
Engineering of redox-triggered polymeric lipid hybrid nanocarriers for selective drug delivery to cancer cells†
B. Siva
Lokesh‡
ac,
Suresh
Ajmeera‡
ade,
Rajat
Choudhary
 ac,
Sanjaya Kumar
Moharana
bc,
C. S.
Purohit
bc and
V. Badireenath
Konkimalla
*ac
ac,
Sanjaya Kumar
Moharana
bc,
C. S.
Purohit
bc and
V. Badireenath
Konkimalla
*ac
aSchool of Biological Sciences, National Institute of Science Education and Research, HBNI, Jatni, Odisha 752050, India. E-mail: badireenath@niser.ac.in; Tel: +91-674-249 42 11
bSchool of Chemical Sciences, National Institute of Science Education and Research, HBNI, Jatni, Odisha 752050, India
cHomi Bhabha National Institute, Training School Complex, Anushakti Nagar, Mumbai 400094, India
dHasselt University, Institute for Materials Research (IMO), Nano-Biophysics and Soft Matter Interfaces (NSI), Wetenschapspark 1, 3590 Diepenbeek, Belgium
eIMEC, associated lab IMOMEC, Wetenschapspark 1, 3590 Diepenbeek, Belgium
First published on 18th December 2024
Abstract
Tunable redox-sensitive polymeric-lipid hybrid nanocarriers (RS-PLHNCs) were fabricated using homogenization and nanoprecipitation methods. These nanocarriers were composed of novel redox-cholesterol with disulfide linkages and synthesized by conjugating cholesterol with dithiodipropionic acid via esterification. Berberine (BBR) was loaded into the fabricated nanocarriers to investigate the selective uptake of BBR by cancer cells as well as its release and enhanced cytotoxicity. The optimized BBR nanocarriers BBR NP-17 and -18 exhibited a spherical shape and uniform distribution, with a particle size of 124.7 ± 1.2 nm and 185.2 ± 1.6 nm and a zeta potential of −5.9 ± 2.5 mV and −20.3 ± 1.1 mV, respectively. These NCs released >80% BBR in a simulated intracellular tumor microenvironment (TME), while only 30%–45% was released under normal physiological conditions. The accelerated drug release in the TME was due to disulfide bond cleavage and ester bond hydrolysis in the presence of GSH and acidic pH, whereas under normal conditions, the NCs remained stable/undissociated. Cellular uptake studies confirmed enhanced BBR uptake in GSH-rich cancer cells (H1975) compared with normal cells (BEAS-2B and HEK293A). Following uptake, compared with the free form of the drug, the optimized nanocarriers displayed significant selective cytotoxicity and apoptosis in cancer cells by notably downregulating anti-oxidant (NFE2L2, HO-1, NQO1, and TXRND1) and anti-apoptotic (MCL-1) genes while upregulating pro-apoptotic genes (PUMA and NOXA). This resulted in increased oxidative stress, thereby inducing selective apoptosis in the GSH-rich lung cancer cells. These results suggest that the synthesized novel NCs hold great potential for specifically delivering drugs to cancer cells (with a reduced environment) while sparing normal cells, thus ensuring safe and efficient cancer therapy.
1. Introduction
Nanoscale drug delivery systems (NDDSs) have emerged as a pivotal advancement in circumventing various pitfalls of traditional drug-delivery systems. This technological advancement has been proven to be a valuable carrier of several drugs, nucleic acids, and proteins in combating several cancers by overcoming biological barriers. Lipid- and polymer-based nanocarriers have been widely explored in the past decade. Commercial examples of these nanocarriers include DOXIL® (doxorubicin-loaded PEGylated liposome), DaunoXome® (daunorubicin liposome), DepoCyt® (cytarabine liposome), Abraxane (paclitaxel albumin nanoparticle), and Oncaspar® (polymer protein conjugate of L-asparaginase). These nanocarriers offer several advantages in drug delivery, including enhancement in solubility, permeability, and bioavailability; protection from enzymatic degradation and harsh gastrointestinal conditions; increased half-life; and mitigation of off-target effects induced by toxic drug molecules.1 Despite their promising potential in treating several cancers, these systems suffer from drawbacks such as poor entrapment, drug loading, and premature drug release.2 Therefore, combining the two systems may endow the obtained material with the positive attributes of both systems, which would aid in overcoming their individual limitations.Polymeric lipid hybrid nanocarriers (PLHNCs) with a size of 100–200 nm are ideal candidates to facilitate the effective delivery of different drugs into cancer cells via the enhanced permeability and retention effect (EPR). These systems consist of a polymer core surrounded by a lipid layer, which enables the effective loading of hydrophobic and hydrophilic drugs in the polymer core and lipid matrix.3 These novel systems offer several advantages for drug delivery; however, premature leakage of the drugs in the blood circulation and their unintended release in healthy cells often result in side effects, posing a significant challenge. Thus, to address these challenges and enhance the precise release of drugs in tumor sites, various unique factors prevailing in the endogenous tumor microenvironment (TME), such as pH, glutathione, and reactive oxygen species, have been explored to deliver drugs in response to a specific stimulus.
TME-responsive nanocarriers have attracted considerable attention in cancer therapy compared to external stimuli-responsive nanocarriers (e.g., magnetic, light, and ultrasound), given that they eliminate the need for additional instruments for drug release at the target site, making them more cost-effective. The prevailing endogenous environment of tumor cells/tissues differs from that of normal cells, aiding the design of specialized nanocarrier systems to release drugs selectively in response to TME, avoiding healthy cells. Intrinsic tumor endogenous stimuli such as the extra and intracellular pH (pH 6.4 to 4.5 due to lactic acidosis),4 several overexpressing proteins/enzymes, reductases (overexpressed in a hypoxia environment),5,6 and intracellular reductive environment (elevated levels of GSH, i.e., about 2–10 mM GSH concentration in tumor cells and 2–20 μM in healthy cells)7 are the potential targets that are being explored. Due to these distinct microenvironment characteristics, many researchers have designed intelligent nanoparticles to deliver drugs in response to endogenous stimuli. For instance, the inclusion of pH-sensitive groups such as Schiff base,8,9 hydrazone bonds,10,11 imine bonds,12 and ester bonds13 in nanoformulations enables the release of drugs in response to the tumor acidic pH. Additionally, the presence of nitroimidazole (NI)14 and azobenzene (Azo)6 derivatives trigger drug release in response to overexpressed biological reductases (e.g., nitroreductases) in the hypoxic tumor and the incorporation of disulfide bonds in nanoparticle systems enable them to respond to the increased GSH levels in the intracellular TME.15,16
Berberine (BBR), a natural isoquinoline phytochemical, is recognized for its diverse pharmacological activities, including anti-cancer, anti-inflammatory, antimicrobial, and antioxidant activities. However, despite its promising therapeutic potential, BBR is categorized as a class IV drug due to its poor solubility, low bioavailability, and limited cellular permeability. Moreover, the short half-life of BBR is a major limitation, which decreases its therapeutic efficacy. Accordingly, to fulfill therapeutic requirements, higher doses of BBR have been employed; however, this can lead to adverse effects such as anorexia, stomach upset, diarrhea, and constipation, potentially compromising its therapeutic advantages.17–20
Recently, several nanoparticle delivery systems have been developed to minimize the limitations and enhance the therapeutic efficacy of BBR in cancer (albumin nanoparticles,17 nano-lipid carriers,19 and chitosan NPs21). However, the selective release of BBR from nanosystems at the cancer cell target remains less explored. Thus, to address the selectivity problem and potentiate the therapeutic effectiveness, we aimed to synthesize nanomaterials responsive to internal stimuli such as pH and GSH using biocompatible lipids (cholesterol,22 DPPC,23 and stearic acid24) and polymers (PEG25 and PNIPAM26) for the loading and delivery of BBR in GSH-rich cancer cells.
Therefore, in this study, a novel cholesterol lipid containing a disulfide (–S–S–) bond as a stimuli-responsive material was synthesized. This bio-reducible lipid was incorporated into redox-sensitive polymeric lipid hybrid nanocarriers (RS-PLHNCs) with BBR loaded in the nanocarrier system. The objective was to investigate the superiority of the synthesized nanocarrier system compared to the free form of BBR in enhancing cancer cell selectivity and killing efficiency in non-small cell lung cancer cells (NSCLC; H1975 cells) in vitro, while also comparing its selectivity on normal lung and embryonic kidney cells (BEAS-2B and HEK293A cells). The efficacy of the nanoformulation was also dependent on its lipid composition, particle size, zeta potential, and the physicochemical characteristics of the loaded drug for its release and therapeutic activity. Therefore, different RS-PLHNCs with varying lipid compositions were prepared and studied.
2. Experimental
2.1. Materials
Berberine (BBR) and stearic acid were purchased from M.P. Biomedicals. Sodium azide (NaN3), 4-toluenesulfonyl chloride (TsCl), 3,3′-dithiodipropionic acid (DTPA), triphenylphosphine (PPh3), 4-dimethylaminopyridine (DMAP), and Pluronic F-127 (PF-127) were procured from Sigma-Aldrich. Span-80, potassium hydroxide (KOH), absolute ethanol, dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and L-glutathione reduced (GSH) were purchased from Himedia. The dialysis membrane (10 kDa) was obtained from Invitrogen, poly-(N-isopropyl acrylamide) from TCI Chemicals, polyethylene glycol (PEG) (M.wt. 2000 g mol−1) from Alfa Aesar, cholesterol (CL) from bioWORLD, acetic anhydride from Pallav Chemicals, toluene from SRL, and dipalmitoyl phosphatidylcholine (DPPC) from Avanti Polar Lipids.All the solvents used to synthesize the modified lipids and polymers were purchased from Spectrochem, including dichloromethane (DCM), diethyl ether, dimethyl formamide (DMF, dry), methanol (MeOH), tetrahydrofuran (THF), n-hexane, pyridine (dry), ethyl acetate, and acetonitrile.
2.2. Synthesis of redox-sensitive disulfide cholesterol (cholesterol-dithiodipropionate)
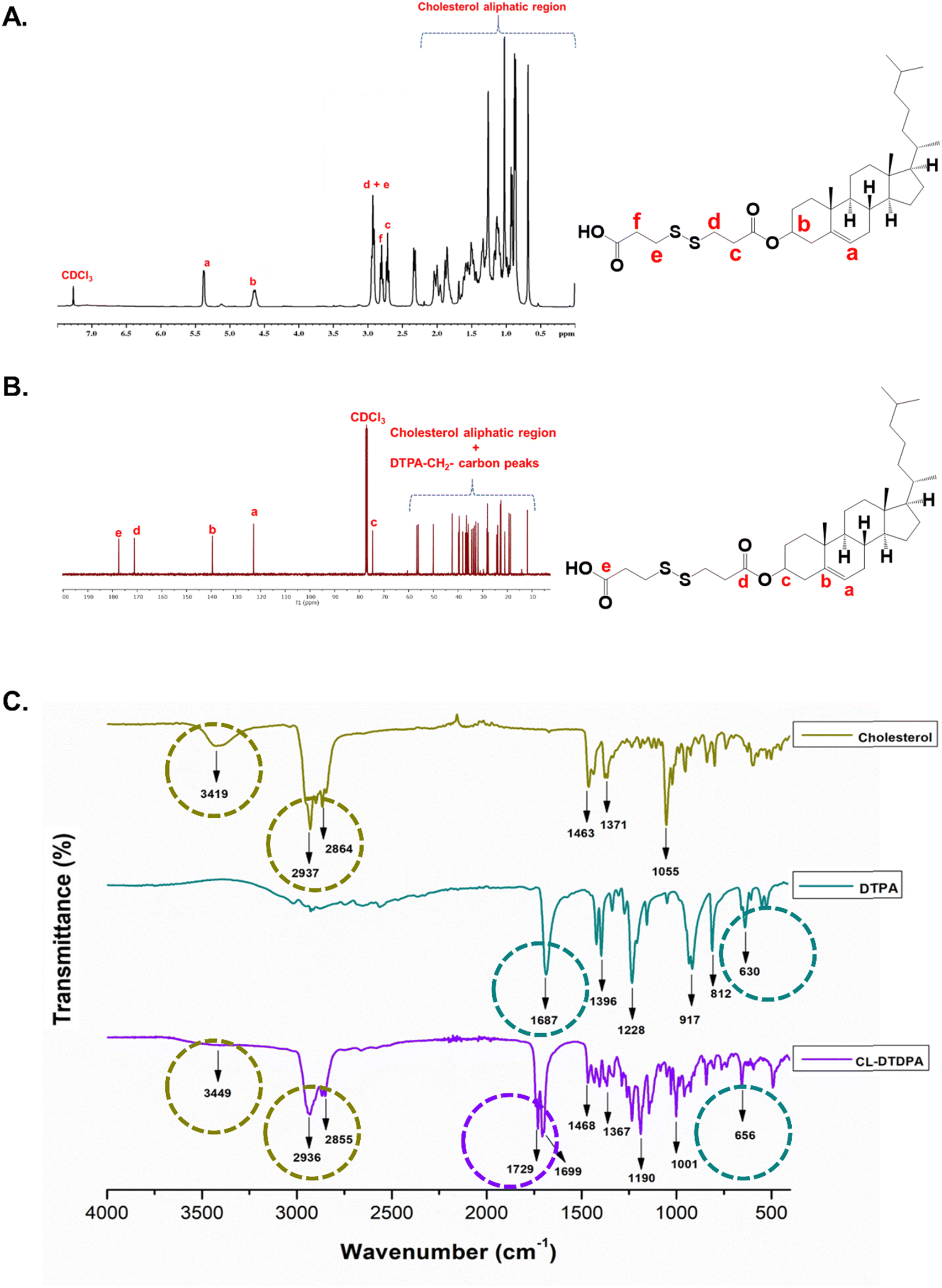
Cholesterol-dithiodipropionate (CL-DTDPA) was synthesized following a reported method with slight modifications.27 Briefly, 3,3′-dithiodipropionic acid (1 g, 4.755 mmol) was dissolved in acetic anhydride (75 mL) and stirred for 3–4 h under an N2 atmosphere to obtain dithiodipropionic anhydride (DTDPA). To the above-mentioned DTDPA solution, toluene (10–15 mL) was added and dried under reduced pressure using a rotary evaporator (Heidolph rotary evaporator) three to four times. The obtained pure DTDPA residue was dissolved in dry DCM (50 mL) and admixed with CL (1.838 g, 4.755 mmol) and DMAP (0.290 g, 2.377 mmol). Then, the reaction mixture was continuously agitated at room temperature under inert N2 atmospheric conditions, and simultaneously the progress of the reaction was monitored using TLC. The resulting product was extracted using a DCM/water system. The organic layer was collected, washed several times with water, dried with anhydrous Na2SO4, and evaporated using a rotary evaporator. Finally, the acquired product was purified using silica-gel column chromatography (stationary phase: silica gel (mesh size: 100–200); mobile phase: 10–30% ethyl acetate and hexane), the eluted mobile phase was evaporated, washed with acetonitrile, and dried under reduced pressure to obtain pure CL-DTDPA (CL–COO–SS–COOH).28 The purified CL-DTDPA was further characterized for 1H NMR and 13C NMR to verify the structure. The obtained pure CL-DTDPA (redox-CL) was used for the fabrication of redox-sensitive PLHNCs.The final yield was 80%. 1H NMR (CDCl3, ppm): δ 5.38 (1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 4.65 (1H, –CH–), 2.93(4H, –CH2–SS–), 2.80 (2H, –CH2–COOH), and 2.72 (2H, –CH2–COO–) (Fig. 1A). 13C NMR (CDCl3, ppm): δ 177.94 (–COOH), and 171.02 (–COO–) (Fig. 1B). ESI-MS (m/z): C33H54O4S2 – 579.35 [M + H]+ (Fig. S2A, ESI†).
CH–), 4.65 (1H, –CH–), 2.93(4H, –CH2–SS–), 2.80 (2H, –CH2–COOH), and 2.72 (2H, –CH2–COO–) (Fig. 1A). 13C NMR (CDCl3, ppm): δ 177.94 (–COOH), and 171.02 (–COO–) (Fig. 1B). ESI-MS (m/z): C33H54O4S2 – 579.35 [M + H]+ (Fig. S2A, ESI†).
 | ||
| Fig. 1 1H NMR (A), 13C NMR (B), and FTIR (C) spectra of cholesterol-dithiodipropionate (CL-DTDPA). | ||
2.3. Synthesis of polyethylene glycol diamine (PEGDA)
The final yield was 98%. 1H NMR (d6-DMSO, ppm): 7.77 (CH–), 7.48 (CH–), 4.09 (–CH2–O–), 3.31–3.66 (–CH2–CH2–O–), and 2.4 (–CH2) (Fig. S1A, ESI†). 13C NMR (d6-DMSO, ppm): 145.41 (C), 132.8 (CH–), 130.59 (CH–), 128.03 (C), 70.3 (–CH2–CH2–O–), and 68.3 (–CH2–O–) (Fig. S1B, ESI†).
The final yield was 80%. 1H NMR (d6-DMSO, ppm): δ 3.46–3.67 (–CH2–CH2–O), and 3.37 (–CH2–N3) (Fig. S1C, ESI†). 13C NMR (d6-DMSO, ppm): δ 70.2 (–CH2–CH2–O), 69.67 (–CH2–O–), and 50.44 (–CH2–N3) (Relevant Information (Fig. S1D, ESI†)).
The final yield was 93%. 1H NMR (CDCl3, ppm): δ 3.46–3.67 (–CH2–CH2–O), 2.86 (–CH2–NH2), 1.65 (–NH2) (Fig. S1E, ESI†). FTMS - probe ESI (m/z): Average mass distribution centered around 2000 for PEG, for PEG diamine, calculatedd-1999.0389 and found-1999.2291 [M + H]+ (Fig. S2B, ESI†). ΔM 44.02 Da was observed between two peaks, indicating ethylene glycol (–CH2–CH2–O–) fragments.
2.4. Preparation of redox-sensitive polymeric–lipid hybrid nanocarriers
The redox-sensitive polymeric–lipid hybrid nanocarriers (RS-PLHNCs) were fabricated using homogenization and nanoprecipitation methods.30 Typically, the PNIPAM and PEGDA co-polymers were dissolved in 10 mL triple-distilled water containing PF-127 as a stabilizer. To the above-mentioned homogenous aqueous phase, 1 mL of ethanolic lipid solution containing CL-DTDPA, DPPC, stearic acid, and Span-80 (75 μL) was added drop-wise under high homogenization. The nanosuspension was further homogenized for another 30 min, followed by gentle stirring until traces of the organic phase were removed. The prepared blank redox nanocarriers (RS-PLHNCs) were further optimized for particle size and stability by varying the composition of the excipients used in the aqueous and organic phases during the preparation (given in Table 1). BBR-loaded redox nanocarriers were prepared similarly to the blank nanocarriers, in which BBR was added to the aqueous phase of the nanocarrier preparation.| Batch no # | Redox-cholesterol (mg) | Stearic acid (mg) | DPPC (mg) | PNIPAM (mg) | BBR (mg) | PS (nm) | PDI |
|---|---|---|---|---|---|---|---|
| 1 | 15 | 10 | 10 | 20 | — | 212 ± 1.29 | 0.15 ± 0.01 |
| 2 | 15 | 10 | 10 | — | — | 246 ± 1.48 | 0.17 ± 0.04 |
| 3** | 15 | — | 10 | 20 | — | 151 ± 2.82** | 0.12 ± 0.03** |
| 4 | 15 | 10 | — | 20 | — | 233 ± 3.36 | 0.25 ± 0.01 |
| 5 | 20 | 10 | 10 | 20 | — | 236 ± 3.37 | 0.26 ± 0.03 |
| 6 | 15 | 20 | 10 | 20 | — | 314 ± 9.22 | 0.36 ± 0.06 |
| 7*** | 15 | 10 | 20 | 20 | — | 210 ± 4.59*** | 0.18 ± 0.05*** |
| 8 | 20 | 20 | 20 | 20 | — | 222 ± 0.64 | 0.25 ± 0.03 |
| Composition for the fabrication of berberine-loaded redox-sensitive PLHNCs (BBR-loaded RS-PLHNCs) | |||||||
| 17** | 15 | - | 20 | 20 | 1 | 125 ± 1.21** | 0.15 ± 0.02** |
| BBR NP-17 | |||||||
| 18*** | 15 | 10 | 20 | 20 | 1 | 185 ± 1.56** | 0.15 ± 0.02*** |
| BBR NP-18 | |||||||
| Constants | |||||||
| PEGDA = 20 mg and PF-127 = 100 mg | |||||||
2.5. Characterization of BBR-loaded RS-PLHNCs
Transmission electron microscopy (TEM) was used to evaluate the shape and surface morphology of the optimized RS-PLHNCs. In brief, the BBR-loaded nanosuspension was deposited on copper mesh (TEM grid) by placing 10 μL of sample and incubated for 45 min and negatively stained using 2% uranyl acetate. The prepared sample was dried and visualized under TEM at a voltage of 200 kV (Model: JEOL-JEM F200).30
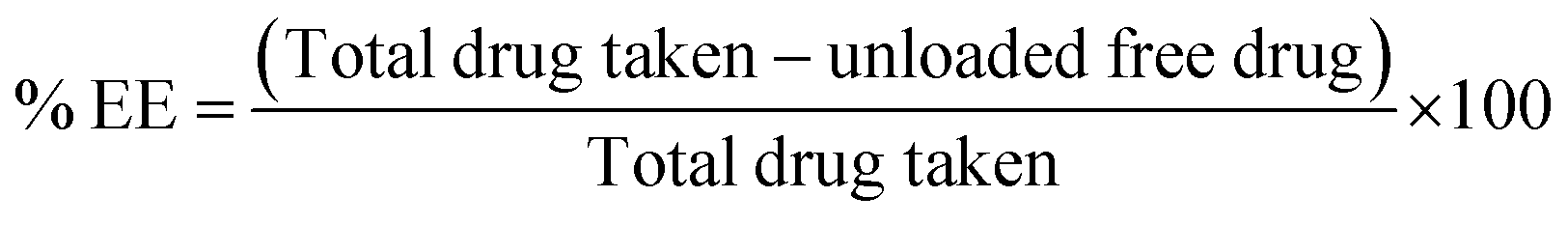
The kinetics of the BBR release from the nanocarrier system was further studied using the following models: zero-order, first-order, Higuchi, Hixson-Crowell, and Korsmeyer-Peppas models.25
2.6. In vitro cell-based assays
2.6.6.1. Flow cytometry (quantitative analysis). The cellular uptake of the free form of BBR or nanoformulation was quantified by flow cytometry.42 H1975, BEAS-2B, and HEK293A cells (1 × 106 cells per well) were seeded in a 6-well plate and incubated for 24 h at 37 °C under 5% CO2. Following cell adherence, the BBR nanoparticles and free form of BBR were added to the corresponding wells (at IC50 values equivalent to BBR nanoparticles) and incubated for 4 h, 12 h, and 18 h. Following incubation, the cells were washed with ice-cold PBS (3 times), harvested, and centrifuged at 3000 g for 10 min. Finally, the cells in each sample were resuspended in 400 μL PBS for FACS analysis. The cellular uptake of BBR was quantified using the fluorescence mean intensity by gating 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells (n = 3) (λex = 488 nm and λem = 585 nm).
000 cells (n = 3) (λex = 488 nm and λem = 585 nm).
2.6.6.2. Confocal laser scanning microscopy (CLSM). CLSM was used to analyze the intracellular distribution of the free form of BBR, BBR-loaded RS-PLHNCs, and blank nanocarriers.43 H1975 lung cancer cells, BEAS-2B normal lung cells, and HEK293A normal embryonic kidney cells (1 × 105 cells per well) were seeded on a cover slip. Post 24 h incubation, the formulations/free form of BBR were added to the cells with the determined IC50 values respective to BBR nanoparticles and incubated for 4 h and 18 h with PBS as a control. Post-treatment, the cells were washed (3×) with ice-cold PBS and fixed with 4% w/v paraformaldehyde. The cells were mounted onto glass slides using Flouromount-G media (Invitrogen) and observed under CLSM (λex = 405 nm and λem = 550 ± 50 nm).
2.6.7.1. Apoptosis study by flow cytometry. To study apoptosis, BEAS-2B, HEK293A, and H1975 cells were seeded in a 6-well plate with 1 × 106 cell density and incubated at 37 °C under a 5% CO2 incubator for 24 h. After adherence, the cells were treated with IC50 values of BBR-loaded nanocarrier (BBR NP-17 and NP-18), free form of BBR (FR-17 and FR-18), blank nanoparticle (BLK NP-17 and BLK NP-18) for 4, 12, and 18 h. After treatment, the cells were collected and washed twice with cold PBS, and then centrifuged at 1500 rpm for 5 min at 4 °C. The resulting pellet was resuspended in 1X binding buffer containing 5 μL each of fluorescein isothiocyanate (FITC)-labeled Annexin V (λex = 488 nm and λem = 518 nm) and propidium iodide (PI) (λex = 561 nm and λem = 616 nm) for 20 min under dark condition at room temperature. Finally, the stained cells were detected in a flow cytometer with 104 gating cells. In the apoptosis dot plot, Q1 (Annexin V-FITC-/PI+) indicates necrosis, and Q3 (Annexin V-FITC-/PI-) normal live cell population, whereas Q2 (Annexin V-FITC+/PI+) and Q4 (Annexin V-FITC+/PI-) represent late and early apoptosis, respectively (Fig. S10, ESI†). The BD FACSDivaTM software was employed to determine the %apoptotic cells.40 The assay was performed in triplicate.
2.6.7.2. Annexin V staining for apoptosis. CLSM was used to analyze the apoptotic cells stained with Annexin V-FITC upon treatment with the free form of BBR, BBR-loaded RS-PLHNCs, and blank nanocarriers.43 H1975 lung cancer cells, BEAS-2B, HEK293A, and H1975 cells were seeded with a density of 1 × 105 cells per well on a cover slip. Post 24 h incubation, the formulations/free form of BBR were added to the cells with the respective determined IC50 values to BBR nanoparticles and incubated for 4 h and 18 h with PBS as a control. Post-treatment, the cells were washed (3×) with ice-cold PBS and stained with Annexin V-FITC for 15 min under dark conditions at room temperature. After staining, the cells were fixed with 4% w/v paraformaldehyde, and the coverslips were mounted onto glass slides using Flouromount-G media (Invitrogen) and observed under CLSM (λex = 488 nm and λem = 550 ± 50 nm).
2.6.8.1. RNA isolation. In seven groups, RNA was isolated from H1975 cells (1 × 106 cells per well) treated with IC50 values of the BBR-loaded nanocarrier (BBR NP-17 and NP-18), free form of BBR (FR-17 and FR-18), and blank nanoparticle (BLK NP-17 and BLK NP-18). Post 24 h, the cell pellets were harvested and resuspended in Trizol (500 μL, Invitrogen). Then, 0.2 mL of chloroform was added to the suspension and vortexed vigorously under ice-cold conditions. The cell suspension was centrifuged at 12
000 × g; 4 °C for 15 min; the supernatant (aqueous phase) was further aspirated carefully and transferred into a fresh tube. 500 μL of isopropanol was added to the supernatant, incubated for 10 min, and centrifuged at 12000 × g for 10 min at 4 °C. Further, the pellet was washed with 75% ethanol by centrifuging at 12000 × g for 10 min at 4 °C. The obtained RNA pellet was air-dried and dissolved in 20 μL of DEPC-treated nuclease-free water. The concentration and purity of RNA were estimated using NanoDrop (ONEC, Thermo Fisher Scientific), and its quality was checked by 1% agarose gel electrophoresis. Good-quality RNA (260/280 ratio = 1.9–2.0) with two visible bands was considered for cDNA synthesis. For reverse transcription, 2 μg of total RNA was used through the Reverse Transcription Kit (Maxima First Strand cDNA kit, Thermo Fisher Scientific) in a Thermal Cycler (Veriti, Applied Biosystems) following the manufacturer's instructions.
2.6.8.2. Quantitative reverse transcription polymerase chain reaction (qRT-PCR) study. A real-time PCR study was performed to measure the mRNA expression levels of PUMA, NOXA, MCL-1, NFE2L2, NQO1, TXRND1, HMOX1, and GAPDH (housekeeping gene) using iTaq Universal SYBR Green Supermix (BioRad) in QuantStudio5 (Applied Biosystems). 2 μL of prepared cDNA was used for 10 μL reaction, with 5 μL of SYBR, 1 μL of nuclease-free water, and 1 μL (0.25 μM) of forward and reverse primer each. PCR was performed using the following temperature cycle: initial denaturation (95 °C for 20 s), 40 amplification cycles (denaturation at 95 °C, 3 s; annealing at 60 °C, 30 s). The fold change of target mRNA was calculated using the 2−ΔΔCt values obtained from the treated samples compared with the control samples. The forward and reverse primer sequences used are listed in Table S2 (ESI†).
2.7. Statistical analysis
All data were normalized to the control values of each assay and expressed as mean ± SEM. Statistical analysis was performed using one-way ANOVA followed by Dunnett's multiple comparison tests for comparative analysis between multiple groups and two-way ANOVA followed by Turkey's multiple comparison tests between multiple groups with two or more independent variables utilizing GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA, USA). p values of *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 were considered statistically significant.3. Results and discussion
3.1. Synthesis and characterization of redox-cholesterol (CL-DTDPA) and aminated polyethylene glycol (PEGDA)
A hydrophobic dual-responsive compound, redox-cholesterol (CL–S–S–COOH, CL-DTDPA), was prepared by modifying CL with a disulfide-containing monomer (DTPA) via an esterification reaction, as shown in Scheme 1A. In the 1H NMR (Fig. 1A) spectrum, the three triplet peaks in the range of 2.6–3.0 ppm confirmed DTDPA conjugation to CL. The peaks in the 13C NMR spectrum (Fig. 1B) at 177.9 ppm for carboxylic acid (CO) and 171 ppm for ester CO also confirmed the conjugation of DTDPA. According to FTIR, characteristic peaks of CL and DTPA were conserved, with a shift in the peak at 1687 to 1729 cm−1, indicating the conversion of carboxylic acid to an ester functional group and further verifying the successful synthesis of the conjugate (Fig. 1C). Thus, both the NMR and FTIR studies confirmed the synthesis of CL-DTDPA.
 | ||
| Scheme 1 Scheme for the synthesis of cholesterol-dithiodipropionate (redox-sensitive cholesterol) (A) and PEG diamine (B). | ||
The hydrophilic PEG diamine (PEGDA) was synthesized via the step-by-step conversion of PEG to di-tosylated PEG (PEG-OTs), later to PEG diazide (PEG-N3), and finally to PEG diamine (PEG-NH2) (Scheme 1B). The formation of PEG-OTs was confirmed by 1H NMR and 13C NMR (Fig. S1A and B, respectively, ESI†). The two doublet peaks corresponding to benzene protons at 7.77 ppm and 7.48 ppm and the carbon peaks in the range of 125–146 ppm confirm the conversion of PEG-OTs. The absence of benzene peaks in the 1H (Fig. S1C, ESI†) and 13C (Fig. S1D, ESI†) NMR spectra indicate the formation of PEG azide. The shift in the triplet peak from 3.37 ppm (–CH2– adjacent to azide) to 2.86 ppm (–CH2– adjacent to amine) indicated the conversion of PEGD to PEGDA (Fig. S1E, ESI†). Moreover, the FTIR peak (Fig. S1F, ESI†) at 3446 cm−1, corresponding to the N–H symmetrical and asymmetrical stretching of the primary amine groups, was only present in the spectrum of PEGDA (absent in PEGD).44,45 Accordingly, the NMR and FTIR data confirmed the successful synthesis of PEG diamine (PEGDA).
Thus, redox-cholesterol (CL-DTDPA) and PEG diamine (PEGDA) were synthesized and characterized to fabricate the BBR-loaded redox-sensitive PLHNCs.
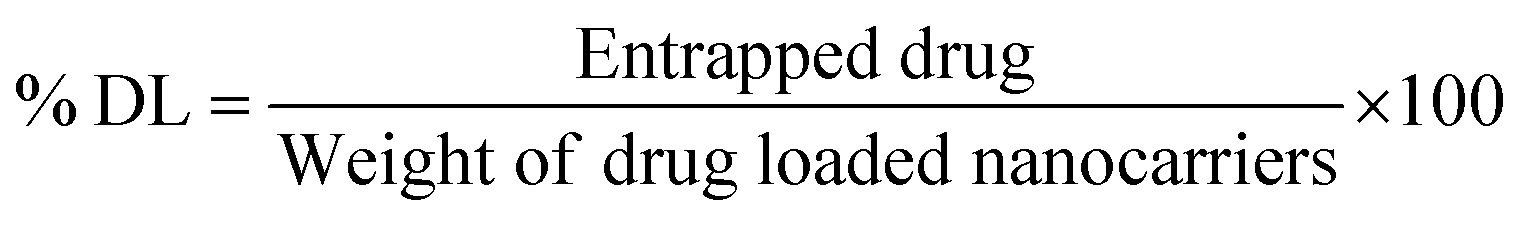
3.2. Fabrication and characterization of berberine-loaded redox-sensitive PLHNCs (BBR-loaded RS-PLHNCs)
In the current work, homogenization and nanoprecipitation methods were employed to fabricate stable homogeneous BBR-loaded RS-PLHNCs with a diameter <200 nm and a PDI of 0.25.46 The fabricated nanocarriers comprised of drug-embedded polymer core layer encased by redox lipid to prevent premature drug release under physiological conditions. According to the preliminary study, blank nanocarriers of different polymer and lipid compositions were prepared to obtain the nanoformulation with the optimal particle size, uniformity, and stability (as shown in Table 1). Among the eight batches, batch #3 and #7 had good stability, relatively small particle size, and homogenous distribution. Batch #7, containing stearic acid, displayed a larger particle size than batch #3, as it increased the thickness of the lipid matrix and thereby increased the overall hydrodynamic radius of the particle. In all batches with stearic acid, Span-80 surfactant with higher lipophilicity (HLB value 4.3) was preferred over Tween-80 for nanocarrier stabilization, as it facilitated the miscibility of the highly lipophilic stearic acid in the aqueous phase by reducing the interfacial tension between the aqueous and lipid phase.Finally, the compositions of batches #3 and #7 used to prepare the blank nanocarriers (BLK NPs) were utilized to fabricate BBR-loadedRS-PLHNCs (Batch 17 and 18), with minor changes in their composition, respectively. In BBR NP-17, the amount of DPPC increased from 10 to 20 mg to equally maintain the DPPC concentration in BBR NP-18 to further examine the influence of stearic acid on the physicochemical characteristics and stability of the nanocarrier. The optimized BBR-loaded RS-PLHNCs showed %EE and %DL of 90.52 ± 4.68 and 1.12 ± 0.06 in BBR NP-17 and 95.42 ± 3.51 and 1.05 ± 0.05 in BBR NP-18, respectively. The increased encapsulation of BBR in BBR NP-18 is ascribed to the presence of the lipophilic stearic acid in the lipid matrix, which may increase the protection of the polymeric layer in which the drug was embedded and further prevent the leakage of the drug into the dispersion phase than BBR NP-17.
The optimized nanoformulations (BBR NP-17 and 18) exhibited nearly spherical uniform nanosized particles (Fig. 2A and B). The appearance of bright portions in the image indicates the presence of a polymeric core, while a thin lining was observed around the periphery of the core, which is the lipid shell coating. During the evaporation process during the synthesis of the nanocarrier, the polymer formed the core, entrapping the drug, and the lipids self-assembled, surrounding the polymeric core.47 These spherical particles of BBR NP-17 and 18 showed an average particle size of 124.7 ± 1.21 and 185.17 ± 1.56 nm and PDI of 0.153 ± 0.02 and 0.152 ± 0.02, respectively, according to dynamic light scattering (DLS). Alongside their particle size, both formulations exhibited a negative zeta potential (−5.91 ± 2.54 and −20.03 ± 1.07 mV) due to the coating of the negatively charged lipid matrix (DPPC, stearic acid, and CL-DTDPA mixture) on the surface of the polymer core (Fig. 2A and B).48 These features are essential for particle retention and uptake by EPR in cancer cells for its activity.
 | ||
| Fig. 2 Characterization of BBR-loaded redox-sensitive PLHNCs. TEM images show the morphology of the fabricated nanoparticles and DLS graphs illustrating the particle size, PDI, and zeta potential of BBR NP-17 (A) and 18 and (B). FTIR spectra of the free form of BBR, blank, and BBR-loaded NPs (C). | ||
FTIR was performed to analyze the incorporation of BBR in the nanocarrier system and to detect incompatibilities/interactions between the BBR and excipients or among the excipients used in the formulation (Fig. 2C and Fig. S3B, ESI†). According to the FTIR data, the primary absorption peaks for the free form of BBR were observed at 3209 cm−1 (–OH stretching vibration of methoxy group), 2906 and 2852 cm−1 (–CH stretching vibration), 1632 cm−1 (–C–N stretching vibration), 1505 cm−1 (–CC stretching vibration) and 1102 cm−1 (–C–O–C stretching vibration).17,49,50 Comparing the FTIR spectra of the free form of BBR, physical mixture, BLK NPs, and BBR NPs (summarized in Table 2), no new characteristic peaks were detected. Only negligible shifts in the characteristic peaks were noted among them, suggesting the association of weak interactions between the drug and excipients. In BBR NPs, the BBR-specific characteristic peaks were not clearly distinguished. This may be due to the overlapping of the peaks with the excipients or BBR incorporated in the polymer core, whose characteristic peak intensities may be negligible. This data confirms the absence of potential chemical interactions between the drug and excipients or among the excipients. However, the incorporation of BBR in the nanocarrier system was not clearly addressed. Therefore, we performed a UV spectroscopy-based study to confirm the loading of BBR in the nanocarrier system.
| Characteristic peak (wavenumber cm−1) | ||||||
|---|---|---|---|---|---|---|
| Free form of BBR (i) | 3209 | 2906, 2852 | — | 1632 | 1505 | 1102 |
| Functional group (i) | Intermolecular –OH (methoxy group | –CH stretching | –C–N stretching | –CC stretching |
–C–O–C stretching | |
| Physical mixture (ii) | 3340 | 2885 | 1729 | 1640 | 1552 | 1102 |
| BLK NPs (iii) | 3369 | 2885 | 1732 | 1644 | 1552 | 1097 |
| BBR NPs (iv) | 3360 | 2885 | 1737 | 1644 | 1547 | 1102 |
| Functional group (ii), (iii) and (iv) | –OH stretching | –CH stretching | –CO stretching (ester or acid) |
–CO stretching (amide group) |
–NH bending (amide group) | –C–O–C stretching |
| Inference | PF-127/DPPC/CL–S–S–COOH/PNIPAM | (ii) and (iii) PNIPAM/PF-127/PEGDA/CL–S–S–COOH/DPPC/stearic acid (iv) BBR/PNIPAM/PF-127/PEGDA/CL–S–S–COOH/DPPC/stearic acid | CL–S–S–COOH/DPPC | PNIPAM | PNIPAM | (ii) PF-127/PEG diamine/DPPC (iv) BBR/PF-127/PEG diamine/DPPC |
The UV spectroscopy data displayed the characteristic peaks for BBR incorporated in BBR NP-17 and NP-18 at wavelengths of 260 nm (peak A), 345 nm (peak B), and 424 nm (peak C), which are similar to that of the free form of BBR, while no peaks were observed in BLK NP-17 and 18 (Fig. S3A, ESI†). The results from both UV and FTIR spectroscopy confirmed the successful encapsulation of BBR into the redox nanocarrier system without any specific covalent modifications/interactions.
3.3. Reduction-triggered nanocarrier disassembly and BBR release from the nanocarriers
The redox/pH-sensitive behaviors of the optimized nanocarriers (BBR NP-17 and BBR NP-18) were studied by observing the changes in their morphology, particle size, and distribution of nanocarriers under tumor microenvironment-simulated conditions. L-Glutathione (GSH, 10 mM) was used as a mimic for the reductive microenvironment51,52 and a pH of 4.5–5.0 to mimic the intracellular endo-lysosomal acidic microenvironment, given that they are the crucial parameters that prevail in the tumor microenvironment.53 Following 24 h incubation in pH 7.4 buffer with 10 mM GSH, the DLS data presented a significant increase in the hydrodynamic radius in both batches from 125.8 ± 0.7 to 321.1 ± 8.7 nm (BBR NP-17) and 195.4 ± 1.9 to 381.2 ± 7.3 nm (BBR NP-18), while no remarkable changes in particle size were evidenced under the condition of pH 7.4 buffer without GSH (Fig. 3A(i) and B(i)), respectively. Additionally, aggregation of the nanocarriers with an alteration in their shape was observed using TEM, while no morphological changes were displayed in the nanocarriers incubated in pH 7.4 buffer without GSH (Fig. 3A(ii) and B(ii)), respectively. Similarly, BBR NP-17 and BBR NP-18 had a non-homogenous particle distribution (with higher PDI values of 0.22 ± 0.014 to 0.48 ± 0.02 and 0.16 ± 0.01 to 0.4 ± 0.03, respectively) in the GSH environment, while negligible deviations were observed under normal conditions. These changes were mainly attributed to the reduction and cleavage of the disulfide bonds (–S–S–) present in the redox-cholesterol (CL-S-S-COOH), which destabilized the lipid matrix coated on the surface of the polymer core.54,55 Similarly, the pH responsiveness of the nanocarriers upon exposure to acidic pH (pH 4.5) resulted in increased aggregation and a complete change in their shape with a substantial increase in their particle size to 457.8 ± 30 nm (BBR NP-17) and 315.6 ± 10.4 nm (BBR NP-18) and PDI to 0.55 ± 0.01 (BBR NP-17) and 0.53 ± 0.04 (BBR NP-18), which is ascribed to the hydrolysis of the ester bonds in acidic pH (Fig. 3A(i) and B(i)), respectively.56,57 Analogous results with the morphology, larger particle size, and PDI were observed upon the combination of pH and reduction dual stimuli (Fig. 3A(ii) and B(ii)), respectively. On the contrary, only a slight decrease in the zeta potential was observed under all the exposure conditions.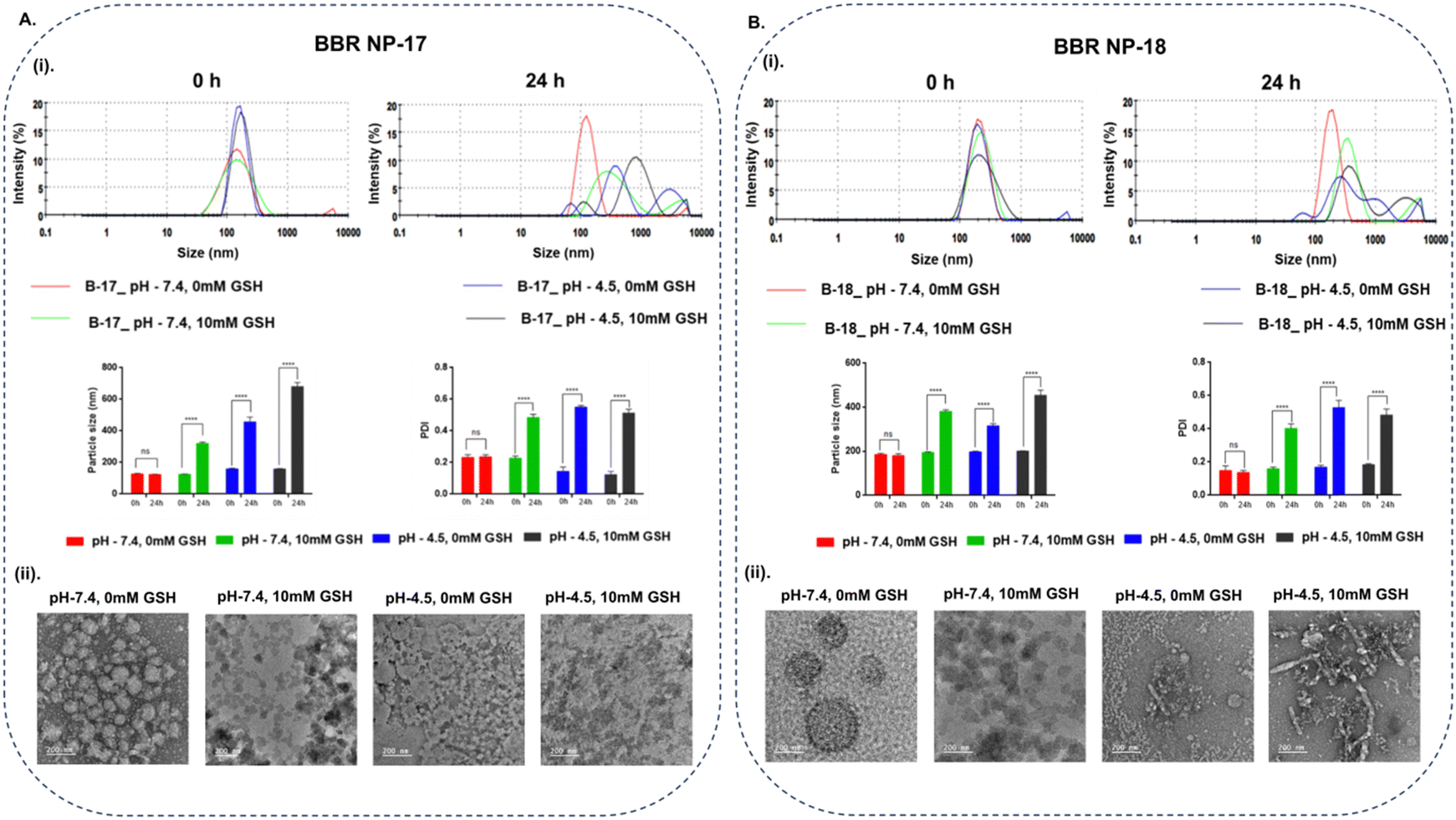 | ||
| Fig. 3 Redox-responsive study of BBR NP-17 (A) and BBR NP-18 (B) in normal and tumor-simulated conditions. Particle size distribution and their respective graphs for both nanoparticles after incubation for 0 and 24 h in normal physiological and tumor-simulated conditions (A (i) and B (i)), respectively. Data is presented as mean ± S.D, (n = 3). Two-way ANOVA was used to compare the deviations in the particle size and PDI of the nanocarriers in normal and tumor-simulated conditions. Differences in p values, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, were considered statistically significant and ns was considered non-significant. TEM images of BBR NP-17 and 18 (A (ii) and B (ii)) after incubation at 0 and 24 h in normal physiological and tumor-simulated conditions, illustrating the changes in the nanoparticle morphology and aggregation in response to the redox-sensitive environment, respectively. | ||
 | ||
| Fig. 4 In vitro release studies. Graphical representation of dissolution studies in normal physiological (pH 7.4 buffer, without GSH) and simulated TME (pH 7.4 with 10 mM GSH, pH 4.5 and pH 4.5 with 10 mM GSH) for BBR NP-17 and NP-18 (A (i and ii)) (data are represented as mean ± S.D, n = 3). Schematic representation of drug release from the redox-sensitive nanocarriers under normal and simulated TME (B). | ||
Further, the BBR-loaded RS-PLHNCs were evaluated for drug release at lysosomal pH (pH 4.5) and complete tumor intracellular microenvironment (pH 4.5 and GSH). At pH 4.5, with and without GSH, the burst release of BBR with >50% release within 2 h and >80% release within 4 h in BBR NP-17 and 8 h in BBR NP-18 was observed. At pH 4.5, the observed accelerated release may be due to the hydrolysis of the ester bonds present in the redox cholesterol (ester linkage between CL and DTPA) and further destabilization of the nanocarriers followed by the release of BBR.58 The remarkably rapid release of the nanocarriers observed under both acidic and GSH conditions, was triggered by the cleavage of the dual stimuli of nanocarriers (i.e., disulfide bonds in response to GSH redox stimuli and ester linkages to acidic pH stimuli) (Fig. 4A and B), respectively.
In all the dissolution media, BBR NP-18 showed a significant, sustained BBR release rate in comparison to BBR NP-17 due to the presence of stearic acid in the lipid composition, which increased the lipophilicity and mechanical strength of the nanocarrier system, resulting in a decrease in the diffusion of the drug from the nanocarrier system.
In summary, the experimental findings indicate that the release of BBR from RS-PLHNCs is influenced by a combination of diffusion and erosion mechanisms, with the release behavior being affected by microenvironmental factors such as pH and the presence of reducing agents such as GSH.
3.4. Storage and colloidal stability studies
Stability studies were performed to assess the long-term storage of the prepared nanoformulation. In this study, stability assessments were conducted for 180 days at 4 °C. The optimized BBR-loaded nanocarriers (BBR NP-17 and 18) were free from aggregation or precipitation with only negligible changes in their particle size and distribution during their initial 60-day storage period. Extending the study to 180 days revealed contrasting behaviors, where BBR NP-17 showed reduced homogeneity and increased particle size, whereas batch 18 demonstrated increased uniformity and size, indicating superior colloidal stability and dispersion (Table 3 and Fig. S5, ESI†). The decreased stability of BBR NP-17 might be due to its zeta potential value being near zero. In contrast, the zeta potential of batch 18 is >−20 mV, contributing to good steric repulsion and colloidal stability. Previous studies suggest that nanocarriers with a lower zeta potential aggregate due to the reduced steric repulsion between particles.32 The higher negative charge of the nanoparticles in batch 18 can be attributed to the addition of stearic acid having carboxylic groups, contributing to their good physical stability during storage.| Time (days) | BBR NP-17 | ||
|---|---|---|---|
| Particle size (nm) | PDI | ZP (mV) | |
| 0 | 124.7 ± 1.2 | 0.153 ± 0.02 | −5.91 ± 2.54 |
| 60 | 156.7 ± 1.7 | 0.146 ± 0.03 | −5.73 ± 0.68 |
| 180 | 173.2 ± 4.7 | 0.366 ± 0.06 | −2.68 ± 0.43 |
| BBR NP-18 | |||
| 0 | 185.2 ± 1.6 | 0.153 ± 0.02 | −20.03 ± 1.07 |
| 60 | 171.3 ± 0.8 | 0.120 ± 0.01 | −23.93 ± 1.1 |
| 180 | 177.7 ± 2.2 | 0.139 ± 0.03 | −25.63 ± 4.29 |
3.5. In vitro cell-based assays
The redox sensitivity hypothesis of the BBR-loaded RS-PLHNCs was validated by investigating the specific killing efficiency of the synthesized BBR-loaded RS-PLHNCs in both normal cells (with lower GSH) and cancer cells (with higher GSH). For this purpose, normal (BEAS-2B and HEK293A) and cancer (H1975) cells were chosen to evaluate the redox sensitivity and therapeutic killing efficiency of the fabricated BBR-loaded NPs and compared to the free form of BBR.Redox-responsive cytotoxicity. The cytotoxic effects of the free form of BBR, BBR NP-17, and BBR NP-18 were evaluated by the MTT assay in BEAS-2B, HEK293A, and H1975 cells. As depicted in Fig. 5A and B, all three formulations exhibited dose-dependent inhibitory effects in these cell lines. The IC50 values of the respective formulations are reported in Table 4 and Fig. S6(B (i & ii)) (ESI†).
 | ||
| Fig. 5 Effect of redox-sensitive NCs on cancer versus normal cells. %Cell viability of BEAS-2B, HEK293A, and H1975 cells after BBR NP-17 and 18 treatments for 24 and 72 h (A). Effect of the free form of BBR versus BBR NPs on cancer cells. %Cell viability of H1975 cells after treatment with BBR in free form and nanoparticulate forms (BBR NP-17 and 18) for 24 and 72 h (B). All values are expressed as mean ± SEM (n = 5). Two-way ANOVA was used to compare the difference in %cell viability between BEAS-2B and H1975, as well as between HEK293A and H1975 following treatment. Additionally, two-way ANOVA was conducted to compare the %cell viability differences in H1975 cells treated with free-form of BBR and BBR nanoparticulate forms (BBR NP-17 and NP-18). Differences in p-values of *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 were considered statistically significant and ns was considered non-significant. Abbreviations: H: H1975 (lung cancer cell line). BBR NP-17-H: H1975 cells treated with BBR NP-17 nanocarriers, BBR NP-18-H: H1975 cells treated with BBR NP-18 nanocarriers, and BBR FR-H- refers to H1975 cells treated with free form of BBR. B: BEAS-2B (normal lung epithelial cells). BBR NP-17-B: BEAS-2B cells treated with BBR NP-17 nanocarriers and BBR NP-18-B: BEAS-2B cells treated with BBR NP-18 nanocarriers. K:HEK293A (normal human embryonic kidney cells). BBR NP-17-K: HEK293A cells treated with BBR NP-17 nanocarriers and BBR NP-18-K: HEK293A cells treated with BBR NP-18 nanocarriers. | ||
| After 24 h incubation | ||||||
|---|---|---|---|---|---|---|
| Batch no | IC50 of NPs (μM) | IC50 of free form of BBR (μM) | ||||
| BEAS-2B | HEK293A | H1975 | BEAS-2B | HEK293A | H1975 | |
| BBR NP-17 | 7.7 ± 1.2 | 14.6 ± 1.1 | 6.8 ± 1.1 | 52.8 ± 1.1 | 47.1 ± 1.2 | 46.2 ± 1.1 |
| BBR NP-18 | 27.5 ± 1.1 | 133.3 ± 1.3 | 8.5 ± 1.1 | |||
| After 72 h incubation | ||||||
| BBR NP-17 | 7.7 ± 1.2 | 13.1 ± 1.0 | 6.8 ± 1.1 | 40.6 ± 1.2 | 22.1 ± 1.1 | 59.6 ± 1.2 |
| BBR NP-18 | 27.5 ± 1.1 | 133.3 ± 1.3 | 8.5 ± 1.1 | |||
In terms of redox sensitivity (Fig. 5A(i) and (iii)), the BBR-loaded RS-PLHNCs (BBR NP-17 and 18) displayed significant cytotoxicity effects at all concentrations except 3.125 μM at 24 h in H1975 cancer cells compared to BEAS-2B cells. After 72 h incubation (Fig. 5A(ii) and (iv)), all concentrations showed significant differences in cytotoxicity except 50 μM and 25 μM of BBR NP-17 and 50 μM of BBR NP-18. The predominant difference in inhibition may be due to the elevated levels of GSH and decreased pH conditions in the cancer cells, which cleaved the disulfide and ester bonds present in the modified redox-cholesterol of the lipid matrix. Further, following erosion of the lipid matrix, BBR was released into the cytoplasm, thereby inducing an increase in cytotoxicity. In contrast, lower cytotoxicity was observed in the BEAS-2B and HEK293A cells owing to their low GSH levels, resulting in the slower diffusion or release of the drug from the nanocarrier system.
Furthermore, the presence of positively charged aminated polyethylene glycol diamine in the core of the nanocarrier system may have contributed to the increased cytotoxicity. Upon degradation of the lipid matrix, this component may become exposed, potentially leading to binding with the negatively charged membranes, thereby inducing an increase in the release of BBR and cytotoxic effects.54
Comparing both batches, BBR NP-18 showed a ∼3.23- and 15.73-fold decrease in IC50 in BEAS-2B and HEK293A cells after 24 h incubation, while that of BBR NP-17 was only ∼1.13- and 2.14-fold, respectively. After 72 h incubation, the BBR NP-18-treated group showed ∼3.85- and 3.27-fold reduction in IC50 values in BEAS-2B and HEK293A cells, while only ∼1.20- and 2.11-fold in the case of BBR NP-17, respectively (Table 4). The enhanced selectivity of BBR NP-18 is likely due to the incorporation of stearic acid in the lipid matrix, which increased the lipophilicity, slowed the release of BBR, and prevented rapid degradation, which is consistent with the results observed in the in vitro release studies (Section 3.3).
Advantage of the nanoparticle form of BBR over free form of BBR. Both optimized redox-sensitive nanocarriers exhibited notably high cytotoxicity in H1975 cancer cells compared to the free form of BBR after 24 and 72 h of treatment (Fig. 5B(i)–(iv)). This significant cytotoxicity observed in the group treated with the BBR nanocarriers may be due to the enhanced penetration and sustained release of BBR from the nanocarriers, which was also observed in the cellular uptake studies. The difference in cytotoxicity induced by the free form of BBR was observed to be non-significant in the normal and cancer cells, possibly due to the non-specific release and penetration of the drug in both cells.
A biocompatibility study was performed on the blank nanocarriers (BLK NP-17 and 18) to assess their independent cytotoxicity. Here, different concentrations (3.125 to 50 μM with respect to BBR concentration used in cytotoxicity studies) were used to treat the BEAS-2B, HEK293A, and H1975 cells for 24 and 72 h incubation periods. As demonstrated in Fig. S7 (ESI†), greater than 75% of the cells were observed to be viable at all concentrations except 50 μM concentration for both 24 and 72 h incubation periods. These findings indicate the absence of significant toxic effects on healthy cells (<50 μM), confirming good biocompatibility and ensuring a safe carrier for the delivery of drugs to cancer cells. Also, this study confirmed that the cytotoxicity induced by the nanocarriers is solely due to the selective release of BBR into the cytoplasm of the cancer cells, which further induced enhanced cytotoxicity.
To validate the BBR-induced cytotoxicity observed in the MTT assay, the cellular viability was further assessed by examining the cell numbers in the presence of BBR nanocarriers (IC50) and free form of BBR (IC50 values of BBR NP-17 and 18 nanocarriers were used). Notably, treatment with BBR NP-17 and BBR NP-18 resulted in a reduction of 56.54% and 48.73% in the number of cells, whereas BBR FR-17 and BBR FR-18 exhibited a lower reduction of 78.95% and 72.16%, respectively (Fig. S8, ESI†). According to the data obtained from both the MTT assay and cell counting, the IC50 for BBR-NP 17 and 18 was determined to be 6.79 ± 1.1 μM and 8.47 ± 1.1 μM, respectively. These findings contribute to the comprehensive understanding of the efficacy of the BBR nanocarrier formulations in inducing cytotoxic effects for potential therapeutic applications.
 | ||
| Fig. 6 Cellular uptake of BBR-loaded RS-PLHNCs and free form of BBR in BEAS-2B, HEK293A, and H1975 cells. Histograms showing the intensity of uptake for blank NP, free form of BBR, and BBR NPs (batch 17 and 18) at 4 h, 12 h, and 18 h and their quantitative mean fluorescence intensity (MFI) (n = 3) (λex = 488 nm; λem = 585 nm) (A). Confocal microscopy images showing intracellular fluorescence of BBR (λex = 405 nm; λem = 550 ± 50 nm) (B). The values were statistically examined using a two-way ANOVA test. Statistical significance: *p < 0.05, **p < 0.01, and ***p < 0.001 and ns- non-significant. Abbreviation: BLK NP-17 (blank nanoparticle equivalent with IC50 of BBR NP-17i.e. 6.8 μM); BLK NP-18 (blank nanoparticle equivalent with IC50 of BBR NP-18i.e. 8.5 μM); BBR FR-17 (free form of BBR equivalent with IC50 of BBR NP-17i.e. 6.8 μM); BBR FR-18 (free form of BBR equivalent with IC50 of BBR NP-18i.e. 8.5 μM); BBR NP-17 (BBR-loaded RS-PLHNCs with an IC50 dose of 6.8 μM); and BBR NP-18 (BBR-loaded RS-PLHNCs with an IC50 dose of 8.5 μM). | ||
Furthermore, CLSM was used to confirm and visualize the intracellular distribution of the internalized free form of BBR and BBR-nanoparticulate form. Post-treatment of BBR NP-17 and NP-18 for 4 and 18 h, the fluorescence intensity was found to be lower in the normal cells compared to the cancer cells. Moreover, the fluorescence intensity of the free form of BBR (BBR FR-17 and FR-18) was significantly lower across all cell lines compared to the nanoparticulate formulation of BBR (Fig. 6B), which is consistent with the flow cytometry results that RS-PLHNCs remarkably enhanced the uptake of BBR. This observation is likely due to the limited permeability of the free form of BBR, which was overcome by its encapsulation within nanoparticles, thereby enhancing its cellular uptake and fluorescence signal. Therefore, our findings suggest that loading BBR in RS-PLHNCs follows greater selectivity and preferential uptake in tumor cells, bypassing the cell membrane and inducing increased cytotoxicity.
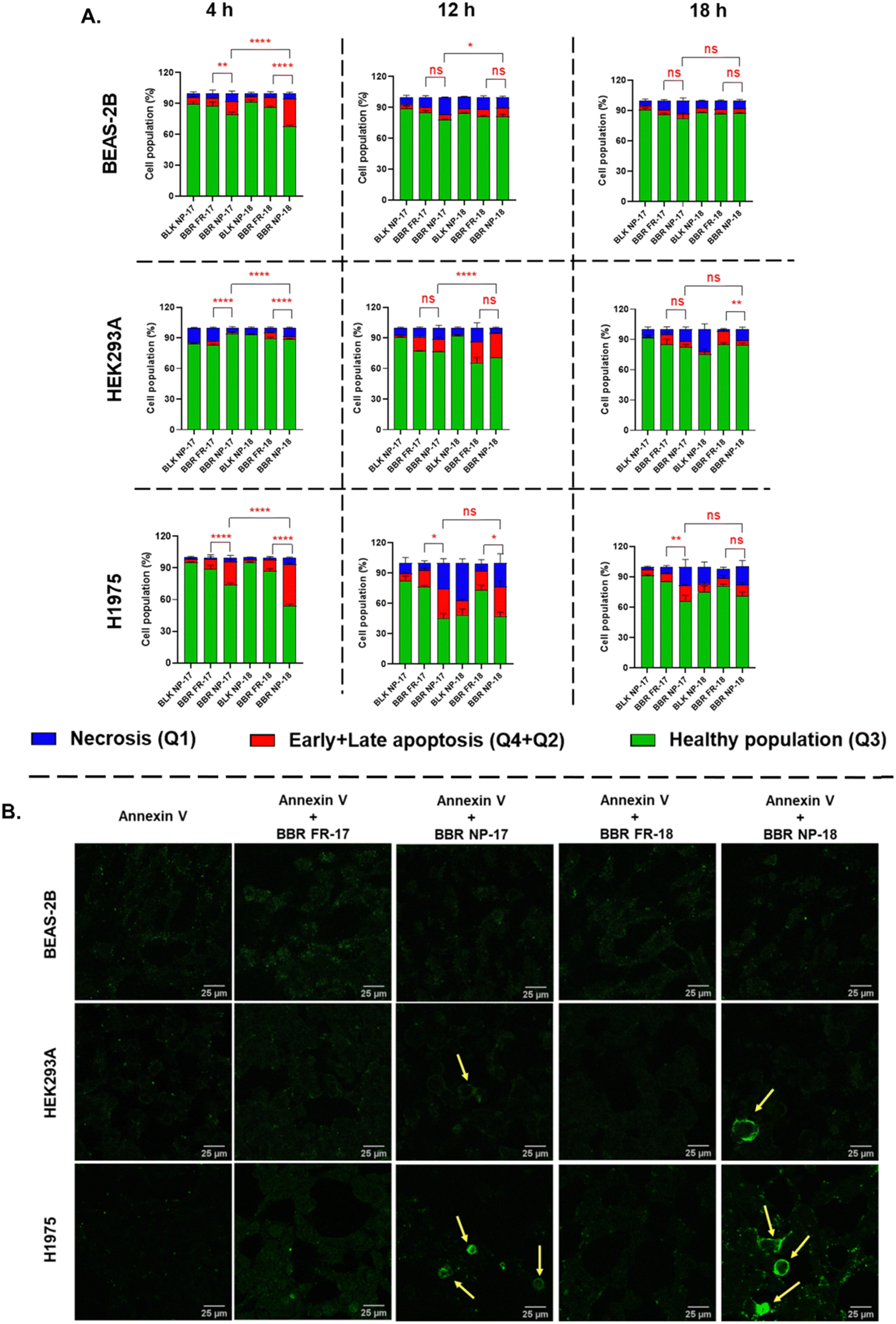 | ||
| Fig. 7 Apoptosis study of free form of BBR and its nanoparticulate form (BBR NP-17 and 18) in BEAS-2B, HEK293A, and H1975 cells assessed via flow cytometry and confocal microscopy. Flow cytometry analysis of the %apoptotic cells determined by Annexin V-FITC/PI staining, following treatment at 4, 12, and 18 h (A). Representative confocal images for apoptosis in Annexin V-FITC-stained BEAS-2B, HEK293A, and H1975 cells after 18 h of treatment (λex = 488 nm; λem = 550 ± 50 nm) (B). (Yellow arrow indicates apoptotic cells). The values were statistically examined to assess apoptotic cell death (%early+late apoptosis) by two-way ANOVA test. Statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and ns- non-significant. | ||
During apoptosis, the breakdown of membrane asymmetry leads to the relocation of phosphatidylserine (PS) to the outer membrane layer, which can be detected by Annexin V-FITC staining.65 We further validated our findings by evaluating the apoptotic potential of RS-PLHNCs loaded with the BBR drug, which was visualized using CLSM. Staining with Annexin V-FITC revealed clear signs of apoptosis when the H1975 cells were incubated with BBR NP-17 and BBR NP-18 (Fig. 7B). Strong green fluorescence-labeled cells were observed compared to the free form of BBR, where Annexin V-FITC was used as a negative control. In addition, upon treatment with BBR NP-17 and NP-18 in BEAS-2B and HEK293A, the green fluorescence intensity was lower, clearly indicating less apoptosis. These findings indicate that the BBR-loaded RS-PLHNCs follow higher cellular uptake, preferentially inducing apoptotic cell death in cancer cells over normal cells, possibly due to the effective BBR release rate inside the cancer cells, which was further validated by gene expression studies.66
The relative mRNA expression of NFE2L2 was significantly downregulated by ∼0.1 fold and ∼0.3-fold in BBR NP-17 and BBR NP-18, respectively. In contrast, an ∼0.9 fold and ∼0.7-fold change was observed in BBR FR-17 and BBR FR-18 compared to the control group, respectively (Fig. 8A). NAD(P)H: quinone oxidoreductase-1 is a phase detoxifying enzyme and overexpressed in ∼60% of NSCLC cases.71,72 NQO1 and HMOX1 mRNA expression was found to be significantly downregulated by ∼0.3 fold and ∼0.4 fold and ∼0.6 fold and ∼0.5 fold in the nanoparticulate forms of BBR-17 and BBR-18 compared to the control group, respectively. Upon treatment with BBR FR-17 and BBR FR-18, the NQO1 and HMOX1 mRNA expression was unaltered (Fig. 8B and C), respectively. Among the NRF2 target redox genes, TXRND1 plays a key role in the NRF2/KEAP1 axis, with poor survival in a cohort of NSCLC patients.73 The significantly downregulated mRNA expression of the TXRND1 gene was observed with ∼0.3 fold and ∼0.3 fold change in BBR NP-17 and BBR NP-18, and ∼1.1 fold and ∼0.8 fold change in BBR FR-17 and BBR FR-18 compared to the control group, respectively (Fig. 8D). In addition, BBR NP-17 and BBR NP-18 were found to significantly upregulate the above-mentioned genes compared to the free form of BBR.
 | ||
| Fig. 8 Nanoparticulate form of BBR promotes apoptosis and lowers oxidative stress in NSCLC (H1975 cells). Relative mRNA expression of Oxidative stress markers (A), NFE2l2 (B), NQO1 (C), HMOX1 and TXRND1 (D). Pro-apoptotic markers PUMA (E) and NOXA (F). Anti-apoptotic marker MCL-1 (G). The data are represented as mean ± SEM (n = 4), and statistical significance was determined using one-way ANOVA test. Statistical significance was denoted as follows: *p < 0.05, **p < 0.01, and ***p < 0.001 and “ns” indicating non-significant differences. | ||
Berberine inhibits cell proliferation and induces apoptosis in breast, lung, and liver cancer via the BCL-2/BAX pathway.74,75 Based on previous studies, we aimed to evaluate the effect of the nanoparticulate BBR on apoptotic markers such as PUMA, NOXA, and MCL-1. The mRNA expression of PUMA and NOXA was significantly upregulated upon treatment with BBR NP-17 and BBR NP-18 by ∼7.3 fold and ∼4.4-fold, whereas an ∼3.2 fold and ∼2.0-fold difference was observed in the BBR FR-17 and BBR FR-18 groups compared with the control group (Fig. 8E and F), respectively. MCL-1 is a key anti-apoptotic marker of the BCL-2 family, which is often overexpressed in tumors, promoting cell survival and drug resistance. Thus, targeting holds promise for cancer therapy due to its central role in blocking apoptosis via the mitochondrial pathway.76,77 Upon treatment with BBR NP-17 and BBR NP-18, the significant downregulation of MCL-1 by ∼0.1-fold and ∼0.2-fold was observed compared to the control group treatment, respectively (Fig. 8G). Additionally, BBR NP-17 and BBR NP-18 resulted in the significant upregulation of PUMA and NOXA and down-regulation of MCL-1, respectively, compared to BBR FR-17 and BBR FR-18, respectively. These results suggest that BBR NP-17 has better therapeutic efficacy than BBR NP-18 by increasing oxidative stress and inducing apoptosis.
4. Conclusion
In the current study, bioreducible redox-sensitive polymeric lipid hybrid nanocarriers (RS-PLHNCs) were successfully fabricated and characterized. The optimized monodispersible BBR-loaded nanocarriers (BBR NP-17 and 18) demonstrated acceptable particle sizes (124 and 185 nm) with good encapsulation efficiency (>80%), respectively. These novel nanocarriers displayed accelerated BBR release (>80% in 24 h) on exposure to tumor-relevant pH (pH 4.5–5.5) and reduction environment (10 mM GSH) compared to physiological conditions (30–45% in 24 h). This increased release was due to the reduction of the disulfide bonds in the GSH reductive environment (disulfide bonds present in modified cholesterol of lipid matrix layer surrounding the polymer core) and hydrolysis of the ester bonds in acidic pH, leading to the disassembly of the nanocarriers, and thereby increasing the release of BBR. These dual-stimuli-triggered nanocarriers exhibited selective cytotoxicity against H1975 (NSCLC) cells over normal BEAS-2B and HEK293A cells. Moreover, they displayed several-fold increased cytotoxicity than the free form of BBR. This enhanced killing efficiency may be due to the increased penetration of the nanocarriers owing to the EPR effect compared to the free form of the drug, as observed in the cellular uptake studies. Several of the underlying mechanisms for the enhanced anti-cancer activity are due to the downregulation of NFE2L2, HO-1, NQO1, and TXRND1 gene expression and decreased GSH levels, leading to oxidative stress, thereby causing apoptosis of the cancer cells. From the cancer therapy perspective, the prepared redox-responsive nanocomposite material has immense potential to precisely deliver similar bioactive compounds by enhancing their pharmaceutical properties and preventing non-selective toxic effects, thereby improving the treatment outcome.Abbreviations
| BBR | Berberine |
| CL | Cholesterol |
| %DL | %Drug loading |
| DPPC | 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine |
| DTDPA | Dithiodipropionate |
| DTPA | 3,3′-Dithiodipropionic acid |
| %EE | %Entrapment efficiency |
| PDI | Polydispersity index |
| PEG | Polyethylene glycol |
| PNIPAM | P-(N-isopropyl acrylamide) |
| MTT | (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) |
| RS-PLHNCs | Redox-sensitive polymeric lipid hybrid nanocarriers. |
Author contributions
B. Siva Lokesh: conceptualization, methodology, investigation, data curation, formal analysis, writing – original draft, preparation. Suresh Ajmeera: conceptualization, methodology, investigation, data curation, formal analysis, writing – original draft, preparation. Rajat Choudhary: conceptualization, methodology, investigation, data curation, formal analysis, writing – original draft, preparation. Sanjaya Kumar Moharana: characterization and analysis of synthesized lipids and polymers. C. S. Purohit: formal analysis and supervision of synthesized lipids and polymers. V. Badireenath Konkimalla: conceptualization, formal analysis, supervision, resources, funding acquisition, project administration, writing – original draft, writing – review & editing.Data availability
All data pertaining to the work done are included in the uploaded ESI.†Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors thank the full financial support by intramural funding (DPR) received from National Institute of Science Education and Research (NISER), Department of Atomic Energy (DAE), Government of India (GoI). B.S.L., R.C., and S.K.M., would like to acknowledge NISER, DAE, GoI for the research fellowship. The authors would also like to acknowledge the support provided by TEM imaging facility, Centre for Interdisciplinary Sciences (CIS), NISER, for their assistance in TEM imaging and Dr Tathagata Mukherjee, Somlata Khamaru, and Kshyama Subhadarsini Tung, School of Biological Sciences (SBS), NISER, during FACS analysis. The authors also acknowledge Dr Amit Jaiswal, IIT-Mandi, India, for helping with the HEK293A cell line for in vitro cell based studies. The authors are thankful to acknowledge Dr Soumendra Rana, IIT-Bhubaneswar, India, for mass characterization.References
- F. Raza, H. Zafar, X. You, A. Khan, J. Wu and L. Ge, J. Mater. Chem. B, 2019, 7, 7639–7655 RSC.
- L. Sun, H. Liu, Y. Ye, Y. Lei, R. Islam, S. Tan, R. Tong, Y.-B. Miao and L. Cai, Signal Transduction Targeted Ther., 2023, 8, 418 CrossRef CAS PubMed.
- S. Krishnamurthy, R. Vaiyapuri, L. Zhang and J. M. Chan, Biomater. Sci., 2015, 3, 923–936 RSC.
- C. Corbet and O. Feron, Nat. Rev. Cancer, 2017, 17, 577–593 CrossRef CAS PubMed.
- D. C. Singleton, A. Macann and W. R. Wilson, Nat. Rev. Clin. Oncol., 2021, 18, 751–772 CrossRef PubMed.
- S. Son, N. V. Rao, H. Ko, S. Shin, J. Jeon, H. S. Han, V. Q. Nguyen, T. Thambi, Y. D. Suh and J. H. Park, Int. J. Biol. Macromol., 2018, 110, 399–405 CrossRef CAS PubMed.
- Y. Chi, X. Yin, K. Sun, S. Feng, J. Liu, D. Chen, C. Guo and Z. Wu, J. Controlled Release, 2017, 261, 113–125 CrossRef CAS PubMed.
- R. Yan, X. Liu, J. Xiong, Q. Feng, J. Xu, H. Wang and K. Xiao, RSC Adv., 2020, 10, 13889–13899 RSC.
- X. Peng, P. Liu, B. Pang, Y. Yao, J. Wang and K. Zhang, Carbohydr. Polym., 2019, 216, 113–118 CrossRef CAS PubMed.
- D. D. Gurav, A. S. Kulkarni, A. Khan and V. S. Shinde, Colloids Surf., B, 2016, 143, 352–358 CrossRef CAS PubMed.
- J. Yu, H. Deng, F. Xie, W. Chen, B. Zhu and Q. Xu, Biomaterials, 2014, 35, 3132–3144 CrossRef CAS PubMed.
- M. G. Adimoolam, N. Amreddy, M. R. Nalam and M. V. Sunkara, J. Magn. Magn. Mater., 2018, 448, 199–207 CrossRef CAS.
- Q. Chen, H. Ding, J. Zhou, X. Zhao, J. Zhang, C. Yang, K. Li, M. Qiao, H. Hu, P. Ding and X. Zhao, RSC Adv., 2016, 6, 17782–17791 RSC.
- Y. Li, A. Lu, M. Long, L. Cui, Z. Chen and L. Zhu, Acta Biomater., 2019, 83, 334–348 CrossRef CAS PubMed.
- J. Wang, G. Su, X. Yin, J. Luo, R. Gu, S. Wang, J. Feng and B. Chen, Biomed. Pharmacother., 2019, 120, 1–8 Search PubMed.
- B. Lu, F. Xiao, Z. Wang, B. Wang, Z. Pan, W. Zhao, Z. Zhu and J. Zhang, ACS Biomater. Sci. Eng., 2020, 6, 4106–4115 CrossRef CAS PubMed.
- F. A. Younis, S. R. Saleh, S. S. A. El-Rahman, A.-S. A. Newairy, M. A. El-Demellawy and D. A. Ghareeb, Sci. Rep., 2022, 12, 17431 CrossRef CAS PubMed.
- A. Agnarelli, M. Natali, M. Garcia-Gil, R. Pesi, M. G. Tozzi, C. Ippolito, N. Bernardini, R. Vignali, R. Batistoni, A. M. Bianucci and S. Marracci, Sci. Rep., 2018, 8, 10599 CrossRef PubMed.
- Y. S. Loo, T. Madheswaran, R. Rajendran and R. J. Bose, J. Drug Delivery Sci. Technol., 2020, 57, 101756 CrossRef CAS.
- K. Zou, Z. Li, Y. Zhang, H. Y. Zhang, B. Li, W. L. Zhu, J. Y. Shi, Q. Jia and Y. M. Li, Acta Pharmacol. Sin., 2017, 38, 157–167 CrossRef CAS PubMed.
- M. Farahani, F. Moradikhah, I. Shabani, R. K. Soflou and E. Seyedjafari, J. Drug Delivery Sci. Technol., 2021, 61, 102134 CrossRef CAS.
- C. Hald Albertsen, J. A. Kulkarni, D. Witzigmann, M. Lind, K. Petersson and J. B. Simonsen, Adv. Drug Delivery Rev., 2022, 188, 114416 CrossRef CAS PubMed.
- Z. Li, W. Qiao, C. Wang, H. Wang, M. Ma, X. Han and J. Tang, Drug Delivery, 2020, 27, 736–744 CrossRef CAS PubMed.
- T. J. Chirayil and G. S. Vinod Kumar, Int. J. Nanomed., 2022, 17, 5099–5116 CrossRef CAS PubMed.
- B. Siva Lokesh, P. Haloi and V. B. Konkimalla, J. Drug Delivery Sci. Technol., 2023, 80, 104101 CrossRef CAS.
- K. Thananukul, C. Kaewsaneha, P. Opaprakasit, N. Zine and A. Elaissari, Sci. Rep., 2022, 12, 1–14 CrossRef PubMed.
- X. Ma, E. Özliseli, Y. Zhang, G. Pan, D. Wang and H. Zhang, Biomater. Sci., 2019, 7, 634–644 RSC.
- P. Xue, D. Liu, J. Wang, N. Zhang, J. Zhou, L. Li, W. Guo, M. Sun, X. Han and Y. Wang, Bioconjugate Chem., 2016, 27, 1360–1372 CrossRef CAS PubMed.
- R. Otter, N. A. Henke, C. Berac, T. Bauer, M. Barz, S. Seiffert and P. Besenius, Macromol. Rapid Commun., 2018, 39, e1800459 CrossRef PubMed.
- N. Tahir, A. Madni, A. Correia, M. Rehman, V. Balasubramanian, M. M. Khan and H. A. Santos, Int. J. Nanomed., 2019, 14, 4961–4974 CrossRef CAS PubMed.
- X. Zhao, H. Guo, H. Bera, H. Jiang, Y. Chen, X. Guo, X. Tian, D. Cun and M. Yang, ACS Appl. Mater. Interfaces, 2023, 15, 4441–4457 CrossRef CAS PubMed.
- R. Ghaffari, N. Eslahi, E. Tamjid and A. Simchi, ACS Appl. Mater. Interfaces, 2018, 10, 19336–19346 CrossRef CAS PubMed.
- P. Haloi, B. S. Lokesh, S. Chawla and V. B. Konkimalla, Drug Delivery, 2023, 30, 2184307 CrossRef PubMed.
- N. S. Thakur, N. Mandal, G. Patel, S. Kirar, Y. N. Reddy, V. Kushwah, S. Jain, Y. N. Kalia, J. Bhaumik and U. C. Banerjee, Nanomedicine, 2021, 33, 102368 CrossRef CAS PubMed.
- Y. Chen, M. Su, Y. Li, J. Gao, C. Zhang, Z. Cao, J. Zhou, J. Liu and Z. Jiang, ACS Appl. Mater. Interfaces, 2017, 9, 30519–30535 CrossRef CAS.
- R. B. KC, B. Thapa and P. Xu, Mol. Pharm., 2012, 9, 2719–2729 CrossRef CAS PubMed.
- L. Chen, X. Zhou, W. Nie, Q. Zhang, W. Wang, Y. Zhang and C. He, ACS Appl. Mater. Interfaces, 2016, 8, 33829–33841 CrossRef CAS PubMed.
- P. Yang, L. Zhang, T. Wang, Q. Liu, J. Wang, Y. Wang, Z. Tu and F. Lin, OncoTargets Ther., 2020, 13, 8055–8067 CrossRef CAS.
- I. Rahman, A. Kode and S. K. Biswas, Nat. Protoc., 2006, 1, 3159–3165 CrossRef CAS.
- P. Haloi, R. Choudhary, B. S. Lokesh and V. B. Konkimalla, Immunol. Lett., 2024, 267, 106854 CrossRef CAS.
- V. M. Lu, F. Crawshay-Williams, B. White, A. Elliot, M. A. Hill and H. E. Townley, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 132–143 CrossRef CAS.
- H. Li, Y. Cui, J. Liu, S. Bian, J. Liang, Y. Fan and X. Zhang, J. Mater. Chem. B, 2014, 2, 3500–3510 RSC.
- N. Tahir, A. Madni, V. Balasubramanian, M. Rehman, A. Correia, P. M. Kashif, E. Mäkilä, J. Salonen and H. A. Santos, Int. J. Pharm., 2017, 533, 156–168 CrossRef CAS PubMed.
- X. Yang, D. Salado-Leza, E. Porcel, C. R. González-Vargas, F. Savina, D. Dragoe, H. Remita and S. Lacombe, Int. J. Mol. Sci., 2020, 21, 1–20 Search PubMed.
- M. Das, D. Bandyopadhyay, R. P. Singh, H. Harde, S. Kumar and S. Jain, J. Mater. Chem., 2012, 22, 24652 RSC.
- E. Jaradat, E. Weaver, A. Meziane and D. A. Lamprou, Mol. Pharm., 2023, 20, 6184–6196 CrossRef CAS PubMed.
- R. S. Fernandes, R. G. Arribada, J. O. Silva, A. Silva-Cunha, D. M. Townsend, L. A. M. Ferreira and A. L. B. Barros, Pharmaceutics, 2022, 14, 2394 CrossRef CAS PubMed.
- N. Vikas, A. K. Mehata, M. K. Viswanadh, A. K. Malik, A. Setia, P. Kumari, S. K. Mahto and M. S. Muthu, Biomacromolecules, 2023, 24, 4989–5003 CrossRef PubMed.
- S. Comincini, F. Manai, M. Sorrenti, S. Perteghella, C. D’Amato, D. Miele, L. Catenacci and M. C. Bonferoni, Pharmaceutics, 2023, 15, 1078 CrossRef CAS PubMed.
- R. Bhanumathi, K. Vimala, K. Shanthi, R. Thangaraj and S. Kannan, New J. Chem., 2017, 41, 14466–14477 RSC.
- Y. Ding, Y. Dai, M. Wu and L. Li, Chem. Eng. J., 2021, 426, 128880 CrossRef CAS.
- B. Niu, K. Liao, Y. Zhou, T. Wen, G. Quan, X. Pan and C. Wu, Biomaterials, 2021, 277, 121110 CrossRef CAS PubMed.
- X. Pang, Y. Jiang, Q. Xiao, A. W. Leung, H. Hua and C. Xu, J. Controlled Release, 2016, 222, 116–129 CrossRef CAS PubMed.
- W. Zhou, H. Yu, L. J. Zhang, B. Wu, C. X. Wang, Q. Wang, K. Deng, R. X. Zhuo and S. W. Huang, Nanoscale, 2017, 9, 17044–17053 RSC.
- Y. Sun, X. Du, J. He, J. Hu, M. Zhang and P. Ni, J. Mater. Chem. B, 2017, 5, 3771–3782 RSC.
- L. Sun, X. Zhang, J. An, C. Su, Q. Guo and C. Li, RSC Adv., 2014, 4, 20208–20215 RSC.
- L. Hu, P. Zhang, X. Wang, X. Cheng, J. Qin and R. Tang, Carbohydr. Polym., 2017, 178, 166–179 CrossRef CAS PubMed.
- H. Lee, K. Lee and G. P. Tae, Bioconjugate Chem., 2008, 19, 1319–1325 CrossRef CAS PubMed.
- K. R. Gajbhiye, R. Salve, M. Narwade, A. Sheikh, P. Kesharwani and V. Gajbhiye, Mol. Cancer, 2023, 22, 1–44 CrossRef PubMed.
- H. Thai, C. Thuy Nguyen, L. Thi Thach, M. Thi Tran, H. Duc Mai, T. Thi Thu Nguyen, G. Duc Le, M. Van Can, L. Dai Tran, G. Long Bach, K. Ramadass, C. I. Sathish and Q. Van Le, Sci. Rep., 2020, 10, 1–15 CrossRef PubMed.
- C. A. Labarrere and G. S. Kassab, Front. Nutr., 2022, 9, 1–25 Search PubMed.
- H. J. Forman, H. Zhang and A. Rinna, Mol. Aspects Med., 2009, 30, 1–12 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- T. E. Ali, M. A. Assiri, M. N. Alqahtani, A. A. Shati, M. Y. Alfaifi and S. E. I. Elbehairi, RSC Adv., 2023, 13, 18658–18675 RSC.
- R. C. Taylor, S. P. Cullen and S. J. Martin, Nat. Rev. Mol. Cell Biol., 2008, 9, 231–241 CrossRef CAS PubMed.
- D. Y. Zhang, Y. Zheng, H. Zhang, J. H. Sun, C. P. Tan, L. He, W. Zhang, L. N. Ji and Z. W. Mao, Adv. Sci., 2018, 5, 1800581 CrossRef PubMed.
- J. D. Hayes, A. T. Dinkova-Kostova and K. D. Tew, Cancer Cell, 2020, 38, 167–197 CrossRef CAS PubMed.
- M. J. Iqbal, A. Kabeer, Z. Abbas, H. A. Siddiqui, D. Calina, J. Sharifi-Rad and W. C. Cho, Cell Commun. Signaling, 2024, 22, 1–16 CrossRef PubMed.
- M. Rojo de la Vega, E. Chapman and D. D. Zhang, Cancer Cell, 2018, 34, 21–43 CrossRef CAS PubMed.
- A. A. Zimta, D. Cenariu, A. Irimie, L. Magdo, S. M. Nabavi, A. G. Atanasov and I. Berindan-Neagoe, Cancers, 2019, 11, 1–26 CrossRef PubMed.
- B. Madajewski, M. A. Boatman, I. Martinez, J. H. Carter and E. A. Bey, Genes, 2023, 14, 607 CrossRef CAS PubMed.
- J. Wu, S. Li, C. Li, L. Cui, J. Ma and Y. Hui, Redox Biol., 2021, 47, 102170 CrossRef CAS PubMed.
- M. Delgobo, R. M. Gonçalves, M. A. Delazeri, M. Falchetti, A. Zandoná, R. Nascimento das Neves, K. Almeida, A. C. Fagundes, D. P. Gelain, J. I. Fracasso, G. B. de Macêdo, L. Priori, N. Bassani, A. J. R. Bishop, C. M. Forcelini, J. C. F. Moreira and A. Zanotto-Filho, Free Radicals Biol. Med., 2021, 177, 58–71 CrossRef CAS PubMed.
- Q. Chen, Y. Hou, D. Li, Z. Ding, X. Xu, B. Hao, Q. Xia, M. Li and L. Fan, Ann. Transl. Med., 2022, 10, 485 CrossRef CAS PubMed.
- Y. Zhu, N. Xie, Y. Chai, Y. Nie, K. Liu, Y. Liu, Y. Yang, J. Su and C. Zhang, Front. Pharmacol., 2022, 13, 1–19 Search PubMed.
- H. Wang, M. Guo, H. Wei and Y. Chen, J. Hematol. Oncol., 2021, 14, 1–18 CrossRef PubMed.
- E. Munkhbaatar, M. Dietzen, D. Agrawal, M. Anton, M. Jesinghaus, M. Boxberg, N. Pfarr, P. Bidola, S. Uhrig, U. Höckendorf, A.-L. Meinhardt, A. Wahida, I. Heid, R. Braren, R. Mishra, A. Warth, T. Muley, P. S. P. Poh, X. Wang, S. Fröhling, K. Steiger, J. Slotta-Huspenina, M. van Griensven, F. Pfeiffer, S. Lange, R. Rad, M. Spella, G. T. Stathopoulos, J. Ruland, F. Bassermann, W. Weichert, A. Strasser, C. Branca, M. Heikenwalder, C. Swanton, N. McGranahan and P. J. Jost, Nat. Commun., 2020, 11, 4527 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01236d |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |