
Nano-bio interactions of Gum Arabic-stabilized lanthanide-based upconverting nanoparticles: in vitro and in vivo study†
Hana
Mirmajidi
 a,
Hyojin
Lee
b,
Niepukolie
Nipu
b,
Jith
Thomas
c,
Zuzana
Gajdosechova
d,
David
Kennedy
d,
Jan A.
Mennigen
*b and
Eva
Hemmer
*a
a,
Hyojin
Lee
b,
Niepukolie
Nipu
b,
Jith
Thomas
c,
Zuzana
Gajdosechova
d,
David
Kennedy
d,
Jan A.
Mennigen
*b and
Eva
Hemmer
*a
aDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, 10 Marie-Curie Private, Ottawa (ON) K1N 6N5, Canada. E-mail: ehemmer@uottawa.ca
bDepartment of Biology, University of Ottawa, 20 Marie-Curie Private, Ottawa (ON) K1N 6N5, Canada. E-mail: jan.mennigen@uottawa.ca
cBureau of Chemical Safety, Food and Nutrition Directorate, Health Products and Food Branch, Health Canada, 251 Sir Frederick Banting Driveway, Tunney's Pasture, Ottawa (ON) K1A 0K9, Canada
dMetrology, National Research Council Canada, 1200 Montreal Road, Ottawa (ON) K1A 0R6, Canada
First published on 7th November 2024
Abstract
Lanthanide-based nanoparticles (Ln-NPs) are highly valued for their unique optical and magnetic properties, making them useful in various scientific fields, including materials science and biomedicine. This study investigated the use of Gum Arabic (GA), a natural, non-toxic biopolymer, as capping agent for Ln-NPs to enhance their biocompatibility and chemical and colloidal stability. Specifically, Er3+/Yb3+ co-doped NaGdF4 Ln-NPs were modified with GA, followed by their characterization with respect to upconversion properties and in vitro as well as in vivo toxicity. Herein, widely used ligand-free and polyacrylic acid (PAA)-capped Ln-NPs were used as reference materials. Importantly, the GA-modified Ln-NPs exhibited superior stability in aqueous and biologically relevant media, as well as relatively lower cytotoxicity across multiple cell lines, including U-87 MG, HEPG2, and J774A.1. In vivo studies using zebrafish embryos confirmed the minimal toxicity of GA-capped Ln-NPs. Despite overall low non-specific cellular uptake, hyperspectral imaging and inductively coupled plasma mass spectrometry confirmed the colocalization of the Ln-NPs as a function of their surface chemistry in both cell models and zebrafish. The results suggest GA as an effective surface-stabilizing agent for Ln-NPs, paving the way for future functionalization with targeting agents.
Introduction
Lanthanide-based nanoparticles (Ln-NPs) are well-known for their excellent optical and magnetic properties, which render them valuable for application in fields like materials science, chemistry, physics, and biomedicine. For instance, Ln-NPs are continuously attracting interest due to their emission of UV-visible light under near-infrared (NIR) excitation (so-called upconversion) as well as NIR-triggered NIR emission.1–3 Particularly for biomedical applications, the use of NIR light has been demonstrated beneficial over UV-visible light used by conventional optical probes, due to reduced photobleaching, deeper tissue penetration depth, and lower background fluorescence from biological media and tissues. Consequently, thanks to their distinct luminescence, upconverting Ln-NPs (UCNPs) have been proposed for therapeutic treatments, including photothermal and photodynamic therapy, as well as photoinduced drug delivery, while NIR emitters show promise for diagnostic applications like biolabeling, biosensing, and bioimaging.4–8To effectively use these inorganic nanomaterials in biological contexts, surface modification and functionalization ensuring dispersion in aqueous media and biocompatibility are essential.9,10 Polyethylene glycol (PEG), polyacrylic acid (PAA), polyethyleneimine (PEI), and their derivatives in addition to lipid-based micelle encapsulation are among the most popular types of polymers used for Ln-NP surface modification.11–16 However, some of these commonly used polymers may come with drawbacks, such as chemical instability, a tendency for agglomeration, or concerns about toxicity and immune response.17–23 Alternatively, natural biopolymers such as chitosan, cellulose, or other polysaccharides have been proposed as candidates for Ln-NP functionalization towards biomedical applications.16,24–31 The presence of glucose molecules in the structure of polysaccharide-based biopolymers may decrease protein binding on the Ln-NP surface, therefore reducing the triggering of immune responses in vitro or in vivo. Still, the exploration of natural polymers for straightforward Ln-NP functionalization to induce colloidal stability and biocompatibility remains limited. In light of this, we here assess the suitability of biopolymer Gum Arabic (GA) as a biocompatible capping agent for Ln-NPs.
Gum Arabic is a complex polysaccharide derived from the Acacia tree, which exhibits a highly branched structure and is characterized by its glycoprotein nature.32 GA is renowned for its non-toxic, biocompatible properties, and Food and Drug Administration (FDA) approval for human consumption.33 The rich composition of diverse functional groups, including hydroxyl and carboxyl groups, underpins its wide use in the food industry.34,35 In nanotechnology, GA has been utilized to modify the surface of nanomaterials like boron nitride nanotubes, metal (Au, Ag, Pt) nanoparticles, chitosan-based particles, and iron oxide magnetic nanoparticles, serving as a stabilizer to prevent aggregation and enhance biocompatibility.36–44 For instance, the remarkable stability of GA-capped gold nanoparticles (GA-AuNPs) in different physiologically relevant pH was reported by de Barros et al., who further demonstrated the suitability of the GA-AuNPs as non-toxic contrast agent in X-ray CT imaging.45 As another example, stable colloidal suspensions of GA-functionalized magnetite nanoparticles (GA-MNPs) were successfully used for therapeutic nanoplatforms.46 Recently, we demonstrated the suitability of GA as a stabilizing agent in Ln-NP-based aqueous inks for printing applications.47 These examples showcase the promising features of GA in enhancing the stability and biocompatibility of various nanoparticle formulations. Although GA has been proven as an effective and highly biocompatible capping agent, it has not yet been evaluated for its use among Ln-NPs for biomedical applications.
Prior to any biomedical application, it is essential to assess nano-bio interactions and potential toxicity to ensure safe application. In general, the interaction of nanomaterials, including Ln-NPs, with biological systems is influenced by factors such as size, surface charge, protein corona formation, as well as colloidal and chemical stability.48–52 Given the large variety of Ln-NPs in terms of size, ranging from a few nanometres to several hundreds of nanometres, and surface chemistry, much effort has been undertaken to assess the cytotoxicity and biodistribution of Ln-NPs. For instance, the surface chemistry of the Ln-NPs plays a crucial role when it comes to interactions with cells, e.g., cellular uptake and toxicity responses.53–55 Still, the understanding of nano-bio interactions of Ln-NPs remains limited, and findings about their chemical stability and cytotoxicity can be ambiguous. Therefore, the establishment and understanding of the relationship between Ln-NP structure and surface chemistry on the one side and their interaction with biological matter on the other remains a challenge towards the materials’ successful in vitro and in vivo applications.20,56,57 It is the goal of this study to help closing this gap by exploring GA as a promising capping agent for Ln-NPs. Specific attention was paid to dispersion as well as chemical and optical stability in biologically relevant media. Nano-bio interactions were assessed at a cellular level and towards zebrafish, which is a widely used model in biomedical research, particularly in areas such as toxicology, neuroactivity, and developmental studies.58 Moreover, the GA-modified Ln-NPs based on NaGdF4 co-doped with Er3+ and Yb3+ to endow upconversion and NIR emission under 980 nm excitation underwent comprehensive characterization in comparison to ligand-free and PAA-capped Ln-NPs. Overall, GA-modified Ln-NPs exhibited improved stability and resistance to agglomeration in biological media and exhibited relatively lower cytotoxicity in various cell lines (human glioblastoma cells U-87 MG, human hepatocyte cell line HEPG2, and mouse macrophages J774A.1) and minimal impact on zebrafish development.
Experimental details
Chemicals
Gadolinium oxide (Gd2O3, 99.999%), erbium oxide (Er2O3, 99.99%), and ytterbium oxide (Yb2O3, 99.9%), trifluoroacetic acid (CF3COOH, TFA, 98%), sodium trifluoroacetate (CF3COONa, Na-TFA, 98%), oleic acid (CH3(CH2)7CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH2)7COOH, 90%), oleylamine (CH3(CH2)7CHCH(CH2)7CH2NH2, 70%), 1-octadecene (CH3(CH2)15CHCH2, 90%), sodium citrate dihydrate (HOC(COONa)(CH2COONa)2·2H2O, Na-Cit, 99%), poly(acrylic acid) ((C3H4O2)n, product #323667, PAA), Gum Arabic (from Acacia tree, spray dried, product #51198, GA), and chloroform (> 99.8%) were purchased from Sigma Aldrich (Oakville, ON, Canada). Ethanol (99%) was purchased from Commercial Alcohols Inc (Toronto, ON, Canada). Acetone, hexane (analytical grade), toluene (99.8%), nitric acid (HNO3), and hydrochloric acid (HCl, 36.5%) were purchased from Fischer Scientific (Waltham, MA, USA). All chemicals were used as received.
CH(CH2)7COOH, 90%), oleylamine (CH3(CH2)7CHCH(CH2)7CH2NH2, 70%), 1-octadecene (CH3(CH2)15CHCH2, 90%), sodium citrate dihydrate (HOC(COONa)(CH2COONa)2·2H2O, Na-Cit, 99%), poly(acrylic acid) ((C3H4O2)n, product #323667, PAA), Gum Arabic (from Acacia tree, spray dried, product #51198, GA), and chloroform (> 99.8%) were purchased from Sigma Aldrich (Oakville, ON, Canada). Ethanol (99%) was purchased from Commercial Alcohols Inc (Toronto, ON, Canada). Acetone, hexane (analytical grade), toluene (99.8%), nitric acid (HNO3), and hydrochloric acid (HCl, 36.5%) were purchased from Fischer Scientific (Waltham, MA, USA). All chemicals were used as received.
Microwave-assisted synthesis of oleate-capped β-NaGdF4:Er3+, Yb3+ nanoparticles
The synthesis of hexagonal (β-) phase Ln3+-doped NaGdF4 nanoparticles (Ln-NPs) was performed by microwave-assisted thermal decomposition of lanthanide trifluoroacetate precursors, [Ln(TFA)3] (Ln = Gd3+, Yb3+, and Er3+).31,59 In brief, for the synthesis of β-NaGdF4 NPs co-doped with 20 mol% Yb3+ and 2 mol% Er3+, 0.625 mmol of [Ln(TFA)3] were prepared by mixing Gd2O3 (0.244 mmol), Yb2O3 (0.0625 mmol), and Er2O3 (0.00625 mmol) in a 50 mL three-neck round-bottom flask. A 10 mL amount of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 TFA-to-H2O mixture was added, and the slurry was refluxed at 95 °C until it became clear and dried at 60 °C overnight. Subsequently, 1.875 mmol of Na-TFA (3:1 Na+-to-Ln3+ ion ratio) was added to the synthesized [Ln(TFA)3] along with 2.5 mL of oleic acid, 2.5 mL of oleylamine, and 5 mL of 1-octadecene. This mixture was degassed at 120 °C under vacuum for 30 min. Subsequently, the degassed precursor solution was transferred to a 35 mL microwave vessel, purged with N2, and tightly sealed. The vessel was inserted into a CEM Discover SP microwave reactor. The precursor solution was heated to 270 °C for 1 s, followed by rapid cooling to 250 °C, the temperature at which the reaction was held for 10 min, and subsequent cooling to 50 °C. Following the synthesis, the reaction mixture containing the synthesized particles was washed with a 1:3 hexane-to-ethanol mixture and centrifuged at RCF = 8346 g for 10 min. The product was then washed with a 1:3 toluene-to-acetone mixture and centrifuged using the same conditions. After purification, the oleate (OA)-capped particles were redispersed and stored in 5 mL of toluene. NaF as a potential by-product can be removed by redispersing the particles in 5 mL of ethanol prior to the addition of 5 mL of water, followed by precipitation via centrifugation. Obtained particles were washed one more time with 10 mL of ethanol before being stored in 5 mL of toluene and further use in the synthesis of core/shell NPs.
:1 TFA-to-H2O mixture was added, and the slurry was refluxed at 95 °C until it became clear and dried at 60 °C overnight. Subsequently, 1.875 mmol of Na-TFA (3:1 Na+-to-Ln3+ ion ratio) was added to the synthesized [Ln(TFA)3] along with 2.5 mL of oleic acid, 2.5 mL of oleylamine, and 5 mL of 1-octadecene. This mixture was degassed at 120 °C under vacuum for 30 min. Subsequently, the degassed precursor solution was transferred to a 35 mL microwave vessel, purged with N2, and tightly sealed. The vessel was inserted into a CEM Discover SP microwave reactor. The precursor solution was heated to 270 °C for 1 s, followed by rapid cooling to 250 °C, the temperature at which the reaction was held for 10 min, and subsequent cooling to 50 °C. Following the synthesis, the reaction mixture containing the synthesized particles was washed with a 1:3 hexane-to-ethanol mixture and centrifuged at RCF = 8346 g for 10 min. The product was then washed with a 1:3 toluene-to-acetone mixture and centrifuged using the same conditions. After purification, the oleate (OA)-capped particles were redispersed and stored in 5 mL of toluene. NaF as a potential by-product can be removed by redispersing the particles in 5 mL of ethanol prior to the addition of 5 mL of water, followed by precipitation via centrifugation. Obtained particles were washed one more time with 10 mL of ethanol before being stored in 5 mL of toluene and further use in the synthesis of core/shell NPs.
Microwave-assisted synthesis of oleate-capped β-NaGdF4:Er3+, Yb3+/NaGdF4 core/shell nanoparticles (Ln-NPs-OA)
An undoped NaGdF4 shell was grown onto the Ln3+-doped core Ln-NPs by subjecting the Ln-NPs to a second microwave-assisted treatment, adding [Gd(TFA)3] as shell precursor. 0.625 mmol of the shell precursor was prepared as described above, using Gd3+ as sole lanthanide. Subsequently, 1.875 mmol of Na-TFA (3:1 Na+-to-Gd3+ ratio), 4 mL of 1-octadecene, and 5 mL of oleic acid were added to the shell precursor. The reaction mixture was degassed under vacuum at 120 °C for 30 min. Approximately 50 mg of core NPs was firstly dispersed in 1 mL of 1-octadecene (note: in contrast to the core synthesis, no oleylamine was used to grow the NaGdF4). Subsequently, this 1 mL of 1-octadecene containing the core Ln-NPs and 9 mL of the degassed shell precursor solution were transferred to a 35 mL microwave vessel, purged with N2, and tightly sealed. The vessel was inserted into a CEM Discover SP microwave reactor, and the mixture was heated to 250 °C and held for 10 min under stirring. The cooling and washing procedure as well as storage of the obtained OA-capped core/shell NPs (Ln-NPs-OA) were as described above for the core NPs.
Surface modification with Gum Arabic (Ln-NPs-GA)
A 0.4% w/w Gum Arabic (GA) solution was prepared by dissolving 100 mg of GA in 25 mL of distilled water. The solution was kept under vigorous magnetic stirring at room temperature for 1 h before use. 2 mL of a 5 mg mL−1 suspension of ligand-free Ln-NPs in water (Ln-NPs-LF; see ESI† for details of ligand removal from Ln-NPs-OA)60 was mixed with 2 mL of the previously prepared GA solution and left stirring overnight at room temperature, followed by centrifugation at RCF = 8346 g for 10 min. The recovered pellet was dispersed in water and washed with acetone (5:1 ratio of acetone-to-water). The final Ln-NPs-GA were dispersed in 5 mL of water. In addition to Ln-NPs-LF, PAA-capped Ln-NPs (Ln-NPs-PAA; see ESI† for details)61 were used as reference samples for comparison with the here proposed Ln-NPs-GA.
Characterization
To confirm the crystalline phase of the NaGdF4:Er3+, Yb3+/NaGdF4 Ln-NPs, X-ray powder diffraction (XRD) analysis was performed using a Rigaku Ultima IV Diffractometer or a Bruker D8 Endeavor (Cu Kα, λ = 1.5401 Å), operating at 44 kV and 40 mA (step size: 0.02°, scan speed: 1° min−1). Therefore, the Ln-NPs were deposited on a glass slide from their suspension and let dry prior to the analysis. The size and morphology of the Ln-NPs were assessed by transmission electron microscopy (TEM) using an FEI Tecnai G2 F20 FETEM microscope operating at 120 kV. Therefore, the samples were dispersed from their suspensions onto a 300-mesh copper TEM grid coated with a formvar/carbon film. Size distributions (mean size ± standard deviation) of the samples were derived from TEM images using ImageJ (U. S. National Institutes of Health, Bethesda, MA, USA) and OriginPro (OriginLab Corporation, Northampton, MA, USA) software. The respective size distributions were obtained analysing 200 particles per sample.The presence of surface capping agents, including OA, GA, and PAA was confirmed using a Nicolet 6700 FT-IR Fourier-transform infrared (FT-IR) spectrometer (Thermo Fisher Scientific) with an attenuated total reflection (ATR) mode. FT-IR spectra were obtained by performing 128 scans with a resolution of 4 cm−1 using a diamond crystal. Thermogravimetric analysis (TGA) was carried out using a TGA Q500/Discovery (TA Instruments) under N2 atmosphere and at a heating rate of 10 °C min−1. In order to evaluate the colloidal and optical stability of the Ln-NPs in biologically relevant media, 200 μL of stock suspensions (1 mg mL−1) of Ln-NPs-GA, Ln-NPs-LF, and Ln-NPs-PAA were stored for up to 48 h at room temperature at a non-controlled atmosphere, with 1.8 mL of the following biological solutions: water, cell culture medium, and 10% of fetal bovine serum in water (FBS). The suspensions containing the Ln-NPs-GA, Ln-NPs-LF, or Ln-NPs-PAA were characterized with respect to upconversion emission by photoluminescence spectroscopy and dispersion stability by dynamic light scattering (DLS) at the day of preparation (day 1) as well as 24 h (day 2) and 48 h (day 3) of incubation. DLS and zeta-potential (ζ) measurements were carried out using a Malvern Zeta Sizer Nano-ZS equipped with a He–Ne laser (633 nm). Investigation of the upconversion and NIR emission of the Ln-NPs in suspension (toluene for Ln-NPs-OA; water, cell culture media, or FBS for Ln-NPs-LF, -GA, and -PAA) was carried out in a quartz cuvette (1 cm optical path) using a QuantaMaster 8075-21 spectrofluorometer from HORIBA Scientific, equipped with double grating emission monochromators, a red-extended photomultiplier detector R13456 PMT (185 to 980 nm), and a liquid nitrogen cooled InAs detector (1000 to 3450 nm). The continuous excitation of the Yb3+/Er3+-doped Ln-NPs was performed using a 980 nm continuous-wave laser diode (max. power density: 6.7 W cm−2). Inductively coupled plasma-optical emission spectroscopy (ICP-OES) was used to determine the Gd3+, Er3+, and Yb3+ ion concentration in the Ln-NP suspensions used for in vitro and in vivo investigations (vide infra). Therefore, an Agilent ICP-OES spectrometer (nebulizer flow of 0.7 L min−1, plasma flow 12 L min−1, auxiliary flow 1 L min−1) was used. The samples were prepared by digesting 10 μL of the Ln-NPs-GA, Ln-NPs-LF, and Ln-NPs-PAA stock solutions with 0.5 mL of HNO3 and 1.5 mL of HCl at 40 °C overnight, followed by the dilution with distilled water to 5 mL. The amount of NaGdF4:Er3+,Yb3+/NaGdF4 NPs internalized by zebrafish was determined using the ICP-OES under same conditions as described above. For each group, nine zebrafish were individually exposed to surface modified Ln-NPs in a 96-well plate. After exposure, zebrafish were pooled into three vials, each containing three zebrafish. These pooled samples were individually analysed using ICP-OES to assess Ln-NP uptake. The uptake values from these three samples were then averaged to produce the reported average values ± standard deviation. The uptake of Ln-NPs by cells was determined using an inductively coupled plasma mass spectrometer (ICP-MS, triple quadrupole 8800, Agilent). Due to the small amount of biomass following incubation with Ln-NPs, only one uptake value was obtained per sample. The samples (cells and zebrafish) were first dried on a hot plate at 80 °C for 2 h, followed by further thermal treatment at 350 °C overnight in air atmosphere using a Barnstead Thermolyne 1400 furnace (Barnstead/Thermolyne Corp).
In vitro cytotoxicity
:1 mixture of water and cell medium. 100 μL of these diluted samples were added to the corresponding wells containing 100 μL of medium and cells resulting in final concentrations of 250 μg mL−1 to 2.5 μg mL−1. To the remaining six wells, 100 μL of medium was added as a control (no Ln-NPs). Cells were then incubated with the respective Ln-NPs for 24 h. The workup consisted of adding a 50 μL PBS solution of MTT (2.5 mg mL−1) to each well, followed by incubation for 3 h. After 3 h, the medium was aspirated from all wells, leaving purple formazan crystals in those wells with viable cells. To each well, 150 μL of DMSO was added and plates were agitated manually to dissolve the crystals. 100 μL of each well was then transferred to a fresh plate to remove scattering from precipitated Ln-NPs. Plates were analysed using a plate reader (FLUOstar Omega, BMG Labtech) to determine the absorbance of each well at 570 nm. The cell viability was calculated using eqn (1).62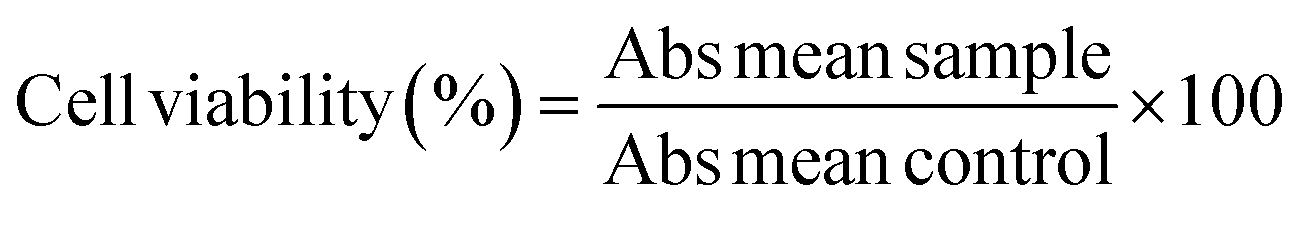 | (1) |
:100 with cell culture medium and subsequently incubated overnight with the cells. After 24 h, the medium was aspirated, the cells were washed once with 150 μL of PBS, and 100 μL of NR medium was added to each well. The plates were then incubated for 2 h after which the NR medium was aspirated from the wells. Cells were washed with 150 μL of PBS, and then 150 μL of destain solution (50% ethanol, 49% water, 1% acetic acid) was added to each well. Plates were shaken for 2 min to extract the NR from the cells prior to measuring the absorbance at 540 nm of each well by using a plate reader (FLUOstar Omega, BMG Labtech).
In vivo studies using zebrafish
Zebrafish embryos were generated from wild-type zebrafish (AB line) housed in a recirculating system (multi-linking system with water treatment unit and palletised centralised life Support system, Techniplast, Montreal, QC, Canada) at the University of Ottawa's Bioscience Aquatics Facility. The fish were bred in 1.7 L sloped breeding tanks (Techniplast). Eggs from all breeders were collected, washed, and maintained in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM MgSO4, 0.33 mM CaCl2; pH 7.5 in double-distilled water without methylene blue) at 28 °C until further processing. Upon assessment of viability and developmental shield stage of embryos using a light microscope (Olympus LS, Toronto, ON, Canada) and camera (Lumenera Corporation, Ottawa, ON, Canada), embryos that developed normally were randomly distributed into 96-well plates (Fisher Scientific) and exposed to graded concentrations of Ln-NPs (mass concentration in water: 0 [control], 0.125, 0.25, and 0.5 mg mL−1). Each well in the 96-well plate contained a total exposure volume of 100 μL E3 medium. Embryos were exposed to the Ln-NPs under static exposure conditions for 5 days post-fertilization (dpf), during which they were maintained at 28 °C, with a photoperiod of 14 h light/10 h dark. For the experimental replicate, each type of Ln-NPs was tested in 2 to 3 replicate plates, with a sample size of n = 16–24 embryos per treatment group. Developmental toxicity assessments (survival, hatching success, and deformity) were conducted from 1 to 5 dpf. Survival was recorded daily by assessing all surviving embryos, both hatched and unhatched. Hatching was recorded daily by calculating the percentage of hatched embryos among all surviving embryos. All data recorded were converted to percentage values per plate and analysed using exposure plates as replicates. Larval behaviour was evaluated at 5 dpf with a light/dark locomotor assay analysing the total distance movement (the detection threshold was set at 20 mm) using the Viewpoint ZebraBox and ZebraLab software (Viewpoint Life Sciences, Lyon, France T) as previously described.64 The locomotor assay was performed for zebrafish exposed to 0.125 and 0.25 mg mL−1 of Ln-NPs-GA, Ln-NPs-LF, and Ln-NPs-PAA. The highest concentration-exposed groups were removed due to overt mortality or delayed hatching. In all cases, the sample size was n = 7–13 larvae. All procedures followed the Canadian Council on Animal Care (CACC) animal care guidelines and were approved by the University of Ottawa's Animal Care and Use Program (protocol # BL-3057).Cell and zebrafish imaging by hyperspectral microscopy
Hyperspectral data from cells and zebrafish embryos incubated with the Ln-NPs were obtained with a custom-build hyperspectral microscope (IMA Upconversion by PhotonEtc, Montreal, QC, Canada) equipped with a continuous wave 980 nm laser diode, an inverted optical microscope (Nikon, Eclipse Ti-U), a microscope objective (20×, NA = 0.6), a broadband camera for colour imaging of samples at the microscope stage, a set of galvanometer mirrors, a monochromator (Princeton Instruments SP-2360), and an EMCCS camera for detection of visible emission (Princeton Instruments ProEM). The upconversion emission spectra obtained on this instrument were non-corrected for the sensitivity of the detector. The same inverted optical microscope was used for brightfield imaging of cells and zebrafishes.Statistical analysis
All data were graphed and analysed using OriginPro and GraphPad Prism Version 9 (GraphPad Software Inc., La Jolla, CA, USA). Zebrafish mortality and hatching data were analysed using a mixed ANOVA statistical test, considering within-subject effects (time) and between-subject effects (treatment concentrations), as well as their interaction for each type of surface-modified Ln-NP. Degrees of freedom (df) indicate the number of independent values that can vary in estimating statistical parameters. The ANOVA F-statistic (F) measures the ratio of variance explained by the treatment to the variance within groups, and the p-value (P) quantifies the probability that any observed differences occurred by chance under the null hypothesis. P values less than 0.05 were considered statistically significant. A Greenhouse–Geisser correction was used to adjust for a lack of sphericity in the data. In cases of significant effects as determined by a P < 0.05 cut-off in the omnibus test, Sidak's multiple comparisons were conducted, and significant differences compared to control assessed for each specific timepoint of the exposure (P < 0.05). Locomotor data was first compiled and graphed as average values ± the standard error of the mean (S.E.M) for each treatment concentration to visualize temporal traces for each Ln-NP type tested. Light and dark period data after the 20 min dark acclimation phase were then averaged for each individual larva, and data was analysed by two-way ANOVA probing treatment and lighting condition as main factors as well as their interaction. In cases of significant effects in the omnibus test (P < 0.05), Sidak's multiple comparisons were conducted, and significant differences between groups were assessed (P < 0.05). Cell viability data are based on triplicate measurements and error bars represent the standard deviation.Results and discussion
Synthesis, structural, and optical characterization of the Ln-NPs
Oleate-capped core/shell nanoparticles (Ln-NPs-OA) composed of a β-NaGdF4 core co-doped with 20 mol% Yb3+ and 2 mol% Er3+ and an outer undoped NaGdF4 protecting shell were successfully synthesized using a microwave-assisted technique.31,59 The morphology and size of core and core/shell Ln-NPs-OA were examined by transmission electron microscopy (TEM). As shown in Fig. 1(A) and (B), the monodisperse spherical particles exhibited a narrow size distribution with an overall size of ca. 10 nm, whereas the diameter of the Ln3+-doped core was ca. 5 nm (Fig. S1A, ESI†), leading to a shell thickness in the order of 2.5 nm. Such small particle size is of interest for the investigation of nano-bio interactions as smaller nanoparticles exhibit a higher surface-to-volume ratio, which potentially enhances their interaction with biological systems. Moreover, smaller nanoparticles can more easily penetrate cellular membranes and are more likely to be internalized by cells through endocytosis. However, in some cases, nanoparticle agglomeration can also facilitate cellular uptake by creating larger clusters that enhance interactions with cell surfaces. Thus, both particle size and agglomeration may play significant roles in determining the efficiency of cellular uptake, depending on the specific conditions of the system.65,66 XRD analysis confirmed that the Ln-NPs-OA crystallized in the hexagonal (β) phase of NaGdF4 (Fig. S1B, ESI†). Multiple batches of Ln-NPs-OA were prepared to ensure enough material for the subsequent surface modification and assessment of nano-bio interactions. Phase and morphology of each batch of Ln-NPs-OA were analysed by XRD and TEM confirming comparable structural properties. The morphology and size were retained upon surface modification (Ln-NPs-GA, Ln-NPs-LF, and Ln-NPs-PAA) (Fig. 1(A) and (B)-2–4). Upon excitation at 980 nm, the Ln-NPs-OA exhibited upconversion emission, showing the characteristic Er3+ emission bands in the green (535–570 nm) and red (635–685 nm) spectral regions ascribed to the 2H11/2 → 4I15/2, 4S3/2 → 4I15/2, and 4F9/2 → 4I15/2 Er3+ f–f transitions (Fig. 1(C)).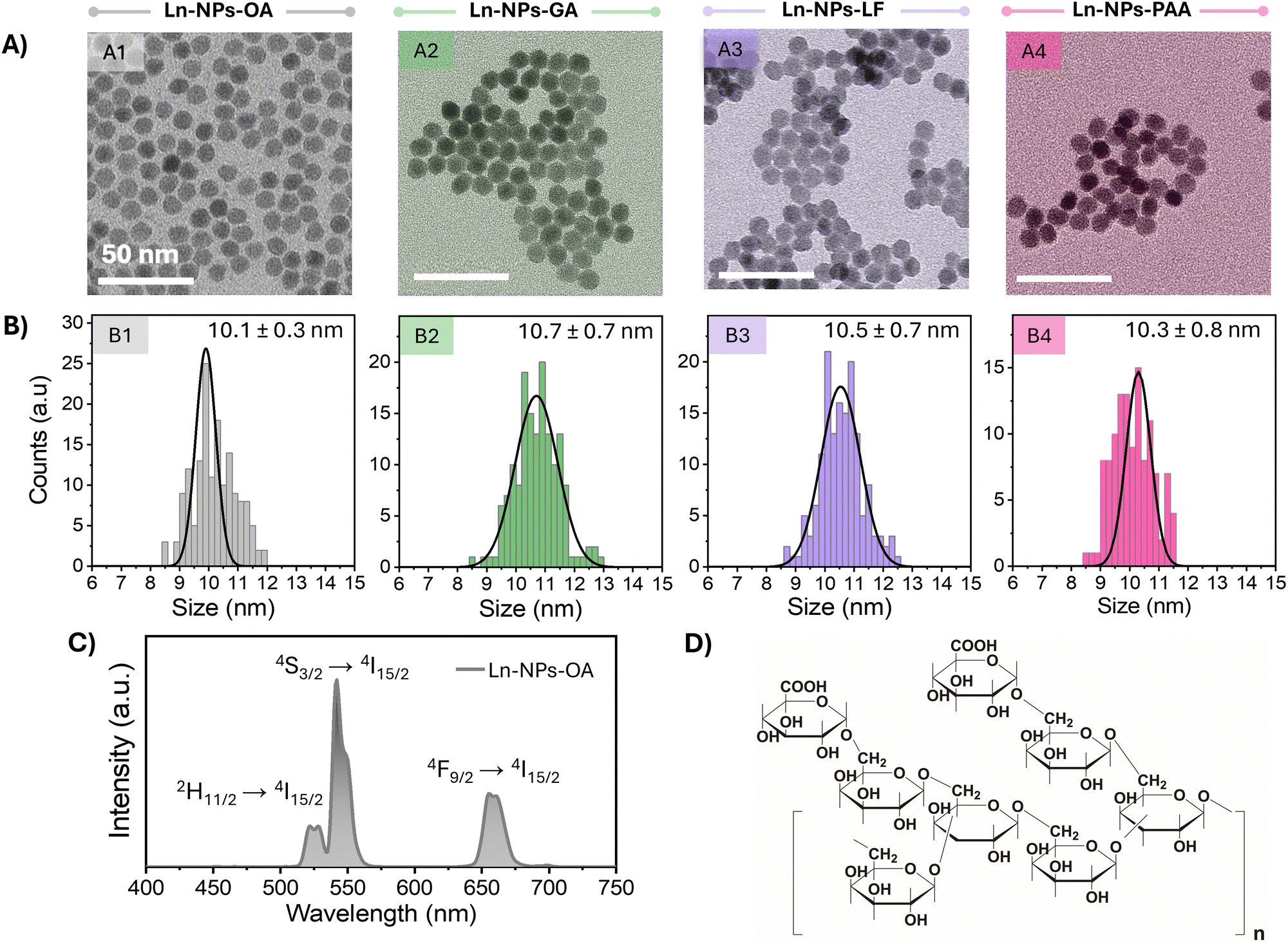 | ||
| Fig. 1 (A) TEM images of NaGdF4:Er3+,Yb3+/NaGdF4 core/shell Ln-NPs with various surface chemistries used in this study: (A1) as-prepared oleate-capped NPs (Ln-NPs-OA) used for further surface modification, (A2) Ln-NPs capped with Gum Arabic (Ln-NPs-GA), (A3) ligand-free Ln-NPs (Ln-NPs-LF), and (A4) Ln-NPs capped with polyacrylic acid (Ln-NPs-PAA). Scale bars are 50 nm. (B) Size distributions of the four types of Ln-NPs. The numbers represent the average particle size ± standard deviation. (C) Upconversion emission spectrum of Ln-NPs-OA dispersed in toluene under 980 nm excitation (laser power density: 6.7 W cm−2, particle concentration: 5 mg mL−1). (D) Chemical structure of Gum Arabic. Redrawn from ref. 67. | ||
Water dispersibility is a prerequisite for the use of lanthanide-based nanostructures in many of their proposed applications. Motivated by the reported biocompatibility, potential for further functionalization, and capability to endow various types of nanomaterials with dispersion and chemical stability, the oleate groups anchored to the surface of the as-prepared Ln-NPs-OA were replaced by Gum Arabic (GA). Therefore, in a first step, the oleate groups, rending Ln-NPs-OA hydrophobic, were removed following a previously established protocol under acid conditions (see ESI† for details).60 The acidic treatment did not compromise the structural integrity of the Ln-NPs (Fig. 1(A) and (B)) and resulted in positively charged Ln-NPs-LF (Table 1). When GA is dissolved in water at pH 6, the carboxy groups of the galactose and arabinose monomers present in the chemical structure of GA (Fig. 1(D)) undergo deprotonation, allowing for binding to the positively charged surface of the Ln-NPs-LF.68 The successful modification of the Ln-NPs-LF with GA was confirmed by Fourier transform infrared (FT-IR) spectroscopy (Fig. S2, ESI†) and thermogravimetric analysis (TGA) (Fig. S3, ESI†). The change of the ζ-potential from +36 mV for Ln-NPs-LF to –22 mV for Ln-NPs-GA dispersed in water provided further evidence for the presence of GA on the surface of the Ln-NPs (Table 1). In addition, surface modification of the Ln-NPs with PAA groups,61 being widely applied when seeking dispersibility in aqueous media, was performed to obtained Ln-NPs-PAA as control sample for the assessment of the stability, cytotoxicity, and potential toxic effects towards zebrafish. Successful modification with PAA groups was confirmed by FT-IR (Fig. S2, ESI†), TGA analysis (Fig. S3, ESI†), and ζ-potential measurements (Table 1). Photoluminescence spectra of the Ln-NPs dispersed in water confirmed their upconverting properties (Fig. S4, ESI†). Comparable emission intensities were obtained for Ln-NPs-PAA and Ln-NPs-PAA-GA, suggesting that both capping agents provide effective shielding against water-induced quenching of the upconversion emission. Conversely, Ln-NPs-LF exhibited lower emission intensity, which can be ascribed to the lack of surface protection. The direct exposure of the Ln-NPs to water molecules led to substantial non-radiative energy transfer to O–H bonds and quenching of the Er3+ emission.
| Ln-NPs-GA | Ln-NPs-LF | Ln-NPs-PAA | |
|---|---|---|---|
| ζ-Potential | −22.6 ± 0.2 mV | +19.6 ± 0.4 mV | −29.3 ± 0.5 mV |
| Cell line | Gd3+ [μg/106 cells] | Uptake [%] | Gd3+ [μg/106 cells] | Uptake [%] | Gd3+ [μg/106 cells] | Uptake [%] |
|---|---|---|---|---|---|---|
| HepG2 | 0.3 | 0.07 | 5.8 | 1.17 | 12.4 | 2.5 |
| U-87 MG | 1.8 | 0.4 | 6.3 | 1.27 | 27.6 | 5.5 |
| J774A.1 | 8.5 | 1.7 | 27.4 | 5.5 | 72.1 | 14.4 |
Chemical and colloidal stability of surface-modified Ln-NPs
For biomedical applications, chemical and colloidal stability in biologically relevant media is a prerequisite. Therefore, the chemical and colloidal stability was assessed directly by TEM and DLS as well as indirectly by monitoring the upconversion emission of the Ln-NPs upon their storage in water, cell culture medium, and FBS. TEM analysis confirmed that the morphology of Ln-NPs-GA after 48 h of incubation in any of these media was retained, and no major agglomerations were observed (Fig. 2(A)). Conversely, particularly Ln-NPs-PAA, but also Ln-NPs-LF, were at risk of aggregation, irrespective of the medium in which they were dispersed (Fig. S5A and S6A, ESI†). In agreement with the observations by TEM, the hydrodynamic size of Ln-NPs-GA remained almost unchanged, indicating better colloidal stability when compared to Ln-NPs-LF and Ln-NPs-PAA (Fig. 2(B) and Fig. S5A, B, S6A, B), ESI.† The good stability in both cell culture medium and FBS underscores the potential of GA as stabilizing agent for Ln-NPs. The observed colloidal stability can be attributed to the dynamic motion of the highly branched polysaccharide chains within GA, which facilitates strong steric resistance between individual particles.43 In contrast to Ln-NPs-GA, the hydrodynamic size of Ln-NPs-LF and Ln-NPs-PAA underwent a more pronounced increase in all tested media, with Ln-NPs-LF being the least stable, as expected due to the lack of any stabilizing capping agents (Fig. S5B and S6B, ESI†). The aggregation of Ln-NPs-LF can be attributed to the bridging flocculation phenomenon between negatively charged proteins from biological media and the positively charged nanoparticle surface.69 Compared to Ln-NPs-LF, capping with PAA reduced the degree of aggregation, yet, not as efficiently as GA. In addition, the surface charge of Ln-NPs-GA incubated in FBS and cell medium did not undergo any major changes compared to Ln-NPs-GA in water (Fig. 2(C)), remaining negative, which suggests the stable anchoring of GA at the Ln-NP surface. Importantly, while Ln-NPs-GA and Ln-NPs-PAA (Fig. 2(C) and Fig. S6C, ESI†) exhibited a negative surface charge irrespective of the chosen medium, a significant change from positive to negative was observed for Ln-NPs-LF upon transfer of the uncapped Ln-NPs-LF from water to cell medium and FBS, respectively (Fig. S5C, ESI†) (all fresh Ln-NP-LF dispersions exhibited a pH of 7, irrespective of the medium). This change in surface charge indicated that the uncapped Ln-NPs-LF were prone to the build-up of surface modification due to the presence of proteins in cell medium and FBS. Herein, the binding of anionic plasma proteins induced the negative surface charge, opposed to a positive surface charge due to positively charged Ln3+ ions at the nanoparticle surface when the Ln-NPs-LF are dispersed in pure water. This transformation illustrates how biological environments can influence the surface properties of nanoparticles and must be kept in mind for the further assessment of cytotoxicity (dispersion of Ln-NPs in cell medium) and interaction with zebrafish (dispersion of Ln-NPs in water) (vide supra).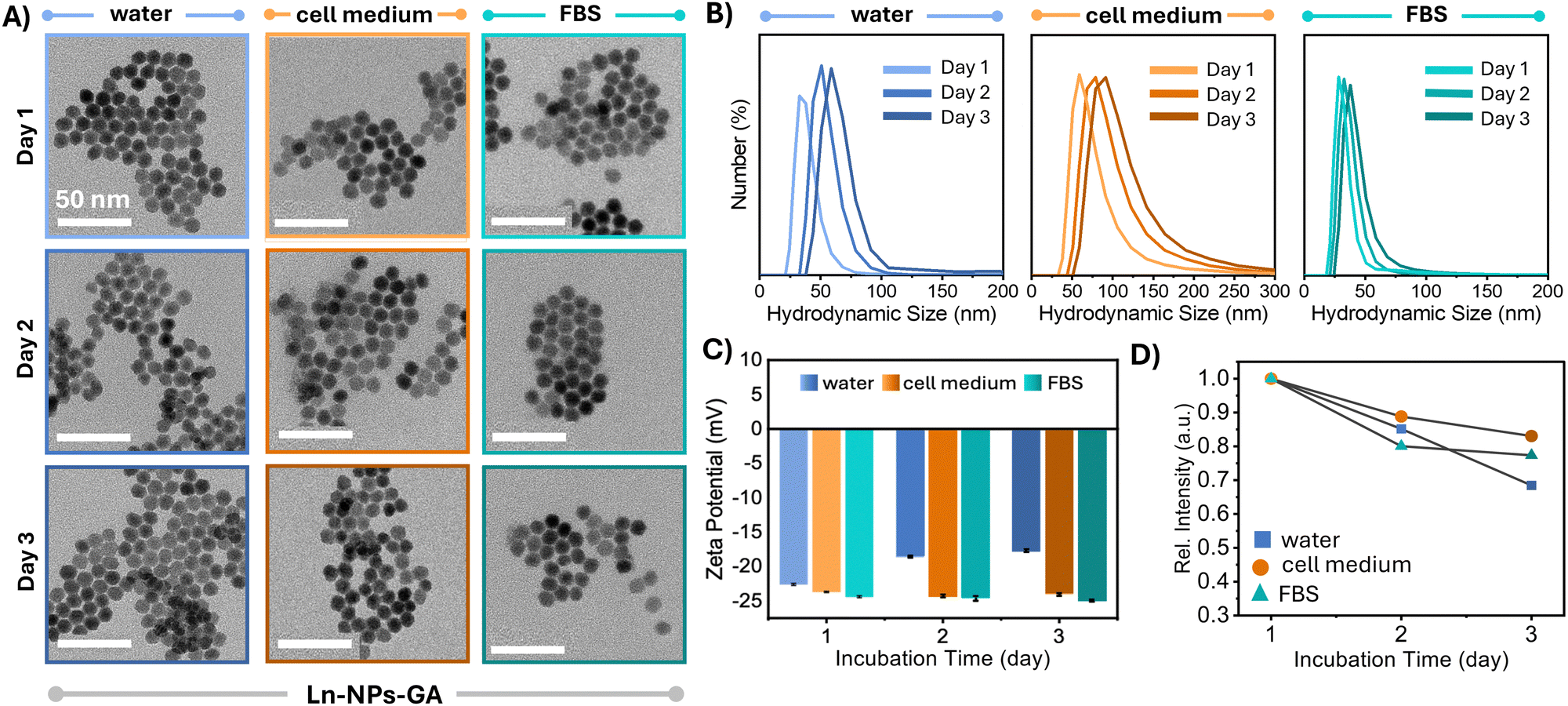 | ||
| Fig. 2 Analysis of the colloidal and optical stability of Ln-NPs-GA dispersed in water (blue), cell culture medium (orange), and FBS (teal). (A) TEM images of Ln-NPs-GA recorded on freshly prepared Ln-NPs-GA (day 1) as well as after storage for 24 h (day 2) and 48 h (day 3); scale bars: 50 nm. (B) DLS curves and (C) zeta potential of Ln-NPs-GA dispersed in water, cell culture medium, and FBS, respectively, recorded at days 1, 2, and 3 of storage in the respective media. DLS data revealed a relatively minor increase in the hydrodynamic diameter of the Ln-NPs-GA from ca. 33 to 60 nm in water (PDI: 0.17), ca. 60 to 90 nm in cell medium (PDI: 0.23), and ca. 28 to 38 nm in FBS (PDI: 0.39). All zeta potential data are presented as mean value ± standard deviation based on triplicate measurements taken for each sample in each of the three solvents at each time point. Error bars, representing the standard deviation, derived from these triplicate measurements. (D) Change in the overall upconversion emission intensity of the Ln-NPs-GA as a function of storage in water, cell culture medium, and FBS. Data represent the integrated intensity in the 510 to 695 nm spectral range (spectra are shown in Fig. S7, ESI†). | ||
Upconversion spectroscopy complemented the results obtained by TEM and DLS, with similar trends observed for the evolution of the emission intensity as a function of storage time. Importantly, the overall emission intensity of Ln-NPs-GA was retained at 70% and higher after 48 h in water and cell culture medium as well as FBS, respectively (Fig. 2(D) and Fig. S7A, ESI†). In contrast, the emission intensity of Ln-NPs-LF in water, cell culture medium, and FBS dropped by 52%, 46%, and 36%, respectively (Fig. S5D and S7B, ESI†). For Ln-NPs-PAA, a similar loss in upconversion intensity was observed, i.e., a decrease by 57%, 45%, and 38% upon storage in water, FBS, and cell culture medium, respectively (Fig. S6D and S7C, ESI†). This decrease in upconversion luminescence over time can primarily be ascribed to Ln-NP agglomeration. The stability of Ln-NPs in aqueous media is known to depend on their surface modification. Without strong steric or electrostatic stabilization, Ln-NPs tend to agglomerate to minimize surface energy, especially when surface ligands degrade or are insufficient to prevent aggregation. This agglomeration reduces colloidal stability and thus the number of excited particles in a dispersed state, thereby diminishing luminescence. Alternatively, partial dissolution of Ln-NPs might result in loss of photoluminescence.17 As the integrity of the Ln-NPs was confirmed by TEM (Fig. 2(A) and Fig. S5A, S6A, ESI†), agglomeration is most likely the cause of the observed drop in emission intensity. Noteworthy, Ln-NPs-GA further exhibited brighter NIR emission at ca. 1550 nm, ascribed to the Er3+ ion's 4I13/2 → 4I15/2 transition, than their ligand-free and PAA-capped counterparts (Fig. S8, ESI†).
Overall, surface modification with GA led to improved chemical and colloidal stability when compared to conventional capping agent PAA or ligand-free Ln-NPs. This improvement in stability may be ascribed to the polysaccharide nature of GA causing steric repulsion between the Ln-NPs.43 Additionally, the presence of small amounts of proteins in GA (∼2%) may contribute to its stronger and more stable binding to the nanoparticle surface, further enhancing stability.70,71 Some studies have highlighted that Ln-NPs may suffer from instability in aqueous environments due to their high surface reactivity and large surface-to-volume ratios, which can lead to aggregation or degradation.53 Given that such instability poses significant challenges for the biomedical application of Ln-NPs, the demonstrated enhancement of the chemical and dispersion stability of Ln-NPs while retaining good photoluminescence, renders GA a promising capping agent.
In vitro cytotoxicity
When nanoparticles are introduced into the human body for potential biomedical applications, they engage with cells within various organs and interact with proteins in the bloodstream, potentially leading to cytotoxic effects. Factors such as surface charge, nanoparticle size, hydrophobicity level, and surface moieties have been identified as crucial determinants of the cytotoxicity associated with nanoparticles.72 This is because the particle surface will determine their recognition by the cells, uptake as well as fate once inside the cells. Consequently, different surface functionalization can result in different toxicity profiles for the same nanoparticle composition. With that in mind, the cytotoxicity of the prepared Ln-NPs-GA was evaluated in comparison to Ln-NPs-PAA and Ln-NPs-LF. Cellular assays were performed on three cell lines, i.e., HepG2, U-87 MG, and J774A.1. The selection of these cell lines was based on their frequent use in other research, facilitating interstudy comparisons. HepG2 cells serve as a representative model for liver-related studies, as the liver is a common target for the localization of various nanomaterials in vivo. U-87 MG cells, a widely utilized human glioblastoma cell line, are commonly employed in brain cancer research, particularly aggressive malignant gliomas, the predominant and highly aggressive primary form of brain tumour. J774A.1 macrophage cells were chosen due to their heightened susceptibility to nanoparticle toxicity, attributed to their often more pronounced uptake compared to other cell lines. The MTT assay was employed to measure mitochondrial enzymatic activity, while the NR assay was used to determine cell viability based on the cells’ ability to absorb and retain lysosomal staining dye.As shown in Fig. 3, overall, Ln-NPs-GA exhibited higher cell viability compared to the reference samples in both the MTT and NR assays across all tested cell lines and up to the highest tested Ln-NP concentrations. Based on the MTT assay, cell viability of HepG2 and U-87 MG cells incubated with up to 250 μg mL−1 of Ln-NPs-GA for 24 h was close to or higher than 90% (Fig. 3(A) and (B)). In contrast, some cytotoxicity effects were observed for J774A.1 cells (Fig. 3(C)). Cell viability of more than 80% was retained for incubation with up to 1 μg mL−1 of Ln-NPs-GA but dropped to ca. 45% at the highest tested concentration of 25 μg mL−1, showing higher sensitivity to Ln-NPs-GA exposure than HepG2 and U-87 MG cells. Similar trends as a function of NP concentration were observed for Ln-NPs-LF and Ln-NPs-PAA, yet with overall lower cell viability compared to Ln-NPs-GA. In HepG2 and U-87 MG cell lines, cell viability dropped to 58% upon incubation with 250 μg mL−1 of ligand-free and PAA-capped Ln-NPs. Cytotoxic effects were most pronounced towards J774A.1 cells, exhibiting lower viability when compared to HepG2 and U-87 MG cells. Notably, both Ln-NPs-LF and Ln-NPs-PAA induced significant cell death in J774A.1 cells, with cell viabilities as low as 5% at the highest tested concentrations (up to 25 μg mL−1), while capping with GA induced improved cytocompatibility. The lower viabilities in case of J774A.1 cells can be attributed to the macrophages’ inherent role in engulfing foreign particles, facilitated by their surface receptors that recognize and bind to nanoparticles, leading to higher internalization.73 Consequently, macrophages typically exhibit greater nanoparticle uptake compared to other cell lines, leading to increased cytotoxicity.
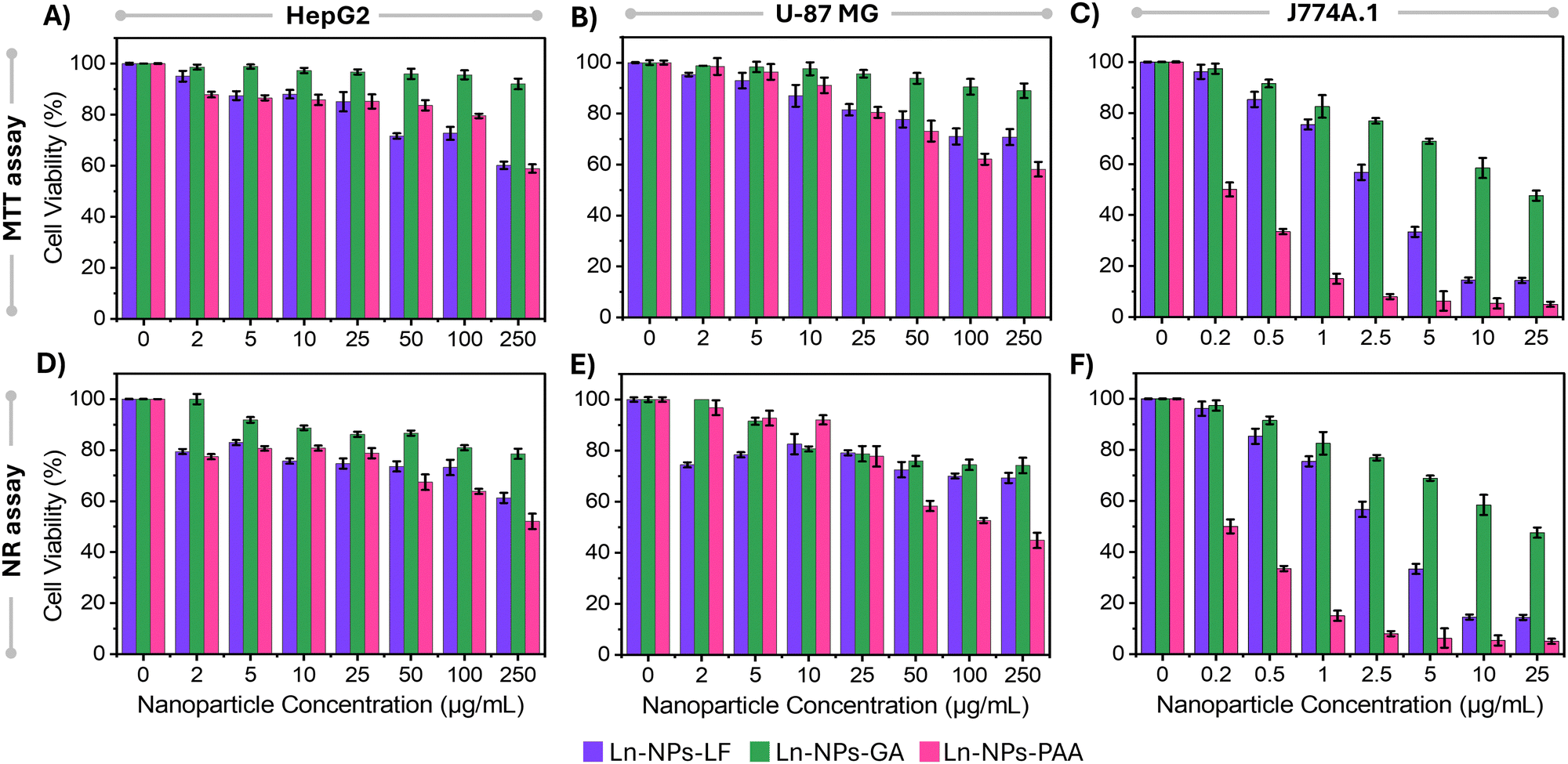 | ||
| Fig. 3 Cytotoxicity effects of Ln-NPs on HepG2, U-87 MG, and J774A.1 cell lines as a function of surface chemistry. All cells were treated for 24 h with different concentrations of Ln-NPs. Assessment of the cell toxicity in (A) HepG2, (B) U-87 MG, and (C) J774A.1 cell lines induced by Ln-NPs-GA (green), Ln-NPs-LF (purple), and Ln-NPs-PAA (pink) using the MTT assay. Quantification of cell toxicity in (D) HepG2, (E) U-87 MG, and (F) J774A.1 cell lines exposed to Ln-NPs-GA (green), Ln-NPs-LF (purple), and Ln-NPs-PAA (pink) by using the NR assay. Note the different x-axes in case of J774A.1 cells. Cell viability data are based on triplicate measurements and error bars represent the standard deviation. | ||
The NR assay unveiled a decrease in cell viability irrespective of the NP surface chemistry and cell type (Fig. 3(D)–(F)). However, the overall viability remained higher for Ln-NPs-GA when compared to Ln-NPs-LF and Ln-NPs-PAA. HepG2 and U-87 MG cell lines displayed approximately 74% and 78% viability when exposed to 250 μg mL−1 of Ln-NPs-GA, while the viability of these cells was significantly reduced when exposed to Ln-NPs-PAA, decreasing to 52% and 44%, respectively (Fig. 3(D) and (E)). Incubation with Ln-NPs-LF induced a similar trend in HepG2 cells with viabilities close to those of HepG2 cells exposed to Ln-NPs-PAA (Fig. 3(D)). Conversely, the viability of U-87 MG was found to be rather independent of NP concentration when incubated with Ln-NPs-LF (Fig. 3(E)). Their viability was comparable to that of cells exposed to Ln-NPs-GA at concentrations higher than 10 μg mL−1. As already observed in the MTT assay, the results of the NR assay confirmed that cell viability was the most affected by surface chemistry when exposing the different Ln-NPs to J774A.1 cells (Fig. 3(F)). Overall, based on MTT and NR assays, Ln-NPs-GA exhibited lower cytotoxicity compared to Ln-NPs-LF and Ln-NPs-PAA, as evidenced by consistently higher cell viability observed in both assays across the tested concentrations in HepG2, U-87 MG, and J774A.1 cell lines.
To better understand the observed trends in cell viability, ICP-MS analysis was performed on the cells treated with the various Ln-NPs to assess the internalization of Ln-NPs in the cells. The estimated numbers of Ln-NPs per cell are presented in Table S1 (see ESI† for details about the estimation). Irrespective of the chosen cell line, the cellular uptake of the Ln-NPs was low, yet most pronounced for Ln-NPs-PAA, followed by Ln-NPs-LF, and least in the case of Ln-NPs-GA (Table 1). In addition, the highest level of Ln-NP uptake was determined for J774A.1 cells, while lower uptake was observed for U-87 MG and HepG2 cells. Thus, cell viability correlated with Ln-NP uptake, whereas more uptake induced higher levels of cytotoxicity. In general, while different cell lines have different intrinsic uptake behaviour, colloidal stability and ζ-potential play an important role in the cellular uptake of nanoparticles. The ζ-potential of all surface-modified Ln-NPs in cell medium showed a negative surface charge (in the range of −20 to −25 mV for Ln-NPs-PAA and Ln-NPs-GA, ca. −15 mV for Ln-NPs-LF). Therefore, differences in cellular uptake are less likely to be attributed to variations in surface charge among the nanoparticles. Instead, the varying degrees of colloidal stability may have significantly influenced nanoparticle uptake by cells. Aggregation of NPs results in overall larger sized nanoparticle clusters that may block or interact abnormally with cellular processes. Again, Ln-NPs-PAA as well as Ln-NPs-LF exhibited less colloidal stability compared to Ln-NPs-GA. Consequently, the resultant nanoparticle aggregation can induce the observed cytotoxic effects. Similar findings were reported by Okuda-Shimazaki et al., who demonstrated the importance of the aggregation state and showed that larger aggregates of TiO2 NPs induced higher cytotoxicity than smaller ones.74
In contrast to our findings unveiling cytotoxic effects in case of PAA-capping, Ho et al. demonstrated that ultra-small (ca. 1.5 nm in diameter) bare Gd2O3 NPs were toxic to both NCTC1469 normal and U-87 MG tumour-cell lines, while no toxic behaviour was found towards DU145, NCTC1469, and U-87 MG cell lines upon PAA-capping of these NPs (concentrations up to 500 μM Gd).75 Similarly, Rafique et al. observed by MTT assay that PAA-coated NaYF4:Yb3+,Er3+ UCNPs of ca. 60 nm in size were less cytotoxic compared to their bare counterparts when exposed to HeK293, HeLa, A549, and SCC7 cell lines (particle concentration: 1000μg mL−1, incubation time: 12h).76 Bietar et al. studied the toxicity of relatively large, sub-100 nm sized, silica-coated LiYF4:Yb3+,Tm3+ UCNPs using the Alamar Blue assay. The silica-coated UCNPs exhibited a negative surface charge and were found in the cytoplasm of NIH3T3 and LLC-PK1 cell, yet, not inducing cytotoxicity at concentrations up to (100 mg mL−1).77 Alternatively, coating of LiYF4-based UCNPs with a lipid-bilayer was proposed by Rojas-Gutierrez et al.78 Relatively low uptake was found compared to uncapped UCNPs, along with a minimal cytotoxicity towards human lung cancer cells A549 at concentrations ranging from 12.5 to 100 μg mL−1. These studies differ in several critical factors that influence toxicity, including surface chemistry, the type of toxicity assay and cell lines used, and nanoparticle size and morphology. Each of these variables can significantly impact the observed cytotoxic effects, whereas size and morphology play a major role when comparing these results with our findings. To recall, the spherical Ln-NPs investigated in this study had an average diameter of ca. 10 nm, opposed to significantly smaller Gd2O3 or larger non-spherical NaYF4 as well as LiYF4 Ln-NPs.
Hyperspectral imaging of cells
The uptake of Ln-NPs by cells was further assessed by hyperspectral imaging (HSI). HSI is a mapping technique that allows for the association of spatial information (in x and y dimensions) with spectral features (in z dimension) of a sample over a certain region of interest (ROI).79 Based on ICP-MS data, the estimated number of internalized Ln-NPs per cell was low. However, ICP-MS can only provide an average value over all cells in the investigated sample (Table 1). Conversely, HSI allows for the assessment at a cellular level by localized mapping of the characteristic Er3+ upconversion emission triggered by 980 nm excitation. In agreement with the lowest uptake estimated via ICP-MS, no photoluminescence could be detected from HepG2 cells by HSI (with emission from Ln-NPs potentially uptaken by the cells below the detection limit). In contrast, the higher uptake of Ln-NPs by J774A.1 and U-87 MG cells resulted in a strong green emission signal from not all but multiple cells assessed via HSI (Fig. 4(A) and (B)). This indicated a heterogenous distribution of the Ln-NPs among the treated cells, with some cells incorporating a larger number of Ln-NPs compared to others. The spectral fingerprints extracted from the ROI unambiguously identified the origin of the observed signal as upconversion emission from the Er3+/Yb3+ co-doped Ln-NPs, confirming the uptake of Ln-NPs-GA, -LF, and -PAA by J774A.1 and U-87 MG cells (Fig. 4(C)). Moreover, hyperspectral mapping revealed variations in emission intensity as a function of surface chemistry. In J774A.1 cells, Ln-NPs-LF (Fig. 4(B2) and (C2)) and Ln-NPs-PAA (Fig. 4(B3) and (C3)) displayed brighter Er3+ emission compared to Ln-NPs-GA (Fig. 3(B1) and (C1)). In U-87 MG cells, Ln-NPs-LF showed the highest emission intensity (Fig. 4(B5) and (C5)). These findings are in good agreement with the results of ICP-MS-based uptake studies, i.e., enhanced uptake in the case of Ln-NPs-LF and Ln-NPs-PAA.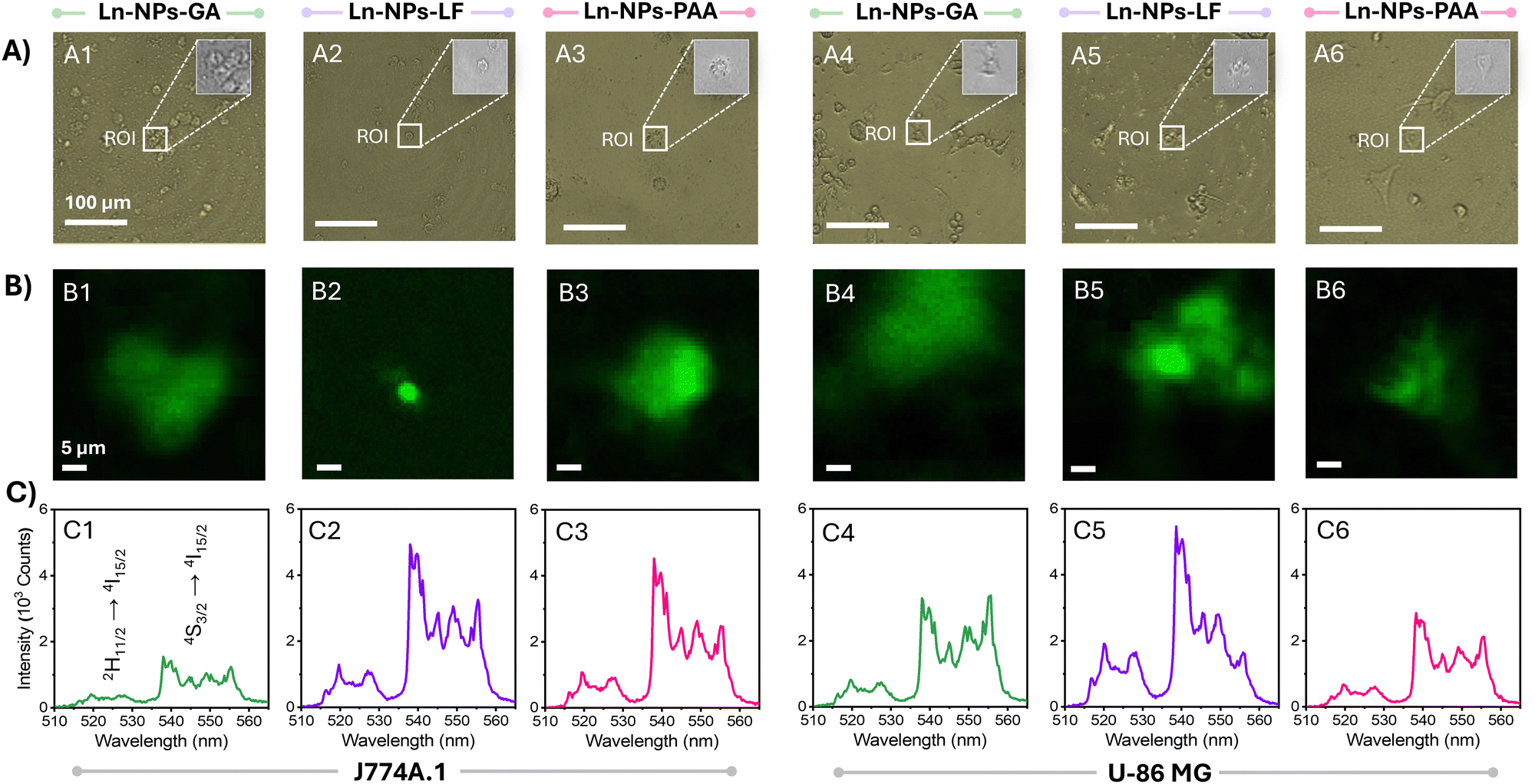 | ||
| Fig. 4 (A) Bright field optical micrographs of (1–3) J774A.1 and (4–6) U-87 MG cells incubated with (1/4) Ln-NPs-GA, (2/5) Ln-NPs-LF, and (3/6) Ln-NPs-PAA taken using a 20× objective. Scale bars are 100 μm. Ln-NP concentration: 500 μg mL−1; incubation time: 24 h. The insets show the magnified region of interest (ROI), marked with a white frame. (B) Corresponding hyperspectral images of the ROI (false colour) showing the spatial distribution of the green Er3+ upconversion emission (spectral range: 510–568 nm) of Ln-NPs colocalized with (1–3) J774A.1 and (4–6) U-86 MG cells. Scale bars are 5 μm. Excitation wavelength: 980 nm; powder density: 4 × 105 W cm−2. (C) Spectral profiles extracted from the corresponding ROI, showing the spectral signature specific to the Er3+ ions doped into the Ln-NPs. No emission was found for the HepG2 cell line. | ||
Zebrafish in vivo studies
Following the promising results of the in vitro assessment of Ln-NPs-GA unveiling relatively good cytocompatibility and nano-bio interactions, we further assessed in vivo impacts of GA-capped Ln-NPs in zebrafish. Herein, the developmental and behavioural effects of Ln-NPs-GA compared to Ln-NPs-LF and Ln-NPs-PAA upon exposure to embryo-larval zebrafish were investigated. Specifically, we examined the impacts on survival, development, and locomotor behaviour following developmental exposure to the Ln-NPs with different surface chemistries.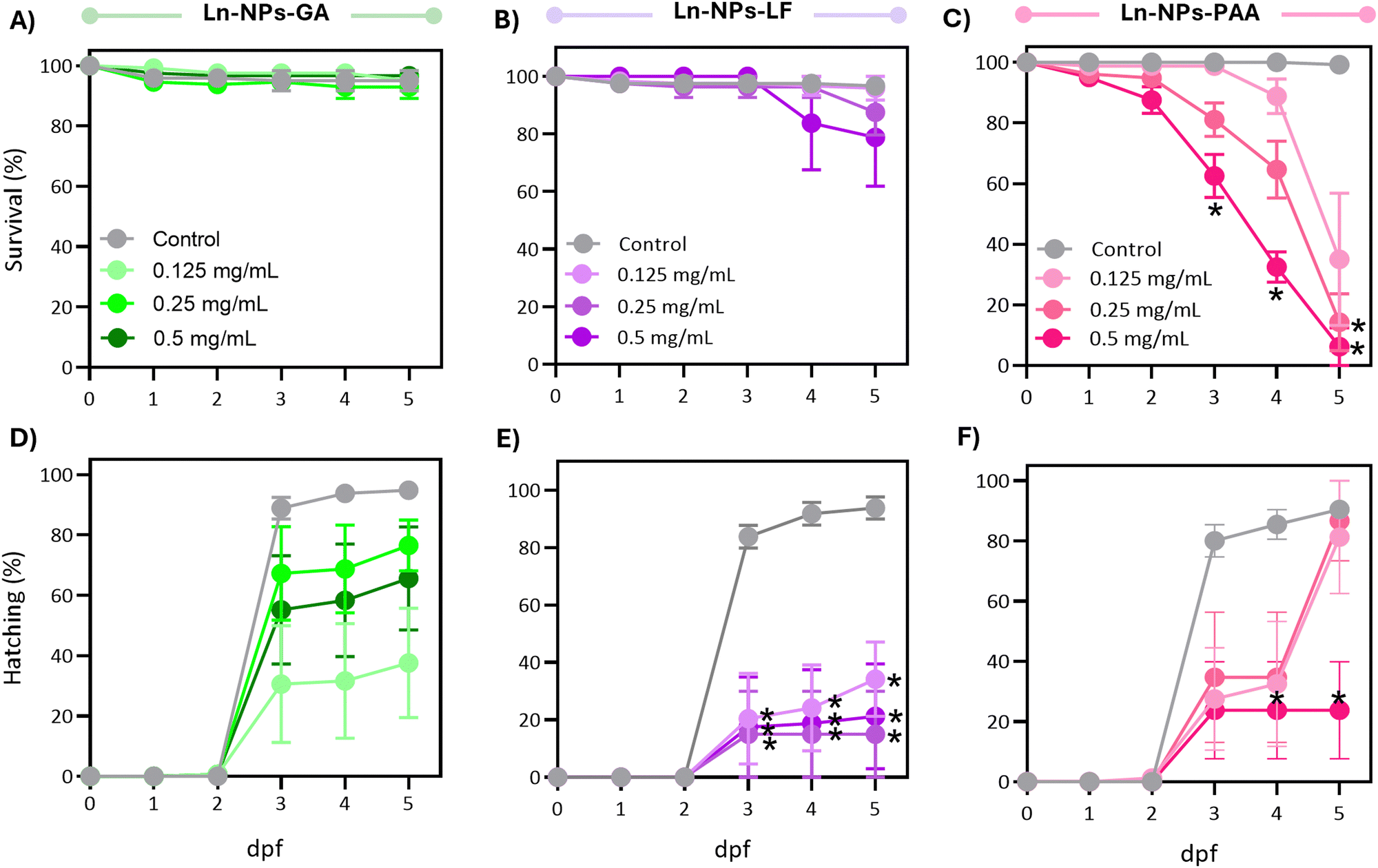 | ||
| Fig. 5 (A)–(C) Survival and (D)–(F) hatching rate over days post fertilization (dpf) in zebrafish embryos statically exposed to (A), (D) Ln-NPs-GA, (B), (E) Ln-NPs-LF, and (C), (F) Ln-NPs-PAA, respectively. Data are expressed as an average percentage ± S.E.M from 5 replicate studies. Within each replicate, n = 16–24 were used per treatment concentration to assess daily survival. Data were analysed using a mixed-ANOVA omnibus test (treatment, time, interaction) with Greenhouse–Geisser correction. Sidak's multiple comparison tests were conducted in cases where the omnibus test was significant. The statistical significance cut-off was set at P < 0.05 in all cases. Asterisks indicate timepoint-specific difference to control groups (P < 0.05). | ||
When considering sublethal effects, Ln-NPs-GA exposure resulted in differences in hatching over time compared to the controls (Fig. 5(D)), which were, however, found to be marginally significant (P > 0.05). In contrast, exposure to Ln-NPs-LF significantly affected hatching over time (Fig. 5(E)), with significant delays in hatching compared to control at all concentrations tested between 3 and 5 dpf (P < 0.05). Exposure to Ln-NPs-PAA induced a significantly lower hatching rate only in embryos exposed to the highest tested concentration of 0.5 mg mLs compared to the control group at 4–5 dpf (P < 0.05) (Fig. 5(F)). The observed decrease in hatching rate upon exposure to Ln-NPs-LF and Ln-NPs-PAA also indicated increased toxicity, likely due to lower colloidal stability, which may lead to chorion canal blockage upon internalization.81 The embryonic stage is crucial for the development of aquatic animals, with the embryonic chorion acting as an efficient barrier against exogenous pollutants.82 Although the pore size of chorion canals (approximately 0.6–0.7 μm) is larger than the size of the NPs, the effect of the chorion on NP transport and subsequent biological toxicity may become complicated when NPs agglomerate or interact with chorion surface proteins, and both passage and retention of nanoparticles of various size and chemical composition have been reported.80 As shown in Fig. 5(C), before hatching (<3 dpf), there is no significant mortality increase in Ln NPs-PAA exposed versus control groups, while survival significantly dropped post-hatching at 3 dpf. Indeed, by 5 dpf, Ln-NPs-PAA resulted in complete mortality, indicating a high level of toxicity associated with this surface modification. Post-hatching, larvae become more vulnerable due to the absence of the chorion's protective barrier, resulting in greater Ln-NP absorption through the skin, gills, mouth, and olfactory epithelium. This increased sensitivity is compounded by higher energetic demands from organogenesis, development, and swimming, making Ln-NPs-PAA more toxic. The observed toxicity of Ln-NPs-PAA following hatching is likely not solely due to the chorion's properties but could also result from increased exposure through oral, olfactory, or skin routes post-hatching. Thus, the toxicity could be a complex interplay of enhanced uptake and the organism's developmental vulnerabilities, rather than a direct consequence of chorion-mediated transport mechanisms alone.
To further investigate potential effects of exposure time and embryo development on the biodistribution of the Ln-NPs, HSI was performed 5 dpf on zebrafish exposed to Ln-NPs-GA and Ln-NPs-LF (Fig. 6). As Ln-NPs-PAA resulted in complete mortality by 5 dpf, the group exposed to those Ln-NPs was excluded from further HSI. Interestingly, different biodistribution was observed for Ln-NPs-GA and Ln-NPs-LF. While both types of Ln-NPs accumulated predominantly in the head and gut regions (Fig. 6(A1-3) and (B1-2)), as already observed at 1 dpf, an additional presence of Ln-NPs-LF was noted in the tail region (Fig. 6(B3)). This difference in biodistribution might be related to (i) Ln-NP surface charge and (ii) dispersion stability and the related formation of agglomerations. It must also be kept in mind that Ln-NP uptake by zebrafish at 5 dpf could occur through the gills, mouth, and olfactory epithelium during ingestion and gas exchange at this early larval stage,84 while uptake at 1 dpf primarily occurs through interactions with the chorion, potentially influenced by factors such as surface charges and the pore size of the chorion. This opens additional pathways for Ln-NP entry into the organism at different time points, whereas the variability in entry routes likely influences the subsequent distribution and accumulation patterns within the zebrafish.
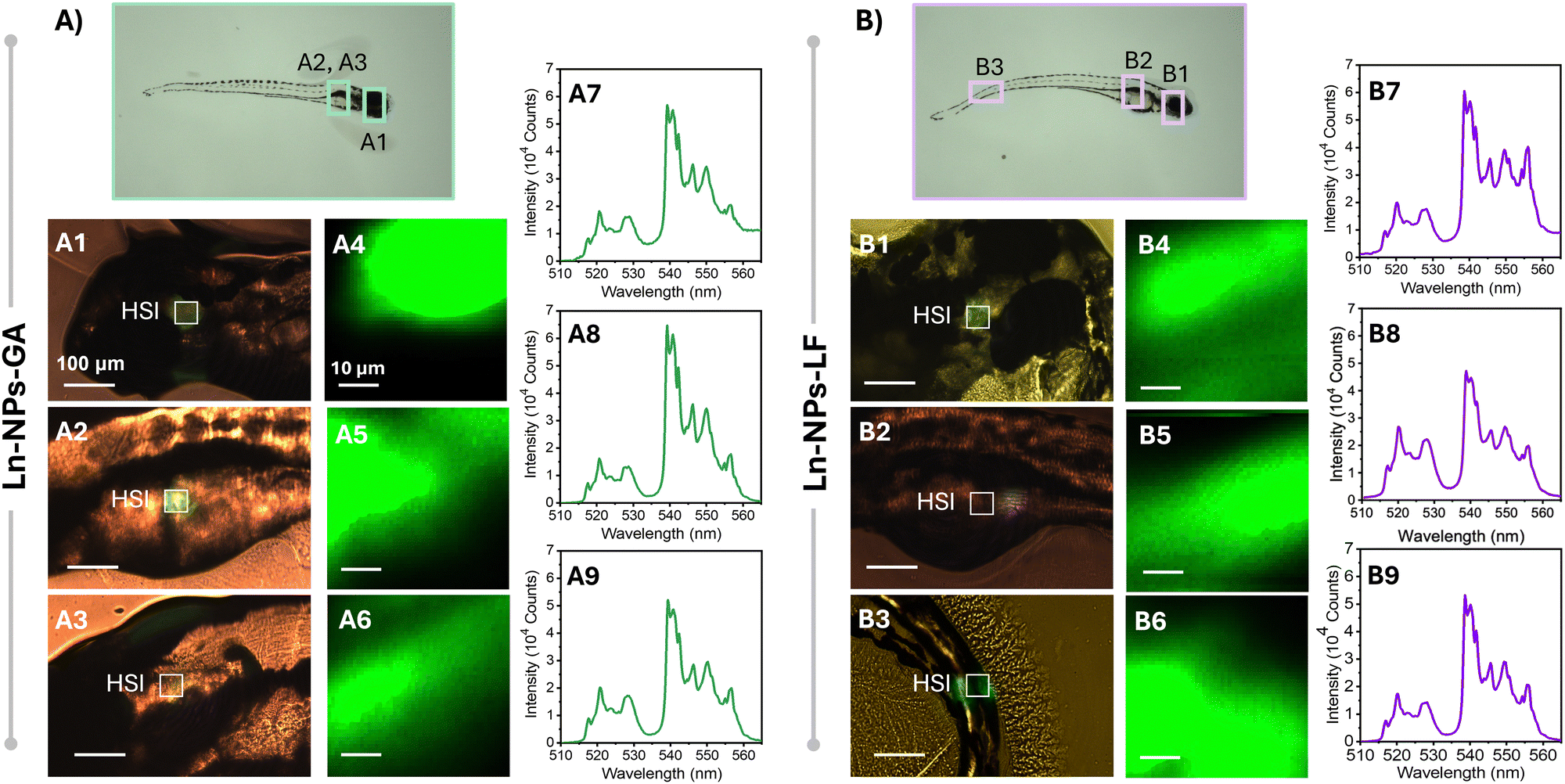 | ||
| Fig. 6 Bight field optical microscopy image of different parts of zebrafish 5 dpf (concentration of Ln-NPs: 0.5 mg mL−1) exposed with (A1-3) Ln-NPs-GA and (B1-3) Ln-NPs-LF, respectively. (A1) Head, (A2-3) gut, as well as (B1) head, (B2) gut, and (B3) tail. Images were taken using a 20× objective. Scale bars are 100 μm. Hyperspectral images recorded at the HSI-marked regions of interest of zebrafish exposed to (A4-6) Ln-NPs-GA and (B4-6) Ln-NPs-LF showing the green emission channel (510–568 nm) of Er3+ doped into the Ln-NPs. Scale bars are 5 μm. (A7-9, B7-9) Spectral profiles of the corresponding areas in (A1-3) and (B1-3), showing the spectral signature specific to the Er3+ dopant ions. | ||
Early work on in vivo zebrafish toxicity of Ln-NPs by Jang et al. showed toxic effects in zebrafish embryos upon exposure to ca. 17 nm sized β-NaYF4:Ce3+,Tb3+ NPs, surface-modified with a combination of lipids.85 Similar to our findings, dose-dependent developmental abnormalities were observed upon incubation with Ln-NPs. Dose-dependent in vivo abnormality was further observed by Wang et al. who injected LaF3:Yb3+,Er3+ Ln-NPs modified with a SiO2 shell (core: 7–10 nm in diameter, shell thickness: ca. 3 nm) through the chorion into the yolk sac.86 While no acute toxicity was observed at a Ln-NP dose of 200 μg mL−1, malformations and other developmental abnormalities, such as shortened larval body length, were noted. Additionally, the hatching time and success rate decreased with increasing concentration of Ln-NPs.
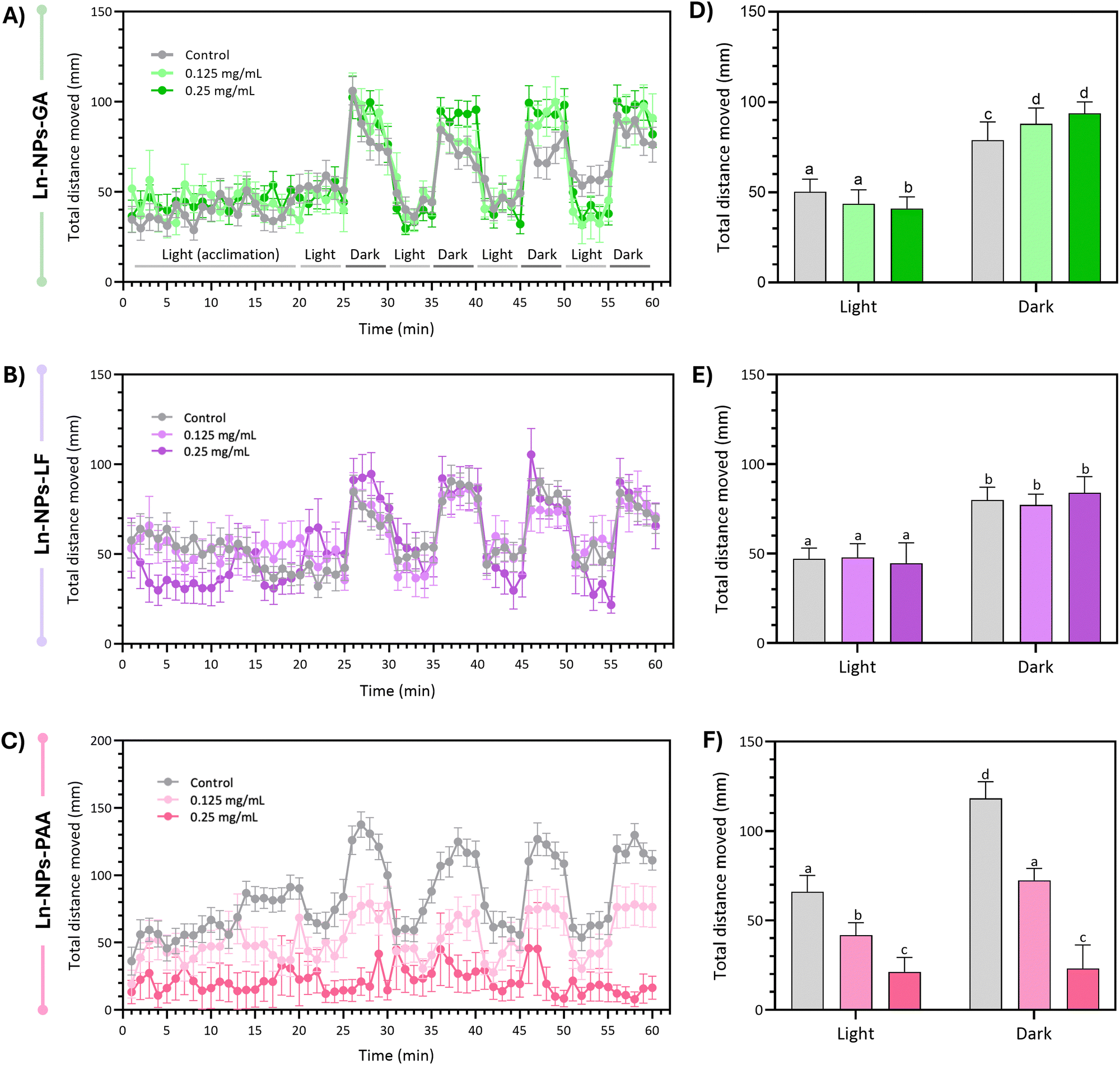 | ||
| Fig. 7 Average large distance locomotion ± S.E.M. by minute and lighting condition of 5 dpf larvae following static exposure to different concentrations of (A) Ln-NPs-GA, (B) Ln-NPs-LF, and (C) Ln-NPs-PAA. Average large-distance locomotion in post-acclimation phase light and dark phases (± S.E.M.) in larvae following static exposure to different concentrations of (D) Ln-NPs-GA, (E) Ln-NPs-LF, and (F) Ln-NPs-PAA. Data were analysed using a two-way ANOVA omnibus test (treatment, lighting condition, interaction). Sidak's multiple comparison tests were conducted in cases where omnibus test was significant. Statistical significance cut-off was set at P < 0.05 in all cases. Different letters, a to d, indicate statistically significant differences between groups (P < 0.05). Groups with the same letter are not significantly different from each other. | ||
A recent study by Khan et al. reports developmental toxicity, negative effects on survival and hatching, as well as locomotory behaviour in zebrafish exposed to ligand-free NaYF4:Er3+,Yb3+ UCNPs of ca. 25–30 nm in size (up to 100 mg L−1, 5 dpf).90 The stark contrast to our results for Ln-NPs-LF might be ascribed to the larger particle size used in the reported study. On the other hand, Yogendra et al. conducted a study exposing zebrafish larvae to similar nanomaterials, namely, Ce3+,Tb3+-doped BaF2 NPs.91 The authors reported no significant effect on the locomotive behaviour at a low NP concentration of 10 mg L−1. And although hypo-locomotive behaviour was observed in developing zebrafish exposed to 150 mg L−1 of NPs, prolonged exposure for 4 days did not completely cease the larval locomotion. Our results further aligned with other studies on different types of nanomaterials, such as polystyrene NPs and carbon nanotubes.92–94
Zebrafish have gained increasing popularity as in vivo model for the assessment of Ln-based nanoparticles for biomedical applications. Examples for imaging applications include PEG-modified upconverting nanorods of various aspect ratios that were microinjected directly into the blood vessel of transgenic zebrafish or microinjected PEGylated UCNPs to monitor neuronal uptake of a transgenic zebrafish with tagged motor neurons in the spinal cord.95,96 Zebrafish have further been used as in vivo model for Ln-NP-based bioanalytical imaging platforms visualizing the distribution of, e.g., H2S, reactive oxygen species (ROS), or β-galactosidase activity.97–99 With the development of UCNPs for optogenetic applications, zebrafish were employed as an animal model to demonstrate in vivo control over cation influx and manipulation of the ion-mediated signalling of apoptosis in living zebrafish, while several studies report the successful imaging of the in vivo distribution of metal cations, such as Zn2+, K+, and Ca2+.100–103 These are excellent examples demonstrating the potential of Ln-NPs for biomedical applications in addition to showcasing the importance of zebrafish as an animal model. In this context, our study provides complementary insights into the fundamental aspects of nano-bio interactions of Ln-NPs with zebrafish, including toxicity, biodistribution, and behaviour.
Conclusions
In this study, capping with Gum Arabic (GA) is proposed as a straightforward and low-cost approach to endow Ln-NPs with dispersibility in biologically relevant media, chemical as well as optical stability, and biocompatibility. Potential toxicity effects of the resultant GA-capped NaGdF4:Er3+,Yb3+/NaGdF4 core/shell Ln-NPs (Ln-NPs-GA) were assessed both in vitro and in vivo. Toxicity assessments using MTT and NR assays across a range of nanoparticle concentrations showed that Ln-NPs-GA were non-toxic towards U-87 MG, HEPG2 cell lines up to the highest tested concentration of 250 μg mL−1. J774A.1 cells exhibited mild toxicity upon incubation with more than 5 μg mL−1 of Ln-NPs-GA. Importantly, irrespective of the cell line, Ln-NPs-GA showed better cytocompatibility than ligand-free and PAA-capped Ln-NPs. Interestingly, our results indicate that surface modification with PAA, a commonly used molecule to render Ln-NPs water-dispersible, indeed comes with the risk of toxicity. These in vitro results were further confirmed by comprehensive in vivo studies with zebrafish. Time- and dose-dependent assessment of zebrafish survival, hatching rate, deformity, and behaviour verified the minimal toxicity of Ln-NPs-GA.The hyperspectral imaging (HSI) technique was employed to visualize the uptake of the Ln-NPs by cells and zebrafish. Herein, the spatially resolved detection of the characteristic spectral features of the Er3+ upconversion emission unveiled colocalization of the Ln-NPs within the cells as well as zebrafish. Comparing GA-capped and ligand-free Ln-NPs, different biodistributions were observed, further demonstrating the role of surface chemistry in nano-bio interactions. ICP unveiled an overall low uptake of the Ln-NPs-GA by the investigated cell lines, pointing towards a lack of non-specific cellular uptake. This, together with the variety of functional groups on GA polymeric chains, can open multiple possibilities to endow Ln-NPs-GA with targeting capabilities, potentially enhancing the precision of their delivery to target cells.
Overall, Ln-NPs-GA have been identified as interesting nanoplatforms to be further explored for various biomedical applications or fundamental in vitro as well as in vivo studies.
Author contributions
H. Mirmajidi: data curation, conceptualization, formal analysis, visualization, writing – original draft. H. Lee, N. Nipu, and J. Thomas: data curation, formal analysis, visualization, writing – original draft. Z. Gajdosechova: data curation. D. Kennedy: data curation, methodology, funding acquisition, writing – review & editing. J. A. Mennigen: methodology, visualization, funding acquisition, supervision, writing – review & editing. E. Hemmer: conceptualization, methodology, funding acquisition, supervision, writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.† Any additional information can be obtained from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support provided by the University of Ottawa, the Canada Foundation for Innovation (CFI, JELF-34800, JELF-148035), the Ontario Research Fund (ORF-34800, ORF-35238), the National Research Council Canada (NRC, QSP 045-1, NBR3-586), and the Natural Sciences and Engineering Research Council of Canada (NSERC, RGPIN-2016-04830, RGPIN-2023-03985, RGPIN-2017-05290). We thank Dr. Emille M. Rodrigues for her contribution and guidance at the early stage of the project. Schemes shown in the ToC figure were made in BioRender (https://www.biorender.com).References
- F. Auzel, Chem. Rev., 2004, 104, 139–173 CrossRef CAS PubMed.
- K. Malhotra, D. Hrovat, B. Kumar, G. Qu, J. V. Houten, R. Ahmed, P. A. E. Piunno, P. T. Gunning and U. J. Krull, ACS Appl. Mater. Interfaces, 2023, 15, 2499–2528 CrossRef CAS.
- A. Puccini, N. Liu and E. Hemmer, Nanoscale, 2024, 16, 10975–10993 RSC.
- G. Y. Chen, H. L. Qju, P. N. Prasad and X. Y. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- Z. Zhang, Q. Han, J. W. Lau and B. Xing, ACS Mater. Lett., 2020, 2, 1516–1531 CrossRef CAS.
- A. A. Ansari, A. K. Parchur and G. Chen, Coord. Chem. Rev., 2022, 457, 214423 CrossRef CAS.
- X. Zhu, H. Zhang and F. Zhang, Acc. Mater. Res., 2023, 4, 536–547 CrossRef CAS.
- Y. Li and G. Chen, Adv. NanoBiomed Res., 2022, 2, 2200092 CrossRef CAS.
- S. Wilhelm, M. Kaiser, C. Würth, J. Heiland, C. Carrillo-Carrion, V. Muhr, O. S. Wolfbeis, W. J. Parak, U. Resch-Genger and T. Hirsch, Nanoscale, 2015, 7, 1403–1410 RSC.
- H. T. T. Duong, Y. Chen, S. A. Tawfik, S. Wen, M. Parviz, O. Shimoni and D. Jin, RSC Adv., 2018, 8, 4842–4849 RSC.
- L. Zhang, D. Jin and M. H. Stenzel, Biomacromolecules, 2021, 22, 3168–3201 CrossRef CAS PubMed.
- E. Hemmer, N. Venkatachalam, H. Hyodo and K. Soga, Adv. Mater. Sci. Eng., 2012, 748098 Search PubMed.
- H. Zhang, X. Wang, R. Jin and Q. Su, Giant, 2022, 12, 100130 CrossRef CAS.
- K. Malhotra, B. Kumar, P. A. E. Piunno and U. J. Krull, ACS Appl. Mater. Interfaces, 2024, 16, 35985–36001 CrossRef CAS.
- L. Zhang, C. Chen, S. S. Tay, S. Wen, C. Cao, M. Biro, D. Jin and M. H. Stenzel, ACS Appl. Mater. Interfaces, 2021, 13, 16142–16154 CrossRef CAS.
- A. E. Guller, A. Nadort, A. N. Generalova, E. V. Khaydukov, A. V. Nechaev, I. A. Kornienko, E. V. Petersen, L. Liang, A. B. Shekhter, Y. Qian, E. M. Goldys and A. V. Zvyagin, ACS Biomater. Sci. Eng., 2018, 4, 3143–3153 CrossRef CAS PubMed.
- S. F. Himmelstoß and T. Hirsch, Part. Part. Syst. Charact., 2019, 36, 1900235 CrossRef.
- V. Bastos, P. Oskoei, E. Andresen, M. I. Saleh, B. Rühle, U. Resch-Genger and H. Oliveira, Sci. Rep., 2022, 12, 3770 CrossRef CAS PubMed.
- N. K. Iversen, S. Frische, K. Thomsen, C. Laustsen, M. Pedersen, P. B. L. Hansen, P. Bie, J. Fresnais, J.-F. Berret, E. Baatrup and T. Wang, Toxicol. Appl. Pharmacol., 2013, 266, 276–288 CrossRef CAS.
- M. F. Torresan and A. Wolosiuk, ACS Appl. Bio Mater., 2021, 4, 1191–1210 CrossRef CAS PubMed.
- M. Hosseinifard, N. Jurga, J. C. Brandmeier, Z. Farka, A. Hlaváček, H. H. Gorris, T. Grzyb and A. Ekner-Grzyb, Chemosphere, 2024, 347, 140629 CrossRef CAS PubMed.
- L. Shi, J. Zhang, M. Zhao, S. Tang, X. Cheng, W. Zhang, W. Li, X. Liu, H. Peng and Q. Wang, Nanoscale, 2021, 13, 10748–10764 RSC.
- H. Wang, Y. Wang, C. Yuan, X. Xu, W. Zhou, Y. Huang, H. Lu, Y. Zheng, G. Luo, J. Shang and M. Sui, npj Vaccines, 2023, 8, 169 CrossRef CAS.
- B. Du, S. P. Han, F. F. Zhao, K. H. Lim, H. W. Xi, X. J. Su, H. C. Yao and J. Zhou, Nanomedicine, 2016, 12, 2071–2080 CrossRef CAS.
- S. Li, S. Cui, D. Yin, Q. Zhu, Y. Ma, Z. Qian and Y. Gu, Nanoscale, 2017, 9, 3912–3924 RSC.
- X. Jiang, J. B. Mietner, C. Harder, R. Komban, S. Chen, C. Strelow, U. Sazama, M. Fröba, C. Gimmler, P. Müller-Buschbaum, S. V. Roth and J. R. G. Navarro, ACS Appl. Mater. Interfaces, 2023, 15, 5687–5700 CrossRef CAS PubMed.
- S. D. Topel, S. Balcioglu, B. Ateş, M. Asilturk, Ö. Topel and M. B. Ericson, Mater. Today Commun., 2021, 26, 101829 CrossRef CAS.
- G. Chen, Y. Wu, K. Jin, H. Lu, M. Tao, T. Wang, J. Zhang, X. Zhu, J. Liu and Y. Zhang, ACS Appl. Bio Mater., 2021, 4, 7542–7553 CrossRef CAS.
- C. Cao, C. Hong, Y. Li, G. Li and G. Jiang, Z. Anorg. Allg. Chem., 2020, 646, 1607–1610 CrossRef CAS.
- D. Przybylska and T. Grzyb, J. Alloys Compd., 2023, 933, 167708 CrossRef CAS.
- E. M. Rodrigues, N. D. Calvert, J. C. Crawford, N. Liu, A. J. Shuhendler and E. Hemmer, Small, 2022, 18, 2107130 CrossRef CAS.
- A. Hassani, M. M. S. Azarian, W. N. Ibrahim and S. A. Hussain, Sci. Rep., 2020, 10, 17808 CrossRef CAS PubMed.
- U. S. G. Center for Food Safety and Applied Nutrition (CFSAN), FDA Grants Citizen Petition on Acacia (Gum Arabic) as a Dietary Fiber, https://www.fda.gov/food/cfsan-constituent-updates/fda-grants-citizen-petition-acacia-gum-arabic-dietary-fiber, (accessed 18/07/2024).
- M. E. Vuillemin, F. Michaux, A. A. Adam, M. Linder, L. Muniglia and J. Jasniewski, Food Hydrocolloids, 2020, 107, 105919 CrossRef CAS.
- P. Shao, J. Feng, P. Sun, N. Xiang, B. Lu and D. Qiu, Int. Food Res., 2020, 137, 109376 CrossRef CAS PubMed.
- G. Ciofani, S. Del Turco, A. Rocca, G. de Vito, V. Cappello, M. Yamaguchi, X. Li, B. Mazzolai, G. Basta, M. Gemmi, V. Piazza, D. Golberg and V. Mattoli, Nanomedicine, 2014, 9, 773–788 CrossRef CAS.
- V. Kattumuri, K. Katti, S. Bhaskaran, E. J. Boote, S. W. Casteel, G. M. Fent, D. J. Robertson, M. Chandrasekhar, R. Kannan and K. V. Katti, Small, 2007, 3, 333–341 CrossRef CAS PubMed.
- M. Eskandari-Nojehdehi, H. Jafarizadeh-Malmiri and A. Jafarizad, Z. Phys. Chem., 2018, 232, 325–343 CrossRef CAS.
- A. M. Gamal-Eldeen, D. Moustafa, S. M. El-Daly, E. A. El-Hussieny, S. Saleh, M. Khoobchandani, K. L. Bacon, S. Gupta, K. Katti, R. Shukla and K. V. Katti, J. Photochem. Photobiol., B, 2016, 163, 47–56 CrossRef CAS PubMed.
- P. Lodeiro, E. P. Achterberg, J. Pampín, A. Affatati and M. S. El-Shahawi, Sci. Total Environ., 2016, 539, 7–16 CrossRef CAS.
- M. B. Elamin, S. M. A. Ali, A. Chrouda, L. M. Alhaidari, R. M. Alrouqi, A. T. Al-Shammari, A. A. Alsuqiani and N. Jaffrezic-Renault, Polym. Adv. Technol., 2023, 34, 3407–3414 CrossRef CAS.
- C. Tan, J. Xie, X. Zhang, J. Cai and S. Xia, Food Hydrocolloids, 2016, 57, 236–245 CrossRef CAS.
- L. Zhang, F. Yu, A. J. Cole, B. Chertok, A. E. David, J. Wang and V. C. Yang, AAPS J., 2009, 11, 693–699 CrossRef CAS PubMed.
- R. H. Huseen, A. A. Taha, I. Q. Ali, O. M. Abdulhusein and S. M. H. Al-Jawad, Mod. Phys. Lett. B, 2021, 35, 2150411 CrossRef CAS.
- H. Ribeiro de Barros, M. B. Cardoso, C. Camargo de Oliveira, C. R. Cavichiolo Franco, D. de Lima Belan, M. Vidotti and I. C. Riegel-Vidotti, RSC Adv., 2016, 6, 9411–9420 RSC.
- M. F. Horst, D. F. Coral, M. B. Fernández van Raap, M. Alvarez and V. Lassalle, Mater. Sci. Eng., C, 2017, 74, 443–450 CrossRef CAS PubMed.
- C. Homann, E. M. Rodrigues, P. Orsini, K. Savard, C.-B. Togola, M.-M. de Denus-Baillargeon, M. Massabki and E. Hemmer, Opt. Mater.: X, 2024, 21, 100290 CAS.
- E. Hemmer, F. Vetrone and K. Soga, MRS Bull., 2014, 39, 960–964 CrossRef CAS.
- M. I. Saleh, B. Rühle, S. Wang, J. Radnik, Y. You and U. Resch-Genger, Sci. Rep., 2020, 10, 19318 CrossRef CAS PubMed.
- Z. Gerelkhuu, Y. I. Lee and T. H. Yoon, Nanomaterials, 2022, 12, 3470 CrossRef CAS.
- L. Liang, A. V. Everest-Dass, A. B. Kostyuk, Z. Khabir, R. Zhang, D. B. Trushina and A. V. Zvyagin, Cells, 2022, 11, 3644 CrossRef CAS PubMed.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- A. Gnach, T. Lipinski, A. Bednarkiewicz, J. Rybka and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1561–1584 RSC.
- P. A. Rojas-Gutierrez, S. Bhuckory, C. Mingoes, N. Hildebrandt, C. DeWolf and J. A. Capobianco, ACS Appl. Nano Mater., 2018, 1, 5345–5354 CrossRef CAS.
- E. Hemmer, T. Yamano, H. Kishimoto, N. Venkatachalam, H. Hyodo and K. Soga, Acta Biomater., 2013, 9, 4734–4743 CrossRef CAS.
- D. M. Samhadaneh, G. A. Mandl, Z. Han, M. Mahjoob, S. C. Weber, M. Tuznik, D. A. Rudko, J. A. Capobianco and U. Stochaj, ACS Appl. Bio Mater., 2020, 3, 4358–4369 CrossRef CAS PubMed.
- H. Oliveira, A. Bednarkiewicz, A. Falk, E. Fröhlich, D. Lisjak, A. Prina-Mello, S. Resch, C. Schimpel, I. V. Vrček, E. Wysokińska and H. H. Gorris, Adv. Healthcare Mater., 2019, 8, 1801233 CrossRef CAS PubMed.
- M. Adhish and I. Manjubala, Heliyon, 2023, 9, e14557 CrossRef PubMed.
- I. Halimi, E. M. Rodrigues, S. L. Maurizio, H.-Q. T. Sun, M. Grewal, E. M. Boase, N. Liu, R. Marin and E. Hemmer, J. Mater. Chem. C, 2019, 7, 15364–15374 RSC.
- N. Liu, N. Gobeil, P. Evers, I. Gessner, E. M. Rodrigues and E. Hemmer, Dalton Trans., 2020, 49, 16204–16216 RSC.
- H. Zhang, Y. Li, I. A. Ivanov, Y. Qu, Y. Huang and X. Duan, Angew. Chem., Int. Ed., 2010, 49, 2865–2868 CrossRef CAS.
- T. Mirmajidi, F. Chogan, A. H. Rezayan and A. M. Sharifi, Int. J. Pharm., 2021, 596, 120213 CrossRef CAS.
- G. Repetto, A. del Peso and J. L. Zurita, Nat. Protoc., 2008, 3, 1125–1131 CrossRef CAS PubMed.
- M. Kalyn, H. Lee, J. Curry, W. Tu, M. Ekker and J. A. Mennigen, Environ. Pollut., 2023, 326, 121479 CrossRef CAS PubMed.
- L. Shang, K. Nienhaus and G. U. Nienhaus, J. Nanobiotechnol., 2014, 12, 5 CrossRef.
- E. C. Cho, Q. Zhang and Y. Xia, Nat. Nanotechnol., 2011, 6, 385–391 CrossRef CAS PubMed.
- K. Cissé, D. Gassama, A. A. Diagne and M. Badji, Am. J. Mater. Sci. Eng., 2020, 8, 17–21 Search PubMed.
- F. Weinbreck, R. H. Tromp and C. G. de Kruif, Biomacromolecules, 2004, 5, 1437–1445 CrossRef CAS.
- T. L. Moore, L. Rodriguez-Lorenzo, V. Hirsch, S. Balog, D. Urban, C. Jud, B. Rothen-Rutishauser, M. Lattuada and A. Petri-Fink, Chem. Soc. Rev., 2015, 44, 6287–6305 RSC.
- P. A. Williams, in Encyclopedia of Food and Health, ed. B. Caballero, P. M. Finglas and F. Toldrá, Academic Press, Oxford, 2016, pp. 283–289 DOI:10.1016/B978-0-12-384947-2.00364-0.
- A. M. Chockalingam, H. K. R. R. Babu, R. Chittor and J. P. Tiwari, J. Nanobiotechnol., 2010, 8, 30 CrossRef CAS PubMed.
- R. Abbasi, G. Shineh, M. Mobaraki, S. Doughty and L. Tayebi, J. Nanopart. Res., 2023, 25, 43 CrossRef CAS PubMed.
- M. Claudia, Ö. Kristin, O. Jennifer, R. Eva and F. Eleonore, Toxicology, 2017, 378, 25–36 CrossRef CAS PubMed.
- J. Okuda-Shimazaki, S. Takaku, K. Kanehira, S. Sonezaki and A. Taniguchi, Int. J. Mol. Sci., 2010, 11, 2383–2392 CrossRef CAS PubMed.
- S. L. Ho, H. Cha, I. T. Oh, K.-H. Jung, M. H. Kim, Y. J. Lee, X. Miao, T. Tegafaw, M. Y. Ahmad, K. S. Chae, Y. Chang and G. H. Lee, RSC Adv., 2018, 8, 12653–12665 RSC.
- R. Rafique, S. H. Baek, C. Y. Park, S.-J. Chang, A. R. Gul, S. Ha, T. P. Nguyen, H. Oh, S. Ham, M. Arshad, H. Lee and T. J. Park, Sci. Rep., 2018, 8, 17101 CrossRef PubMed.
- K. Bietar, S. Chu, G. Mandl, E. Zhang, N. Chabaytah, R. Sabelli, J. A. Capobianco and U. Stochaj, RSC Adv., 2024, 14, 8695–8708 RSC.
- P. A. Rojas-Gutierrez, D. Bekah, J. Seuntjens, C. DeWolf and J. A. Capobianco, ACS Appl. Bio Mater., 2019, 2, 4527–4536 CrossRef CAS.
- E. M. Rodrigues and E. Hemmer, Anal. Bioanal. Chem., 2022, 414, 4269–4279 CrossRef CAS.
- S. Böhme, M. Baccaro, M. Schmidt, A. Potthoff, H.-J. Stärk, T. Reemtsma and D. Kühnel, Environ. Sci.: Nano, 2017, 4, 1005–1015 RSC.
- W. Bai, Z. Zhang, W. Tian, X. He, Y. Ma, Y. Zhao and Z. Chai, J. Nanopart. Res., 2010, 12, 1645–1654 CrossRef CAS.
- Z.-Y. Chen, N.-J. Li, F.-Y. Cheng, J.-F. Hsueh, C.-C. Huang, F.-I. Lu, T.-F. Fu, S.-J. Yan, Y.-H. Lee and Y.-J. Wang, Int. J. Mol. Sci., 2020, 21, 2864 CrossRef CAS PubMed.
- B. Bauer, A. Mally and D. Liedtke, Int. J. Mol. Sci., 2021, 22, 13417 CrossRef CAS PubMed.
- Y. Lu, Y. Zhang, Y. Deng, W. Jiang, Y. Zhao, J. Geng, L. Ding and H. Ren, Environ. Sci. Technol., 2016, 50, 4054–4060 CrossRef CAS PubMed.
- G. H. Jang, M. P. Hwang, S. Y. Kim, H. S. Jang and K. H. Lee, Biomater., 2014, 35, 440–449 CrossRef CAS PubMed.
- K. Wang, J. Ma, M. He, G. Gao, H. Xu, J. Sang, Y. Wang, B. Zhao and D. Cui, Theranostics, 2013, 3, 258–266 CrossRef CAS.
- R. Chackal, T. Eng, E. M. Rodrigues, S. Matthews, F. Pagé-Lariviére, S. Avery-Gomm, E. G. Xu, N. Tufenkji, E. Hemmer and J. A. Mennigen, Front. Pharmacol., 2022, 13, 822111 CrossRef CAS PubMed.
- W. Tu, R. Martínez, L. Navarro-Martin, D. J. Kostyniuk, C. Hum, J. Huang, M. Deng, Y. Jin, H. M. Chan and J. A. Mennigen, Environ. Sci. Technol., 2019, 53, 13427–13439 CrossRef CAS.
- R. Martínez, W. Tu, T. Eng, M. Allaire-Leung, B. Piña, L. Navarro-Martín and J. A. Mennigen, Chemosphere, 2020, 256, 127080 CrossRef.
- I. A. Khan, T. Yu, Y. Li, C. Hu, X. Zhao, Q. Wei, Y. Zhong, M. Yang, J. Liu and Z. Chen, Ecotoxicol. Environ. Saf., 2024, 284, 116905 CrossRef CAS PubMed.
- Y. N. Chouryal, S. Nema, R. K. Sharma, H. L. Kewat, A. Pandey, P. Ghosh and Y. Bhargava, Biomater. Sci., 2020, 8, 6730–6740 RSC.
- J. A. Pitt, J. S. Kozal, N. Jayasundara, A. Massarsky, R. Trevisan, N. Geitner, M. Wiesner, E. D. Levin and R. T. Di Giulio, Aquat. Toxicol., 2018, 194, 185–194 CrossRef CAS.
- A. F. Pedersen, D. N. Meyer, A.-M. V. Petriv, A. L. Soto, J. N. Shields, C. Akemann, B. B. Baker, W.-L. Tsou, Y. Zhang and T. R. Baker, Environ. Pollut., 2020, 266, 115090 CrossRef CAS.
- J. Cheng, E. Flahaut and S. H. Cheng, Environ. Toxicol. Chem., 2007, 26, 708–716 CrossRef CAS PubMed.
- L. Fu, B. Shi, S. Wen, M. Morsch, G. Wang, Z. Zhou, C. Mi, M. Sadraeian, G. Lin, Y. Lu, D. Jin and R. Chung, Acta Biomater., 2022, 147, 403–413 CrossRef CAS PubMed.
- L. Fu, M. Morsch, B. Shi, G. Wang, A. Lee, R. Radford, Y. Lu, D. Jin and R. Chung, Nanoscale, 2017, 9, 13683–13692 RSC.
- F. Wang, C. Zhang, X. Qu, S. Cheng and Y. Xian, Biosens. Bioelectron., 2019, 126, 96–101 CrossRef CAS.
- F. Wang, X. Qu, D. Liu, C. Ding, C. Zhang and Y. Xian, Sens. Actuators, B, 2018, 274, 180–187 CrossRef CAS.
- D. Jiang, Q. Tan, Y. Shen, M. Ye, J. Li and Y. Zhou, Spectrochim. Acta, Part A, 2023, 292, 122411 CrossRef CAS PubMed.
- X. Ai, L. Lyu, Y. Zhang, Y. Tang, J. Mu, F. Liu, Y. Zhou, Z. Zuo, G. Liu and B. Xing, Angew. Chem., Int. Ed., 2017, 56, 3031–3035 CrossRef CAS PubMed.
- J. Peng, W. Xu, C. L. Teoh, S. Han, B. Kim, A. Samanta, J. C. Er, L. Wang, L. Yuan, X. Liu and Y.-T. Chang, J. Am. Chem. Soc., 2015, 137, 2336–2342 CrossRef CAS PubMed.
- Z. Yang, K. Y. Loh, Y.-T. Chu, R. Feng, N. S. R. Satyavolu, M. Xiong, S. M. Nakamata Huynh, K. Hwang, L. Li, H. Xing, X. Zhang, Y. R. Chemla, M. Gruebele and Y. Lu, J. Am. Chem. Soc., 2018, 140, 17656–17665 CrossRef CAS PubMed.
- J. Liu, L. Pan, C. Shang, B. Lu, R. Wu, Y. Feng, W. Chen, R. Zhang, J. Bu, Z. Xiong, W. Bu, J. Du and J. Shi, Sci. Adv., 2020, 6, eaax9757 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, including ligand removal (Ln-NPs-LF) and PAA-capping (Ln-NPs-PAA); additional structural analysis by TEM, XRD, FT-IR, and TGA; additional colloidal and optical stability of Ln-NPs-LF and Ln-NPs-PAA; NIR-emission spectra of the Ln-NPs; estimation of cellular and zebrafish uptake; hyperspectral imaging of zebrafish; zebrafish deformity. See DOI: https://doi.org/10.1039/d4tb01579g |
| This journal is © The Royal Society of Chemistry 2025 |