
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermo-responsive hydrogel via sustained Co-delivery of TA and PDGF to modulate the diabetic microenvironment and accelerate diabetic wound healing†
Jisun Kim ab,
Ki Wan Bongb,
Jung-Kyo Choc and
Soo-Chang Song*ac
ab,
Ki Wan Bongb,
Jung-Kyo Choc and
Soo-Chang Song*ac
aCenter for Biomaterials, Biomedical Research Institute, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea. E-mail: scsong@kist.re.kr
bDepartment of Chemical and Biological Engineering, Korea University, Seoul 02841, Republic of Korea
cNexgel Biotech, Co., Ltd., Hanam, 12939, Republic of Korea
First published on 19th May 2025
Abstract
In modern society, the need for diabetic wound healing is increasing due to the increase in the number of diabetic patients. In particular, chronic inflammation is a major problem in diabetic wounds due to excessive accumulation of reactive oxygen species (ROS). Therefore, it is essential to remove ROS and promote angiogenesis for effective diabetic wound healing. In this study, we developed a thermo-responsive poly(organophosphazene) hydrogel system (TSP–TP) designed to deliver antioxidants and growth factors for a long period of time. The TSP–TP hydrogel stably loads and continuously releases tannic acid (TA) and platelet-derived growth factor (PDGF) through various physical interactions. Effective ROS scavenging induced macrophage polarization and alleviated chronic inflammation, while the sustained release of PDGF promoted angiogenesis, ultimately maximizing wound healing efficacy in a diabetic mouse model. Based on these results, the proposed TSP–TP hydrogel demonstrates synergistic effects through sustained delivery of antioxidants and growth factors, demonstrating a promising system with high applicability in diabetic wound treatment.
1. Introduction
As the number of diabetic patients increases worldwide, delayed healing of chronic wounds has become a serious medical problem.1,2 Unlike the normal healing process, diabetic wounds have difficulty in healing because the immune balance is disrupted by high levels of reactive oxygen species (ROS), and the production of growth factors that induce angiogenesis is suppressed in a hyperglycemic environment.3,4 As a result, the wound remains in the inflammatory phase. The persistent inflammatory response induces neutrophil hyper-activation and protease secretion, which worsens the wound microenvironment and leads to a vicious cycle in which tissue regeneration is delayed due to insufficient nutrient and oxygen supply.5,6 Therefore, effectively removing ROS and promoting angiogenesis are considered to be key strategies for diabetic wound healing.7Macrophages play an important role in the wound healing process, and in particular, polarization from M1 macrophages to M2 macrophages promotes tissue regeneration and angiogenesis.8–10 However, in diabetic wounds, the high glucose and high oxidative stress inhibit the polarization from M1 to M2, resulting in persistent inflammation and delayed healing.11–13 Recent studies have demonstrated that removing ROS enhances wound healing by fostering macrophage polarization from M1 to M2.14,15 Accordingly, modulating the immune environment by reducing ROS has been attracting attention as an important strategy in diabetic wound healing. This strategy is particularly effective in diabetic wound healing, the use of antioxidants to modulate the immune environment has gained attention. Polyphenol-based antioxidants possess potent ROS-scavenging capabilities, but their delivery is challenged by low stability, limited targeting, and reduced bioavailability.16 In particular, tannic acid (TA) is a plant-derived polyphenol with high water solubility and biocompatibility, and has been widely studied as an effective ROS scavenger due to its strong antioxidant and anti-inflammatory properties17,18 Biomaterials utilizing TA have the potential to promote tissue regeneration and improve wound healing through anti-inflammatory effects and reduction of oxidative stress.19 However, TA has limited bioavailability due to its short half-life, and it is difficult to act stably in a wound environment for a long time.20 Therefore, the development of suitable biomaterials that can effectively deliver TA for a sufficient period of time and increase its stability is essential.
Meanwhile, angiogenesis is an important factor for tissue repair in diabetic wound healing. PDGF being particularly vital as it is released in the early stages to induce the migration of various cells such as monocytes, neutrophils, and fibroblasts, and to enhance fibroblast proliferation, thereby increasing ECM production.21,22 Moreover, PDGF significantly contributes to the development of mature blood vessels, offering potential benefits in the treatment of diabetic wounds.23 PDGF has been used as an FDA-approved local growth factor due to these properties.24 Existing PLGA nanoparticle25 or collagen sponge26 based growth factor delivery systems have short release periods and initial growth factor loss issues, indicating that the growth factor may be lost before it can effectively act.27,28 Specifically, in the diabetic wound environment, high ROS alters the structure of growth factor receptors, while the overexpression of MMPs and proteases rapidly degrades proteins such as growth factors, complicating effective delivery.29 To overcome this and effectively treat diabetic wounds, the development of a delivery system that can release growth factors slowly and long-term while alleviating inflammation is necessary.
Hydrogels are widely used as biomaterials due to their excellent biocompatibility, mechanical properties, and water retention.30,31 In particular, thermo-responsive hydrogels can be converted to a gel state at body temperature, providing a scaffold similar to the extracellular matrix (ECM) at the site of injury, thereby promoting differentiation, migration, and proliferation of cells required for tissue regeneration.32,33 These properties are advantageous in effectively filling three-dimensional damaged areas and helping tissue regeneration, and also show great potential in delivering therapeutic agents locally. The hydrogel based DDS systems are mainly based on entrapping and releasing drugs within the network.34 Therefore, conventional hydrogels still have limitations in designing delicate networks, as drug release mainly depends on their degradation rate, network size, and swelling.35
To overcome these limitations, developing a stable and long-term drug delivery system by inducing various physical interactions between drugs and polymer chains is necessary. Introducing various interactions such as hydrogen bonding, electrostatic interactions, and hydrophobic interactions can increase drug loading efficiency while strongly binding the drug to the hydrogel.34 However, existing hydrogels have limitations in loading various drugs because the sites that can interact with drugs are limited.36,37 Poly(organophosphazene) hydrogels are promising systems capable of satisfying the requirements for multi-drug delivery by providing sites for interaction with various drugs and enhancing the flexibility of drug delivery. Hydrophobic drugs can be stably loaded through hydrophobic interaction with the hydrogel network,38 and by introducing anionic functional groups, ionic interaction can be formed with drugs with cationic properties to achieve effective drug delivery.32 These properties can be a promising approach for delivering and controlling multiple drugs. Recently, hydrogel systems that simultaneously deliver antioxidants and growth factors have been developed, attracting attention as a strategy that can simultaneously alleviate oxidative stress and induce tissue regeneration. For example, systems that simultaneously deliver curcumin and VEGF,39 or polydopamine and EGF,40 have been reported, but there are limitations such as an initial burst release due to the absence of specific binding functional groups between the hydrogel and the drug, difficulty in controlling drug release, and the low mechanical properties of hydrogel, resulting in low stability in the body. The TSP–TP hydrogel developed in this study can easily load drugs at low temperatures based on its thermo-responsive properties, and is effective for local drug delivery due to immediate gelation at body temperature. Importantly, the functional groups within the hydrogel enable stable encapsulation of antioxidants (TA) and growth factors (PDGF) via hydrogen bonding and ionic interactions, respectively, without the need for additional chemical or physical modifications. This characteristic allows for long-term and controlled release of therapeutic agents while preventing initial burst release. These structural and chemical characteristics provide significant advantages in terms of hydrogel stability and drug release control compared to conventional systems.
In this study, we developed drug delivery system for diabetic wound healing by stably loading TA and PDGF into a thermo-responsive poly(organophosphazene) hydrogel (TSP–TP) (Scheme 1). TA forms hydrogen bonds with PEG in the hydrogel. At the same time, the carboxyl group forms an ionic interaction with the cationic PDGF. The two substances are stably loaded into the hydrogel by this noncovalent interaction, enabling sustained release. This dual delivery system can relieve inflammation by removing ROS due to TA and promote angiogenesis and tissue regeneration through continuous and slow-released PDGF. The hydrogel exists in solution at room temperature, making it easy to apply to deep and irregular wound areas, and transforms into a stable gel at body temperature. It effectively prevents moisture loss from the wound and acts as a scaffold for tissue formation. The system accelerates healing by (1) alleviating ROS levels, (2) inducing polarization toward M2 macrophages, and (3) promoting angiogenesis. The TSP–TP hydrogel designed in this study provides a promising new approach to treat diabetic wounds by simultaneously and continuously delivering antioxidants and growth factors.
