
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular hydrogels derived from 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h2_char_2009.gif) :1-[α/aza]-pseudopeptides: design, structural analysis and self-assembly in solution, solid, and gel states†
:1-[α/aza]-pseudopeptides: design, structural analysis and self-assembly in solution, solid, and gel states†
Mohamed I. A.
Ibrahim‡
 *abc,
Jacques
Bodiguel
a,
Guillaume
Pickaert
a,
Loïc
Stefan
a,
Koichi
Matsuo
b and
Marie-Christine
Averlant-Petit‡
*a
*abc,
Jacques
Bodiguel
a,
Guillaume
Pickaert
a,
Loïc
Stefan
a,
Koichi
Matsuo
b and
Marie-Christine
Averlant-Petit‡
*a
aUniversité de Lorraine, CNRS, LCPM, F-54000 Nancy, France. E-mail: marie.averlant@univ-lorraine.fr
bResearch Institute for Synchrotron Radiation Science, HiSOR, Hiroshima University, Higashi-Hiroshima, 739-0046, Japan
cNational Institute of Oceanography and Fisheries, NIOF, Cairo, Egypt
First published on 7th May 2025
Abstract
Synthesis of innovating supramolecular hydrogels based on low molecular weight gelators (LMWGs < 2000 Da) has gained significant interest within the scientific research community due to their wide-ranging potential applications. Most supramolecular hydrogels, induced by a self-assembly process, are stabilized via non-covalent interactions. Recently, our research group has investigated the gelation properties of new low molecular weight hydrogelators (LWMHGs) based on Fmoc-functionalized 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-[α/aza]-azapeptides. This study presents the synthesis and characterization of two Fmoc-N-functionalized 2:1-[α/aza]-trimers: Fmoc-FazaFA (1) and Fmoc-D-FazaFA (2). Both molecules have demonstrated their capability to form hydrogels at pH 7.0 and 10.0, and the analysis of their monomeric states in solution using spectroscopic techniques (including NMR and FTIR) and in the crystal state (using X-ray diffraction) revealed a β-turn conformation. Molecular dynamics simulations were used for 3D model determination. Additionally, the structure of both molecules in the gel state was examined by circular dichroism (CD) experiments. The findings have confirmed that intermolecular hydrogen bonding and π-stacking between the aromatic moieties are the primary driving forces behind the self-assembly and hydrogelation phenomena. Rheology measurements validated the solid-like behavior of both hydrogels (G′ > G′′ within the studied frequency range). Finally, the self-assembly is emphasized through the fibrous structure observed by SEM imaging.
:1-[α/aza]-azapeptides. This study presents the synthesis and characterization of two Fmoc-N-functionalized 2:1-[α/aza]-trimers: Fmoc-FazaFA (1) and Fmoc-D-FazaFA (2). Both molecules have demonstrated their capability to form hydrogels at pH 7.0 and 10.0, and the analysis of their monomeric states in solution using spectroscopic techniques (including NMR and FTIR) and in the crystal state (using X-ray diffraction) revealed a β-turn conformation. Molecular dynamics simulations were used for 3D model determination. Additionally, the structure of both molecules in the gel state was examined by circular dichroism (CD) experiments. The findings have confirmed that intermolecular hydrogen bonding and π-stacking between the aromatic moieties are the primary driving forces behind the self-assembly and hydrogelation phenomena. Rheology measurements validated the solid-like behavior of both hydrogels (G′ > G′′ within the studied frequency range). Finally, the self-assembly is emphasized through the fibrous structure observed by SEM imaging.
Introduction
The demand for hydrogels has increased rapidly in the last few decades, particularly hydrogels derived from small organic molecules which have gained extensive interest in supramolecular chemistry. Synthesis and design of low molecular weight organic molecules with the ability to gel aqueous solvents have become the goal of many research groups due to their possible promising applications in tissue engineering, drug release, and environmental remediation for example.1–5 The hydrogelation phenomenon reflects the equilibrium state between the tendency of the molecules to dissolve or to aggregate and depends on the hydrophobic/hydrophilic balance of the gelator molecules. Thus, they self-assemble in the aqueous phase to form a three-dimensional network that captures aqueous molecules leading to hydrogels. The self-assembly of small molecules leads to physical gels stabilized through noncovalent low energy interactions (including hydrogen bonds, van der Waals interactions, electrostatic forces, π–π and hydrophobic interactions, etc.).6Supramolecular hydrogels have been extensively studied, with diverse gelator structures categorized into small organic molecules,7–19 inorganic–organic hybrids,20–23 nucleobase derivatives,24–28 saccharides,29–33 and peptide-based systems. Among these, peptide-based low-molecular-weight hydrogelators (LMWHGs) are particularly promising due to their biocompatibility, tunability, and responsiveness to external stimuli.34–37 Various types of LMWHGs based on amino acids and peptides have emerged over the past few decades. Some LMWHGs are formulated with ionic amino acids, while others use non-polar amino acids, functionalized amino acids with aromatic groups like ferrocene, fluorene, naphthalene, and pyrene, or amino-acid-based bolaamphiphiles.38–47 Additionally, a significant number of LMWHGs have been designed using short linear or cyclic peptide sequences.19,48–56 Further research into peptide-based LMWHGs has resulted in peptide derivatives featuring capped N- and C-terminals, functionalized peptides with aromatic or photoresponsive groups, and peptide bolaamphiphiles, among others.57–70 While previous studies have explored various peptide-based LMWHGs, challenges remain in achieving enhanced mechanical stability, controlled self-assembly, and resistance to enzymatic degradation.
Our group has, for decades, been interested in the rational design of synthesis and conformational analysis of pseudopeptides and peptide mimics, particularly, the pseudopeptidic bis-nitrogen compounds such as N-aminopeptides,71,72 and hydrazinopeptides.73,74 Our interest has extended to synthesize a new family of bis-nitrogen compounds called azapeptides; here at least one of the amino acids’ α-carbon is replaced with a nitrogen atom in the peptide sequence.75–77 Various features of azapeptides have been highlighted including their ability to adopt stable conformation (mainly β-turn) in solution and crystal states and their ability to self-assemble and form stable organogels.78 More recently azapeptides containing lysine residues were used as bioadditives and effectively improved the membrane performance for CO2 separation.75
Since then, our group has investigated the capacity of the azapeptides to form hydrogels. In this regard, the current research aims to design and characterize a novel class of LMWHGs based on 2:1-[α/aza]-azapeptides, representing the first reported formulation of hydrogels from this family. By developing a straightforward strategy to synthesize Fmoc-2:1-[α/aza]-trimers from their Boc analogues, this work explores their self-assembly behavior, mechanical properties, and structural organization. The unique β-turn conformation of azapeptides, stabilized by hydrogen bonding, is expected to enhance hydrogel stability and enzymatic resistance, making them promising candidates for future biomedical applications such as tissue engineering and cell culture.
Results and discussion
In this study, a simple and an efficient strategy was proposed through which two Fmoc-azapeptides from the corresponding Boc-analogues belong to the family 2:1-[α/aza]-azapeptide series and were successfully synthesized, with yields of 76% for compound 1 and 74% for compound 2 (Scheme 1). The prepared Fmoc-members could effectively self-assemble in a wide range of pH leading to hydrogels, in contrast to their Boc-analogues. The data obtained from the different spectroscopic techniques were analyzed in order to study the structure, conformation and characteristic features of the two Fmoc-molecules in solid, solution and gel states. Interpretation of these results will be detailed in the next sections.
 | ||
| Scheme 1 Stepwise synthesis of 1 and 2 from the corresponding Boc-precursors. | ||
Structure and conformation of molecules in solution
The conformations of both molecules were investigated by conducting 2D NMR experiments at the same samples’ concentrations (Fig. S5 and S6, ESI†). Fig. 1 summarizes ROE correlations observed in 2D-ROESY, reflecting the spatial proximity between involved atoms. Strong ROE correlations between CαH (Phe or D-Phe) and NH (azaPhe) and moderate correlation between the NH (azaPhe) and NH (Ala) indicate that compounds 1 and 2 adopt βII-turn conformation.79,80 Based on the structural studies of acyclic series, we can suggest that the change in absolute configuration of Cα(Phe) from (S) in 1 to (R) in 2 led to a change from β-conformation to β′-conformation, subsequently compound 2 adopts βII′-turn conformation.77 This finding was confirmed by the X-ray study of compound 2 (see below). Furthermore, compounds 1 and 2 revealed the presence of a weak correlation between NH(Phe or D-Phe) and NH(azaPhe), suggesting that molecules 1 and 2 might adopt βI- and βI′-turn conformation, respectively.79,80
 | ||
| Fig. 1 ROE correlations of β-turn in compound 1 (left) and β′-turn conformation in compound 2 (right); (4.0 mmol L−1, DMSO-d6). | ||
In order to determine the NH protons involved in hydrogen bonding, 1D-NMR experiments in mixtures of polar/nonpolar solvents were performed. Fig. 2 shows that the chemical shifts of the NH protons of Phe, D-Phe and azaPhe in both compounds are highly sensitive to the addition of DMSO-d6, with Δδ values of 2.04, and 2.44 ppm for molecule 1, and 2.21 and 2.27 ppm for molecule 2. In contrast, the NH protons of the Ala residues are weakly affected, showing Δδ values of 0.05 and 0.049 for 1 and 2, respectively. Regarding the OH protons in both compounds, it was difficult to follow the variations of their chemical shift due to the broadening of the peaks (Fig. S7, ESI†). These results reveal that the NH proton of Ala may be involved in the intramolecular hydrogen bond.81 Interestingly, the aromatic protons suffer the deshielding effect when increasing DMSO-d6 percentage, indicating that the conformation in chloroform might be stabilized by π-stacking between the aromatic moieties (Fig. S8, ESI†).
 | ||
| Fig. 2 Chemical shift-variations (δ) of NH protons for: (a) 1 and (b) 2 as a function of % [CDCl3/DMSO-d6] mixtures (300 MHz, 4 mmol L−1). | ||
NMR experimental observations suggest that both molecules 1 and 2 adopted β-turn conformation in solution stabilized by the intramolecular hydrogen bond [NH(Ala)–CO(Fmoc)] closing a 10-atom pseudocycle.
 | ||
| Fig. 3 FT-IR spectra of the NH stretching region for 1 (a) and 2 (b), and the CO stretching region for 1 (c) and 2 (d) at dilute concentrations in CDCl3. | ||
FTIR spectra of compounds 1 and 2 clearly show that NH and OH groups are partially involved in hydrogen bonding. Indeed, in the stretching vibration area of those two functions (between 3200 and 3500 cm−1), the spectra exhibit rather sharp bands over 3400 cm−1 (3435 cm−1 for 1 and 3432 cm−1 for 2) typical of free groups. Below 3400 cm−1, broad signals, with multiple shoulders, are attributed to the bonded NH groups. In the case of compound 1 (Fig. 3a), the main band located at 3369 cm−1 bears at least one shoulder (3331 cm−1), whereas compound 2 exhibits at least three bands corresponding to bonded groups (3368, 3349 and 3275 cm−1). In addition, regarding the intensity ratio between free and bonded OH/NH, compound 2 seems to have more hydrogen-bonded functions compared to compound 1 (Fig. 3b).
The experimental infrared spectra (black line Fig. 3c and d), the CO stretching vibrations of compounds 1 and 2 show in both cases four main visible bands. Deconvolution of these spectra gives rise to seven bands (blue lines in Fig. 3c and d) found using second derivative method, along with the fitted spectra (red line in Fig. 3c and d). Assignments were achieved by studying FTIR spectra of precursors or derivatives of compounds 1 and 2 such as Boc-azaPhe-Ala-OMe, Boc-Phe-azaPhe-Ala-OMe or Acetyl-Phe-azaPhe-Ala-OMe, (Fig. S9, ESI†). Thus, the band found at 1762 cm−1 for both compounds was assigned to free carbonyl of the carboxylic acid group. Dimerization of carboxylic acid units can often be observed and give rise to a vibrator located at 1744 and 1738 cm−1 for compound 1 and 2, respectively.82 Frequencies of free urethane CO of the Fmoc group are found at 1717 cm−1 for 1 and 1719 cm−1 for 2, whereas bonded vibration's frequencies are both shifted to 1702 cm−1. It is noteworthy to notice, when looking at intensity ratios of free and bonded states bands, that the urethane carbonyl is more involved in hydrogen bonding in the case of compound 2 than in compound 1. This result is consistent with previous observations made on NH/OH stretching vibrations. Considering CO Phe and CO azaPhe vibrators, the corresponding bands are overlapped at 1675 and 1672 cm−1 for compounds 1 and 2, respectively, while two separate bands are found for the H-bonded carbonyls vibrators at 1651/1633 cm−1 and 1650/1632 cm−1 for compounds 1 and 2 respectively and 1614 cm−1 for bonded AzaPhe in 2. Unfortunately, we were unable to assign unambiguously those vibrators. The presence of bands at 1675/1672 cm−1 in compounds (1) and (2), respectively, along with 1651/1633 cm−1 (1) and 1650/1632 cm−1 (2), suggests a β-turn structure in solution, consistent with NMR findings.
Molecular dynamic calculations
The above experimental data have shown that both compounds are structured with β-tuns. They also have suggested mobility of the peptides’ backbones. In order to investigate the different possible conformation(s) and their occurrences in 1 and 2, molecular dynamic calculations were carried out according to the protocol described in the experimental section.The AMBER suite program was used to run molecular modelling calculations and analyze the trajectories. Dihedral angles were measured along the trajectories of molecular dynamic simulations for molecules 1 and 2. According to classical values in peptides and proteins (Table S1, ESI†), the torsion angles measured in oligomers 1 and 2 indicate that both molecules adopt distorted β- and β′-turn conformations. However, no hydrogen bonds stabilizing a beta turn were found in the calculations, while a 7-atom pseudocycle stabilized by a CO(azaPhe)/OH(Ala) intramolecular hydrogen bond was clearly found in 61% and 33% of the conformations adopted by 1 and 2, respectively (Fig. 4). Interestingly, the deviation of torsion angles compared to optimum values is smaller for compound 2 than for compound 1 (Table S2, ESI†). All these observations show the impact of the chirality of Phe/D-Phe, since it reduces the possibility of hydrogen bond occurrence by about 50% in the (R,S) compound. This predicted hydrogen bond could not be verified in NMR spectroscopy due to the broadening of the OH signals; however, the modelling method is consistent with the infrared results that showed extended broad bound bands in both compounds 1 and 2. The difference in modelling and NMR results may be related to the experimental conditions in both techniques.
 | ||
| Fig. 4 Selected frame for molecules 1 (left) and 2 (right) from the molecular dynamic simulations, showing the new intramolecular hydrogen bond. | ||
Structure and conformation analysis by X-ray crystallography
Only compound 2 gave single crystals suitable for X-ray crystallographic analysis which were grown by slow evaporation from EtOH. Compound 2 crystallizes in space group P1 with two conformers in the asymmetric unit as shown in Fig. 5a (two half-asymmetric units in the unit cell (Z = 2)), (Table S3, ESI†). X-ray results revealed interatomic distance between CO(Fmoc) and NH(Ala) of 3.060 Å and 3.049 Å for conformers 2A and 2B, respectively (Table S4, ESI† and §)which are adequate values for i, i + 3 intramolecular hydrogen bond interactions83 closing 10 atoms pseudocycle as shown in Fig. 5b. Moreover, the distances between the oxygen atom CO of residue i (Fmoc) and Cα of residue i + 3 (Ala) of both conformers were less than 7 Å which supports the existence of β-turn conformation.84 Furthermore, each conformer is stabilized by orthogonal π-stacking between the phenyl groups of D-Phe and azaPhe. The two conformers (2A and 2B) are antiparallel in the asymmetric unit (Fig. 5a). They present two intermolecular hydrogen bonds between CO(Fmoc) of one conformer and (NH)azaPhe of the other conformer with a distance of 3.0 Å between the O and N atoms and, and bond angles of 175.84° and 175.10° measured for both hydrogen bonds (Table S5, ESI†). Finally, Fig. 5b shows that the two conformers in the asymmetric unit are stabilized by intermolecular orthogonal π-stacking between the benzyl groups (D-Phe and azaPhe) of one conformer and the fluorenyl group of the other conformer. | ||
| Fig. 5 Crystal molecular structures of: (a) asymmetric unit of 2 and (b) conformers 2A (top) and 2B (bottom); and the packing mode in 2: (c) side view, and (d) top view. Models are represented in capped sticks. The hydrogen bonds are marked as dotted lines colored in purple and orange for intermolecular while in green for intramolecular interactions. The H atoms, except those of the NH and OH groups, have been omitted for clarity (a), (c) and (d). In (c) and (d) the asymmetric unit cell is represented as light grey boxes. | ||
Whilst the values of the torsion angle |ω| range from 175.16° to 179.87° confirming Z-configuration for all the peptide bonds (Table 1), the torsion angles (ϕ, ψ) help in determining the nature of folding.79 The torsion angles showed values of: (ϕD-Phe = 63.87°, ψD-Phe = −125.12°) and (ϕazaPhe = −83.31°, ψazaPhe = −8.2°) for conformer 2A and (ϕD-Phe = 62.96°, ψD-Phe = −122.06°) and (ϕazaPhe = −87.58°, ψazaPhe = −7.68°) for conformer 2B. These angles are close to classical βII′-turn in peptides, confirming that the insertion of aza-amino acid in the peptide sequence supports the β-folding, which is stabilized through the intramolecular hydrogen bond of i, i + 3 type.79,80
| Conformer | Residue (i + 1) | Residue (i + 2) | ||||
|---|---|---|---|---|---|---|
| Φ (°) | Ψ (°) | Ω (°) | Φ (°) | ψ (°) | ω (°) | |
| 2A | 63.87 | −25.12 | 175.16 | −3.31 | −8.20 | −78.66 |
| 2B | 62.96 | −22.06 | 175.36 | −7.58 | −7.68 | −79.87 |
| βII′-turn | 60 | −120 | 180 | −80 | 0 | 180 |
Concerning the molecular packing of compound 2, three of the four carbonyl groups are oriented up and down perpendicularly for intermolecular hydrogen bonds, while the fourth carbonyl group is involved in intramolecular hydrogen bonds (Fig. 5c and d). In addition, the packing is reinforced by perpendicular and parallel π-stacking interactions between each fluorenyl ring of 2A or 2B and the two phenyl rings of D-Phe and azaPhe residues of 2B or 2A, respectively. Finally, intermolecular hydrogen bonds between NH(D-Phe), CO(D-Phe), CO(Ala), and OH(carboxylic) of conformer 2A with CO(Ala), OH(carboxylic), NH(D-Phe), and CO(D-Phe) of two molecules of conformer 2B, respectively, so that each molecule of one conformer forms 4 intermolecular hydrogen bonds with 2 molecules of the other conformer (Fig. 5c and d). The interatomic distances between these hydrogen bonds are 2.949 Å, 2.668 Å, 2.980 Å and 2.687 Å, while the bond angles are 162.02°, 165.22°, 161.40° and 162.28°, respectively (Table S6, ESI†). Therefore, the arrangement of the carbonyl groups supports the formation of a supramolecular structure held via an antiparallel hydrogen-bonding network plus the stacking between aromatic moieties leading to an extended β-sheet conformation.85,86
Characterization of LMWHGs
:1-[α/aza]-trimers form more stable hydrogels due to their β-turn conformation, which enhances hydrogen bonding and structural integrity. These hydrogelators might exhibit increased resistance to enzymatic degradation, making them promising for biomedical applications.90 Additionally, π–π stacking and intermolecular interactions reinforce the supramolecular structure, differentiating them from Fmoc-modified native peptides.
 | ||
| Fig. 6 Supramolecular hydrogels from 1 and 2 at pH 7.0 and pH 10.0, (1.0% w/w). | ||
Hydrogel characterization in this study was conducted at pH 7.0, as it aligns with physiological conditions relevant to medical applications. A combination of spectroscopy techniques (IR, CD, and fluorescence), rheometry, and scanning microscopy was used to examine the influence of amino acid chirality on the hydrogels’ physicochemical, mechanical, and thermal properties, as well as their morphology.
 | ||
| Fig. 7 Oscillatory stress sweep (OSS) experiments for hydrogels at pH 7.0 from (a) 1 and (b) 2, (c = 2.0% w/w, ω = 0.63 rad s−1, T = 25 °C); and the oscillatory frequency sweep experiments (OFS) for hydrogels at pH 7.0 from (c) 1 and (d) 2, (c = 2.0% w/w, σ = 1.5 Pa, T = 25 °C). | ||
In the current study, the dynamic moduli G′ and G′′ for both hydrogels exhibited weak frequency dependence (Fig. 7c and d), suggesting entangled network structures. The moduli strongly increase at higher frequencies, which is most likely due to the gel instability resulting from gel thickening associated with the displacement of gel water.94,95 These findings suggest that the two hydrogels exhibit typical solid-like rheological behavior, with G′ dominating G′′ over the studied oscillation frequency range.45,93,96 Unexpectedly, both hydrogels exhibited two crossover points in the oscillatory stress sweep, suggesting distinct viscoelastic regimes and structural transitions. The first crossover point marks a shift in the viscoelastic properties from elastic to viscous, indicating the initial breakdown of the gel network. The second crossover point suggests further structural rearrangements, potentially driven by sol–sol transitions, variations in cross-linking density, or reorganization of intermolecular interactions such as hydrogen bonding and π–π stacking. While these transitions provide insights into the dynamic behaviour of the gel, their detailed investigation was beyond the scope of this study, which primarily aimed to determine the linear viscoelastic region (LVR). Then, both hydrogels at pH 7.0 were subjected to a shear stress ranging from 0.6 to 6.25 Pa for 2.0 minutes to induce deformation. Subsequently, the gel recovery from the deformed state was monitored at 0.01 Pa as a function of time (15 minutes) using an oscillatory time sweep (OTS) experiment (Fig. S11, ESI†).
The plotted data show a fast recovery behavior after removing the applied shear stress (G′ > G′′). The gel strength is fully recovered with the time span of <100 s for gel 1 and <40 s for gel 2. These results reflect a high re-healing property after the deformation of both hydrogels. The faster recovery of compound 2 shows again the impact of the chirality of the CαPhe.
Despite small changes at the molecular scale, differences are observed in the mechanical properties of the obtained gels. The structuring of molecular assemblies of both hydrogels was then investigated in greater detail, particularly at the macromolecular level: CD, IR, fluorescence spectroscopy experiments were carried out for that purpose.
Structuration and conformation analysis of hydrogels
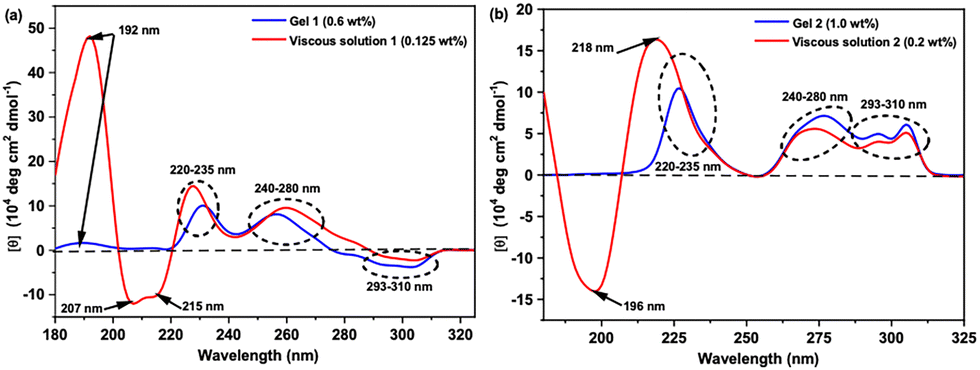 | ||
| Fig. 8 Normalized CD spectra of gel and viscous states for hydrogels from pH 7.0 of: (a) 1 and (b) 2. | ||
In the gel state (∼MGCs) of 1 (9.9 mmol L−1, 0.6 wt%) and 2 (16.5 mmol L−1, 1.0% w/w), it was difficult to have detectable CD signals in the range of 180–215 nm, except the positive maximum shallow peak at 192 nm. The low response signals in the CD spectra in the range 180–215 nm in the gel state of both 1 and 2 are due to the saturation effect observed in the UV-absorption spectra in this region (Fig. 8). In the range 215–350 nm, the CD spectra of both hydrogelators in solution or gel states are very similar and show the same interactions, but they exhibit opposite signs of CD signals within the range 290–310 nm. Indeed, the CD spectra of hydrogelators 1 and 2 at pH 7.0 in viscous or gel states show two successive positive maxima peaks in the range of 220–235 nm, indicative of π–π stacking of the aromatic phenylalanine residues, and 240–280 nm which can be assigned to phenylalanine residues with tertiary structure or interactions between phenyl and fluorenyl groups.52,106 Finally, the presence of the Cotton effect with opposite maxima signs for 1 and 2 in the range 293–310 nm was assigned previously to the π → π* transition of the fluorenyl groups by Xu and coworkers.100,107,108 The difference in the Cotton effect associated with the fluorenyl groups from negative maxima in 1 to positive maxima in 2 implies that the Fmoc-groups in the two gels might have different chiral orientations in the formed fibril self-assembled nanostructures which may be attributed to the difference in the chirality of the Phe/D-Phe amino acid.45
Based on CD results, we could suppose that both hydrogelators (1 and 2) can self-assemble at pH = 7.0 into fibrils in which the molecules orient themselves in a supramolecular structure displaying CD signatures characteristic of classical β-sheet arrangements stabilized by π–π stacking between the aromatic moieties. This supramolecular organization is still conserved in the gel state as most of the CD signals (215–310 nm) are the same in both normalized spectra of viscous and gel states, assuming a β-sheet self-assembly of the gelator molecules within the fibrils in the gel state. Additionally, changing the chirality of the first amino acid from L-Phe in 1 to D-Phe in 2 reflected an opposite Cotton effect sign in the ranges 180–215 nm and 195–310 nm, which changes the chirality of the orientation of Fmoc-groups in the self-assembled fibril structure yielding two opposite orientations of handedness of the β-sheet structure.95,106
 | ||
| Fig. 9 Concentration-dependent fluorescence spectra of (a) Fmoc-FazaFA (1) and (b) Fmoc-D-FazaFA (2); and temperature-dependent fluorescence spectra of (c) Fmoc-FazaFA (1) and (d) Fmoc-D-FazaFA (2) at 25 °C and 90 °C; (λex = 265 nm). | ||
Temperature-dependent fluorescence experiments were performed (i.e., heating ramp from 25 °C to 90 °C at a 5° min−1) for each hydrogel at pH 7.0 and a concentration of 0.7% (w/w). Fig. 9c and d show only the lowest and highest temperature measurements for clarity to focus on changes in the both gelators’ behaviors upon heating. At high temperature (90 °C, sol state), the two spectra show distinct blue shifts at higher wavelengths, suggesting that most of the intermolecular interactions (π-stacking and hydrogen bonds) are broken and the gelator molecules move freely in solution. Hydrogel 2 shows a bigger red shift than 1 proposing that the arrangement of aromatic moieties is better in gel 2 than for hydrogel 1. This result is consistent with the rheological measurements as 2 shows more mechanical strength than 1 and XRD data. The fluorescence study clearly emphasized that for both compounds, hydrogel formation is induced by same intermolecular forces namely fluorenyl and phenyl moieties’ π-stacking interactions. Moreover, we believe that the intercalation of phenyl rings and Fmoc-group via the π-stacking interactions plays a key role in the stabilization of the extended β-sheet structures at the tested pH values.
Morphological study
HR-SEM was used to image the freeze-dried hydrogels from gelators 1 and 2 at pH 7.0. The SEM images of the lyophilized gels for both gelators displayed a structure of randomly arranged thin fibers (Fig. 10). | ||
| Fig. 10 SEM images for dried gels from: (a) 1 and (b) 2 at pH 7.0. | ||
Fig. 10 shows that both hydrogels display fibrous structures with non-uniform thickness, indicating similar morphology for hydrogels from 1 and 2. This suggests that hydrogelator molecules self-organize in the same way, forming one-dimensional fibrils that align laterally, creating thicker fibers. This interconnected 3D fibrous structure traps water, forming hydrogels.109
Experimental section
Chemicals and reagents
All chemicals and reagents were purchased from commercial suppliers (Sigma-Aldrich, Fluka, Merck or Alfa-Aesar). Dry CH2Cl2 was obtained by distillation over P2O5 under an argon atmosphere, MeOH was purchased in anhydrous form, and other reagent-grade solvents were used without further purification as received. Buffer solutions (pH 4.0 = citric acid/sodium hydroxide/hydrogen chloride, pH 7.0 = potassium dihydrogen phosphate/disodium hydrogen phosphate, pH 10.0 = borax/sodium hydroxide) were purchased from Fluka company. Reactions were monitored by thin-layer chromatography (TLC) using aluminum-backed silica gel plates. TLC spots were viewed under UV light or/and by heating the plate after treatment with a staining solution of phosphomolybdic acid. Flash chromatography was carried out on silica gel 60 (0.04–0.063 μm Mesh ASTM). All yields were calculated for pure isolated products. Electron spray ionization mass spectra (ESI-MS) were recorded with a Bruker MicroTof-Q HR spectrometer at the “Service commun de Spectrométrie de Masse”, Faculté des Sciences et Technologies, Vandoeuvre-lès-Nancy, France.Synthesis of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1-[α/aza]-trimers
:1-[α/aza]-trimers
Compounds 1 and 2 were synthesized at good yields by carrying out three additional steps on the general strategy that has been reported previously.75,77 Fmoc-products 1 and 2 were obtained from the corresponding Boc-analogues in three consecutive steps.113
General methyl ester deprotection procedure
To a solution of methyl ester-protected trimers (Boc-FazaFAOMe or Boc-D-FazaFAOMe) (1.0 equiv., 2.0 mmol) in CH3CN (10.0 mL), a solution of 1 N NaOH (2.0 equiv., 4.0 mmol) was added at 0 °C. The mixture was vigorously stirred for 6–10 h at room temperature until reaction completion (as monitored by TLC). The aqueous phase was cooled to 0 °C and acidified with 2.0 N HCl (pH 2.0) and extracted with CH2Cl2 (3 × 10.0 mL). The combined organic layers were dried over MgSO4, filtered, and evaporated in a vacuum to give the corresponding carboxylic acid in quantitative yield which was used in the next step without further purification.General Boc-deprotection procedure
To the Boc-deprotected compounds obtained from the previous step (1.0 equiv., 2.0 mmol), EtOAc/3.0 M HCl (10.0 equiv., 20.0 mmol) was added. The mixture was stirred for about 3–5 h at room temperature for reaction completion without any control by TLC. The solution was concentrated under vacuum, and the excess of HCl was co-evaporated with DCM (4 times), affording the corresponding hydrochloride salt compounds as a white solid in quantitative yield.Synthesis of Fmoc-N-functionalized 2:1-[α/aza]-trimers
The crude residue (deprotected trimer) from last step (1.0 equiv., 2.0 mmol) was dissolved in 5.0 mL H2O with the help of DIPEA (1.0 equiv., 2.0 mmol). Then, a solution of re-crystallized Fmoc-OSu in 5.0 mL of ACN (0.95 equiv., 1.9 mmol) was added in one portion (Note S1, ESI†). The pH of the reaction mixture drops due to the liberation of the free N-hydroxy-succinimide, so the pH of the mixture should maintain a pH of 8.0–9.0 by the controlled addition of DIPEA. The reaction was stirred overnight for completion monitoring by TLC (20% EtOH: 80% DCM). The solution was concentrated, poured into 30.0 mL of 1.5 M HCl in a separatory funnel, and then the product was extracted by ethyl acetate (10.0 mL × 3 times). The organic layer was washed with water, saturated NaCl, dried over MgSO4, and then the solvent was evaporated under vacuum and the resulting product was crystallized from hexane, yielding the corresponding Fmoc-product (1 or 2) as a white solid.
Characteristic properties
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–NH, fluorenyl), 156.53 (HN–N–C = O), 170.96 (OC–NH), 174.49 (OC–OH). IR (CDCl3) ṽmax = 3331 cm−1, 3369 cm−1, 3435 cm−1 (NH), 1633 cm−1, 1651 cm−1, 1675 cm−1, 1702 cm−1, 1717 cm−1, 1744 cm−1, 1762 cm−1 (CO). HRMS (ESI) (m/z) for [C35H34N4O6Na]: calculated 629.2376; found, 629.2398 [M + Na]+.
C–NH, fluorenyl), 156.60 (HN–N–CO), 170.82 (OC–NH), 174.68 (OC–OH). IR (CDCl3) ṽmax = 3275 cm−1, 3349 cm−1, 3368 cm−1, 3432 cm−1 (NH), 1632 cm−1, 1650 cm−1, 1672 cm−1, 1702 cm−1, 1719 cm−1, 1738 cm−1, 1762 cm−1 (CO). HRMS (ESI) (m/z) for [C35H34N4O6Na]: calculated 629.2376; found, 629.2384 [M + Na]+.
C–NH, fluorenyl), 156.53 (HN–N–C = O), 170.96 (OC–NH), 174.49 (OC–OH). IR (CDCl3) ṽmax = 3331 cm−1, 3369 cm−1, 3435 cm−1 (NH), 1633 cm−1, 1651 cm−1, 1675 cm−1, 1702 cm−1, 1717 cm−1, 1744 cm−1, 1762 cm−1 (CO). HRMS (ESI) (m/z) for [C35H34N4O6Na]: calculated 629.2376; found, 629.2398 [M + Na]+.
C–NH, fluorenyl), 156.60 (HN–N–CO), 170.82 (OC–NH), 174.68 (OC–OH). IR (CDCl3) ṽmax = 3275 cm−1, 3349 cm−1, 3368 cm−1, 3432 cm−1 (NH), 1632 cm−1, 1650 cm−1, 1672 cm−1, 1702 cm−1, 1719 cm−1, 1738 cm−1, 1762 cm−1 (CO). HRMS (ESI) (m/z) for [C35H34N4O6Na]: calculated 629.2376; found, 629.2384 [M + Na]+.
Hydrogels preparation and minimum gelation concentrations (MGCs)
Stock solutions of 1 and 2 were prepared by dissolving an exact weight of 1 or 2 in ethanol (10.0% w/w), and then buffer solution (i.e., pH 4.0, pH 7.0 or pH 10.0) was added dropwise to a final weight corresponding to the desired concentration (w/w%). The mixtures were mixed by handshake until obtaining homogenous solutions and then the tubes were maintained at room temperature till transparent hydrogels were formed. To determine the MGC, a gel sample of 2.0% (w/w) was further diluted by buffer solution, handshake and then kept at room temperature to see the possibility of the gelation process. The previous step was repeated continuously until reaching the concentration below which no gelation occurs, which is considered as the MGC.87Structure analysis and characterization techniques
Molecular dynamic calculations
Molecular calculations were conducted on 1 and 2. Dynamics simulations were carried out over 5 ns, with time steps of 2 femtoseconds, in an explicit solvent (dimethyl sulfoxide; DMSO box). These simulations were performed without any constraints under periodic boundary conditions, at 300 K, and constant pressure (1.0 atm). The AMBER 12 suite of molecular dynamics simulation programs was employed for this purpose.114,115 Molecules were constructed using MarvinSketch (ChemAxon), and since they contain non-canonical amino acids, Antechamber and Xleap from the AMBER program suite were used. Molecular simulations were run using the general AMBER force field (GAFF) and amino acid force fields (ff99SB). A trajectory of 25000 structures was generated and analysed using the Ptraj AMBER suite program.
Rheology measurements
All hydrogels from Fmoc-FazaFA and Fmoc-D-FazaFALG were prepared directly in 7 mL glass tubes and left overnight (∼12 h) at room temperature to obtain stable gels.112,116 All rheological experiments were performed on Advanced Rheometer-AR2000 (TA instruments) operating in oscillatory mode with a 20 mm parallel plate geometry with serrated surfaces to prevent sliding due to the liquid film expelled by samples (diameter gap was adjusted to 1000 μm).117 To keep the sample hydrated by minimizing the solvent evaporation, a solvent trap was used and the atmosphere within the sample chamber was saturated with water.118 In order to determine the linear viscoelastic region (LVR), each gel sample (2.0% w/w, pH 7.0) was subjected to oscillatory stress sweep (OSS) experiments; (G′ and G′′ were measured as a function of oscillatory stress (σ = 0.6–505 Pa) at a constant angular frequency of ω = 0.628 rad s−1.118,119 Then a dynamic oscillatory time sweep was performed for 15 minutes with an angular frequency of ω = 0.628 rad s−1, and applied stress of σ = 1.5 Pa was chosen within the LVR from the OSS experiment.120 After that, each sample was subjected to an oscillatory frequency sweep (OFS) experiment over a frequency range of ω = 0.1–628.3 rad s−1 and the applied stress of σ = 1.5 Pa obtained from the OSS step at 25 °C.85 Repeated measurements on fresh samples (three times) were also carried out to ensure reproducibility.Fluorescence emission spectroscopy
Fluorescence spectra were recorded on a Fluorolog FL3-222 spectrofluorometer (HORIBA Jobin Yvon, LONGJUMEAU, France) equipped with a 450 W Xenon lamp, a thermo-stated cell compartment (25 °C), and a UV-visible photomultiplier R928 (HAMAMATSU, Japan). Excitation beam is diffracted by a double ruled grating SPEX monochromator (1200 grooves per mm blazed at 330 nm). Emission beam is diffracted by a double ruled grating SPEX monochromator (1200 grooves per mm blazed at 500 nm). All spectra were recorded in 4 faces of 1.0 cm path-length quartz cuvettes. The fluorescence emission spectra were recorded for a series of concentrations from 1.0% (w/w) (i.e., gel state) to 0.025% (w/w) (i.e., sol state) for both hydrogels (1 and 2) in phosphate buffer solution (pH 7.0) by excitation at 265 nm (slit width of 2.0 nm), and the emission spectra were recorded from 250 to 600 nm.CD spectroscopy
The CD spectra of viscous solutions and gels (at MGCs) for 1 and 2 in phosphate buffer (pH 7.0) were monitored on a Chirascan-plus qCD spectrometer equipped with a temperature control and reported in molar ellipticity [ε]; spectra were recorded in the wavelength range of 180–350 nm at 25 °C. Samples were loaded into a 0.1 mm path length quartz cover slip cuvette and spectra were measured at 50 nm min−1 with a 1.0 nm step size and a 1.0 nm bandwidth, taking two averages. Additional synchrotron radiation circular dichroism (SRCD) experiments were carried out at the Research Institute for Synchrotron Radiation Science (HiSOR; Hiroshima University, Japan) including temperature- and concentration-dependent experiments; however, the results are not presented in the current work while all findings will be detailed in the following study regarding the kinetics and thermodynamics of hydrogels’ formation.Morphological study
Lyophilized dried gel samples of hydrogels from 1 and 2, prepared in phosphate buffer (pH 7.0), were inspected at the nanoscale level using high-resolution field emission scanning electron microscopy (HR-SEM) (JEOL JSM-649OLV, France, high vacuum, acceleration voltage of 5.0 kV, large field detector) after treatment by gold coating.Conclusions
This study opens up new possibilities for creating supramolecular hydrogels using pseudopeptides, focusing on the azapeptides family. The research explores the synthesis and characterization of two newly developed Fmoc-N-functionalized 2:1-[α/aza]-trimers: Fmoc-FazaFA (1) and Fmoc-D-FazaFA (2). Spectroscopic and X-ray techniques showed that in their monomeric states, both molecules adopt a β-turn conformation. When transitioning to the gel state, various spectroscopic methods indicated that both hydrogelators could self-assemble into a supramolecular β-sheet structure at different pH values. The driving forces for this self-assembly and hydrogelation were found to be intermolecular hydrogen bonding and π-stacking among aromatic groups as verified by fluorescence spectroscopy. This self-assembly process was confirmed by observing a fibrous structure through SEM imaging. Rheological tests demonstrated that both hydrogels exhibited solid-like behavior, as G′ was greater than G′′ across the studied frequency range. The findings suggest that changing the chirality of the initial amino acid from (S) in 1 to (R) in 2 has little impact on the physicochemical properties in both solution and gel states, indicating similar behavior for both hydrogels.
Author contributions
Mohamed I. A. Ibrahim: methodology, investigation, data curation, formal analysis, writing and reviewing. Jacques Bodiguel: methodology, data curation, and reviewing. Guillaume Pickaert: validation and reviewing. Loïc Stefan: validation and reviewing. Koichi Matsuo: methodology, data analysis, and reviewing. Marie-Christine Averlant-Petit: conceptualization, supervision, validation, writing and reviewing.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. C. Averlant-Petit and Loïc Stefan acknowledge the Centre National de la Recherche Scientifique (CNRS) for funding. M. I. A. Ibrahim acknowledges Erasmus+European program and Université de Lorraine for funding, and NIOF for scientific collaboration. The authors thank Université de Lorraine facilities: Ms Mathilde Achard and Mr Olivier Fabre at the APPEL facility of LCPM (https://lcpm.univlorraine.fr/content/plateforme-appel-0), NMR facility (https://crm2.univ-lorraine.fr/en/platforms/nmr-facility) and Emmanuel Wenger at the PMD2X facility (https://crm2.univlorraine.fr/en/platforms/x-ray-diffraction-pmd2x) of CRM2, Mr Philippe Arnoux and Mr Jean-Francois Remy at the SAMPL facility of LRGP (https://lrgp-nancy.cnrs.fr). The authors acknowledge the Research Institute for Synchrotron Radiation Science (HiSOR, Japan) for SRCD measurements (Proposals: 23AG028 and 23BG013).References
- T. Shao, N. Falcone and H.-B. Kraatz, ACS Omega, 2020, 5, 1312–1317 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed.
- J. C. Tiller, Angew. Chem., Int. Ed., 2003, 42, 3072–3075 CrossRef CAS PubMed.
- C. B. P. Oliveira, V. Gomes, P. M. T. Ferreira, J. A. Martins and P. J. Jervis, Gels, 2022, 8, 706 CrossRef CAS PubMed.
- A. K. Das and P. K. Gavel, Soft Matter, 2020, 16, 10065–10095 RSC.
- M. Suzuki, M. Yumoto, H. Shirai and K. Hanabusa, Chem. - Eur. J., 2008, 14, 2133–2144 CrossRef CAS PubMed.
- F. Cai, J.-S. Shen, J.-H. Wang, H. Zhang, J.-S. Zhao, E.-M. Zeng and Y.-B. Jiang, Org. Biomol. Chem., 2012, 10, 1418–1423 RSC.
- M. Bastrop, A. Meister, H. Metz, S. Drescher, B. Dobner, K. Mäder and A. Blume, J. Phys. Chem. B, 2009, 113, 574–582 CrossRef CAS PubMed.
- S. Sun, J. Song, Z. Shan and R. Feng, J. Electroanal. Chem., 2012, 676, 1–5 CrossRef CAS.
- J. Li, K. Fan, L. Niu, Y. Li and J. Song, J. Phys. Chem. B, 2013, 117, 5989–5995 CrossRef CAS PubMed.
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Commun., 2008, 2644–2646 RSC.
- U. K. Das and P. Dastidar, Chemistry, 2012, 18, 13079–13090 CrossRef CAS PubMed.
- N. Maeda, K. Masuda, J. Li, S.-I. Kabashima, I. Yoshikawa and K. Araki, Soft Matter, 2010, 6, 5305–5307 RSC.
- S.-i Kabashima, M. Kageyama, T. Okano, I. Yoshikawa and K. Araki, J. Colloid Interface Sci., 2013, 408, 107–112 CrossRef CAS PubMed.
- Y. Wang, X. Xin, W. Li, C. Jia, L. Wang, J. Shen and G. Xu, J. Colloid Interface Sci., 2014, 431, 82–89 CrossRef CAS PubMed.
- L. Galantini, C. Leggio, A. Jover, F. Meijide, N. V. Pavel, V. H. Soto Tellini, J. V. Tato, R. Di Leonardo and G. Ruocco, Soft Matter, 2009, 5, 3018–3025 RSC.
- L. Li, R. Sun, R. Zheng and Y. Huang, Mater. Des., 2021, 205, 109759 CrossRef CAS.
- N. Mehwish, X. Dou, Y. Zhao and C.-L. Feng, Mater. Horiz., 2019, 6, 14–44 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 Search PubMed.
- M. Suzuki, M. Yumoto, H. Shirai and K. Hanabusa, Org. Biomol. Chem., 2005, 3, 3073–3078 RSC.
- B. N. Ghosh, S. Bhowmik, P. Mal and K. Rissanen, Chem. Commun., 2014, 50, 734–736 Search PubMed.
- M. B. Taraban, M. Weerasekare, J. Trewhella, X. Shi, E. K. Jeong and Y. B. Yu, Biopolymers, 2012, 98, 50–58 CrossRef CAS PubMed.
- J.-J. Wu, M.-L. Cao, J.-Y. Zhang and B.-H. Ye, RSC Adv., 2012, 2, 12718–12723 RSC.
- G. Godeau and P. Barthélémy, Langmuir, 2009, 25, 8447–8450 CrossRef CAS PubMed.
- K. J. Skilling, A. Ndungu, B. Kellam, M. Ashford, T. D. Bradshaw and M. Marlow, J. Mater. Chem. B, 2014, 2, 8412–8417 RSC.
- N. Sreenivasachary and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5938–5943 CrossRef CAS PubMed.
- D. Yuan, X. Du, J. Shi, N. Zhou, J. Zhou and B. Xu, Angew. Chem., Int. Ed., 2015, 54, 5705–5708 CrossRef CAS PubMed.
- P. Hoschtettler, G. Pickaert, A. Carvalho, M.-C. Averlant-Petit and L. Stefan, Chem. Mater., 2023, 35, 4259–4275 Search PubMed.
- Y. Sako and Y. Takaguchi, Org. Biomol. Chem., 2008, 6, 3843–3847 RSC.
- K. Munenobu, T. Hase, T. Oyoshi and M. Yamanaka, Anal. Chem., 2014, 86, 9924–9929 Search PubMed.
- M. J. Clemente, J. Fitremann, M. Mauzac, J. L. Serrano and L. Oriol, Langmuir, 2011, 27, 15236–15247 Search PubMed.
- M. J. Clemente, P. Romero, J. L. Serrano, J. Fitremann and L. Oriol, Chem. Mater., 2012, 24, 3847–3858 CrossRef CAS.
- M. J. Clemente, R. M. Tejedor, P. Romero, J. Fitremann and L. Oriol, RSC Adv., 2012, 2, 11419–11431 RSC.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- W. Y. Seow, G. Salgado, E. B. Lane and C. A. E. Hauser, Sci. Rep., 2016, 6, 32670 CrossRef CAS PubMed.
- P. Tiwari, A. Gupta, D. N. Shukla, A. K. Mishra, A. Basu and A. Dutt Konar, ACS Appl. Bio Mater., 2021, 4, 4119–4130 CrossRef CAS PubMed.
- R. Ahuja, V. Shivhare and A. D. Konar, Macromol. Rapid Commun., 2024, 45, 2400255 CrossRef CAS PubMed.
- X. Fu, N. Wang, S. Zhang, H. Wang and Y. Yang, J. Colloid Interface Sci., 2007, 315, 376–381 CrossRef CAS PubMed.
- S. Cao, X. Fu, N. Wang, H. Wang and Y. Yang, Int. J. Pharm., 2008, 357, 95–99 CrossRef CAS PubMed.
- Y. J. Yang, Chin. Chem. Lett., 2007, 18, 1001–1004 CrossRef.
- A. Pal and J. Dey, Langmuir, 2011, 27, 3401–3408 CrossRef CAS PubMed.
- T. Patra, A. Pal and J. Dey, Langmuir, 2010, 26, 7761–7767 CrossRef CAS PubMed.
- P. Gao, C. Zhan, L. Liu, Y. Zhou and M. Liu, Chem. Commun., 2004, 1174–1175 RSC.
- B. Adhikari, J. Nanda and A. Banerjee, Soft Matter, 2011, 7, 8913–8922 RSC.
- J. Nanda, A. Biswas and A. Banerjee, Soft Matter, 2013, 9, 4198–4208 RSC.
- Z. Sun, Z. Li, Y. He, R. Shen, L. Deng, M. Yang, Y. Liang and Y. Zhang, J. Am. Chem. Soc., 2013, 135, 13379–13386 CrossRef CAS PubMed.
- T. Wang, J. Jiang, Y. Liu, Z. Li and M. Liu, Langmuir, 2010, 26, 18694–18700 CrossRef CAS PubMed.
- N. S. de Groot, T. Parella, F. X. Aviles, J. Vendrell and S. Ventura, Biophys. J., 2007, 92, 1732–1741 CrossRef PubMed.
- L. Adler-Abramovich, M. Reches, V. L. Sedman, S. Allen, S. J. B. Tendler and E. Gazit, Langmuir, 2006, 22, 1313–1320 CrossRef CAS PubMed.
- Z. Xie, A. Zhang, L. Ye and Z.-G. Feng, Soft Matter, 2009, 5, 1474–1482 Search PubMed.
- S. Marchesan, L. Waddington, C. D. Easton, D. A. Winkler, L. Goodall, J. Forsythe and P. G. Hartley, Nanoscale, 2012, 4, 6752–6760 Search PubMed.
- S. Marchesan, C. D. Easton, F. Kushkaki, L. Waddington and P. G. Hartley, Chem. Commun., 2012, 48, 2195–2197 RSC.
- S. Ghosh, S. K. Singh and S. Verma, Chem. Commun., 2007, 2296–2298 RSC.
- J.-B. Guilbaud, E. Vey, S. Boothroyd, A. M. Smith, R. V. Ulijn, A. Saiani and A. F. Miller, Langmuir, 2010, 26, 11297–11303 CrossRef CAS PubMed.
- M. J. Krysmann, V. Castelletto, A. Kelarakis, I. W. Hamley, R. A. Hule and D. J. Pochan, Biochemistry, 2008, 47, 4597–4605 CrossRef CAS PubMed.
- I. W. Hamley, G. D. Brown, V. Castelletto, G. Cheng, M. Venanzi, M. Caruso, E. Placidi, C. Aleman, G. Revilla-López and D. Zanuy, J. Phys. Chem. B, 2010, 114, 10674–10683 Search PubMed.
- A. Desii, F. Chiellini, R. Di Stefano, M. R. Tiné and R. Solaro, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 986–990 Search PubMed.
- C. J. Bowerman, W. Liyanage, A. J. Federation and B. L. Nilsson, Biomacromolecules, 2011, 12, 2735–2745 CrossRef CAS PubMed.
- C. Cao, M. Cao, H. Fan, D. Xia, H. Xu and J. R. Lu, Chin. Sci. Bull., 2012, 57, 4296–4303 CrossRef CAS.
- F. Rodríguez-Llansola, J. F. Miravet and B. Escuder, Chem. Commun., 2009, 7303–7305 RSC.
- C. Berdugo, J. F. Miravet and B. Escuder, Chem. Commun., 2013, 49, 10608–10610 RSC.
- A. Dehsorkhi, I. W. Hamley, J. Seitsonen and J. Ruokolainen, Langmuir, 2013, 29, 6665–6672 CrossRef CAS PubMed.
- H. A. Behanna, J. J. J. M. Donners, A. C. Gordon and S. I. Stupp, J. Am. Chem. Soc., 2005, 127, 1193–1200 CrossRef CAS PubMed.
- Y. Zhang, H. Gu, Z. Yang and B. Xu, J. Am. Chem. Soc., 2003, 125, 13680–13681 CrossRef CAS PubMed.
- Y. Zhang, Z. Yang, F. Yuan, H. Gu, P. Gao and B. Xu, J. Am. Chem. Soc., 2004, 126, 15028–15029 CrossRef CAS PubMed.
- Y. Kuang, Y. Gao and B. Xu, Chem. Commun., 2011, 47, 12625–12627 RSC.
- Y. Lin, Y. Qiao, P. Tang, Z. Li and J. Huang, Soft Matter, 2011, 7, 2762–2769 RSC.
- C.-S. Chen, X.-D. Xu, S.-Y. Li, R.-X. Zhuo and X.-Z. Zhang, Nanoscale, 2013, 5, 6270–6274 Search PubMed.
- I. Maity, D. B. Rasale and A. K. Das, RSC Adv., 2013, 3, 6395–6400 Search PubMed.
- J. Makarević, M. Jokić, L. Frkanec, V. Caplar, N. Sijaković Vujičić and M. Zinić, Beilstein J. Org. Chem., 2010, 6, 945–959 CrossRef PubMed.
- A.-S. Felten, S. Dautrey, J. Bodiguel, R. Vanderesse, C. Didierjean, A. Arrault and B. Jamart-Grégoire, Tetrahedron, 2008, 64, 10741–10753 Search PubMed.
- R.-O. Moussodia, S. Acherar, A. Bordessa, R. Vanderesse and B. Jamart-Grégoire, Tetrahedron, 2012, 68, 4682–4692 Search PubMed.
- I. Bouillon, N. Brosse, R. Vanderesse and B. Jamart-Grégoire, Tetrahedron, 2007, 63, 2223–2234 CrossRef CAS.
- A. Lecoq, G. Boussard, M. Marraud and A. Aubry, Tetrahedron Lett., 1992, 33, 5209–5212 Search PubMed.
- M. I. A. Ibrahim, X. Solimando, L. Stefan, G. Pickaert, J. Babin, C. Arnal-Herault, D. Roizard, A. Jonquières, J. Bodiguel and M.-C. Averlant-Petit, RSC Adv., 2023, 13, 10051–10067 RSC.
- M. I. A. Ibrahim, Z. Zhou, C. Deng, C. Didierjean, R. Vanderesse, J. Bodiguel, M.-C. Averlant-Petit and B. Jamart-Grégoire, Eur. J. Org. Chem., 2017, 4703–4712 CrossRef CAS.
- Z. Zhou, C. Deng, C. Abbas, C. Didierjean, M.-C. Averlant-Petit, J. Bodiguel, R. Vanderesse and B. Jamart-Grégoire, Eur. J. Org. Chem., 2014, 7643–7650 CrossRef CAS.
- M. I. A. Ibrahim, G. Pickaert, L. Stefan, B. Jamart-Grégoire, J. Bodiguel and M.-C. Averlant-Petit, RSC Adv., 2020, 10, 43859–43869 RSC.
- H. J. Lee, H. M. Park and K. B. Lee, Biophys. Chem., 2007, 125, 117–126 CrossRef CAS PubMed.
- M. Thormann and H. J. Hofmann, THEOCHEM, 1999, 469, 63–76 CrossRef CAS.
- F. André, A. Vicherat, G. Boussard, A. Aubry and M. Marraud, J. Pept. Res., 1997, 50, 372–381 CrossRef PubMed.
- A. Pal, Y. K. Ghosh and S. Bhattacharya, Tetrahedron, 2007, 63, 7334–7348 CrossRef CAS.
- W. S. Horne, C. D. Stout and M. R. Ghadiri, J. Am. Chem. Soc., 2003, 125, 9372–9376 CrossRef CAS PubMed.
- H. H. Mantsch, A. Perczel, M. Hollósi and G. D. Fasman, Biopolymers, 1993, 33, 201–207 Search PubMed.
- C. Tang, R. V. Ulijn and A. Saiani, Langmuir, 2011, 27, 14438–14449 CrossRef CAS PubMed.
- J. Montenegro, M. R. Ghadiri and J. R. Granja, Acc. Chem. Res., 2013, 46, 2955–2965 CrossRef CAS PubMed.
- Y. Y. Chen, H. Wang, D. W. Zhang, J. L. Hou and Z. T. Li, Chem. Commun., 2015, 51, 12088–12091 Search PubMed.
- F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679–11691 CrossRef CAS.
- S. Bhuniya, S. M. Park and B. H. Kim, Org. Lett., 2005, 7, 1741–1744 Search PubMed.
- M. Zivec, Z. Jakopin and S. Gobec, Curr. Med. Chem., 2009, 16, 2289–2304 CrossRef CAS PubMed.
- L. Xu, G. Xu, T. Liu, Y. Chen and H. Gong, Carbohydr. Polym., 2013, 92, 516–522 Search PubMed.
- Y. T. Wang, X. Xin, W. Z. Li, C. Y. Jia, L. Wang, J. L. Shen and G. Y. Xu, J. Colloid Interface Sci., 2014, 431, 82–89 CrossRef CAS PubMed.
- M.-O. M. Piepenbrock, N. Clarke and J. W. Steed, Soft Matter, 2011, 7, 2412–2418 RSC.
- G. Cheng, V. Castelletto, C. M. Moulton, G. E. Newby and I. W. Hamley, Langmuir, 2010, 26, 4990–4998 CrossRef CAS PubMed.
- N. Javid, S. Roy, M. Zelzer, Z. M. Yang, J. Sefcik and R. V. Ulijn, Biomacromolecules, 2013, 14, 4368–4376 Search PubMed.
- G. Palui, A. Garai, J. Nanda, A. K. Nandi and A. Banerjee, J. Phys. Chem. B, 2010, 114, 1249–1256 Search PubMed.
- D. Yang, P. Duan, L. Zhang and M. Liu, Nat. Commun., 2017, 8, 15727 Search PubMed.
- S. M. M. Reddy, G. Shanmugam, N. Duraipandy, M. S. Kiran and A. B. Mandal, Soft Matter, 2015, 11, 8126–8140 Search PubMed.
- S. Roy, N. Javid, P. Frederix, D. A. Lamprou, A. J. Urquhart, N. T. Hunt, P. J. Halling and R. V. Ulijn, Chem. – Eur. J., 2012, 18, 11723–11731 Search PubMed.
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. van Esch, S. Santabarbara and R. V. Ulijn, Nat. Chem., 2010, 2, 1089–1094 CrossRef CAS PubMed.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS PubMed.
- R. W. Woody, Circular dichroism: Principles and applications, VCH Publishers, New York, 1994, pp. 473–496 Search PubMed.
- M. Gupta, A. Bagaria, A. Mishra, P. Mathur, A. Basu, S. Ramakumar and V. S. Chauhan, Adv. Mater., 2007, 19, 858–861 Search PubMed.
- K. Wang, J. D. Keasling and S. J. Muller, Int. J. Biol. Macromol., 2005, 36, 232–240 CrossRef CAS PubMed.
- P. Manavalan and W. C. Johnson, Nature, 1983, 305, 831–832 Search PubMed.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, T. Tuttle and R. V. Ulijn, Chem. Commun., 2013, 49, 10587–10589 RSC.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 Search PubMed.
- Z. Yang, H. Gu, Y. Zhang, L. Wang and B. Xu, Chem. Commun., 2004, 208–209 RSC.
- B. Adhikari and A. Banerjee, Soft Matter, 2011, 7, 9259–9266 RSC.
- Y. Yang, L. Wang, J. Wang, P. Gao and B. Xu, J. Mater. Chem., 2010, 20, 2128–2132 RSC.
- D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Soft Matter, 2010, 6, 1971–1980 Search PubMed.
- L. Chen, J. Raeburn, S. Sutton, D. G. Spiller, J. Williams, J. S. Sharp, P. C. Griffiths, R. K. Heenan, S. M. King, A. Paul, S. Furzeland, D. Atkins and D. J. Adams, Soft Matter, 2011, 7, 9721–9727 RSC.
- P. B. W. Ten Kortenaar, B. G. Vandijk, J. M. Peeters, B. J. Raaben, P. J. H. M. Adams and G. I. Tesser, Int. J. Pept. Protein Res., 1986, 27, 398–400 Search PubMed.
- D. A. Case, T. E. Cheatham Iii, T. Darden, H. Gohlke, R. Luo, K. M. Merz Jr, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- D. A. Pearlman, D. A. Case, J. W. Caldwell, W. S. Ross, T. E. Cheatham, S. DeBolt, D. Ferguson, G. Seibel and P. Kollman, Comput. Phys. Commun., 1995, 91, 1–41 Search PubMed.
- J. Raeburn, G. Pont, L. Chen, Y. Cesbron, R. Levy and D. J. Adams, Soft Matter, 2012, 8, 1168–1174 Search PubMed.
- P. Terech, D. Pasquier, V. Bordas and C. Rossat, Langmuir, 2000, 16, 4485–4494 CrossRef CAS.
- N. A. Dudukovic and C. F. Zukoski, Soft Matter, 2014, 10, 7849–7856 Search PubMed.
- Z. Yang, G. Liang and B. Xu, Chem. Commun., 2006, 738–740 Search PubMed.
- S. Sathaye, A. Mbi, C. Sonmez, Y. C. Chen, D. L. Blair, J. P. Schneider and D. J. Pochan, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 34–68 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2383629. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tb02288b |
| ‡ These authors share equal corresponding authorship. |
| § The details of the crystal structure have been submitted to the Cambridge Crystallographic Data Centre (CCDC) – deposition number: 2383629.† Space group: P1 cell: a 10.108Å b 11.218Å c 15.476Å, α 99.653(4)° β 96.494(3)° γ 116.412(4)°, formula: C35H34N4O6, Temperature: 100 K. |
| This journal is © The Royal Society of Chemistry 2025 |