Bioinspired programmable coacervate droplets and self-assembled fibers through pH regulation of monomers†
Received
15th July 2024
, Accepted 10th November 2024
First published on 12th November 2024
Abstract
Phase separation and phase transitions pervade the biological domain, where proteins and RNA engage in liquid–liquid phase separation (LLPS), forming liquid-like membraneless organelles. The misregulation or dysfunction of these proteins culminates in the formation of solid aggregates via a liquid-to-solid transition, leading to pathogenic conditions. To decipher the underlying mechanisms, synthetic LLPS has been examined through complex coacervate formation from charged polymers. Nonetheless, temporal control over phase transitions from prebiotically relevant small organic synthons remains largely unexplored. Herein, we propose utilizing pH modulation to regulate the charge of small molecular building blocks, thereby controlling the LLPS process. Through a bio-inspired, enzyme-mediated pH-regulated reaction, we introduce temporal control over both LLPS and the transition from coacervates to supramolecular polymers. Additionally, by incorporating antagonistic pH modulators, we achieve transient LLPS and further temporal regulation of supramolecular polymer disassembly. Our investigation into pH-regulated LLPS provides a new avenue for exploring the stimuli-responsive, dynamic, and transient nature of LLPS.
Introduction
In cells, in addition to membrane-bound organelles, membraneless organelles (MLOs) play a crucial role in intracellular organization.1 The formation of MLOs results from the liquid–liquid phase separation (LLPS) of RNA and proteins, allowing them to perform multiple roles, such as modulating enzymatic activity, transporting RNA, buffering protein concentration noise, and serving as reaction hubs.2 These MLOs are liquid and highly dynamic, forming transiently to perform functions before redissolving into their constituent building blocks.3 Recent studies indicate that the transient nature of MLOs is regulated by external factors such as crowding, concentration gradients, and biochemical reactions.4 Conversely, the misregulation and malfunction of the protein components of these organelles can lead to neurodegenerative diseases through a liquid-to-solid transition (LST).5 Intrinsically disordered proteins (IDPs) such as α-synuclein, Tau, prion, and FUS undergo a metastable LLPS before forming ordered fibrous or gel structures. Despite these insights, many mechanisms underlying the transient nature of these droplets and their transition to solid states remain poorly understood.
To obtain a detailed understanding of the behavior of MLOs, synthetic LLPS systems have been extensively studied. Early reports focused on the formation of complex coacervate droplets through electrostatic interactions between oppositely charged polymers.6 Significant efforts have been made to achieve reversible complex coacervation by altering pH, enzymatic or chemical reaction networks, temperature, light, and salt concentration.7–13 Conversely, the recent exploration of LLPS from small molecules has garnered significant interest due to their prebiotic relevance.14–19 However, the design of small-molecule-based coacervates remains rare and extremely challenging due to the inherent difficulty in solvent entrapment and finding an optimal balance between the monomer–monomer and monomer–solvent interactions. In addition, achieving control over the transient behavior and temporal phase transitions of small-molecule-based coacervate droplets akin to biological droplets is challenging and has rarely been explored.
In this context, herein, we aim to utilize pH as a tool to regulate the charge of small molecule building blocks and thereby control the LLPS process. Thus, through a bio-inspired enzymatic pH-regulated reaction influencing the formation of coacervate droplets, temporal control over LLPS and coacervate-to-supramolecular-polymer formation is presented. Additionally, by integrating antagonistic pH modulators, we aim to realize transient LLPS and further temporal control over supramolecular polymer disassembly.20–22
Results and discussion
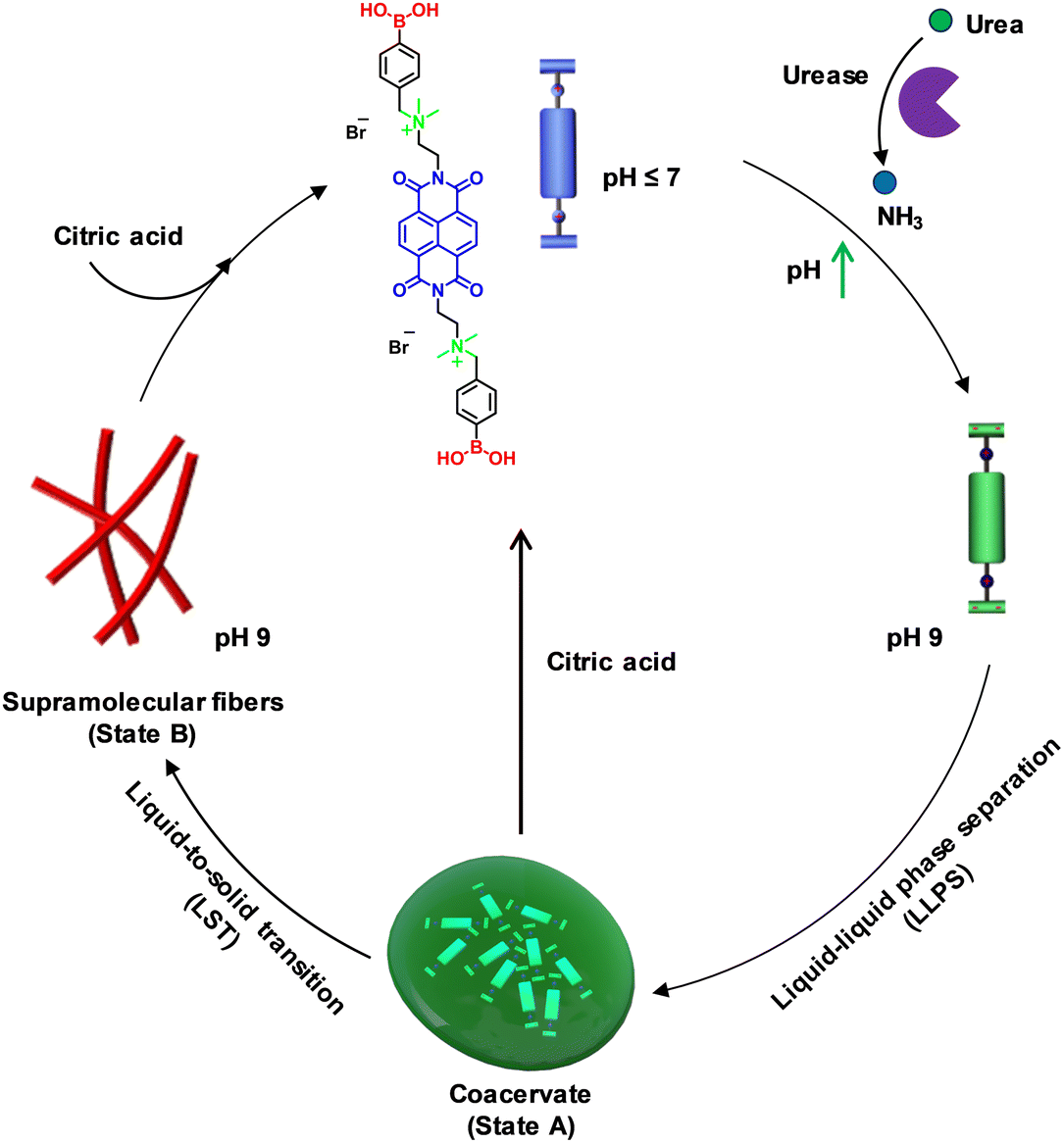
To explore pH-modulated LLPS and supramolecular polymerization, we used the recently reported molecule naphthalene diimide boronic acid (NDBA) from our group. Its molecular design involves an NDI aromatic core bis-functionalized with terminal boronic acid groups through quaternary ammonium spacers.16 Our prior report provided a comprehensive mechanistic elucidation of its phase transition from monomers to liquid coacervate droplets, culminating in supramolecular polymer formation and harnessing the metastable nature of the coacervate droplets to achieve seed-induced structural control over the resultant supramolecular polymer. In contrast, in the present work, we sought to introduce temporal control over pH through an enzymatic reaction to create transient coacervate droplets and facilitate the disassembly of the final supramolecular fibers. This approach distinguishes our work from previous studies by offering stimulus-responsive temporal control over both coacervate droplet formation and fiber disassembly. Notably, the NDBA monomer with a naphthalene diimide backbone is end-functionalized with pH-sensitive boronic acid functional groups that can be utilized to achieve the desired pH regulation of the LLPS formation. At pH values below the pka (∼8.4 ± 0.2) of boronic acid, NDBA would remain in a molecularly dissolved state due to the presence of positively charged quaternary amine groups that engender strong electrostatic repulsion between the monomers. A temporal increase in pH through enzymatic reaction could lead to a temporal LLPS (state A) followed by a supramolecular polymerization process through monomer rearrangement (Scheme 1) to form one-dimensional dynamic fibers (state B). Further, by coupling the system to antagonistic pH modulators, we planned to achieve transient control of the LLPS. Finally, we envisaged that changing the pH of the temporally grown fiber solution would lead to reversible supramolecular polymerization.23–28
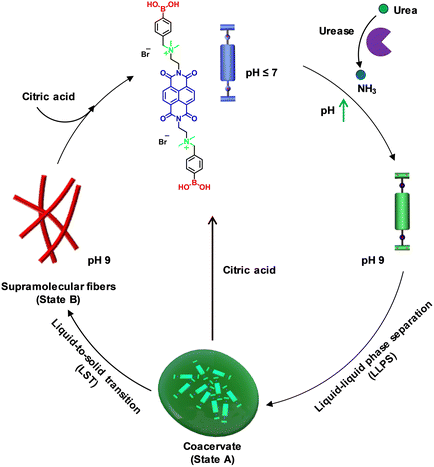 |
| | Scheme 1 Schematic representation of pH-regulated transient liquid–liquid phase separation (LLPS) and temporal supramolecular polymerization and disassembly. | |
pH-Modulated temporal coacervate and fiber formation
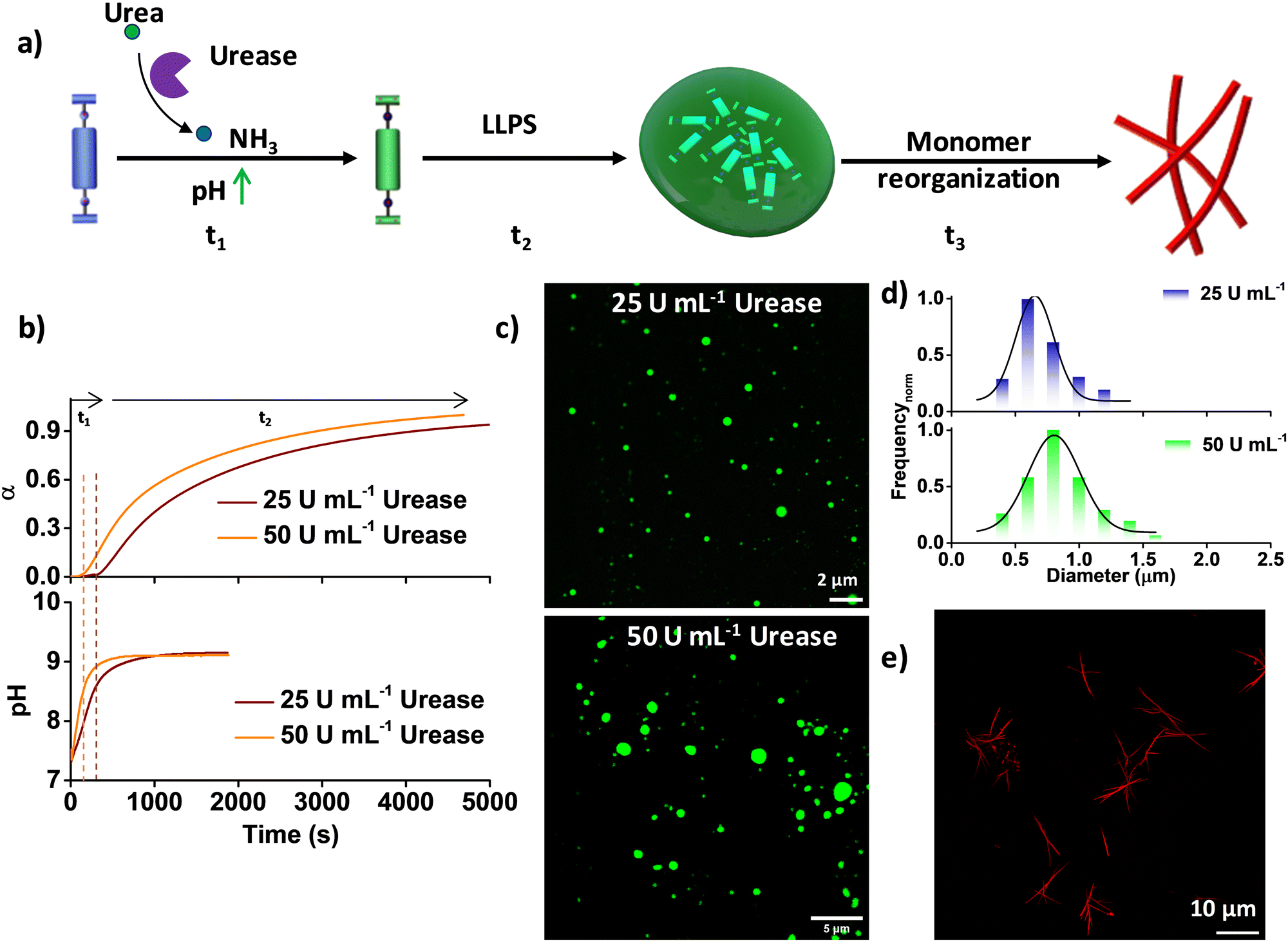
At pH 7, NDBA remains in the monomeric state even after 6 days, as confirmed by its unaltered time-dependent absorption and emission spectra, and at pH 9, it forms coacervate droplets, as reported previously (Fig. S1, ESI†).16 As the present system (monomer) is pH sensitive, we integrated an enzymatic reaction-controlled temporal pH activator to exert additional control over the temporal growth process (Fig. 1a). It is well-established that in the presence of the enzyme urease, urea is converted to ammonia, leading to a temporal increase in the solution pH.20 Leveraging this mechanism, we attempted to achieve pH-triggered temporal LLPS. The formation of LLPS was investigated by probing the spectroscopic changes in the NDI π–π* absorption band at 382 nm, as any conformational rearrangement in the NDI core is known to markedly affect this band. In a solution comprising 50 μM NDBA and 50 mM urea at pH 7, sigmoidal growth was observed upon the addition of different amounts of urease (ranging from 50 U mL−1 to 25 U mL−1), resulting in a gradual increase of the lag-phase of the growth kinetics from 139 ± 20 s to 304 ± 30 s, in stark contrast to the instantaneous growth observed at pH 9 (Fig. 1b). Overlaying the temporal profiles of the growth with the changes in pH shows that the onset of growth is driven by the pH rather than any pre-nucleation lag phase, which resulted in pH-trigged coacervate formation (Fig. 1b). Furthermore, confocal laser scanning microscopy (CLSM) visualization of the LLPS, by making use of the NDI excimer emission in the condensed phase after 2 h confirmed coacervate formation in both cases, with diameters ranging from 0.5 μm to 1.5 μm (Fig. 1c, d and Fig. S2, ESI†). However, size control of coacervate droplets is challenging, as the size depends strongly on parameters such as incubation time and temperature. As anticipated, the transition from coacervate droplets to the fiber state followed a pathway similar to the unbiased route through a molecular rearrangement, as reported previously (Fig. 1e and Fig. S3, S4, ESI†).16 Hence, by regulating the pH through an enzymatic reaction, additional control over temporal LLPS and supramolecular polymerization was achieved.
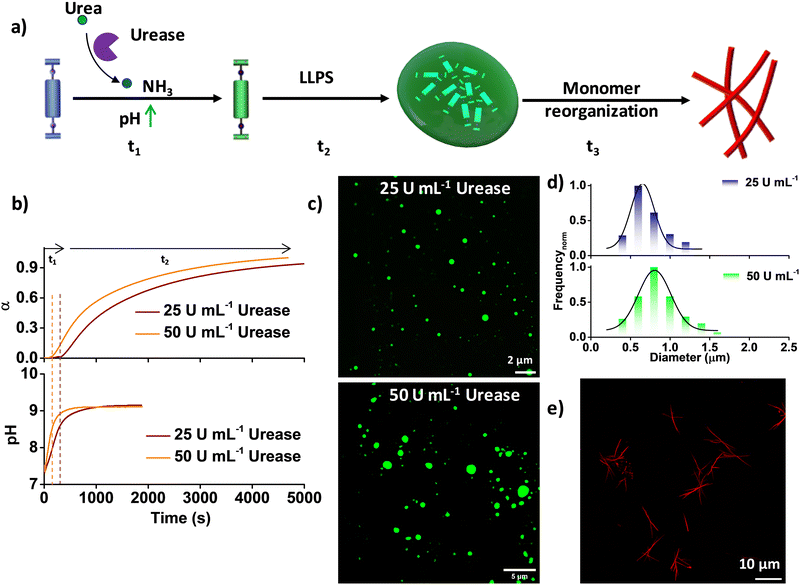 |
| | Fig. 1 Enzymatic-reaction-driven pH-modulated temporal LLPS and supramolecular polymerization. (a) Schematic representation of temporal transformation from the inactive monomer to coacervates through the activated monomer and finally supramolecular polymers with modulation of the pH conditions. (b) pH-Triggered LLPS kinetics (as monitored using the 382 nm absorption band) with different amounts of urease, along with the corresponding pH profiles. (c) Corresponding CLSM images and (d) size (diameter, calculated for 100 droplets using ImageJ software) distribution. (e) CLSM images after 96 h of the temporally grown thermodynamically stable fiber state with 1 μM Nile red. α = extent of LLPS. 50 mM urea, [NDBA] = 5 × 10−5 M, H2O/DMSO, 98/2 (v/v). | |
Transient dissolution of coacervate droplets
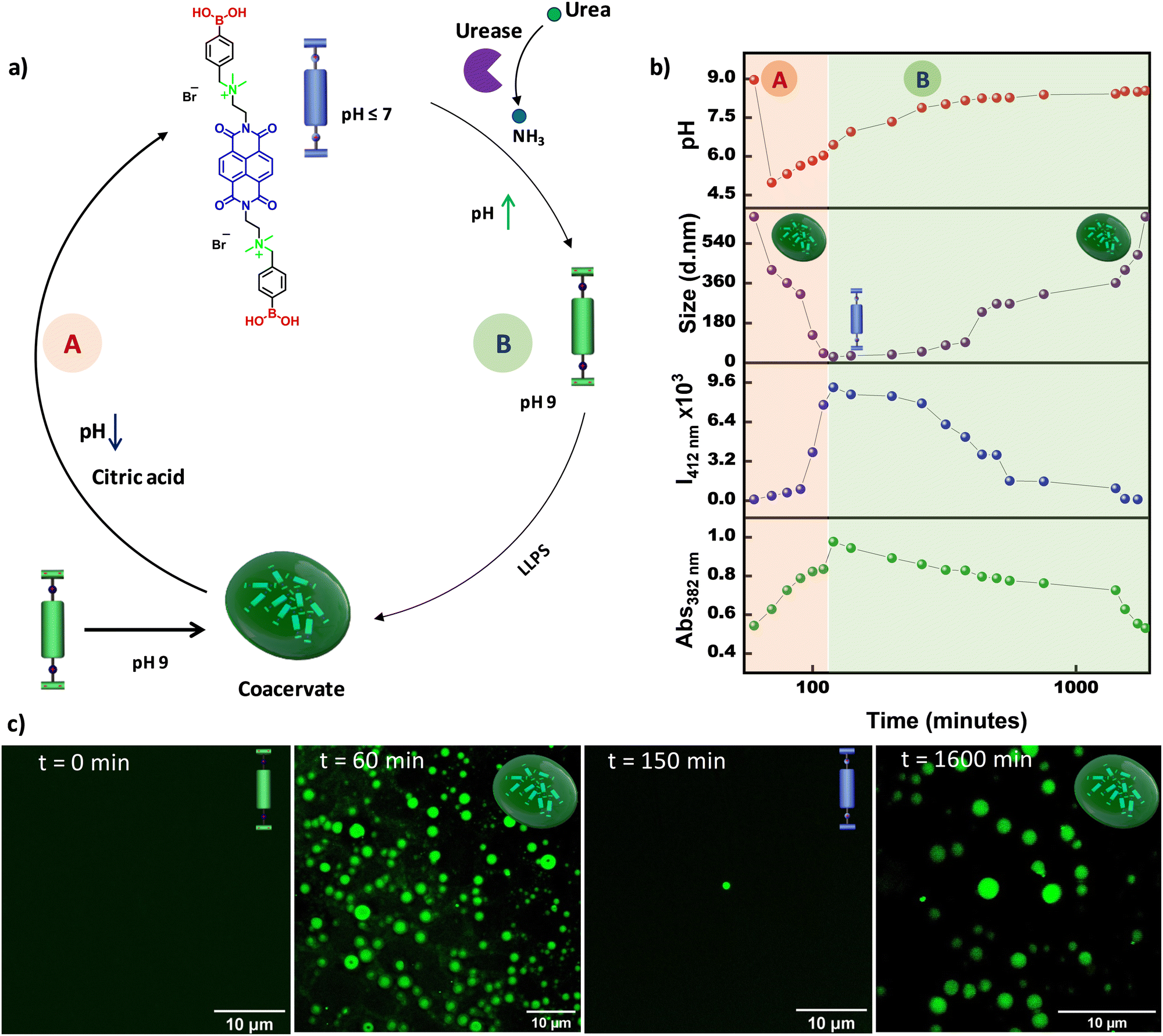
Further, we envisaged increasing the complexity of the system from temporal control to transient control over the LLPS. To this end, we planned to introduce two antagonistic pH modulators into the system; one component would first decrease the pH of the solution and then activate the other component to temporally restore the pH to the initial state by neutralizing the first component. To fulfill these criteria, we selected citric acid,23,28 which is well known as a deactivator, and used the previously explained urea–urease enzyme as an activator. We predicted that the addition of both citric acid and urea–urease into the grown coacervate solution at pH 9 would lead to an initial sudden decrease in pH that would trigger both the dissolution of coacervate droplets and the activation of urease to produce ammonia in the system and temporally increase the pH, which would finally result in LLPS and formation of coacervate droplets (Fig. 2a). Indeed, upon the addition of 15 mM citric acid, 50 mM urea, and 25 U mL−1 urease to a grown (after 60 minutes) coacervate droplet solution at pH 9, an instantaneous decrease in pH from 9 to ∼5.5 was observed, which resulted in a temporal increase in the monomeric 382 nm absorption and 412 nm emission bands, and a decrease in size via dynamic light scattering, indicating temporal dissolution of the droplets (Fig. 2b, marked as A, and Fig. S5, ESI†). This was further confirmed by the CLSM images, which indicated that the coacervate droplets disappeared within ∼2 h (Fig. 2c). It is well known that at lower pH, urease shows comparably better activity. Thus, once the pH decreases to ∼5.5, the urease is activated and temporally increases the pH of the solution (marked as B in Fig. 2b). After ∼150 min, a temporal decrease in the monomeric 382 nm absorption and 412 nm emission bands, along with an increase in the size in the DLS measurements, were observed, indicating temporal liquid–liquid phase separation, which was further confirmed from the CLSM images at various time intervals. This confirmed the reassembly of the coacervate droplets upon a temporal increase in pH. Hence, by coupling citric acid and the urea–urease enzymatic reaction, we achieved the transient dissolution of coacervate droplets.
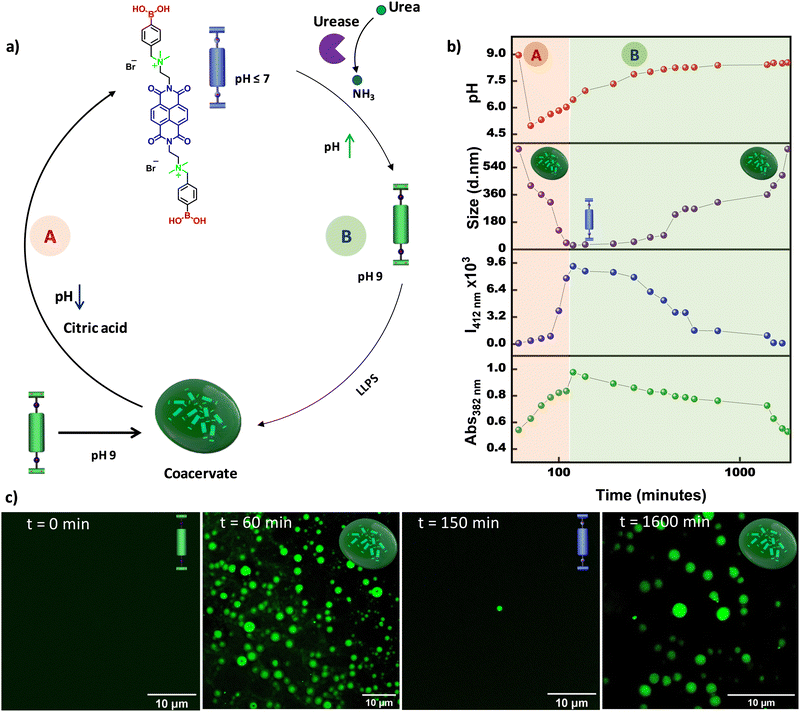 |
| | Fig. 2 Transient LLPS. (a) Schematic representation of the pH-modulated transient dissolution of coacervate droplets. Blue and green representations of the molecules indicate the boronic acid and boronate forms, respectively. (b) Addition of both citric acid and urea–urease results in initial dissolution followed by temporal LLPS of NDBA, which was probed using several spectroscopic techniques: the changes in absorbance (at 382 nm) and emission (at 412 nm), DLS, and the change in the pH of the solution. (c) CLSM images at different time intervals showing the temporal morphological changes, suggesting transient LLPS. A and B represent the dissolution and reformation process of the coacervate droplets, respectively. [NDBA] = 5 × 10−5 M, H2O/DMSO, 98/2 (v/v), 15 mM citric acid, 50 mM urea, and 25 U mL−1 urease. | |
pH-Regulated temporal disassembly of fibers
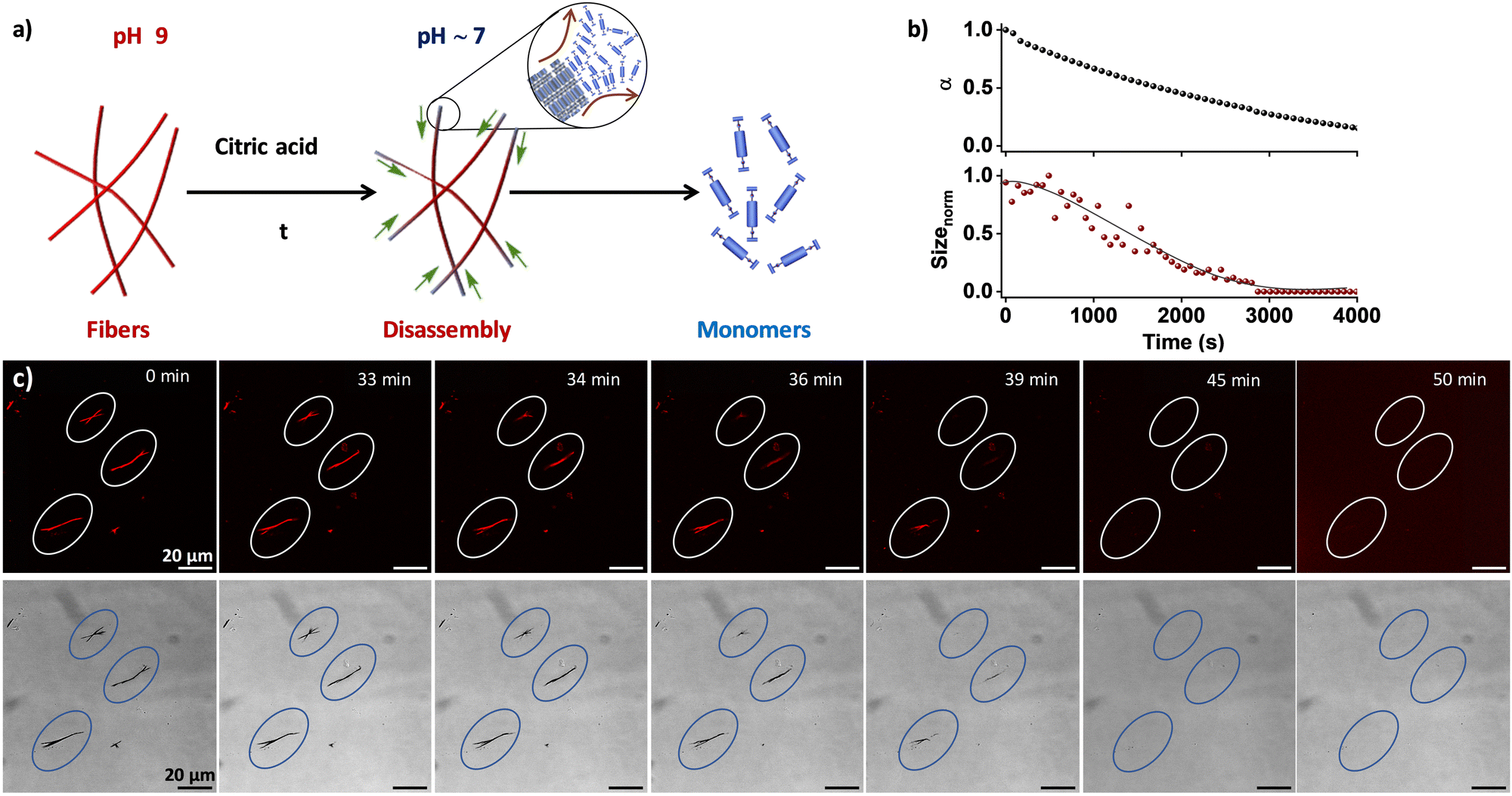
Supramolecular polymers exhibit a distinct advantage over traditional polymers due to their reversible nature, which is a character bestowed by intricate non-covalent interactions involved.29,30 As the present system is pH-sensitive, the reversibility of the temporally grown fibers to monomers was examined under varying pH conditions (Fig. 3a). Upon the addition of 100 mM citric acid23,28 to a pre-grown fiber solution at pH 9, a temporal change in the pH (∼5) of the solution was observed, which resulted in a time-dependent alteration in both the absorption and DLS kinetics (Fig. 3b). Moreover, monitoring of the absorption kinetics at 382 nm, a progressive increase in absorption band intensity over time was observed, indicative of the disintegration of the fiber state into its constituent monomers (Fig. S6, ESI†). This was corroborated by the temporal decrease in size from the DLS kinetics, confirming the fiber-to-monomer transformation within 1 h. This was further confirmed through in situ visualization using CLSM and bright-field imaging techniques. The fibers were found to be stable for 33 ± 5 min after the addition of the 100 mM citric acid (Fig. 3c and Fig. S7, Movies S1, S2, ESI†). However, within the next 7–10 min, an abrupt decrease in fiber length from the edge and finally complete disassembly of the fibers were observed. Through tracing the disassembly 10 individual fibers, the rate of disassembly was calculated to be around 3.0 ± 0.2 μm min−1 (Fig. S8, ESI†). We posit that in the pH-induced disassembly of fibers, the boronate ions of NDBA at the fiber surface are accessible for transformation into neutral boronic acid, leading to disassembly from the edge of the fibers. As a result, the fibers significantly impede the pH alteration of NDBA, potentially serving as an effective form of negative feedback. It is noteworthy that the disassembly of the fibers bypasses the coacervate state, proceeding directly to the monomer state, in line with the metastable nature of the droplet phase. This profound observation imparts valuable insights into the reversible nature of the system. Hence, by tuning the pH of the system, temporal control over disassembly was achieved. However, in situ visualization of the supramolecular disassembly process is rare and can provide valuable insights into the depolymerization pathways and the presence of pathway complexity.31–33 In the context of biomedical research, understanding the disassembly of supramolecular structures can inform the development of drug delivery systems, diagnostic tools, and therapeutic agents, in which controlled disassembly is often crucial for efficacy.
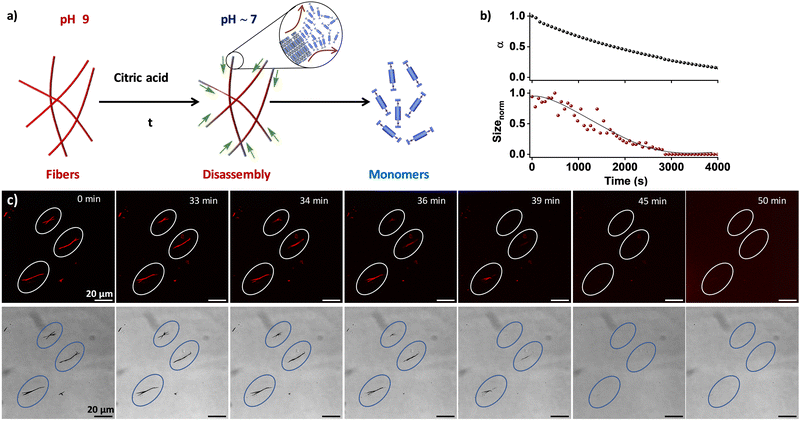 |
| | Fig. 3 Temporal disassembly of supramolecular fibers formed from NDBA. (a) Schematic of the pH-modulated temporal disassembly of the fiber to the monomers. (b) Disassembly of fibers to monomers probed through the temporal change in absorbance (shown as the extent of aggregation) and DLS upon the addition of 100 mM of citric acid to a pre-grown fiber solution. (c) CLSM (top) and corresponding bright-field (bottom) images of the in situ visualization of fiber disassembly upon the addition of 100 mM citric acid. α = extent of aggregation. [NDBA] = 5 × 10−5 M, H2O/DMSO, 98/2 (v/v), 1 μM Nile red. | |
Conclusions
In conclusion, a demonstration of programmable coacervate droplets and fibers was achieved by temporally regulating the pH of the solution through citric acid and urea–urease enzymatic reaction. Initially, additional control over LLPS was introduced using coupling the pH-modulator urea–urease enzymatic reaction ultimately resulting in supramolecular fiber formation through monomer rearrangement. By increasing the complexity of the system and incorporating antagonistic pH regulators (citric acid and urea–urease), transient control over the LLPS was established. Finally, similar to the coacervate droplets, temporal disassembly of the kinetically grown supramolecular fibers was monitored spectroscopically and via in situ visualization using fluorescence microscopy. Overall, utilizing pH as a stimulus, we achieved programmable LLPS and supramolecular polymerization.
Author contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank JNCASR, Department of Science and Technology (DST), Government of India. S. J. G. acknowledges the funding received from Swarna Jayanti Fellowship award (DST/SJF/CSA01/2016-2017) S. P. thanks the INSPIRE and SRISTI-BIRAC for the fellowship and funding, respectively. Authors acknowledge SAMat research facilities and JNCASR for CLSM experiments.
References
- Y. Shin and C. P. Brangwynne, Science, 2017, 357, 6357 CrossRef PubMed.
- A. S. Lyon, W. B. Peeples and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2021, 22, 215–235 CrossRef CAS.
- M. Linsenmeier, M. R. G. Kopp, F. Grigolato, L. Emmanoulidis, D. Liu, D. Zurcher, M. Hondele, K. Weis, U. Capasso Palmiero and P. Arosio, Angew. Chem., Int. Ed., 2019, 58, 14489–14494 CrossRef CAS.
- C. P. Brangwynne, C. R. Eckmann, D. S. Courson, A. Rybarska, C. Hoege, J. Gharakhani, F. Julicher and A. A. Hyman, Science, 2009, 324, 1729–1732 CrossRef CAS.
- S. Alberti, A. Gladfelter and T. Mittag, Cell, 2019, 176, 419–434 CrossRef CAS.
- S. Koga, D. S. Williams, A. W. Perriman and S. Mann, Nat. Chem., 2011, 3, 720–724 CrossRef CAS PubMed.
- C. Love, J. Steinkuhler, D. T. Gonzales, N. Yandrapalli, T. Robinson, R. Dimova and T. D. Tang, Angew. Chem., Int. Ed., 2020, 59, 5950–5957 CrossRef CAS PubMed.
- K. K. Nakashima, J. F. Baaij and E. Spruijt, Soft Matter, 2018, 14, 361–367 RSC.
- C. Donau, F. Späth, M. Sosson, B. Kriebisch, F. Schnitter, M. Tena-Solsona, H. Kang, E. Salibi, M. Sattler, H. Mutschler and J. Boekhoven, Nat. Commun., 2020, 11, 5167 CrossRef CAS PubMed.
- F. Späth, C. Donau, A. M. Bergmann, M. Kränzlein, C. V. Synatschke, B. Rieger and J. Boekhoven, J. Am. Chem. Soc., 2021, 143, 4782–4789 CrossRef PubMed.
- N. Martin, L. Tian, D. Spencer, A. Coutable-Pennarun, J. L. Ross Anderson and S. Mann, Angew. Chem., Int. Ed., 2019, 58, 14594–14598 CrossRef CAS.
- J. Deng and A. Walther, Chem, 2020, 6, 3329–3343 CAS.
- R. W. Lewis, B. Klemm, M. Macchione and R. Eelkema, Chem. Sci., 2022, 13, 4533–4544 RSC.
- M. Abbas, W. P. Lipinski, K. K. Nakashima, W. T. S. Huck and E. Spruijt, Nat. Chem., 2021, 13, 1046–1054 CrossRef CAS PubMed.
- R. Kubota, T. Hiroi, Y. Ikuta, Y. Liu and I. Hamachi, J. Am. Chem. Soc., 2023, 145, 18316–18328 CrossRef CAS PubMed.
- S. Patra, S. Chandrabhas, S. Dhiman and S. J. George, J. Am. Chem. Soc., 2024, 146, 12577–12586 CrossRef CAS.
- M. Kumar, J. N. S. Hanssen and S. Dhiman, ChemSystemsChem, 2024, 6, e202400013 CrossRef CAS.
- A. Jain, S. Kassem, R. S. Fisher, B. Wang, T.-D. Li, T. Wang, Y. He, S. Elbaum-Garfinkle and R. V. Ulijn, J. Am. Chem. Soc., 2022, 144, 15002–15007 CrossRef CAS.
- C. Yuan, A. Levin, W. Chen, R. Xing, Q. Zou, T. W. Herling, P. K. Challa, T. P. J. Knowles and X. Yan, Angew. Chem., Int. Ed., 2019, 58, 18116–18123 CrossRef CAS PubMed.
- S. Dhiman and S. J. George, Bull. Chem. Soc. Jpn., 2018, 91, 687–699 CrossRef CAS.
- A. Das, S. Ghosh, A. Mishra, A. Som, V. B. Banakar, S. S. Agasti and S. J. George, J. Am. Chem. Soc., 2024, 146, 14844–14855 CrossRef CAS PubMed.
- S. Dhiman, A. Sarkar and S. J. George, RSC Adv., 2018, 8, 18913–18925 RSC.
- T. Heuser, E. Weyandt and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 13258–13262 CrossRef CAS PubMed.
- T. Heuser, A.-K. Steppert, C. M. Lopez, B. Zhu and A. Walther, Nano Lett., 2015, 15, 2213–2219 CrossRef CAS.
- L. Heinen, T. Heuser, A. Steinschulte and A. Walther, Nano Lett., 2017, 17, 4989–4995 CrossRef CAS.
- A. Jain, S. Dhiman, A. Dhayani, P. K. Vemula and S. J. George, Nat. Commun., 2019, 10, 450 CrossRef CAS PubMed.
- S. Patra, S. Dhiman and S. J. George, Chem. Mater., 2024, 36, 9460–9468 CrossRef CAS.
- B. Su, T. Chi, W. Chen, S. Xian, D. Liu, C. J. Addonizio, Y. Xiang and M. J. Webber, J. Mater. Chem. B, 2024, 12, 4666–4672 RSC.
- T. Aida and E. W. Meijer, Isr. J. Chem., 2020, 60, 33–47 CrossRef CAS.
- G. Vantomme and E. W. Meijer, Science, 2019, 363, 1396–1397 CrossRef CAS PubMed.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- S. Onogi, H. Shigemitsu, T. Yoshii, T. Tanida, M. Ikeda, R. Kubota and I. Hamachi, Nat. Chem., 2016, 8, 743–752 CrossRef CAS.
- K. Jalani, A. D. Das, R. Sasmal, S. S. Agasti and S. J. George, Nat. Commun., 2020, 11, 3967 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Sushmitha
Chandrabhas
and
Subi J.
George
,
Sushmitha
Chandrabhas
and
Subi J.
George