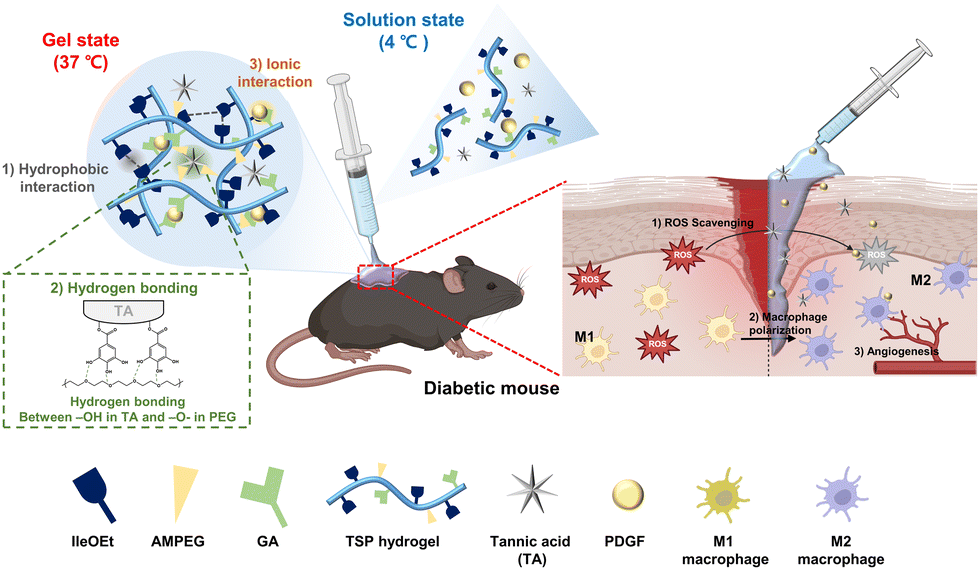 | ||
| Scheme 1 Schematic illustration of the gelation mechanism of a thermo-responsive TSP–TP hydrogel and the wound healing process when the hydrogel is applied to a diabetic wound. | ||
2. Results and discussion
2.1. Characterization of TSP, TSP–T, and TSP–TP hydrogels
Thermo-responsive TSP hydrogel was designed for long term antioxidant and growth factor co-delivery for diabetic wound healing. To enable the long term stable encapsulation of therapeutic molecules, we designed TSP polymer containing functional groups capable of forming physical interactions with the incorporated agents. The synthesis process of TSP polymer was carried out in three steps, as illustrated in Fig. S1 (ESI†). The side chain ratio and molecular weight of the synthesized TSP polymer were analyzed using 400 MHz 1H-NMR and gel permeation chromatography (GPC). 1H-NMR analysis of the TSP polymer indicated that the side chain ratios of isoleucine ethyl ester (IleOEt), α-amino-ω-methoxy-poly(ethylene glycol) (AMPEG), and glutaric acid (GA) were 75%, 15.5%, and 9.5%, respectively (Fig. S1, ESI†). The TSP polymer is mainly composed of hydrophobic IleOEt and hydrophilic AMPEG, which form thermo-responsive physical crosslinks through hydrophobic interactions between IleOEt groups. In addition, hydrogen bonding can occur between AMPEG in TSP hydrogel and TA. In addition, the GA moiety, which is negatively charged by the carboxyl group, induces ionic interactions with positively charged PDGF. It is expected that antioxidants and growth factors can be stably loaded and released for a long period of time through various functional groups in the TSP polymer. The weight average molecular weight (Mw) of the TSP polymer was determined to be 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 542 Da, with a polydispersity index (PDI) of 2.40 through GPC analysis.
542 Da, with a polydispersity index (PDI) of 2.40 through GPC analysis.
To confirm the sol–gel phase transition of TSP hydrogel according to temperature, the morphological changes of the hydrogel were observed at 4 °C, the hydrogel maintained a sol state, but when placed in a 37 °C water bath, it immediately transitioned to a gel state. The temperature dependent gelation occurred immediately. To verify this, the TSP polymer solution was injected into water at 4 °C and 37 °C using a syringe. In contrast to 4 °C, the polymer solution formed a 3D structure immediately at 37 °C (Fig. S2, ESI†). This demonstrates that the temperature dependent sol–gel transition occurs within a few seconds in body temperature, suggesting that gels can be precisely and stably formed at the target site without dispersion when injected into the body.
In addition, the surface structures of TSP, TSP–T, and TSP–TP hydrogels according to temperature change were observed using a cryo-SEM (Fig. 1A). The hydrogel network was not observed at 4 °C. However, as the temperature increased to 37 °C, the polymer chains formed a porous network structure through physical crosslinking. This porous structure is crucial factor in tissue engineering for supporting cell growth by facilitating the diffusion of nutrients and oxygen and maintaining the structural stability of the hydrogel.41 After incorporating the antioxidant and growth factor in TSP hydrogel, the pore size of the TSP–T and TSP–TP hydrogel slightly decreased, but the porous structure was stably maintained without aggregation (Fig. S3, ESI†). This result suggests that TA and PDGF interact with the functional groups of the TSP hydrogel, allowing these molecules to be uniformly loaded without disrupting the existing network structure.
 | ||
| Fig. 1 Thermo-responsive properties of TSP, TSP–T, and TSP–TP hydrogels (A) images depicting the sol–gel transition and cryo-SEM image of the TSP hydrogel after thermal crosslinking. (B) Temperature dependent rheological data for TSP, TSP–T, and TSP–TP hydrogels (10 wt% in DPBS) and merged storage modulus data. | ||
The temperature-dependent sol–gel transition characteristics and the property changes according to the additives mentioned above were clearly evaluated using a rheometer (Fig. 1B, Table S1, ESI†). The TSP hydrogel exhibited a gelation temperature (T0) of 27.13 °C, reached a maximum storage modulus temperature (Tmax) of 44.65 °C, and achieved a maximum storage modulus  of 3037.9 Pa; the storage modulus at 37 °C
of 3037.9 Pa; the storage modulus at 37 °C 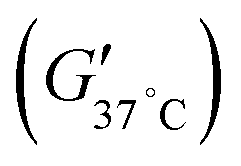 was 1909.1 Pa. For the TSP–T and TSP–TP hydrogels containing additives, the maximum storage moduli were recorded at 3342.8 Pa and 3529.1 Pa, respectively. In particular, the significantly higher maximum storage modulus of the TSP–TP hydrogel is thought to be due to the strong physical interactions mediated by hydrogen bonds and ionic bonds among TA, PDGF, and the TSP hydrogel. This observation is consistent with our previous report that the introduction of physical interactions such as ionic bonding into PPZ-based hydrogels improves their mechanical properties.32 The storage moduli of the TSP–T and TSP–TP hydrogels at 37 °C were measured at 2217.7 Pa and 1075.4 Pa, respectively. The storage modulus of the TSP–TP hydrogel began to increase significantly at 26 °C, providing the mechanical strength necessary to form a stable gel at body temperature. Additionally, when the gel properties were analyzed through a frequency sweep at 37 °C, the TSP hydrogel failed to maintain its gel state at high frequency, whereas the TSP–T and TSP–TP hydrogels maintained stable gel states even under the same conditions. This indicates that the encapsulated TA and PDGF enhance the network density of the TSP–T and TSP–TP hydrogels through various physical interactions with TSP, thereby ensuring their stability at the target site (Fig. S4, ESI†). Consequently, the TSP hydrogel system maintains a sol state at room temperature (below the gelation temperature), making it easy to apply to irregular or deep wound areas, and converts to a gel state at body temperature, allowing it to maintain its shape stably. This thermo-responsive mechanism enables localized delivery of therapeutic substances directly to the wound area, enhancing therapeutic efficacy and minimizing effects on surrounding tissue. Moreover, the TSP hydrogel can not only homogeneously encapsulate bioactive molecules such as antioxidants and growth factors, but also form a dense hydrogel network with enhanced mechanical properties through various physical interactions. Through this, the TSP hydrogel demonstrates its potential as a new material for effective drug delivery systems (DDS).
was 1909.1 Pa. For the TSP–T and TSP–TP hydrogels containing additives, the maximum storage moduli were recorded at 3342.8 Pa and 3529.1 Pa, respectively. In particular, the significantly higher maximum storage modulus of the TSP–TP hydrogel is thought to be due to the strong physical interactions mediated by hydrogen bonds and ionic bonds among TA, PDGF, and the TSP hydrogel. This observation is consistent with our previous report that the introduction of physical interactions such as ionic bonding into PPZ-based hydrogels improves their mechanical properties.32 The storage moduli of the TSP–T and TSP–TP hydrogels at 37 °C were measured at 2217.7 Pa and 1075.4 Pa, respectively. The storage modulus of the TSP–TP hydrogel began to increase significantly at 26 °C, providing the mechanical strength necessary to form a stable gel at body temperature. Additionally, when the gel properties were analyzed through a frequency sweep at 37 °C, the TSP hydrogel failed to maintain its gel state at high frequency, whereas the TSP–T and TSP–TP hydrogels maintained stable gel states even under the same conditions. This indicates that the encapsulated TA and PDGF enhance the network density of the TSP–T and TSP–TP hydrogels through various physical interactions with TSP, thereby ensuring their stability at the target site (Fig. S4, ESI†). Consequently, the TSP hydrogel system maintains a sol state at room temperature (below the gelation temperature), making it easy to apply to irregular or deep wound areas, and converts to a gel state at body temperature, allowing it to maintain its shape stably. This thermo-responsive mechanism enables localized delivery of therapeutic substances directly to the wound area, enhancing therapeutic efficacy and minimizing effects on surrounding tissue. Moreover, the TSP hydrogel can not only homogeneously encapsulate bioactive molecules such as antioxidants and growth factors, but also form a dense hydrogel network with enhanced mechanical properties through various physical interactions. Through this, the TSP hydrogel demonstrates its potential as a new material for effective drug delivery systems (DDS).
The biocompatibility of hydrogels means that they can interact smoothly with cells and tissues without inducing an immune response or inflammation when injected into the body, which is an important factor for tissue regeneration.42 The MTT assay performed in this study showed that the cell viability of fibroblasts exceeded 80% at all concentrations of TSP and TSP–T hydrogels, and the live/dead assay confirmed that most cells remained viable in all TSP, TSP–T, and TSP–TP hydrogels (Fig. S5, ESI†). These results suggest that TSP, TSP–T, and TSP–TP hydrogels are non-cytotoxic and have excellent biocompatibility. Furthermore, they demonstrate that the TSP hydrogels system can be utilized as a platform that can induce a tissue regeneration environment without inflammation. In particular, TSP–TP hydrogels exhibit potential as drug delivery carriers suitable for chronic wound treatment, which requires long-term retention and drug release in the body, based on their excellent mechanical properties, immediate sol–gel transition at body temperature, and biocompatibility.
2.2. Interactions between TSP hydrogel, TA and PDGF
In this study, we fabricated TSP–TP hydrogels through a simple mixing process. As a first step, the optimal concentration of TA inside the TSP hydrogel was determined. When the TSP hydrogel and TA solution were physically mixed to produce the TSP–T hydrogel, aggregation occurred as the TA concentration increased, resulting in non-homogeneous dispersion within the TSP hydrogel (Fig. S6, ESI†). In contrast, at a TA concentration of 0.1 wt%, TA was homogeneously dispersed without aggregation, which is an important condition for delivering TA at the consistent concentration throughout the hydrogel during treatment. Therefore, the optimal TA concentration of the TSP–T hydrogel was set to 0.1 wt% to maintain homogeneous dispersion in the hydrogel and enable application via syringe. Next, to prepare the TSP–TP hydrogel, 1 μg mL−1 of PDGF was physically mixed with the optimized TSP–T hydrogel. In general, the effective PDGF concentration for wound healing is in the range of 5–20 ng mL−1.43,44 Accordingly, the initial loading concentration was set to 1 μg mL−1 so that the PDGF released from the hydrogel could reach the above concentration range, and a release test confirmed that PDGF was sustainably released at an appropriate concentration. In this system, TA and PDGF encapsulated in the TSP hydrogel were stably holding within the hydrogel network through hydrogen bonding and ionic interactions, respectively. To verify if TA formed hydrogen bonds with the hydrogel, Fourier transform infrared (FT-IR) analysis was conducted (Fig. 2A). The results showed that the spectrum of TSP–T hydrogel featured peaks of both TSP hydrogel and TA across the whole wavelength range. Specifically, the hydroxyl group stretching peak, identified as the –OH peak, shifted from 3403 cm−1 to 3389 cm−1 upon TA loading. It is well established that the formation of intra or intermolecular hydrogen bonds leads to weaker chemical bonds and shifts the vibrational frequency to a lower wavenumber region.45–48 Moreover, the C–O–C peak of PEG also shifted when the C–O–C group of PEG, linked with the –OH group of TA, moved from 1103 cm−1 to 1094 cm−1.46 This indicates the successful incorporation of TA into the TSP hydrogel through hydrogen bonding, forming the TSP–T hydrogel. Consequently, it is expected that TA can be stably incorporated into the hydrogel network via hydrogen bonding to provide a sustained antioxidant effect over a long period of time, suggesting that it may be utilized as a particularly useful delivery system in environments requiring long-term treatment, such as chronic wounds.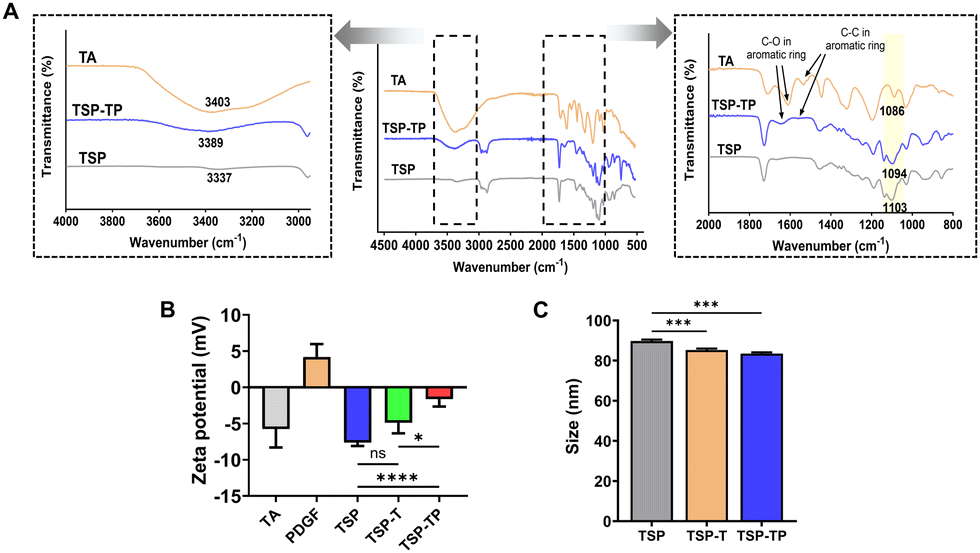 | ||
| Fig. 2 Various interactions between TSP, TA, and PDGF. (A) FT-IR spectra of TA, TSP, and TSP–T. (B) Zeta potential, and (C) particle size analysis utilizing DLS (n = 5 for each group). All quantitative data are expressed as mean ± standard deviation. | ||
Next, the zeta potential was measured to evaluate the ionic binding between the TSP–T hydrogel and PDGF (Fig. 2B). The basic TSP hydrogel showed a negative charge of −7.63 ± 0.41 mV due to the carboxyl group introduced through the binding with GA. In addition, the TSP–T hydrogel introduced TA also showed a negative charge of −5.61 ± 1.60 mV, which is judged to be due to the relatively small amount of TA added, showing no significant difference from the TSP hydrogel. On the other hand, PDGF has a positive charge of +4.18 ± 1.46 mV due to high isoelectric point (pI = 9.5–10.5),49 allowing for natural electrostatic interactions with the negatively charged of TSP hydrogel. After PDGF was encapsulated in the TSP–T hydrogel, the zeta potential of the final TSP–TP hydrogel increased to −1.61 ± 0.92 mV, which is interpreted as the result of the positively charged protein neutralizing the negatively charged surface. This change in potential indicates that the encapsulation was successfully achieved by the ionic bond between TSP–T and PDGF. These results suggest that the electrostatic properties of the TSP hydrogel system are controllable and that PDGF was effectively encapsulated within the hydrogel network through ionic bonding. In particular, this ionic interactions are advantageous of the sustained delivery of growth factors, demonstrating its potential as a delivery system suitable for chronic wound treatment requiring long-term drug release.
Due to the thermo-responsive property, the gel-forming hydrogel gradually decomposes while encapsulating the added bioactive molecules in the form of nanocomposites. These nanocomposites self-assemble into micelle particles through hydrophobic isoleucine ethyl ester and hydrophilic PEG in the polymer side chains.50 Our previous studies have shown that the introduction of growth factors or siRNA into hydrogels leads to a decrease in particle size due to noncovalent interactions.51,52 Therefore, we measured the particle size to clearly demonstrate the interactions between the TSP hydrogel and the added bioactive molecules (TA and PDGF). Measurement of particle sizes revealed that TSP hydrogel had a diameter of 89.74 ± 0.61 nm, while TSP–T and TSP–TP hydrogels measured 85.23 ± 0.62 nm and 83.5 ± 0.52 nm, respectively (Fig. 2C). In particular, the TSP–TP hydrogel exhibited the smallest particle size. This result can be attributed to various interactions, such as hydrogen bonding and ionic interactions that occurred simultaneously. This particle size reduction indicates that TA and PDGF were successfully encapsulated within the TSP hydrogel network. Moreover, nanoparticles with a size of 50–100 nm were formed, which are generally considered to have the potential for cellular uptake via endocytic pathway.53,54 Therefore, the small particle size of TSP–TP hydrogel highlights its potential for efficient intracellular drug delivery. These findings demonstrate that TSP–TP hydrogel can serve as a promising platform for delivery of bioactive molecules.
2.3. Long-term ROS scavenging effect of the TA-containing hydrogels
During the wound healing process, excessive ROS induces oxidative stress in cells, causes an imbalance in immune cells, and creates a chronic inflammatory environment.5,55 Therefore, the effective removal of ROS plays a crucial role in enhancing wound healing. In this study, we evaluated the ROS scavenging effects of TSP hydrogel, TSP–T hydrogel, and TA using the DPPH assay. The scavenging rates were 6.73 ± 5.67%, 64.13 ± 1.21%, and 85.29 ± 2.68% for TSP hydrogel, TSP–T hydrogel, and TA, respectively. As expected, the TSP hydrogel without TA exhibited minimal radical scavenging ability, while the TSP–T hydrogel demonstrated a significantly higher effect due to the encapsulated TA's antioxidant action. However, the antioxidant effect of the TSP–T hydrogel was slightly lower than that of free TA. This result is assumed to be due to the interactions between the phenolic moieties, which are the key antioxidant functional groups of TA, and AMPEG, a component of the TSP hydrogel, which likely reduced the number of available phenolic groups that can react with ROS. Moreover, radical scavenging was visually confirmed as the DPPH solution transitioned from purple to yellow.56 (Fig. 3A). This confirms the successful incorporation and maintenance of antioxidant activity by TA in the TSP hydrogel. Additionally, observing the radical scavenging effect over time revealed no change in the scavenging rates of the TSP hydrogel and TA for 30 min. On the other hand, the TSP–T hydrogel, which encapsulates TA through hydrogen bonding, exhibited a gradual improvement in the ROS scavenging rate from 42.6 ± 0.95% to 64.8 ± 0.58% over 30 min (Fig. 3B). This indicates the potential for long-term ROS scavenging by the TSP–T hydrogel due to continuous TA release. Furthermore, to evaluate the long-term antioxidant effect, the TSP–T hydrogel was immersed in DPBS for 228 h (9.5 days), and radical scavenging activity was measured at specified intervals. As a comparative group, the ability of free TA to maintain the antioxidant effect was evaluated. When TA was treated alone, the ROS scavenging effect decreased from 86.3 ± 2.41% to 9.3 ± 3.90% after 168 h (Fig. S7, ESI†). This shows that the antioxidant effect disappeared very quickly when TA was treated alone. In contrast, the radical scavenging ability of the TSP–T hydrogel increased to 64.4 ± 1.52% at 1 h and 66.1 ± 1.23% at 6 h, peaking at 69.4 ± 0.61% at 12 h and then maintaining a consistent level of about 70% for 9.5 days (Fig. 3C). These results demonstrated that the TSP–T hydrogel system encapsulating TA could effectively remove ROS over a long period of time. In previous studies, various antioxidants have been shown to remove ROS, but their sustained effects were limited.20 On the other hand, although the antioxidant effect of the TSP–T hydrogel is lower than that of free TA, it maintains approximately 70% antioxidant activity over an extended period by stably encapsulating TA through hydrogen bonds, and has the potential to provide sustained antioxidant activity even in chronic wound sites. This suggests that it may have superior therapeutic benefits over existing antioxidant-based treatments. | ||
| Fig. 3 Radical scavenging effect of TSP, TSP–T, and TA. (A) DPPH scavenging effect of TSP, TSP–T, and TA. The image presented is from the DPPH assay conducted at 15 min. (B) Time-dependent ROS scavenging effect of TSP, TSP–T, and TA. (C) Long-term radical scavenging effect of TSP–T hydrogel over 228 h. All quantitative data are presented as mean ± standard deviation (n = 3 for each group). | ||
2.4. Swelling behavior and sustained release of TA and PDGF in vitro
Whether hydrogels injected in vivo can continuously release loaded substances for long periods of time while maintaining structural integrity is one of the most important factors in biomaterials. To evaluate this, the swelling properties of the hydrogels were analyzed. The swelling and degradation behaviors of the hydrogels in an aqueous environment were confirmed through in vitro degradation tests. After 24 h, the TSP hydrogel exhibited greater swelling than other groups, with its weight ratio increasing to 109 ± 3.38%. In contrast, the weights of the TSP–T and TSP–TP hydrogels increased less significantly, to 105 ± 0.62% and 105 ± 3.63% respectively. By 336 h, the TSP hydrogel had swelled to 126 ± 3.40%, the TSP–T hydrogel to 119 ± 3.26%, and the TSP–TP hydrogel to 111 ± 2.76% (Fig. 4A). Notably, the TSP–TP hydrogel maintained a stable shape with minimal swelling, likely due to the additional physical interactions of TA and PDGF with the hydrogel network via hydrogen bonding and ionic bonds, resulting in a denser network. These results suggest that TSP–TP hydrogel maintains its structure integrity over a long period in a physiological environment and is a suitable system for sustained drug delivery. | ||
| Fig. 4 In vitro hydrogels swelling behavior and release profiles of TA and PDGF from TSP–TP hydrogels. (A) Swelling ratios of TSP, TSP–T, and TSP–TP hydrogels when immersed in DPBS over time. (B) Cumulative release profiles of TA and PDGF from the TSP–TP hydrogel over 336 hours. (C) Released, remained, and undetected amounts of TA and PDGF in TSP–TP hydrogel after 336 hours. Quantitative data were calculated using a standard curves and are presented as mean ± standard deviation (n = 5 for each group). | ||
To investigated the effect of he physical interaction between TSP hydrogel and bioactive molecules affect release profile. Release profiles and residual amounts of TA were determined using the Folin–Ciocalteu (FC) assay, which measures phenol and polyphenol compounds through a color change.57 First, the amount of TA released from TSP–T loaded with various concentrations of TA was calculated. Previous research has indicated that TA concentrations exceeding 40 μM can induce cytotoxicity in fibroblasts.55,58 TSP–T containing 1 wt% TA showed a release amount that can induce cytotoxicity, 44.3 ± 5.76 μM on 96 h, and TSP–T hydrogels containing 0.5 wt% and 0.3 wt% TA showed 50.2 ± 2.60 μM and 49.4 ± 3.15 μM on 168 h, respectively. However, the cumulative amount of TA released from the optimized TSP–T hydrogel containing 0.1 wt% TA was 33.2 ± 0.51 μM for 336 h (Fig. S8, ESI†). This value is below the concentration that induces cytotoxicity. Therefore, this hydrogel system suggests the possibility that it can provide an effective antioxidant effect by continuously releasing TA without inducing toxicity to surrounding cells and tissues. The release profiles of TA and PDGF are related to the interaction with TSP depicted Fig. 2. The release of TA and PDGF from TSP–TP was measured at predetermined points to verify whether the initial burst release could be suppressed and sustained release could be achieved (Fig. 4B and C). The cumulative release data showed that there was no initial burst release of both TA and PDGF. The released amount of TA was 9.3% over 336 h. About 36% of the loaded TA remained in the TSP–TP after 336 h. In addition, 6.6% of the loaded PDGF was released over 336 h, and 65% of the encapsulated PDGF remained inside the TSP–TP after 336 h. The reason for the low release rate of PDGF is that both the TSP hydrogel and TA are negatively charged, which form strong ionic bonds with the positively charged PDGF. Therefore, TSP–TP could achieve slow release and long-term retention of TA and PDGF. These results demonstrate that the various physical interactions between TSP hydrogels and antioxidants and growth factors can be utilized to simultaneously deliver two agents without complex scaffold design or additional processes. These results indicate that TSP–TP hydrogels provide a stable and sustained release platform by utilizing multiple physical interactions without requiring additional crosslinking step or complex scaffold fabrication. Unlike previous hydrogel systems that limitation from burst release or rapid degradation,59 the TSP–TP system offers a more controllable and practical approach to providing long-term therapeutic effects. These properties are particularly advantageous in chronic wound healing where long-term availability of antioxidants and growth factors is essential for effective tissue regeneration.
To clarify the release mechanism of TA and PDGF, the release patterns were analyzed using various kinetic models. The correlation coefficients (R2) and release constants (k) derived from different models, including the zero-order, first-order, Higuchi, and Korsmeyer–Peppas models, are summarized in Table 1.38 It was determined that the Korsmeyer–Peppas model best represents the release data for both TA and PDGF. This model is commonly applied to complex release mechanisms, such as diffusion, swelling, and degradation, making it suitable for analyzing substances that are released slowly or maintained for extended periods in vivo.60 Therefore, the use of this model supports the hypothesis that the release of TA and PDGF is driven by the swelling and degradation of the hydrogel. In addition, the diffusion mechanism can be described using the calculated diffusion exponents of 0.78 for TA and 0.56 for PDGF. This diffusional exponent values implied that TA and PDGF are released via a non-Fickian mechanism.61 The non-Fickian release behavior observed for both TA and PDGF suggests that release is controlled not only by diffusion but also by structural deformation of the hydrogel, such as swelling or erosion. Therefore, the release mechanism of the TSP–TP hydrogel provides more sustained and controlled release than previously reported materials. Whereas previous studied systems often reported a rapid hydrogel degradation and initial drug burst release,62,63 the TSP–TP hydrogel enables a more sustained release of the bioactive molecules. This suggests that the TSP–TP system can promote effective chronic wound healing with a single application, through the sustained release of antioxidants and growth factors over 336 h.
| Zero-order | First-order | Higuchi | Kors–peppas | Release mechanism | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| k0 | R2 | k1 | R2 | kH | R2 | n | kp | R2 | ||
| TA | 0.026 | 0.98 | 0.0001 | 0.98 | 0.59 | 0.92 | 0.78 | 3.04 | 0.96 | Non-Fickian |
| PDGF | 0.019 | 0.95 | 0.00006 | 0.91 | 0.45 | 0.99 | 0.56 | 2.49 | 0.99 | Non-Fickian |
2.5. Intracellular ROS scavenging by TA-containing hydrogel
In diabetic wounds, hyperglycemia induces persistent inflammation and excessive ROS accumulation at the wound site.4,40 This increases oxidative stress in the wound microenvironment and ultimately delays wound healing. We hypothesized that this system could effectively induce macrophage differentiation by removing reactive oxygen species (ROS) through the antioxidant effect of TA. TSP–T and TSP–TP hydrogels containing TA have excellent antioxidant properties, which is due to the sustained release of TA. In addition, as shown in Fig. 3, TA loaded in the hydrogels shows long-term, sustained antioxidant effects. Based on this, we estimated that the hydrogels could lower the level of intracellular ROS for a long time without losing their antioxidant effects when applied in the body, and induce macrophage differentiation. To verify this, two groups of hydrogels were prepared. Group 1 used hydrogels prepared immediately before treatment, group 2 used hydrogels soaked in DPBS for 7 days. This experiment was designed to determine whether TA encapsulated in the hydrogels does not lose its antioxidant activity over a long period of time in a physiological environment such as PBS. To determine the level of intracellular ROS scavenging of the hydrogels, RAW 264.7 cells were treated with TSP, TSP–T, and TSP–TP hydrogels, and the intracellular ROS levels were measured by H2DCFDA staining. Bright green fluorescence indicated the presence of ROS within the cell, providing a visual confirmation of ROS levels.64 Analysis of the intracellular ROS levels using confocal microscopy revealed that green fluorescence intensity was lower in macrophages treated with TA-containing hydrogels (TSP–T and TSP–TP) in both group 1 and group 2. Notably, there was no significant difference in the fluorescence intensities between TSP–T and TSP–TP, indicating that PDGF co-encapsulated in the hydrogels did not interfere the release or antioxidant activity of TA loaded in the hydrogel. In contrast, TSP hydrogels without TA exhibited higher green fluorescence intensities (Fig. 5A). This indicates that TA encapsulated in the hydrogels can effectively remove intracellular ROS through its antioxidant effect. As shown in Fig. 2C, the small size of the bioactive material loaded nanocomposites may have facilitated cellular uptake, which is presumed to have contributed to the intracellular ROS scavenging. Notably, the green fluorescence intensity level was still low in the TA-containing hydrogel of group 2, which correlated with Fig. 3C, suggesting that the antioxidant activity of TA can be maintained for a long time even under physiological conditions. Collectively, these results demonstrate that TSP–T and TSP–TP hydrogels can stably encapsulate TA and enable long-term ROS scavenging, contributing to the modulation of the wound microenvironment and potentially promoting tissue regeneration. | ||
| Fig. 5 Long-term ROS scavenging and macrophage differentiation effects of TSP, TSP–T, and TSP–TP hydrogels. Group 1 consisted of freshly prepared hydrogels, while group 2 consisted of hydrogels incubated in DPBS for 7 days. (A) Representative H2DCFDA staining images and fluorescence intensity quantification for both group 1 and group 2 hydrogels (scale bar = 100 μm). (B) Relative gene expression of M1 and M2 markers in RAW264.7 cells treated with hydrogels from both groups. Data are presented as mean ± standard deviation (n = 5 for each group). | ||
2.6. In vitro M1 to M2 transition of macrophages by TA-containing hydrogel
Regulation of ROS levels plays a crucial role in macrophage polarization. Excessive ROS induces oxidative stress in cells, leading to persistent inflammation at the wound site and inhibiting differentiation into M2 macrophages.65 Previous studies have shown that reducing excessive ROS can convert macrophages to the M2 phenotype.55,66 We hypothesized that our system could effectively induce macrophage differentiation by eliminating ROS through the antioxidant effect of TA. To investigate whether the ROS-scavenging effect of the TA-containing hydrogel not only reduces intracellular ROS but also promotes M2 polarization of macrophages, RAW 264.7 cells treated with the hydrogels were harvested, and their mRNA was subsequently extracted for analysis via real-time polymerase chain reaction (RT-PCR) (Fig. 5B). In group 1, macrophages exposed to LPS exhibited a 19.4-fold and 40.7-fold increase in the expression of M1 marker iNOS and TNF-α, respectively, compared to the control. Conversely, in macrophages treated with TSP–T and TSP–TP hydrogels, TNF-α expression decreased by 9.7-fold and 11.67-fold, while iNOS expression decreased by 7.3-fold and 9.1-fold, respectively. Additionally, the expression of the M2 marker IL-10 rose by 29.5-fold and 36.5-fold, and Arg-1 expression increased by 23.8-fold and 21.2-fold, respectively, demonstrating through gene expression that TA in the TSP hydrogel effectively induced M2 macrophage differentiation. This result supports the hypothesis that TA treatment can lower intracellular ROS levels and induce macrophage polarizations. ROS scavenging and the induction of macrophage phenotypic changes by TA were also confirmed in group 2 using hydrogels soaked in DPBS for 7 days. In the group 2, there was a trend of decreasing M1 marker expression and increasing M2 marker expression, similar to group 1. Notably, in group 2, TSP–T hydrogel reduced iNOS expression by 2.62-fold and TNF-α by 2.3-fold compared to the control group, while the TSP–TP hydrogel reduced iNOS and TNF-α expressions by 1.0-fold and 2.4-fold, respectively. Interestingly, both hydrogels containing TA (TSP–T and TSP–TP) showed enhanced antioxidant effects and macrophage polarization in group 2 compared to group 1. To investigate the cause of this enhancement, we compared the TA release levels in the TSP–TP hydrogel between groups 1 and 2 (Fig. S9, ESI†). The results exhibited that the TA release was higher in group 2, compared to group 1. This suggests that the hydrogel in group 2, after being soaked in PBS for 7 days, absorbed water from PBS leading to hydrogel swelling, which decreased their internal structural density and consequently accelerated the degradation process, increasing the release of TA.To complement the gene expression data and verify macrophage polarization at the protein level, immunofluorescence (IF) staining and enzyme-linked immunosorbent assay (ELISA) were performed on RAW264.7 cells after hydrogel treatment (Fig. S10, ESI†). In IF staining, the M1 marker CD86 (green) and the M2 marker CD206 (red) were stained, and the red fluorescence was predominant in the TSP–T and TSP–TP hydrogel groups including TA. This indicates that the polarization toward M2 was induced by the removal of ROS in M1 macrophages by the antioxidant function of the hydrogel. Additionally, ELISA was conducted to qualify cytokine secretion in cell culture medium. The levels of TNF-α and IL-10 in the LPS-treated group were 2826.0 ± 209.28 pg mL−1 and 600.6 ± 385.40 pg mL−1, respectively. The TSP hydrogel without TA showed similar values, indicating that there was no significant difference from the LPS-treated group. However, both TSP–T and TSP–TP hydrogels in groups 1 exhibited a decrease in TNF-α and an increase in IL-10, and there was no significant difference between TSP–T and TSP–TP. Specifically, the TNF-α and IL-10 levels of TSP–T were 1679.1 ± 120.75 pg mL−1 and 1696.6 ± 117.7 pg mL−1, and in TSP–TP, they were 1793.1 ± 209.01 pg mL−1 and 1455.2 ± 42.24 pg mL−1, respectively. This result of TNF-α reduction and IL-10 increase reveal the TA encapsulated in the TSP–T and TSP–TP hydrogel induced the M2 phenotype macrophage polarization. Interestingly, RAW264.7 cell treated with hydrogels from both group 1 and group 2 exhibited similar trends in cytokine secretion. This finding reveals that the TA encapsulated TSP hydrogel system can induce ROS scavenging and macrophage polarization without losing the activity of TA for a long time in a physiological environment. Collectively, TA encapsulated into the hydrogel effectively induces the transition of macrophages from the M1 to M2 phenotype by scavenging ROS. Importantly, this system maintains antioxidant activity under physiological conditions and continuously promotes M2 polarization, indicating its potential to continuously remove excessive ROS present in diabetic wound environments. Therefore, the TSP–TP hydrogel proposed in this study presents a promising therapeutic strategy for chronic wounds, that long term ROS removal is required. By inducing M2 macrophage polarization, this system can regulate the inflammatory microenvironment toward a regenerative environment, offering clinically meaningful implications for the treatment of diabetic chronic wounds.
2.7. Cell migration and angiogenesis effect of hydrogels
In diabetic patients, angiogenesis is suppressed during wound healing at the wound site, and fibroblast migration and proliferation are also inhibited.67,68 To address these issues, we propose using PDGF in this study, a growth factor crucial for wound healing. PDGF is well known to promote cell proliferation, migration, and angiogenesis, and is used as the main active ingredient of Regranex™, an FDA approved diabetic foot ulcer treatment.69 PDGF, the first growth factor released when a wound occurs, participates in all wound healing processes.22,70 Incorporating PDGF into a hydrogel for delivery is hypothesized to enhance cell migration and angiogenesis, thus facilitating effective tissue regeneration. The enhancement of cell migration by PDGF was demonstrated in a migration assay, where 24 hours post-treatment, the scratch area for the TSP–TP group reduced to 58.3 ± 5.26%, a significantly higher migration rate of fibroblasts compared to the TSP and TSP–T hydrogel samples, which showed scratch areas of 82.7 ± 7.78% and 73.8 ± 5.26% respectively, indicating reduced fibroblast migration (Fig. 6A). This suggests that PDGF loaded into the hydrogel was released and acted on the cells to promote migration. This cell migration plays a key role in tissue regeneration, including the reconstruction of the wound site and the production of ECM components.71 Therefore, the PDGF loaded TSP–TP hydrogel demonstrates a promising therapeutic strategy that can induce effective tissue regeneration. | ||
| Fig. 6 Effects of PDGF loaded TSP–TP hydrogels on cell migration and angiogenesis (A) Fibroblast migration assay of TSP, TSP–T, and TSP–TP measured over 24 hours (scale bar = 200 μm). (B) HUVEC cell tube formation assay and quantitative data on node number, branch number, and total tube length of TSP, TSP–T, and TSP–TP at 8 hours (scale bar = 100 μm). Quantitative results were derived using ImageJ software and are presented as mean ± standard deviation (n = 5 for each group). | ||
Neovascularization is a crucial factor for effective wound healing, as the formation of new blood vessels enables the efficient delivery of oxygen and nutrients to the regenerating tissue, thereby accelerating tissue repair.72 To evaluate the in vitro angiogenic effects of PDGF, TSP, TSP–T, and TSP–TP hydrogels, as well as PDGF alone, were cultured with HUVECs, and the formation of tubular structures was observed for 4 and 8 hours (Fig. S11, ESI†). TSP–TP hydrogels loaded with PDGF exhibited faster and more active angiogenic effects than the TSP and TSP–T hydrogel treatment groups after 8 h, which was similar to the effects observed in the PDGF alone treatment group (Fig. 6B). Quantifying angiogenesis revealed significantly more nodes, branches, and a longer total tube length at 8 hours in the TSP–TP hydrogel group. This implies that TSP–TP hydrogel can promote the formation of longer and more complex vascular networks than the control, TSP, and TSP–T (Fig. 6B). Similarly, there was no statistically significant difference between TSP–TP and PDGF alone. These results demonstrate that PDGF retains its activity and structural integrity after encapsulation and release from the hydrogel, suggesting that the original biological function of PDGF is well preserved. This preservation is likely due to the stable encapsulation into the hydrogel through simple ionic interactions without chemical modification. Therefore, the TSP–TP hydrogel system encapsulated with PDGF exhibits the potential to promote tissue regeneration by effectively enhancing cell migration and angiogenesis.
2.8. In vivo diabetic wound healing
Based on the results of the in vitro experiments, we further evaluated the therapeutic effects of TA and PDGF delivered via the hydrogel system on diabetic wound healing using a diabetic mouse model (Fig. 7A). Diabetic wound sites are characterized by chronic inflammation, elevated levels of ROS, and the degradation of ECM and proteins due to MMP overexpression. Furthermore, structural modifications of growth factor receptors can reduce the effectiveness of growth factors.29 We hypothesized that sustained delivery of antioxidants and growth factors through TSP–TP hydrogel could reduce ROS levels to those of normal tissues and enhance the efficacy of growth factors, thus promoting effective wound healing. To clearly verify the synergistic effect of the TSP–TP hydrogel delivering growth factors and antioxidants simultaneously, the control group was set as an untreated group, a TSP-only group, a TSP hydrogel group mixed with TA only (TSP–T), and a TSP hydrogel group mixed with PDGF only (TSP-P). The results showed that on day 7, wound area ratios were 63.9 ± 11.23%, 55.7 ± 13.06%, 51.8 ± 11.84%, and 45.1 ± 11.07% for the control, TSP, TSP–T, and TSP-P groups, respectively. The TSP–TP hydrogel group demonstrated a significantly reduced wound area ratio of 36.4 ± 8.79%, approximately half that of the control group. This reduction is attributed to the antioxidant effects of the loaded TA, which promotes the differentiation of M1 macrophages into M2 types, resolves chronic inflammation, and advances to the normal proliferation phase, thereby enhancing tissue regeneration through PDGF.73 On the 14th day, wound area ratios for the control, TSP, TSP–T, TSP-P, and TSP–TP groups were 20.2 ± 6.83%, 19.7 ± 3.71%, 9.2 ± 2.55%, 12.6 ± 3.93%, and 2.9 ± 1.95%, respectively. The TSP–TP hydrogel group exhibited a significantly higher wound healing rate compared to other groups (p value ≤ 0.05). Specifically, the TSP–T and TSP-P hydrogel groups showed better wound healing effects than the control group, but not as effectively as the TSP–TP hydrogel (Fig. 7B and C). It was confirmed that the rapid wound healing by the TSP–TP hydrogel, due to the synergistic effect of TA and PDGF, was consistent with the in vitro data. Moreover, the TSP–TP hydrogel applied in the body gradually decomposed along with tissue regeneration and disappeared without any side effects.32,74 This indicates that the TSP–TP hydrogel may serve as a potential treatment for diabetic wounds by promoting tissue regeneration and decomposing safely. | ||
| Fig. 7 In vivo tissue regeneration in diabetic wounds and histological analysis. (A) Provides a timeline for the in vivo study. (B) Shows optical images tracking the wound area over time post-treatment with hydrogels (scale bar = 5 mm). (C) Details the quantification of the wound closure rate using ImageJ software. (D) Displays an H&E stained image of the entire wound site (scale bar= 200 μm). (E) Presents H&E and MT staining of the epidermis and dermis regions. Quantifications of (F) wound length, (G) granulation thickness, (H) epidermis thickness, and (I) collagen deposition at data were analyzed using ImageJ software and are presented as mean ± standard deviation (n = 5 for each group). | ||
For detailed analysis of tissue regeneration, histopathological analysis was conducted using H&E staining and MT staining. The wound length in the TSP–TP hydrogel treatment group was 0.71 μm, significantly shorter than those in the control groups (Fig. 7D and F). The TSP–TP group demonstrated superior tissue regeneration, as indicated by granulation tissue and epidermal thickness (Fig. 7E and G and H). Moreover, unlike in the other groups, hair follicles were observed in the regenerated site of the TSP–TP group. In Fig. 7B, hair growth was notable only in the regenerated wound area treated with TSP–TP hydrogel, indicating that the tissue function was restored to a state close to normal skin tissue. Masson's trichrome staining was utilized to examine collagen accumulation in the wound area. Consequently, the TSP–TP hydrogel group exhibited a 68.19% accumulation of collagen in the dermal layer, which was significantly higher than in the other groups (p value < 0.0001) (Fig. 7E and I). This confirmed effective formation of the ECM and successful tissue regeneration. Through this in vivo diabetic mouse model experiment, the hypothesis that the TSP–TP system can effectively release antioxidants and growth factors to promote diabetic wound healing was validated. In addition, due to its thermo-responsive properties, this hydrogel can be easily applied to deep and irregular wound sites by converting into a gel immediately after injection, and its biodegradability naturally decomposes without a separate removal process. These characteristics offer significant advantages for improving patient convenience and treatment efficiency in future clinical applications.
2.9. Immunofluorescence (IF) staining for angiogenesis and macrophage differentiation
Subsequently, we confirmed the effects of angiogenesis and macrophage polarization in the regenerated tissue areas post-treatment with hydrogels using immunofluorescence (IF) staining (Fig. 8A and C). Angiogenesis is crucial for tissue regeneration as it supplies essential nutrients and oxygen to the wound area.72 The in vivo angiogenic impacts of the TSP–TP hydrogel were assessed through immunofluorescence labelling of the angiogenic marker, cluster of differentiation-31 (CD31), and the vascular smooth muscle marker, alpha-smooth muscle actin (α-SMA).75 On day 14 post-treatment, minimal expression of angiogenesis markers CD31 and α-SMA was observed in the control and TSP groups; however, distinct vascular structures were evident in the TSP–T, TSP-P, and TSP–TP treatment groups. Notably, the TSP–TP group exhibited abundant mature vascular structures (yellow arrows) with high fluorescence intensity (Fig. 8A and B). This suggests that the TSP–TP hydrogel effectively delivered antioxidants and growth factors in a diabetic wound environment, reducing ROS and enhancing the influence of growth factors, thereby promoting efficient tissue regeneration. | ||
| Fig. 8 Angiogenic and macrophage polarization effects of TSP–TP hydrogel demonstrated through immunofluorescence analysis. (A) IF images of angiogenic markers, CD31 and α-SMA (scale bar = 100 μm), with arrows highlighting blood vessels. (B) Quantification of fluorescence intensities for CD31 (red) and α-SMA (green). (C) IF images of macrophage markers, iNOS and CD206 (scale bar = 100 μm). (D) Quantification of fluorescence intensities for iNOS (green) and CD206 (red). All quantitative data were analyzed using ImageJ software and are presented as mean ± standard deviation (n = 5 for each group). | ||
Moreover, iNOS and CD206 were selected to mark M1 and M2 macrophage, respectively, and then stained to verify the extent of macrophage polarization in the regenerated area.76 In chronic wounds, the overexpression of M1 macrophages and delayed transition to M2 macrophages impede tissue regeneration; on the other hand, polarization into M2 facilitates tissue regeneration by secreting various cytokines and growth factors that enhance stem cell recruitment and the proliferation and migration of keratinocytes, fibroblasts, and endothelial cells.77 Observations indicated that CD206 expression was virtually non-existent in the control, TSP, and TSP-P groups, but significantly increased in the TSP–T and TSP–TP groups. This implies that the antioxidant properties of the loaded TA effectively reduced ROS levels at the wound site, thereby enhancing immune regulation and tissue regeneration (Fig. 8C and D). These results illustrate that the TSP–TP hydrogel system effectively supports tissue regeneration processes such as angiogenesis and ECM remodeling, typical of normal tissues, even within a chronic wound environment by consistently delivering antioxidants and growth factors.
Based on the in vitro and in vivo results, we demonstrated the diabetic wound healing efficacy of TSP–TP hydrogel. In particular, the antioxidant TA and the growth factor PDGF were stably encapsulated through the physical interaction with the functional groups in the TSP hydrogel and were sustainably released over a long period. The simultaneous release of these bioactive molecules effectively reduced ROS level at the wound site and promoted tissue regeneration by inducing the differentiation of macrophages. Modulation of the wound microenvironment further enhanced the biological activity of the growth factors, leading to effective tissue regeneration through angiogenesis and fibroblast migration. Therefore, the TSP–TP hydrogel has potential as a next-generation treatment material for chronic wound healing by stable co-delivery of TA and PDGF.
3. Experimental
3.1. Materials
All chemical reagents were used as received unless otherwise noted. L-Isoleucine ethyl ester hydrochloride (IleOEt·HCl) and hexachlorocyclotriphosphazene were obtained from Angene Chemical (UK) and Sagechem (China), respectively. 750 Da methoxy poly(ethylene glycol), ethanolamine, glutaric anhydride (GA), 4-(dimethylamino)pyridine (DMAP), tannic acid, and PDGF-BB were sourced from Sigma-Aldrich (USA). 750 Da methoxy poly(ethylene glycol) was converted to 750Da α-amino-ω-methoxy poly(ethylene glycol) (AMPEG). The detailed synthetic method is described in the ESI.† Anhydrous tetrahydrofuran (THF), methanol, and triethylamine (TEA) were sourced from Daejung (Korea). The TEA was distilled in N2 atmosphere and barium oxide (Daejung) was added to the distilled reactor for drying. The Dulbecco's phosphate-buffered saline (DPBS) and Dulbecco's modified Eagle's medium (DMEM) were purchased from Welgene (Korea). 4% paraformaldehyde was procured from Biosesang (Korea). Antibodies for α-SMA and CD31 were purchased from Abcam (UK), and antibodies for iNOS and CD206 were obtained from Invitrogen (USA).3.2. Synthesis of TSP polymer
All procedures were conducted under an N2 atmosphere and a cannula was used to prevent moisture. All chemical reagents were vacuum-dried before use. IleOEt·HCl (128.33 g, 0.65 mol) was dissolved in anhydrous THF (1300 mL) and the solution was stirred in a dry ice bath before adding distilled TEA (400 mL). Subsequently, poly(dichlorophosphazene) (50 g, 0.43 mol) dissolved in anhydrous THF (500 mL) was incorporated to reaction flask. The reaction flask was heated to 45 °C for 24 h. On a subsequent day, the flask was cooled to room temperature, then aminoethanol (6.06 mL, 0.099 mol) and AMPEG (80.9 g, 0.11 mol) were added and reacted at 50 °C for 24 h. The obtained polymer solution was dialyzed using a dialysis membrane (Spectra/Por membrane, MWCO: 12–14 kDa) in methanol for 4 days and distilled water (DW) for another 4 days. The final polymer product was filtered using a syringe filter (Whatman, PP filter, 0.45 μm) and freeze-dried to yield TSP polymer with hydroxyl groups. Finally, the carboxylic acid modified polymer was synthesized by following reactions. The TSP polymer with a hydroxyl group (90.4 g, 0.18 mol) was dissolved in anhydrous THF (1000 mL) and stirred at room temperature. To this polymer solution, glutaric anhydride (14.5 g, 0.12 mol) and DAMP (15.6 g, 0.12 mol) dissolved in anhydrous THF (500 mL) were added. The polymer mixture was stirred at 50 °C for 24 h. After the reaction, the obtained reaction mixture was filtered and evaporated. The polymer product was purified by dialysis in methanol for 4 days and then distilled water for 4 days. The carboxylated TSP polymer was obtained through lyophilization.3.3. Preparation of TSP, TSP–T, TSP-P and TSP–TP hydrogel
TSP hydrogels were prepared by dissolving TSP polymer in DPBS (10 wt%) with stirring at 4 °C. To prepare TSP–T hydrogels, TSP hydrogel (10 wt% in DPBS) was mixed with various concentrations of TA solutions (0.1 wt%, 0.3 wt%, 0.5 wt%, and 1 wt% in DPBS), and the mixtures were sonicated on ice for 30 min. The optimized TSP–T hydrogel was used at 0.1 wt% TA concentration. TSP-P hydrogels were prepared by adding PDGF (1 μg) to the 10 wt% TSP hydrogel (1 mL), gently mixing using a pipette, and incubating for 30 min. Finally, to prepare TSP–TP hydrogel, the same amount of PDGF (1 μg) was added to the optimized TSP–T hydrogel (1 mL), gently mixed by pipette, and incubated for 30 min. All fabrication processes were performed on ice.3.4. TSP polymer characterization
The chemical structure of the TSP polymer was characterized using 1H-NMR spectroscopy (Varian Gemini-400 spectrometer, operating at 400 MHz in Fourier transform mode with CDCl3) and FT-IR (ThermoFisher, USA). TSP polymer samples were prepared at a concentration of 1 wt% in CHCl3 to measure FT-IR. The weight-average molecular weight (Mw) of the TSP polymer was measured using a GPC system (Agilent, 1260 Infinity II) equipped with a refractive index detector and a gel column (Agilent, PLgel 5 μm MIXED-C). THF containing 0.1 wt% tetrabutylammonium bromide served as the mobile phase, and the analysis was conducted at 35 °C with a flow rate of 1 mL min−1. Additionally, polystyrene standard samples with molecular weights of 6570000, 3152000, 885000, 479200, 194500, 75050, 22290, 10330, 4880, 1210, 580, and 162 g mol−1 were used for calibration.
3.5. Cryo-SEM imaging
Cryo-focused ion beam scanning electron microscopy (cryo-FIB/SEM) analysis was performed using a Quanta 3D FEG system (Thermo Fisher Scientific, USA) equipped with an Alto 2500 cryo-transfer system (Ganta, UK). Hydrogel samples were prepared in both solution and gel states. The samples were rapidly submerged in liquid nitrogen, and after 1 second, the freezing chamber was evacuated. They were then transferred to a preparation chamber maintained at approximately −190 °C under a pre-evacuated pressure of 10−5 mbar. A metal coating was applied via plasma sputtering at a 10 mA current for 60 seconds. The coated samples were subsequently placed into the microscope chamber, which was also pre-evacuated to 10−5 mbar and cooled to −190 °C. Cryo-SEM imaging was performed with a 5 keV electron beam and an electron current of 11.8 pA. FIB milling was carried out using a 30 keV gallium ion beam with an ion current of 1 nA. To acquire EDS spectra, the electron beam energy was increased to 15 keV.783.6. Hydrogel characterization
The size and zeta potential of the hydrogel complexes were measured by dynamic light scattering (DLS) (Marlvern, UK). For particle size measurement, hydrogel samples were diluted to 1 wt% in DPBS and measured at 25 °C. For zeta potential, all hydrogel samples, including TA and PDGF, were prepared at 1 wt% concentration in DW and analyzed at room temperature.3.7. Swelling capability of hydrogels
The 100 μL of hydrogel in a liquid state was placed into a Millicell insert (diameter: 12 mm, Millipore) and incubated at 37 °C for 10 min to induce gelation. Subsequently, the hydrogel was immersed in 5 mL of warm DPBS and weighed at 24, 72, 168, 240, and 336 hours. Excess DPBS was removed using tissue paper. The swelling ratio was calculated using the following equation:| Swelling ratio (%) = Wt/W0 × 100 | (1) |
3.8. Rheological characterization of hydrogels
The rheological properties of the TSP, TSP–T, and TSP–TP hydrogels were measured using a rheometer (MCR102, Anton Parr). Hydrogel solutions were prepared at 10 wt% concentration in DPBS. Measurements were conducted using 25 mm parallel plates with a 0.5 mm zero gap. Temperature dependent modulus was evaluated at 1 Hz frequency and 1% strain, with temperatures ranging from 4 °C to 60 °C. Frequency sweeps were conducted at 37 °C with 1% strain, and all modulus data were processed using Anton Parr software from Rheocompass.3.9. MTT assay
NIH3T3 cells were employed for the MTT assay. All hydrogel samples (80 mg) were prepared in 4 mL of DMEM media (without FBS) 24 hours prior to beginning the MTT assay. NIH3T3 cells (2 × 104 cells per well) were seeded in a 96-well plate (SPL, Korea) and incubated for 24 h at 37 °C with 5% CO2. Following the removal of all media, hydrogel solutions were introduced to each well. At this point, hydrogel samples were diluted to concentrations ranging from 20000 μg mL−1 to 100 μg mL−1. After 24 hours, the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solution was added to each well and further incubated for 3 hours at 37 °C with 5% CO2. Dimethyl sulfoxide (150 μL; Daejung, Korea) was then added, and the absorbance was measured at 570 nm using a Flash Multimode Reader (VarioskanTM, Thermo Scientific, USA).79 Cell viability was determined using the following equation:| Cell viability (%) = As/Ac × 100 | (2) |
3.10. Live/dead assay
NIH3T3 cells (1 × 105 cells per well) were seeded in a 24-well plate (SPL, Korea) and incubated for 24 h at 37 °C with 5% CO2. 100 μL of 10 wt% hydrogel solutions were prepared in transwells (pore size 1 μm, Corning) and incubated for 20 minutes at 37 °C. The samples were subsequently placed in wells and incubated for durations of 1, 3, 5, and 7 days. At each time point, the wells were washed with warm DPBS and stained using the live/dead assay kit (Invitrogen, Carlsbad, CA, USA). Staining was performed according to the manufacturer's protocol. The stained cells were imaged using a confocal laser scanning microscope (Carl Zeiss, LMS7000).3.11. DPPH assay
Briefly, a 0.2 mM DPPH solution (Biozoa, Korea) was utilized in a cold condition. All samples were dissolved in methanol. To conduct the assay, 200 μL of the sample and 800 μL of the 0.2 mM DPPH solution were combined in 1.5 mL Eppendorf tubes and vortexed in the dark. After 15 minutes, 200 μL from each tube was transferred to a 96-well plate, and the absorbance was measured at 517 nm. To evaluate long-term antioxidant properties, 100 μL of hydrogel was introduced into a 2 mL Eppendorf tube containing 1 mL DPBS. At designated time points, the DPBS was collected, and the samples were lyophilized. Post-lyophilization, the samples were redissolved in 200 μL of methanol and 800 μL of fresh DPPH solution. Following another 15-minute incubation, 200 μL of the mixed solution was transferred to a 96-well plate, and absorbance at 517 nm was measured using a Flash Multimode Reader (VarioskanTM, Thermo Scientific, USA). The DPPH scavenging percentages were calculated using the following equation:| DPPH scavenging effect (%) = (Ab − As)/Ab × 100 | (3) |
3.12. H2DCFDA staining
RAW 264.7 cells (1 × 105 cells per well) were seeded in 24-well plates (SPL, Korea) and cultured in a cell incubator (37 °C, 5% CO2) for 24 h. After 24 hours, the cells were exposed to DMEM (10% FBS and 1% P/S) containing LPS (500 ng mL−1) for 12 h. Subsequently, all media were replaced with fresh DMEM (10% FBS and 1% P/S) and 100 μL of various TSP, TSP–T, or TSP–TP hydrogels were administered on 24-well inserts (Corning, USA). The hydrogels were gelled for 10 min in a 37 °C incubator and then treated with cells for 24 h under the same conditions (37 °C, 5% CO2). For fluorescence staining, the media were removed and 500 μL of H2DCFDA solution (Invitrogen, USA) was added to each well. After a 30-minute incubation, the cells were washed three times with warm DPBS, and fluorescence was quantified using a confocal laser scanning microscope (Carl Zeiss, LMS7000).3.13. RT-PCR
We utilized the same protocol used for H2DCFDA staining to prepare RAW 264.7 cells. RAW 264.7 cells were treated with each hydrogel sample for 24 h. RNA was extracted using Trizol (InvitrogenTM, USA), and its concentration was determined using a NanoDrop ND-1000 (Thermo Fisher, USA). The RNA was reverse-transcribed into cDNA using Superscript VILO MasterMix (InvitrogenTM, USA). We assessed the expression of M1 macrophage markers (iNOS and TNF-α) and M2 macrophage markers (Arg-1 and IL-10) using a QuantStudioTM 1 RT-PCR instrument (Applied Biosystems, CA). Primers were obtained from Cosmogenetech (Korea), and mRNA expression levels were normalized against the β-actin housekeeping gene. The experiments were conducted in triplicate. Gene expression was quantified using the 2−ΔΔCt method. Details on primer sequences can be found in Table S2 (ESI†).3.14. ELISA assay
TSP–TP hydrogels were prepared in liquid form at 4 °C. 100 μL of the TSP–TP hydrogels were placed in Millicell inserts (diameter: 12 mm, Millipore) and allowed to gel at 37 °C for 10 min. Following gelation, the inserts were submerged in 5 mL of warm DPBS within a 15 mL conical tube (SPL, Korea). The conical tubes containing the hydrogels underwent incubation in a shaking water bath (Vision, Korea) at 37 °C. DPBS was collected at specified times and stored at −20 °C. PDGF-BB release was quantified using an ELISA kit (Human PDGF-BB Immunoassay, R&D Systems, USA) following the manufacturer's protocol. The amount of released cytokines was quantified using RAW264.7 cell culture medium. RAW264.7 cells (1 × 105 cells per well) were cultured in a 24-well plate for 24 h, and then treated with DMEM (with FBS) containing LPS (500 ng mL−1) to induce an inflammatory response for 12 h. Afterwards, the cells were replaced with DMEM (without FBS). Simultaneously, 100 μL of each hydrogel was loaded to a 24-well insert and incubated until gelation. The inserts were transferred to the 24-well plate containing the cells and co-cultured for 24 h. After the culture was completed, the medium was collected and ELISA analysis was performed using a TNF-α ELISA kit (RK00027, ABclonal Technology, USA) and an IL-10 ELISA kit (RK00016, ABclonal Technology, USA), respectively. All experiments were performed according to the manufacturer's protocol.3.15. Folin–Ciocalteu (FC) assay
The TA release profile was determined using the FC assay. The release of TA was established by placing the TSP–T and TSP–TP hydrogel in DPBS, using the same method described in section for ELISA analysis. TSP–T hydrogels loaded with various concentrations of TA (0.1, 0.3, 0.5 and 1 wt%) and TSP–TP with optimized 0.1 wt% TA were used in the experiments. 100 μL of DPBS collected at specified times was transferred to a 2 mL Eppendorf tube, 200 μL of FC solution (Sigma-Aldrich, USA) was added, and the mixture was agitated gently for 10 min. Subsequently, 800 μL of 2% sodium carbonate was added, and the mixture was incubated in the dark at room temperature for 1 hour. The absorbance at 765 nm was measured using a Flash Multimode Reader (Varioskan™, Thermo Scientific, USA).553.16. Cell migration assay
Cell migration was assessed using a scratch assay. NIH3T3 cells (1 × 105 cells per well) were seeded in 24-well plates and cultured at 37 °C in a 5% CO2 atmosphere for 24 h. Post-incubation, 100 μL of TSP, TSP–T, or TSP–TP hydrogels were introduced into 24-well inserts and allowed to gel for 10 min at 37 °C. Following gelation, a scratch was made in the cell monolayer using a 200 μL pipette tip. The prepared hydrogel inserts were subsequently placed in the wells and cells were cultured under standard conditions. Cell migration was periodically monitored using a microscope (Zeiss Axio Vert.A1, Germany) and the wound area was quantitatively analyzed using ImageJ software.3.17. Angiogenesis assay
In vitro angiogenesis was evaluated using a tube formation assay. Human Umbilical Vein Endothelial Cells (HUVECs) cultured in Human Umbilical Vein Endothelial Growth Medium (CEFOgro™, Korea), at passages 5–6, were utilized for the assay. 250 μL of Matrigel (Corning, USA) was added to each well of a 24-well plate to gel, followed by the seeding of HUVECs (1 × 105 cells per well). Subsequently, 24-well inserts containing 100 μL of TSP, TSP–T, or TSP–TP hydrogels and the same amount of PDGF in the hydrogels were added to each well. The plates were incubated until the designated time points were reached. At each time point, HUVECs were stained with Calcein AM (Invitrogen, USA) and imaged using a confocal laser scanning microscope (Carl Zeiss, LSM7000).3.18. In vivo diabetic wound healing
All animal studies were conducted in accordance with the relevant laws and guidelines of the Institutional Animal Care and Use Committee (IACUC) at the Korea Institute of Science and Technology (KIST), and the experimental protocol received approval from the IACUC (2023-086). To establish a diabetic animal model, male C57BL/6J mice aged 4 weeks (DBL, Korea) were injected with streptozotocin (STZ, Glentham Life Science, UK), which is known to induce diabetes. The STZ solution was prepared by dissolving it in a sodium citrate buffer at pH 4.5 (8 mg mL−1, Biosesang, Korea). Mice received intraperitoneal injections of STZ (50 mg kg−1) daily for 7 consecutive days and were provided with 10% sucrose water to prevent sudden hypoglycemia. Blood glucose levels were monitored weekly using a Caresens N blood glucose meter (i-Sens, Korea). Mice with blood glucose levels above 300 mg dL−1 at 3 weeks post-injection were considered to have successfully developed diabetes. Diabetic mice aged 7 weeks, male, n = 5 per group, were randomly assigned to one of 5 groups: control, TSP, TSP–T, TSP-P, and TSP–TP. The hair on the mice's backs was shaved, and wounds were created using a 6 mm biopsy punch. PDMS molds measuring 1 × 1 cm with 8 mm holes were sutured around the wounds. 100 μL of hydrogel samples were applied to the wounds and allowed to gel for 30 seconds. Tegaderm (3 M, USA) was applied to all wounds and bandaged. Hydrogel samples were applied once and left in place for 14 days. Wound closure rates were calculated using ImageJ software. All mice were euthanized by CO2 inhalation on day 14, and tissue samples were collected and preserved in 4% formaldehyde solution.3.19. Analysis of regenerating skin tissue by histological analysis
Collected wound tissue samples were fixed in 4% paraformaldehyde at 4 °C for 2 days. The tissues were then embedded in paraffin, from which blocks were created and subsequently sliced into 5 μm-thick sections using a microtome. The tissue slices were deparaffinized and rehydrated by sequential immersion in xylene, ethanol concentrations of 100%, 90%, 80%, 70%, and DW, respectively. The slices were then stained with hematoxylin and eosin (H&E) and Masson's trichrome (MT).3.20. Immunofluorescence (IF) staining
Hydrogel-treated RAW264.7 cells were prepared following the same protocol as for H2DCFDA staining. After washing with DPBS, cells were fixed with 4% paraformaldehyde for 1 h at room temperature (RT) and blocked with 3% BSA for 1 h at RT. Cells were then incubated overnight at 4 °C with primary antibodies: anti-CD86 (1 μg mL−1, ab220188, Abcam) and anti-CD206 (1:100, PA5-101657, Invitrogen). After washing three times with DPBS, cells were incubated with secondary antibodies: Alexa Fluor 488-conjugated goat anti-mouse IgG (1 μg mL−1, A21121, Invitrogen), Alexa Fluor 555-conjugated goat anti-rabbit IgG (4 μg mL−1, A21428, Invitrogen), and DAPI (1 μg mL−1, 62248, Invitrogen) for 1 h at room temperature. Samples were washed with DPBS, mounted using VECTASHIELD™ anti-fade medium (H-1000, Vector Laboratories), and imaged with a confocal microscope (FV3000, Olympus). Mouse skin samples were collected after 14 days and fixed with 4% formaldehyde at 4 °C for 2 days. The fixed tissues were embedded into paraffin blocks and sectioned to a thickness of 5 μm. The sectioned tissues were then deparaffinized, rehydrated, and subjected to antigen retrieval in retrieval solution (S3022, DAKO, Denmark), heated at 100 °C for 15 minutes. After heating, the samples were washed three times with DPBS (1×, pH 7.4) and blocked with 1% BSA solution for 1 hour at room temperature. The samples were then treated with primary antibodies and incubated overnight at 4 °C. The primary antibodies used included anti-CD31 (1:200, ab28364, Abcam), anti α-SMA (1:100, ab7817, Abcam), anti-iNOS (5 μg mL−1, MA5-17139, Invitrogen), and anti-CD206 (1:100, PA5-101657, Invitrogen). The samples were washed three times with DPBS and subsequently treated with secondary antibodies for 1 hour. The secondary antibodies employed were Alexa Fluor488-conjugated goat anti-mouse IgG (1 μg mL−1, A21121, Invitrogen) and Alexa Fluor 555-conjugated goat anti-rabbit IgG (4 μg mL−1, A21428, Invitrogen). Tissues were washed three times with DPBS and mounted with FluoroshieldTM histological mounting medium (F6057, Sigma) containing DAPI. All samples were imaged using a confocal microscope (Carl Zeiss, LSM7000).
3.21. Statistical data analysis
All quantitative data are presented as mean ± standard deviation. Significant differences were analyzed using one-way analysis of variance (ANOVA) and two-way ANOVA, both supplemented with Tukey's post hoc and multiple comparison tests in GraphPad Prism 9.0. The p-values are represented as * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001, and ns = not significant.4. Conclusions
The key innovation of our study is the sustained delivery of TA and PDGF, whereby TA initially scavenges ROS and alleviates inflammation at the wound site, and then the sustained release of PDGF effectively induces tissue regeneration at the alleviated site. Within this hydrogel, antioxidants and growth factors are securely encapsulated through physical interactions, which not only minimizes the impact on surrounding tissues by reducing the initial burst release but also facilitates a long-term stable release. The antioxidants mobilized from the hydrogel efficiently alleviate ROS, leading to M2-type macrophage differentiation, and the growth factors enhance cell migration and angiogenesis, yielding synergistic effects on wound healing. The TSP hydrogel demonstrates exceptional mechanical properties, biocompatibility, and biodegradability, rendering it ideal for the localized delivery of antioxidants and growth factors. In an in vivo diabetic wound healing model, the TSP–TP hydrogel system significantly accelerated wound closure rates compared to groups in which antioxidants or growth factors were individually loaded into the TSP hydrogel. Moreover, the regenerated skin tissues exhibited properties similar to normal tissues. This suggesting that the TSP–TP hydrogel holds promise as a sustained local delivery system for dual antioxidants and growth factors tailored for diabetic wound healing.Author contributions
Jisun Kim: investigation, methodology, formal analysis, writing – original draft, writing – review & editing. Ki Wan Bong: visualization, writing – review & editing. Jung-Kyo Cho: visualization, writing – review & editing. Soo-Chang Song: conceptualization, visualization, writing – review & editing.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (2021M3E5D9021367). The schematic was created using PowerPoint with some elements from https://BioRender.com.References
- Y. Cai, K. Chen, C. Liu and X. Qu, Bioact. Mater., 2023, 28, 243–254 Search PubMed.
- W. Ouyang, Y. Jia and L. Jin, Am. J. Transl. Res., 2021, 13, 9554 CAS.
- G. Han and R. