hitachi HT7700 electron microscope (Hitachi, Tokyo, Japan) operating at an acceleration voltage of 120 kV. Nanoparticle sizes were determined by analyzing more than 50 nanoparticles from at least 3 distinct areas using ImageJ software (Version 1.54 h, National Institutes of Health, USA).Protein corona formation in human plasma
The use of human plasma was approved by the local ethics committee of The University of Queensland (ethical approval no. #2021/HE000391). Human blood from three healthy anonymous donors, treated with anti-coagulants, was provided by the Australian Red Cross. The blood was aliquoted into 50 mL tubes and centrifuged at 2000g for 15 min at 4 °C. The supernatant was subsequently aliquot into 15 mL tubes and stored at −80 °C for future use.The protein concentration was determined by BCA protein assay (Thermo Scientific, Germany) in accordance with the manufacturer's instructions. Equal volumes of plasma from each donor were mixed.
The samples were centrifuged at 20000g for 1 hour to eliminate any aggregated proteins prior to use. Nanoparticles were incubated at 55% plasma concentration diluted with PBS (pH = 7.4) in three replicates. For consistency, the ratio of total nanoparticles to dilution was maintained at 1 mg mL−1. This dispersion was incubated for 1 h at 37 °C with constant agitation. To obtain the corona–nanoparticle complexes, the samples were separated from excess plasma by centrifugation at 20000g for 1 h. The particle pellet was resuspended in PBS and underwent three centrifugation steps, followed by redispersion in PBS. Subsequently, the pellet was dissolved in 100 μL of urea-thiourea buffer (7 M Urea, 2 M Thiourea, 4% CHAPS) to elute the proteins.
SDS polyacrylamide gel electrophoresis (SDS-PAGE)
For SDS PAGE, 7.5 μL of the protein samples were mixed with 7.5 μL sample buffer (1 mL sample buffer containing 50 μL βME + 950 μL 2xlaemmli buffer). It was then boiled for 10 minutes at 100 °C and an equal sample volume is loaded in 4–20% polyacrylamide gel. Gel electrophoresis in running buffer was performed at 100 V for about 80 minutes. The gel was then washed in milliQ water, stained with Coomassie blue staining for 1 hour and destained overnight in 30% methanol. After that, gel was scanned using a Biorad GS-800 calibrated densitometer scanner. Precision protein standards were used as molecular weight markers.Liquid-chromatography mass-spectrometry (LC-MS) analysis
The protein samples eluted from the nanoparticles underwent a 60-minute incubation at 70 °C after the addition of DTT. Subsequently, iodoacetamide was added, followed by a 15-minute incubation in the dark. Subsequently, 2.5 μL of 12% phosphoric acid was added to each lysate, followed by the addition of 165 μL of S-trap binding buffer (composed of 90% MeOH and a Tris concentration of 100 mM). The solution was then transferred to an S-trap column and centrifuged at 4000g for 1 minute. The samples were subjected to three washes by centrifuging through S-Trap binding buffer (150 μL). Afterward, 1 μg of trypsin dissolved in 50 μL of 50 mM ammonium bicarbonate (pH = 8) was added to the top of the S-traps. The columns were loosely capped and allowed to digest in a clean tube overnight at 37 °C. Following digestion, elution of peptides was carried out using 40 μL of 5% ACN in 0.1% formic acid, followed by subsequent elutions with 50% ACN and 75% ACN in 0.1% formic acid. The dried peptides were then resuspended in 20 μL of A-buffer before undergoing liquid chromatography. An Ultimate 3000 RSLC nano (Thermo Fisher, Germany) HPLC system, together with Buffer A (0.1% formic acid) and Buffer B (80% ACN) were utilized. 2 μL of Buffer A was injected onto a trap column and washed at a rate of 15 μL min−1 before being switched in-line to a resolving C18 nanoEase M/Z CSH 1.7 μm (150 μm × 100 mm) column (Waters). Gradient Buffer B was utilized according to the specified time/%B: 0/8, 4/8, 47/24, 52/40, 54/95, 57/95, 58/8, 60/8. The peptides were introduced into a ThermoFisher Eclipse mass spectrometer (ThermoFisher Scientific, USA) using an electro-spray ionization method. The MS parameters were configured as follows: for MS1, a resolution of 120000 was employed for mass acquisition spanning the range of 400–1100 Da; for MS2, an isolation window of 1.6 Da was utilized, with collision energy set at 30, resolution at 15000, a normalized AGC target of 75%, and a maximum ionization time of 40 ms. ThermoFisher Protein Discoverer software was utilized for post analysis, employing the relevant organism database.
For further bioinformatics analysis, mean and variance statistics were calculated in R, with heatmap plotting and clustering with the pheatmap package.22 We also used the TBtools software23 and UniProt database (https://www.uniprot.org/) for the classification of proteins.
MTT assay
The cytotoxicity of nanoparticles was determined by MTT assay. Briefly, 5 × 104 cells per mL of RAW 264.7 cells were placed in sterile 96-well cell culture plates (100 μL per well) in complete culture medium (DMEM, 10% FBS, 100 U mL−1 penicillin, 100 U mL−1 streptomycin). The cell culturing was carried out at 37 °C in a humidified environment containing 5% CO2 for 24 h. Next, the culture media in the wells was discarded, and serials of different concentrations of nanoparticles (5, 10, 20, 40, 80 μg mL−1) were added to the wells successively. A negative control was established using untreated cells.After incubation of another 24 h, MTT solution (5 mg mL−1, 15 μL) was introduced to each well, followed by an additional incubation period of 4 hours at 37 °C in a 5% CO2 environment. Afterward, the supernatant was discarded, and DMSO (150 μL) was added to fully dissolve the dark blue crystals. A microplate reader (Infinite, Tecan Trading AG, Männedorf, Switzerland) was applied to measure the absorbance of the solution at 565 nm, assessing cell viability.
Cellular uptake of nanoparticle tests
To prepare FITC-labeled MSN-CC, MSN-CC-PO4, MSN-CC-SH, and MSN-CC-PEG2k, a solution of FITC in ethanol (1 mg mL−1, 1 mL) was first prepared and combined with APTES (5 μL). After stirring for 24 hours, 400 μL of the solution was added dropwise to a dispersion of nanoparticles in ethanol (5 mg mL−1, 4 mL). Stirring was continued for an additional 24 hours. Subsequently, the FITC-labeled nanoparticles were washed, dried and obtained.24 To synthesize FITC-labeled MSN-CC-NH2 and MSN-CC-PEI, 50 mg of nanoparticles was dispersed in 5 mL of ethanol containing 0.25 mg FITC. Stirring was conducted for 4 h at 25 °C, followed by centrifuging and washing with ethanol three times.FITC-labeled nanoparticles and a nanoparticle–corona complex were utilized to evaluate the cellular internalization inside RAW 264.7 (purchased from American Type Culture Collection, ATCC). Raw 264.7 cells were cultured following seeding into 24-well plates (1 × 105 cells per mL, 500 μL per well) and incubating for 24 h at 37 °C and 5% CO2. Bare and protein coronated FITC conjugated nanoparticles were treated with the cells (20 μg mL−1) and incubated for 4 h. The media was then removed, and the cells adhered onto the cover slips were visualized via a confocal laser scanning microscope (CLSM).
The cellular uptake study also employed human macrophages differentiated from THP-1 cells (human monocytic leukemia cell line, purchased from ATCC) following stimulation with PMA, which is commonly used to induce macrophage-like differentiation in the THP-1 monocytic cell line. The THP-1 was cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U mL−1 penicillin, and 100 U mL−1 streptomycin in a humidified 5% CO2 atmosphere at 37 °C. Cells were seeded into 24-well plates at a density of 1 × 105 cells per mL (500 μL per well) and differentiated with 25 ng mL−1 PMA for 48 hours, followed by an additional 24-hour incubation without PMA. Subsequently, cells were treated with 20 μg mL−1 of both pristine and protein-coronated FITC-conjugated nanoparticles and incubated for 4 hours. After media removal, the cells adhered to coverslips were visualized using confocal laser scanning microscopy (CLSM).
For CLSM visualization, the cells were fixed with 4% paraformaldehyde for 30 minutes. Subsequently, 0.1% Triton X was used to permeabilize the cells for 10 minutes. Following three washes with PBS, DAPI and phalloidin were employed to stain the nuclei and actin filaments, respectively. The samples were then mounted on glass slides and observed using CLSM (Nikon C2+, Nikon, Tokyo, Japan). FITC-labeled nanoparticles were visualized using 488 nm laser excitation. All observations were conducted in triplicate with appropriate controls.
Statistical analysis
Three experimental replicates were conducted to acquire data. Statistical analysis was carried out utilizing GraphPad Prism 10 (Version 10.2.1, GraphPad Software, Inc, Boston, USA) and Excel (Microsoft, Co, Washington, USA). Data in figures and tables were presented as mean ± standard deviation (SD). Student's t-test was employed for statistical comparison between two groups. Significance was determined at a threshold of p < 0.05.Results
Synthesis and surface functionalization of MSN-CC
TEM images showed MSNs exhibited a highly porous structure with a uniformed size of around 186 nm (Fig. S1 and Table 1, ESI†). Nitrogen sorption analysis revealed that MSN-CC has a BET surface area of 625.01 m2 g−1. The average pore size, determined from the pore size distribution curve (Fig. S2, ESI†), was 37 nm. After surface modification, minimal alteration was observed in their morphology and size under TEM analysis (Fig. S1, ESI†). Verification of surface functionalization was conducted through Fourier-transform infrared spectroscopy (FTIR, Fig. S3, ESI†) and X-ray photoelectron spectroscopy (XPS, Fig. S4, ESI†). Upon amino modification, the FTIR spectra of MSN-CC-NH2 exhibited characteristic peaks at 1640 and 1558 cm−1, corresponding to amine vibrations, MSN-CC-PEI revealed minor bands at 2838 and 2935 cm−1, indicative of CH3 and CH2 bond vibrations, along with absorption bands at 1469 and 1646 cm−1, attributed to C–H and N–H bonds in PEI. Subsequent polyethylene glycol (PEG) modification of MSN-CC-PEG2k manifested methylene C–H asymmetric stretching and bending bands around 2880 and 1450 cm−1, with a discernible C–C signal at approximately 950 cm−1 (Fig. S3, ESI†). XPS analysis further confirmed the surface modification, with nitrogen (N) detected in MSN-CC-NH2, MSN-CC-PEI, and MSN-CC-PEG2k, while sulfur (S) was detected in MSN-CC-SH (Fig. S4, ESI†). The nitrogen concentration in MSN-CC-PEI (8.08%) notably exceeded that of MSN-CC-NH2 (1.90%), indicative of higher nitrogen content in PEI (Table S1, ESI†).| Sample | Particle size by TEM (nm) | Hydrodynamic size by DLS (nm) | Zeta potential (mv) | Polydispersity index (PDI) |
|---|---|---|---|---|
| MSN-CC | 186.6 ± 13.0 | 205.5 ± 3.5 | −12.4 ± 1.3 | 0.048 ± 0.036 |
| MSN-CC with corona | 195.9 ± 11.4 | 276.9 ± 5.6 | −4.4 ± 0.6 | 0.072 ± 0.016 |
| MSN-CC-PO4 | 182.9 ± 9.7 | 248.8 ± 17.6 | −12.5 ± 1 | 0.045 ± 0.016 |
| MSN-CC-PO4 with corona | 189.0 ± 10.9 | 254.5 ± 5.1 | −5.8 ± 0.5 | 0.051 ± 0.029 |
| MSN-CC-NH2 | 188.4 ± 10.6 | 313 ± 3.9 | 12.8 ± 1.4 | 0.271 ± 0.020 |
| MSN-CC-NH2 with corona | 203.8 ± 11.8 | 328.7 ± 5.8 | −4.3 ± 1.1 | 0.137 ± 0.024 |
| MSN-CC-SH | 176.6 ± 12.3 | 250.4 ± 16.2 | −11.6 ± 0.9 | 0.139 ± 0.102 |
| MSN-CC-SH with corona | 198.6 ± 10.1 | 257.6 ± 3.4 | −6.1 ± 1 | 0.073 ± 0.051 |
| MSN-CC-PEI | 195.5 ± 9.5 | 307.1 ± 17.1 | 16.7 ± 0.5 | 0.388 ± 0.008 |
| MSN-CC-PEI with corona | 201.7 ± 11.1 | 452.8 ± 43.1 | −0.5 ± 0.3 | 0.305 ± 0.041 |
| MSN-CC-PEG2k | 196.6 ± 10.3 | 276.6 ± 13.9 | −1.5 ± 1 | 0.126 ± 0.037 |
| MSN-CC-PEG2k with corona | 196.1 ± 12.9 | 231.5 ± 2 | −2.9 ± 2.3 | 0.049 ± 0.009 |
To further verify surface functionalization and quantify the amount of grafted organic groups, TGA analysis of MSNs was conducted (Fig. 2g and Table S2, ESI†). The slight mass loss observed at around 100 °C is attributed to absorbed water and gas. At 900 °C, MSN-CC, as well as MSN-CC modified with -PO4, -NH2, -SH, -PEI, and -PEG2k exhibited a mass loss of 7.8%, 9.9%, 15%, 9.2%, 43.8% and 37.8%, respectively. For MSN-CC, the slight mass loss in the interval 100–900 °C was mainly generated by the condensation of silanol moieties (Si–OH). The weight loss of MSN-CC-PO4 between 300 and 580 °C can be ascribed to the decomposition of the phosphonate groups. MSN-CC-NH2 showed a typical mass loss signal between 280 and 680 °C, which is mainly connected with the decomposition step of the amino groups. For MSN-CC-SH, the weight loss between 150 and 650 °C is taken as an estimate of the total amount of the organic thiol group. Considerable weight loss was observed for MSN-CC-PEI above 100 °C, which was significantly influenced by the PEI modification. The degradation of PEG on MSN-CC-PEG2k was from 160 °C, and the residual mass at 900 °C was 62.2%. The percentage of organics grafted onto nanoparticles in MSN-CC-PO4, MSN-CC-NH2, MSN-CC-SH, MSN-CC-PEI, and MSN-CC-PEG2k was calculated to be 2.1%, 7.2%, 1.4%, 36% and 30%. Since MSN-CC-PEI was modified using electrostatic adhesion methods, we assessed its stability in solution over time. The results indicated that, over a 72-hour period, the zeta potential of MSN-CC-PEI in Milli-Q water ranged from 31.7 ± 1.7 mV to 39.9 ± 3.0 mV, while in PBS, it ranged from 9.25 ± 0.7 mV to 14.1 ± 0.7 mV (Fig. S5 and Table S3, ESI†). The polydispersity index (PDI) values ranged from 0.178 ± 0.019 to 0.341 ± 0.009 in Milli-Q water and from 0.307 ± 0.004 to 0.429 ± 0.023 in PBS, demonstrating the stability of MSN-CC-PEI over time.
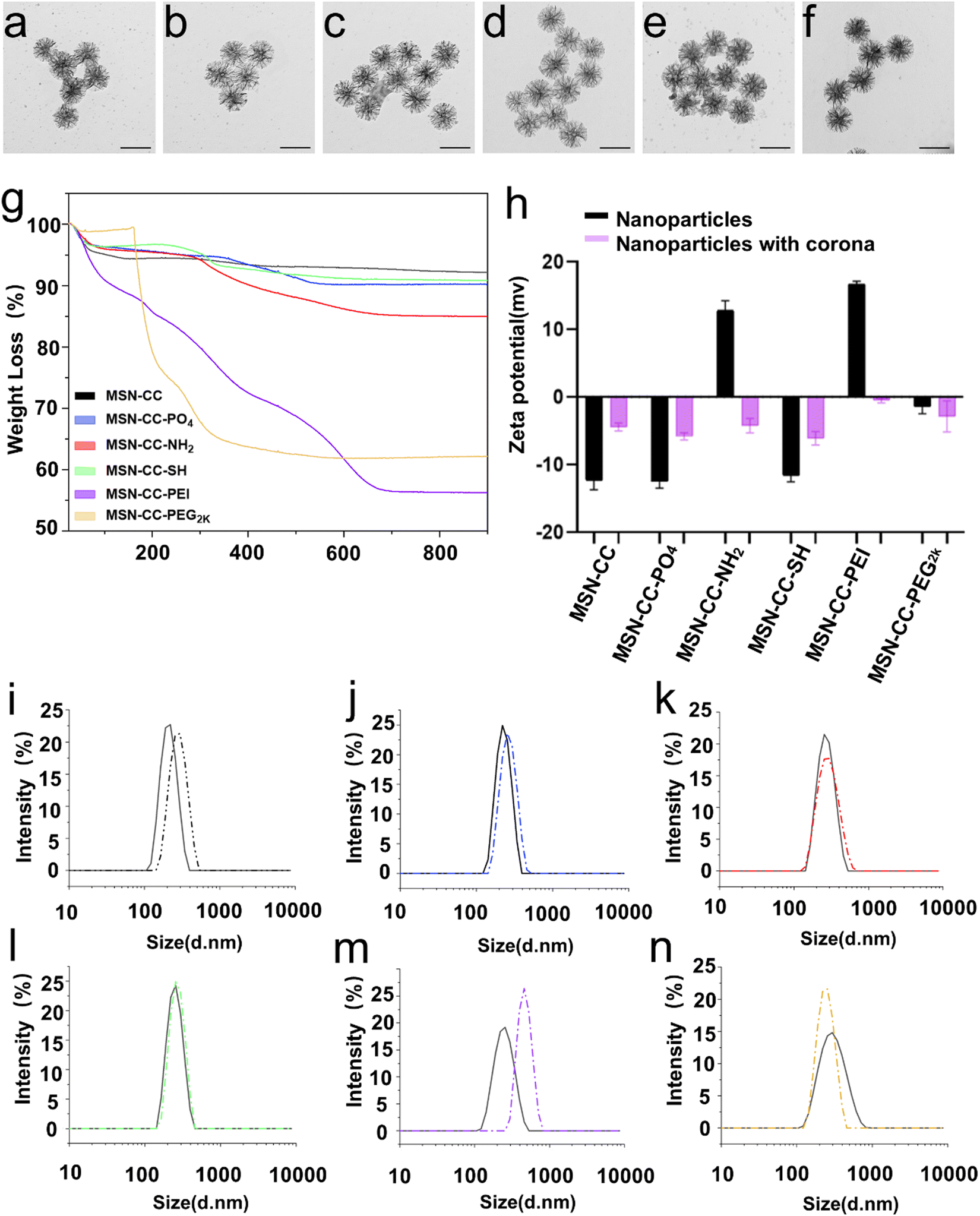 | ||
| Fig. 2 Characterization of surface modifications and the protein adsorption behavior of MSN-CCs. TEM images of corona coated MSN-CC (a), MSN-CC-PO4 (b), MSN-CC-NH2 (c), MSN-CC-SH (d), MSN-CC-PEI (e) and MSN-CC-PEG2k (f), scale bar: 200 nm. Thermogravimetric analysis of MSN-CCs with different surface chemistries (g). Zeta potential (h) and hydrodynamic diameter (i)–(n) of surface modified MSN-CCs (solid lines) and corona coated surface modified MSN-CCs (dashed lines) in PBS measured by DLS. Specifically, (i) MSN-CC, (j) MSN-CC-PO4, (k) MSN-CC-NH2, (l) MSN-CC-SH, (m) MSN-CC-PEI, and (n) MSN-CC-PEG2k. | ||
Characterizations of the nanoparticle–corona complexes
To study the interaction of surface modified MSNs with plasma, we prepared the nanoparticle–corona complexes by incubating the nanoparticles in 55% human plasma (protein concentration in bloodstream). After being treated with plasma, the protein corona can be observed under TEM, resulting in a slightly larger size compared to those without a corona (Fig. 2a–f and Table 1). The particles were further characterized by DLS and zeta-potential measurements (Fig. 2h–n) in PBS buffer (pH = 7.4). After treating with blood plasma, the hydrodynamic diameter of MSN-CC, MSN-CC-PO4, -NH2, -SH, and -PEI, increased, likely due to the adsorption of proteins and salts. In contrast, the hydrodynamic diameter of MSN-CC-PEG2k decreased after being incubated in blood plasma. The zeta potential values for all nanoparticle–protein corona complexes were slightly negative (−0.5 to −6.1 mV, Fig. 2h), irrespective of the significant variances in the original charge of these nanoparticles (−12.5 to +16.7 mV). This finding is consistent with previous studies,25,26 indicating that the protein corona itself largely dictates the surface charge of nanoparticles in blood plasma.Protein identification and classification
To identify and quantify the composition of the protein corona absorbed on surface functionalized nanoparticles, we employed SDS-PAGE and HPLC-MS analyses. SDS-PAGE analysis (Fig. 3a) showed a complex mixture of proteins isolated from the particles. The protein concentrations at approximately 85 kDa and 120 kDa within the MSN-CC-NH2, -PEI, and -PEG2k groups were notably lower in comparison to the remaining three groups. Furthermore, in contrast to other nanoparticles, the protein attachment to MSN-CC-PEG2k exhibited a significant reduction. This phenomenon was consistent with previous reports,27,28 suggesting that the adsorption of proteins around PEGylated nanoparticles occurred to a lesser extent. The polymer modification can establish a hydrophilic layer surrounding the nanoparticles, stabilizing them, reducing non-specific protein adsorption, and prolonging their circulation time in the bloodstream.29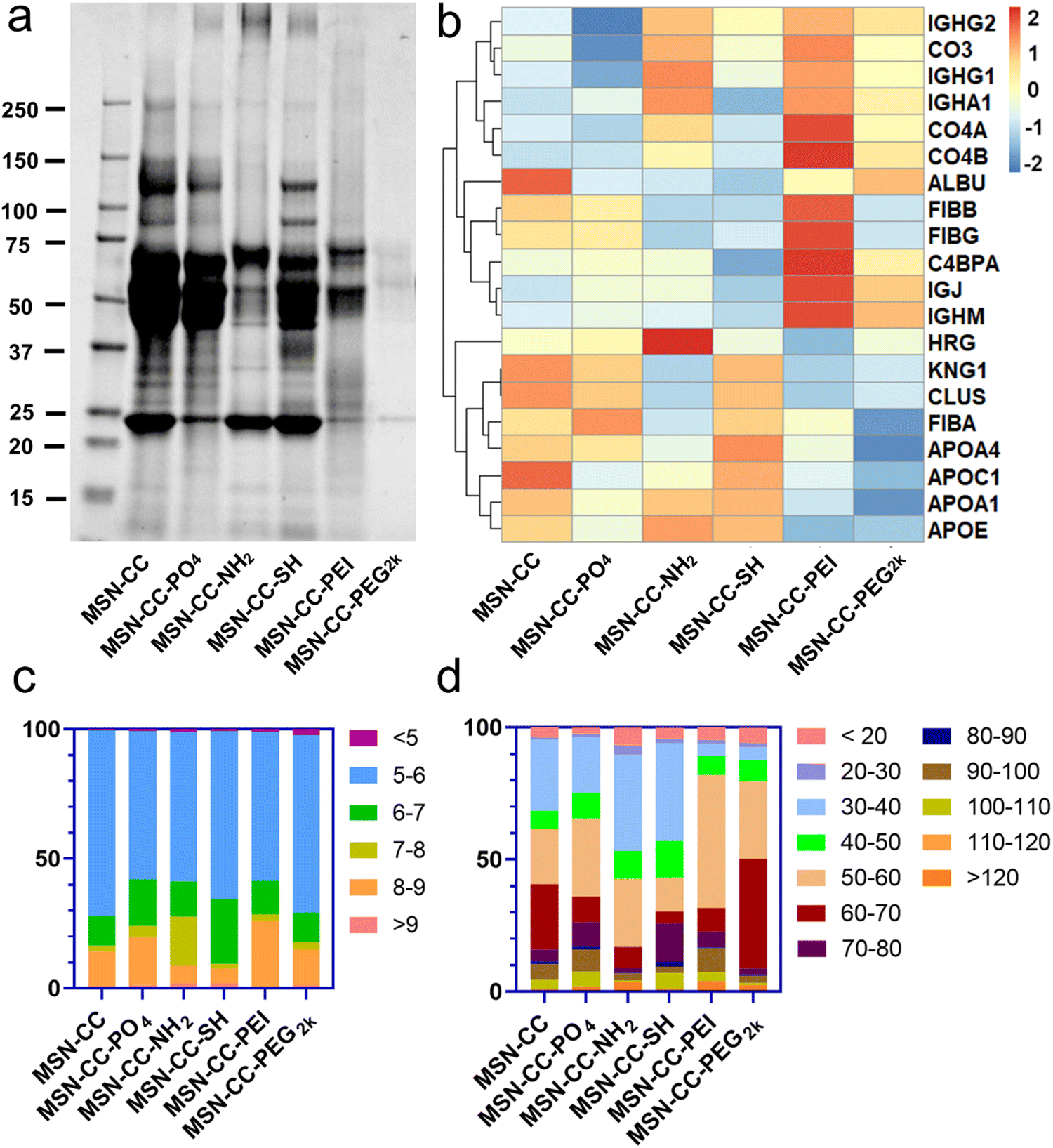 | ||
| Fig. 3 Proteomic analysis of the protein corona components formed on the surface of MSN-CCs with different surface chemistries. (a) Representative SDS-PAGE gel showing protein coronas obtained from MSN-CCs with different surface chemistries after a 1-hour incubation in blood plasma. (b) Twenty notable proteins selected from the top 5% protein pools in the protein corona. (c) Classification of proteins based on their isoelectric point. (d) Classification of proteins based on their molecular weight. | ||
With HPLC-MS, we identified a total of 1866 proteins on these surface functionalized nanoparticles. Approximately 991 of the proteins existed on MSN-CC, 1206 on MSN-CC-PO4, 1238 on MSN-CC-NH2, 1294 on MSN-CC-SH, 946 on MSN-CC-PEI, and 1382 on MSN-CC-PEG2k. The top 5% most abundant proteins were sorted in the protein corona and utilized to generate a heatmap (see Fig. S6, ESI†), illustrating the relative changes in protein abundance across all surface-functionalized nanoparticles. Twenty noteworthy proteins, including clusterin, albumin, histidine-rich glycoprotein, kininogen-1, fibrinogen, complement factors, immunoglobulins, and apolipoproteins, were further singled out from the top 5% protein pools. The corresponding heatmap in Fig. 3b illustrated the comparison of the protein abundance across various surface functionalization groups.
In the MSN-CC group, APOA1, APOA4, CLUS, HRG, and ALBU were found to be most abundantly absorbed on the nanoparticles' surface. For the MSN-CC-PO4 group, high protein absorption was observed for FIBA, APOA4, CLUS, KNG1, and FIBB. Within the MSN-CC-NH2 group, the most abundant proteins were HRG, APOD, APOE, APOA1, and IGHG1. Notably, apolipoproteins such as APOA1, APOE, APOC1, APOA4, and KNG1 exhibited high absorption on MSN-CC-SH. On PEI-modified MSN-CC surfaces, complement factors including C4BPA, CO4A, CO4B, CO3 and immunoglobulins such as IGJ and IGHM were the most abundant. Among the identified 20 proteins, ALBU, IGHM, and IGJ displayed high absorption on the surface of MSN-CC-PEG2k.
With proteomic analysis results of HPLC-MS, we firstly classified proteins by their isoelectric point (pI, Fig. 3c) and molecular weight (mW, Fig. 3d). According to the results, these surface-modified MSN-CC exhibited a preference for absorbed protein based on the pI. More than 70% of the proteins in the corona of the nanoparticles had pI < 7 (about 83.5% for MSN-CC, 75.8% for MSN-CC-PO4, 72.4% for MSN-CC-NH2, 90.5% for MSN-CC-SH, 71.4% for MSN-CC-PEI and 82.2% for MSN-CC-PEG2k). Moreover, these surface functionalized nanoparticles adsorbed proteins with pI between 5 and 6 most abundantly (about 71.7% for MSN-CC, 57.2% for MSN-CC-PO4, 57.6% for MSN-CC-NH2, 64.8% for MSN-CC-SH, 57.6% for MSN-CC-PEI and 68.4% for MSN-CC-PEG2k). The results were consistent with previous literature reports.30 Further analysis was conducted to categorize proteins based on their molecular weight. It was observed that more than 70% of the proteins in the corona exhibited a molecular weight below 70 kDa. Notably, the predominant molecular weight range for proteins absorbed on MSN-CC, MSN-CC-NH2, and MSN-CC-SH was between 30 and 40 kDa. For MSN-CC-PO4, MSN-CC-PEI, and MSN-CC-PEG2k, the predominantly absorbed proteins had a molecular weight ranging from 50 to 60 kDa.
All identified proteins were further systematically categorized based on their physiological functions (Fig. 4), including immunoglobulin, complement, coagulation, lipoprotein, acute phase, tissue leakage, and others. Our bioanalytical analysis revealed a diverse composition of proteins adsorbed onto distinct surface-functionalized nanoparticles. We identified that immunoglobulins, complement, and coagulation-related proteins demonstrated a high affinity for MSN-CC-PEI. In contrast, MSN-CC, MSN-CC-PO4, MSN-CC-NH2, and MSN-CC-SH exhibited strong interactions with coagulation-related proteins and apolipoproteins. While acute phase response proteins are abundant in serum,31 no notable enrichment was detected among these nanoparticles. The tissue leakage proteins were highly enriched on PEG2k modified MSN-CC. This type of protein plays a role in numerous diseases. Meanwhile, the whole amount of these physiologically functional proteins was relatively lower on MSN-CC-PEG2k compared to other groups.
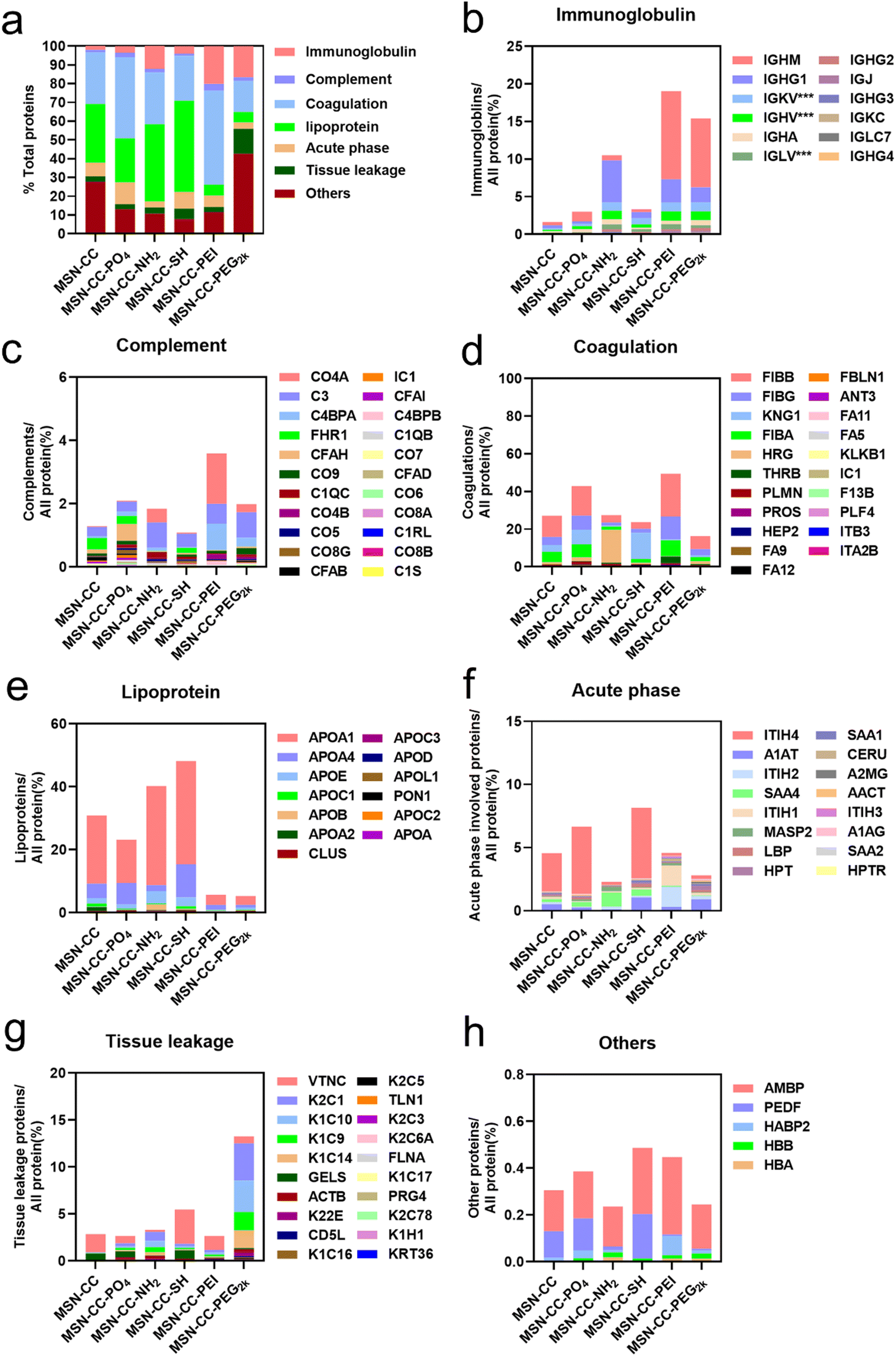 | ||
| Fig. 4 The categorization of corona proteins in protein corona on various MSN-CC based on their physiological functions (a), including immunoglobulin (b), complement (c), coagulation (d), lipoprotein (e), acute phase (f), tissue leakage (g) and others (h). | ||
The macrophage uptake of nanoparticles with or without pre-formed protein corona
Next, we investigated the uptake of nanoparticles by two types of macrophages, both with and without a protein corona, to assess the effects of the protein corona on cellular uptake. The macrophages utilized in this study included the murine macrophage-like cell line RAW 264.7 and THP-1-differentiated macrophages derived from humans. For RAW 264.7 cells, nanoparticles without pre-treatment with blood plasma-specifically, those with positively charged surface modifications, such as MSN-CC-NH2 and MSN-CC-PEI (Fig. 5), demonstrated higher internalization efficiency compared to negatively charged nanoparticles like MSN-CC, MSN-CC-PO4, and MSN-CC-SH in the absence of a protein corona.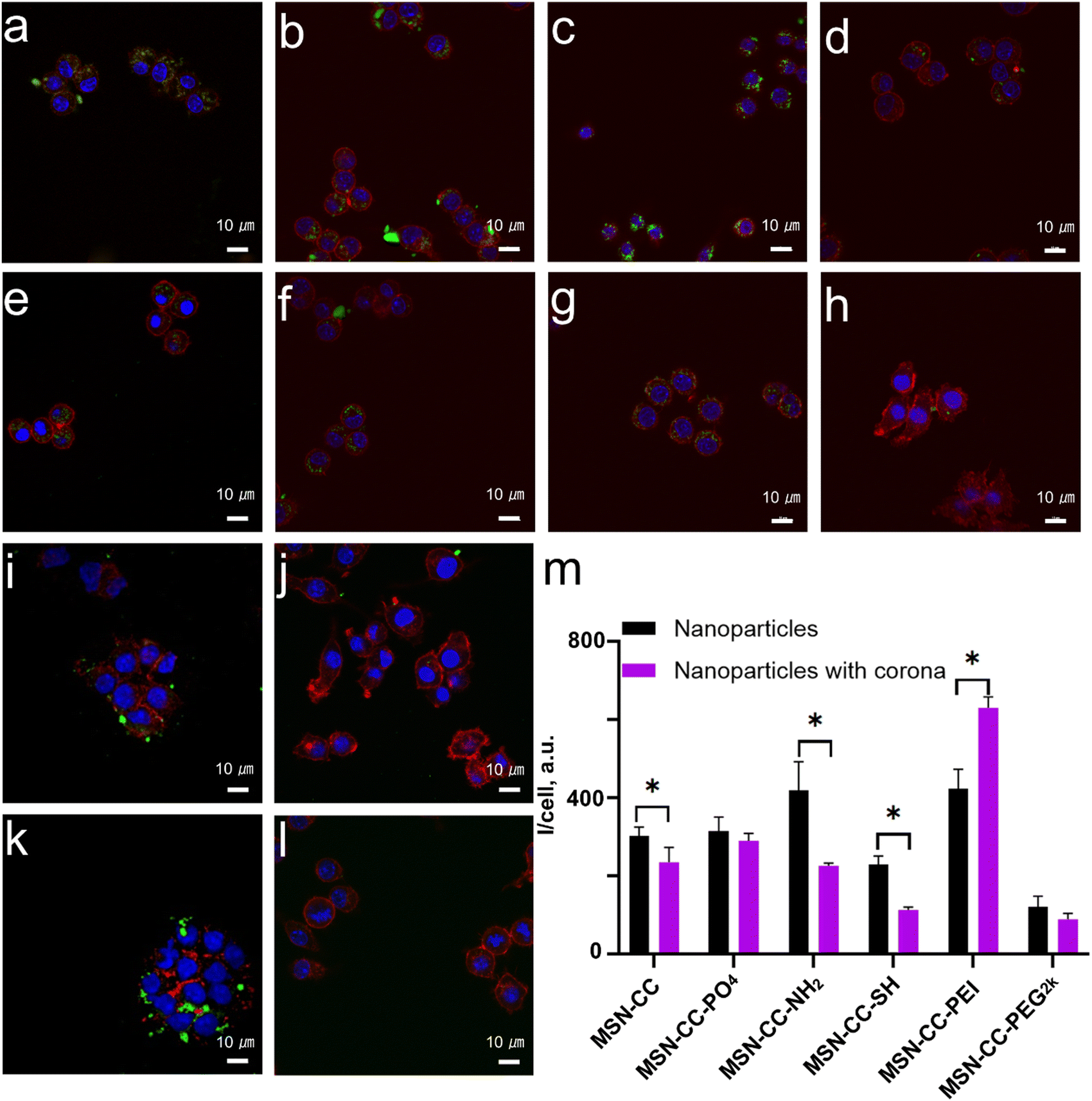 | ||
| Fig. 5 Cellular uptake of uncoated and corona-coated MSN-CCs with different surface functional groups in RAW 264.7 cells. Confocal microscopy images of RAW after being cultured with uncoated MSN-CC (a), MSN-CC-PO4 (b), MSN-CC-NH2 (c), MSN-CC-SH (d), MSN-CC-PEI (i), and MSN-CC-PEG2k (j), alongside their respective corona-coated nanoparticles including MSN-CC (e), MSN-CC-PO4 (f), MSN-CC-NH2 (g), MSN-CC-SH (h), MSN-CC-PEI (k), and MSN-CC-PEG2k (l). Cell nuclei were stained with DAPI (blue), the cytoskeleton was stained with phalloidin (red), and nanoparticles were conjugated with FITC, appearing green. (m) Semi-quantification of cellular uptake of nanoparticles through the average fluorescence intensity per cell. Scale bars: 10 μm. Asterisks (*) indicate significant differences compared to pristine nanoparticles (p < 0.05). | ||
However, upon treatment with blood plasma, which facilitates the formation of a protein corona, a significant reduction in nanoparticle endocytosis was observed for MSN-CC, MSN-CC-NH2, and MSN-CC-SH. Conversely, the presence of a protein corona enhanced the uptake of PEI-modified MSN-CC by macrophages, while the effects on MSN-CC-PO4 and MSN-CC-PEG2k were less pronounced. These cellular uptake experiments were conducted under conditions where the nanoparticle concentration was shown to be relatively safe (Fig. S7, ESI†). This observation indicated the critical role of protein coating in the nanoparticle's internalization, rather than the surface charge.
In THP-1-differentiated macrophages (Fig. 6), the cellular uptake patterns were similar to those observed in RAW 264.7 cells. Positively charged nanoparticles (MSN-CC-NH2 and MSN-CC-PEI) exhibited greater internalization efficiency compared to negatively charged nanoparticles (MSN-CC, MSN-CC-PO4, and MSN-CC-SH) in the absence of a protein corona. Following the formation of the protein corona, the trend in nanoparticle phagocytosis shifted; phagocytosis of MSN-CC, -NH2, and -SH significantly decreased, whereas PEI-modified MSN-CC showed increased cellular uptake. For the other two nanoparticle types, the presence of a protein corona did not result in noticeable changes in cellular uptake. These findings further corroborate our previous conclusion that the surface modifications of nanoparticles, along with the protein corona formed in human plasma, significantly influence macrophage phagocytosis.
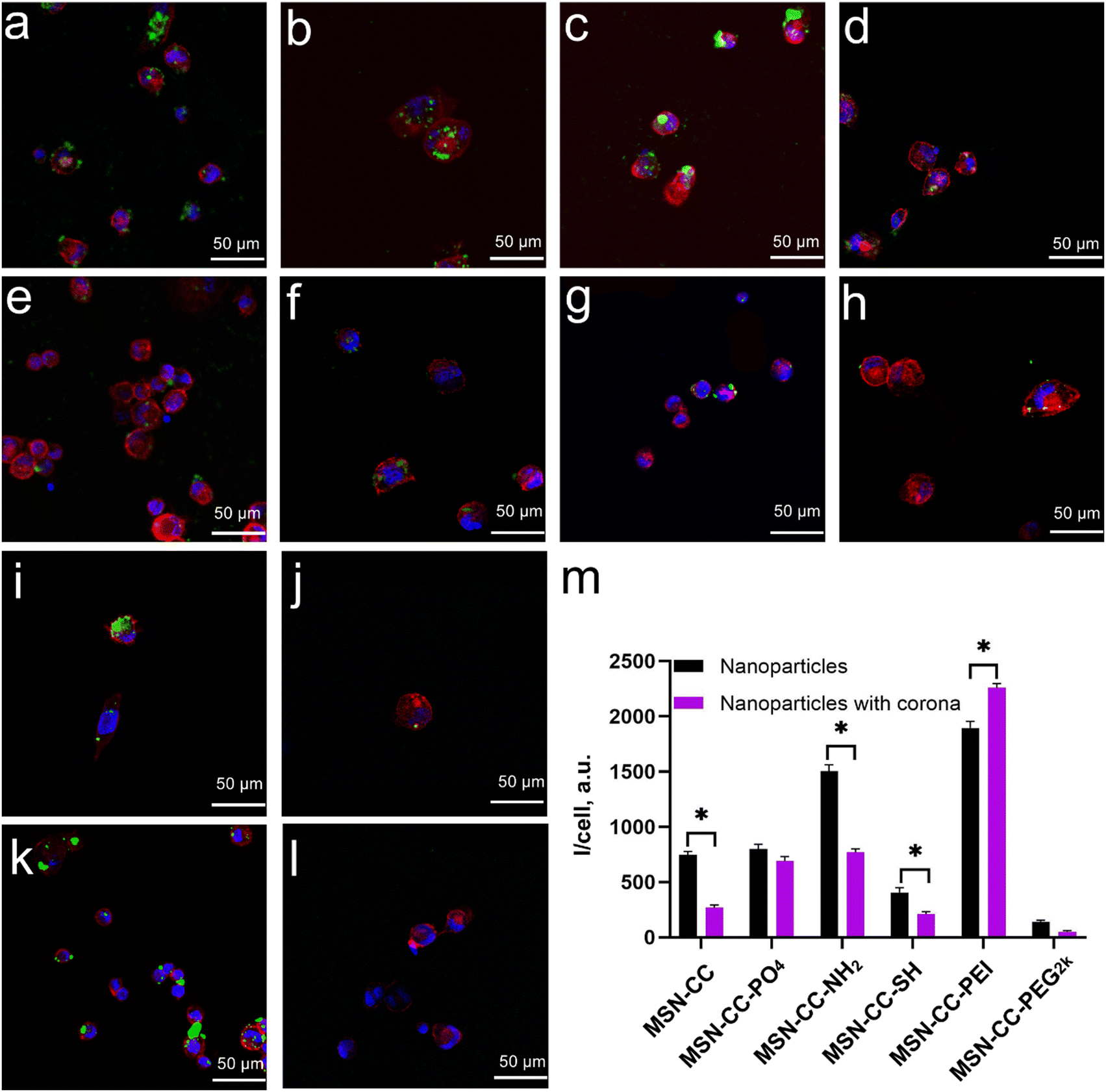 | ||
| Fig. 6 Cellular uptake of uncoated and corona-coated MSN-CCs with different surface functional groups in PMA-stimulated THP-1 macrophages. Confocal microscopy images of THP-1 macrophages after being cultured with uncoated MSN-CC (a), MSN-CC-PO4 (b), MSN-CC-NH2 (c), MSN-CC-SH (d), MSN-CC-PEI (i), and MSN-CC-PEG2k (j), alongside their respective corona-coated nanoparticles including MSN-CC (e), MSN-CC-PO4 (f), MSN-CC-NH2 (g), MSN-CC-SH (h), MSN-CC-PEI (k), and MSN-CC-PEG2k (l). Cell nuclei were stained with DAPI (blue), the cytoskeleton was stained with phalloidin (red), and nanoparticles were conjugated with FITC, appearing green. (m) Semi-quantification of cellular uptake of nanoparticles through the average fluorescence intensity per cell. Scale bars: 10 μm. Asterisks (*) indicate significant differences compared to pristine nanoparticles (p < 0.05). | ||
Discussions
The formation of a protein corona around nanoparticles is a critical factor influencing their nano-bio interactions. Several factors determine the corona formation and composition, including the material composition, surface properties, size, and shape of the nanoparticles, as well as environmental conditions such as pH, temperature, shear stress, and the composition of the biological milieu.13,14 The formation of the protein corona is a dynamic process, involving competition among different proteins. The adsorption of proteins onto nanoparticles is influenced not by the overall charge or the protein's isoelectric point, but rather by short-range interactions between specific charged regions on the proteins and the local charge distribution on the nanoparticles.32,33 Previous studies have demonstrated that surface functional groups,18 shape,19 and pores20 of MSNs significantly affect protein corona formation. Surface functionalization, in particular, plays a pivotal role in protein binding.Studying the composition of the protein corona on MSNs is important, as it directly influences macrophage recognition and uptake. A biomolecular corona enriched with opsonins, such as C3, C4, and IgG, is typically associated with immune cell phagocytosis.34 Additionally, the presence of immunoglobulins can promote the deposition of C3 proteins on nanoparticles, further facilitating immune recognition.35 In contrast, a protein corona enriched with dysopsonins, such as clusterin, apolipoproteins, and human serum albumin, generally reduces immune cell uptake, leading to prolonged nanoparticle circulation in the bloodstream.34
We synthesized a series of MSN-CCs with the same particles size (∼186 nm), introducing surface functionalities via covalent modifications or stable electrostatic interactions. These nanoparticles were designed to be with different electrostatic potentials, including anionic (MSN-CC, MSN-CC-PO4, MSN-CC-SH), cationic (MSN-CC-NH2, MSN-CC-PEI), and electroneutral (MSN-CC-PEG2k) potential. To study the interaction between the surface functionalized nanoparticles and proteins in blood, we incubated the nanoparticles in 55% plasma to imitate the in vivo conditions. Following incubation, all nanoparticles exhibited a slightly negative charge, consistent with previous studies,25,36 likely due to the predominance of negatively charged proteins in the plasma.
Using HPLC-MS, we identified and semi-quantitatively analyzed the protein corona composition of the surface-functionalized nanoparticles, revealing the presence of over 1800 proteins. This is a higher number than solid nanoparticles, which typically identified only hundreds of proteins.31,37 This increased protein adsorption can likely be attributed to the large pore size and surface area of the MSN-CC nanoparticles. Additionally, proteins with a negative charge (pI < 7) at pH 7.4 were predominantly enriched in the corona, providing a plausible explanation for the shift in zeta potential observed after corona formation. Interestingly, despite their positive surface charge, MSN-CC-NH2 and MSN-CC-PEI did not accumulate more negatively charged proteins (pI < 7).
It is noteworthy that the protein adsorption profile of MSN-CC-PEI differed from the other groups. Specifically, we identified that immunoglobulins, complement proteins, and coagulation-related proteins exhibited a high affinity for MSN-CC-PEI (Fig. 3). In contrast, MSN-CC, MSN-CC-PO4, MSN-CC-NH2, and MSN-CC-SH exhibited strong interactions with coagulation-related proteins and apolipoproteins. Meanwhile, the whole amount of these physiologically functional proteins was relatively lower on MSN-CC-PEG2k compared to other groups. Interestingly, despite both MSN-CC-NH2 and MSN-CC-PEI carrying similar positive charges, significant differences were observed in the types of proteins adsorbed. This suggests that charge alone does not solely determine protein adsorption; the structural differences between the -NH2 and -PEI functional groups played a critical role in influencing both the variety and quantity of proteins adsorbed. This observation is supported by previous studies. For instance, Rong Cai et al.25 demonstrated that surface modification had a greater impact on protein corona formation than surface charge in their work on gold nanorods modified with different chemical ligands. Similarly, Keni Yang et al.38 found that variations in protein corona formation, despite similar zeta potentials, influenced cellular uptake behaviors in liposomes with distinct surface modifications. These findings further underscore the importance of surface chemistry in determining nanoparticle–protein interactions.
We tested cellular uptake studies of these nanoparticle–corona complexes using murine and human macrophages. Positively charged MSN-CC-NH2 and MSN-CC-PEI showed significantly higher cellular uptake compared to negatively charged or electroneutral nanoparticles. In particular, the corona on PEI-modified MSN-CC enhanced uptake by macrophages. Conversely, for MSN-CC, MSN-CC-NH2, and MSN-CC-SH, the presence of the protein corona reduced nanoparticle endocytosis. Similarly, MSN-CC-PO4 and MSN-CC-PEG2k also exhibited a decrease in cellular uptake, though the difference before and after corona formation was less pronounced. This variation is likely due to the different compositions of the protein corona on these surface-modified nanoparticles. For example, MSN-CC-PEI had an abundance of immunoglobulins and complement proteins on its surface. Certain proteins, such as C4BPA and C3, are known to promote nanoparticle uptake. Complement protein C3 plays a pivotal role in opsonization by mediating macrophage biorecognition through specific complement receptors (CR1 and CR3, CD11b/CD18).31,39 C4BPA can bind to apoptotic or necrotic cells, marking them for clearance by the immune system.30,40 When these proteins become concentrated on the surface of nanoparticles, macrophage phagocytosis is significantly enhanced.
Our findings demonstrate that the protein corona formed in human plasma significantly influence nanoparticle phagocytosis by macrophages. For example, while nanoparticles modified with amino groups or PEI may exhibit similar surface charges, variations in the composition of the protein corona in biological fluids can markedly affect their uptake by macrophages. Even for nanoparticles with identical surface charges, the composition of the corona must be carefully considered. Additionally, nanoparticles modified with PEG2k benefit from the “stealth” effect of PEG, which reduces protein adsorption in biological fluids and lowers macrophage phagocytosis. A deeper understanding of these bio-nano interactions is essential for advancing the development of nanoparticle-based therapeutics and diagnostics, with broad implications for future clinical applications.
Conclusion
In conclusion, this study underscores the critical importance of surface modification and protein corona formation in determining the biological interactions and fate of nanoparticles, particularly concerning immune cell recognition and clearance. Our findings reveal that while surface charge influences nanoparticle behavior, the composition of the protein corona formed in biological environments can profoundly impact macrophage uptake and phagocytosis. Specifically, for MSN-CC, MSN-CC-SH, and MSN-CC-NH2, the presence of the protein corona resulted in decreased macrophage uptake, whereas PEI-modified MSN-CC exhibited increased uptake. This enhanced cellular uptake of MSN-PEI may be attributed to the high composition of immunoglobulins, complement proteins, and coagulation-related proteins within its protein corona.Additionally, nanoparticles modified with PEG2k demonstrated reduced protein adsorption and lower recognition by immune cells. These insights highlight the necessity of considering the protein corona when designing nanoparticles for drug delivery. As the field of nanotechnology continues to evolve, a deeper understanding of these intricate bio-nano interactions will be essential for the development of more effective and safer nanoparticle-based diagnostic and therapeutic strategies.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare there is no conflict of interest.Acknowledgements
C. X. acknowledges the funding support from the Sydney Horizon Fellow by The University of Sydney. The authors acknowledge the technical support from the Queensland Node of Metabolomics Australia (Lian Liu and Gert H. Talbo), the Australian National Fabrication Facility, and the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, The University of Queensland. Additionally, the authors express gratitude to Dr Tulio Fernandez Medina for his assistance with blood sample preparation and to Mr Zijian Shen for his technical support regarding figure preparation. J.W. would like to acknowledge the scholarship provided by the China Scholarship Council.References
- C. Xu, C. Lei, S. Hosseinpour, S. Ivanovski, L. J. Walsh and A. Khademhosseini, Natl. Sci. Rev., 2022, 9, nwac124 CrossRef CAS PubMed.
- C. Xu, C. Lei, Y. Wang and C. Yu, Angew. Chem., Int. Ed., 2022, 61, e202112752 CrossRef CAS PubMed.
- Y. Liu, X. Quan, J. Li, J. Huo, X. Li, Z. Zhao, S. Li, J. Wan, J. Li, S. Liu, T. Wang, X. Zhang, B. Guan, R. Wen, Z. Zhao, C. Wang and C. Bai, Natl. Sci. Rev., 2023, 10, nwac167 CrossRef CAS PubMed.
- C. Xu, L. Xiao, Y. X. Gao, Y. He, C. Lei, Y. Xiao, W. J. Sun, S. Ahadian, X. T. Zhou, A. Khademhosseini and Q. S. Ye, Nano Res., 2020, 13, 2323–2331 CrossRef CAS.
- C. Xu, C. Lei and C. Z. Yu, Front. Chem., 2019, 7 CAS.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1 Search PubMed.
- M. Mahmoudi, M. P. Landry, A. Moore and R. Coreas, Nat. Rev. Mater., 2023, 1–17, DOI:10.1038/s41578-023-00552-2.
- J. Ren, N. Andrikopoulos, K. Velonia, H. Tang, R. Cai, F. Ding, P. C. Ke and C. Chen, J. Am. Chem. Soc., 2022, 144, 9184–9205 CrossRef CAS PubMed.
- R. Cai and C. Chen, Adv. Mater., 2019, 31, e1805740 CrossRef.
- C. Marques, M. J. Hajipour, C. Marets, A. Oudot, R. Safavi-Sohi, M. Guillemin, G. Borchard, O. Jordan, L. Saviot and L. Maurizi, ACS Nano, 2023, 17, 12458–12470 CrossRef CAS.
- H. Hyun, J. Park, K. Willis, J. E. Park, L. T. Lyle, W. Lee and Y. Yeo, Biomaterials, 2018, 180, 206–224 CrossRef CAS PubMed.
- R. M. Visalakshan, M. N. MacGregor, S. Sasidharan, A. Ghazaryan, A. M. Mierczynska-Vasilev, S. Morsbach, V. Mailander, K. Landfester, J. D. Hayball and K. Vasilev, ACS Appl. Mater. Interfaces, 2019, 11, 27615–27623 CrossRef CAS PubMed.
- Q. Xiao, M. Zoulikha, M. Qiu, C. Teng, C. Lin, X. Li, M. A. Sallam, Q. Xu and W. He, Adv. Drug Delivery Rev., 2022, 186, 114356 CrossRef CAS PubMed.
- V. H. Nguyen and B. J. Lee, Int. J. Nanomed., 2017, 12, 3137–3151 CrossRef CAS.
- S. Hosseinpour, L. J. Walsh and C. Xu, J. Mater. Chem. B, 2020, 8, 9863–9876 RSC.
- S. Hosseinpour, M. N. Gomez-Cerezo, Y. Cao, C. Lei, H. Dai, L. J. Walsh, S. Ivanovski and C. Xu, Pharmaceutics, 2022, 14, 2302 CrossRef CAS PubMed.
- C. Xu, M. Yu, O. Noonan, J. Zhang, H. Song, H. Zhang, C. Lei, Y. Niu, X. Huang, Y. Yang and C. Yu, Small, 2015, 11, 5949–5955 CrossRef CAS.
- A. Kurtz-Chalot, C. Villiers, J. Pourchez, D. Boudard, M. Martini, P. N. Marche, M. Cottier and V. Forest, Mater. Sci. Eng., C, 2017, 75, 16–24 CrossRef CAS PubMed.
- R. Madathiparambil Visalakshan, L. E. González García, M. R. Benzigar, A. Ghazaryan, J. Simon, A. Mierczynska-Vasilev, T. D. Michl, A. Vinu, V. Mailänder and S. Morsbach, Small, 2020, 16, 2000285 CrossRef CAS PubMed.
- C. Vidaurre-Agut, E. Rivero-Buceta, E. Romaní-Cubells, A. M. Clemments, C. S. D. Vera-Donoso, C. C. Landry and P. Botella, ACS Omega, 2019, 4, 8852–8861 CrossRef CAS PubMed.
- T. P. Liu, S. H. Wu, Y. P. Chen, C. M. Chou and C. T. Chen, Nanoscale, 2015, 7, 6471–6480 RSC.
- R. Kolde and M. R. Kolde, R package, 2015, 1, 790 Search PubMed.
- C. Chen, Y. Wu, J. Li, X. Wang, Z. Zeng, J. Xu, Y. Liu, J. Feng, H. Chen, Y. He and R. Xia, Mol. Plant, 2023, 16, 1733–1742 CrossRef CAS PubMed.
- X. Tan, Y. Zhang, Q. Wang, T. Ren, J. Gou, W. Guo, T. Yin, H. He, Y. Zhang and X. Tang, Biomater. Sci., 2019, 7, 2934–2950 RSC.
- R. Cai, J. Ren, Y. Ji, Y. Wang, Y. Liu, Z. Chen, Z. Farhadi Sabet, X. Wu, I. Lynch and C. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 1997–2008 CrossRef CAS.
- Y. Zou, S. Ito, F. Yoshino, Y. Suzuki, L. Zhao and N. Komatsu, ACS Nano, 2020, 14, 7216–7226 CrossRef CAS PubMed.
- S. Zalba, T. L. M. Ten Hagen, C. Burgui and M. J. Garrido, J. Controlled Release, 2022, 351, 22–36 CrossRef CAS PubMed.
- P. Wen, W. Ke, A. Dirisala, K. Toh, M. Tanaka and J. Li, Adv. Drug Delivery Rev., 2023, 198, 114895 CrossRef CAS PubMed.
- S. Y. Fam, C. F. Chee, C. Y. Yong, K. L. Ho, A. R. Mariatulqabtiah and W. S. Tan, Nanomaterials, 2020, 10 Search PubMed.
- Tengjisi, Y. Hui, Y. Fan, D. Zou, G. H. Talbo, G. Yang and C. X. Zhao, J. Colloid Interface Sci., 2022, 606, 1737–1744 CrossRef CAS PubMed.
- K. Saha, M. Rahimi, M. Yazdani, S. T. Kim, D. F. Moyano, S. Hou, R. Das, R. Mout, F. Rezaee, M. Mahmoudi and V. M. Rotello, ACS Nano, 2016, 10, 4421–4430 CrossRef CAS PubMed.
- P. Maffre, K. Nienhaus, F. Amin, W. J. Parak and G. U. Nienhaus, Beilstein J. Nanotechnol., 2011, 2, 374–383 CrossRef CAS.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth, C. Bier, J. Sirirattanapan, W. Mann, L. Treuel, R. Zellner, M. Maskos, H. Schild and R. H. Stauber, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS PubMed.
- F. Chen, G. Wang, J. I. Griffin, B. Brenneman, N. K. Banda, V. M. Holers, D. S. Backos, L. Wu, S. M. Moghimi and D. Simberg, Nat. Nanotechnol., 2017, 12, 387–393 CrossRef CAS PubMed.
- V. P. Vu, G. B. Gifford, F. Chen, H. Benasutti, G. Wang, E. V. Groman, R. Scheinman, L. Saba, S. M. Moghimi and D. Simberg, Nat. Nanotechnol., 2019, 14, 260–268 CrossRef CAS PubMed.
- A. Arcella, S. Palchetti, L. Digiacomo, D. Pozzi, A. L. Capriotti, L. Frati, M. A. Oliva, G. Tsaouli, R. Rota, I. Screpanti, M. Mahmoudi and G. Caracciolo, ACS Chem. Neurosci., 2018, 9, 3166–3174 CrossRef CAS PubMed.
- G. Caracciolo, S. Palchetti, V. Colapicchioni, L. Digiacomo, D. Pozzi, A. L. Capriotti, G. La Barbera and A. Lagana, Langmuir, 2015, 31, 10764–10773 CrossRef CAS PubMed.
- K. Yang, B. Mesquita, P. Horvatovich and A. Salvati, Acta Biomater., 2020, 106, 314–327 CrossRef CAS PubMed.
- L. M. Werner and A. K. Criss, J. Immunol., 2023, 211, 1443–1449 CrossRef CAS.
- A. Moore, R. Weissleder and A. Bogdanov, Jr., J. Magn. Reson. Imaging, 1997, 7, 1140–1145 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01124d |
| This journal is © The Royal Society of Chemistry 2025 |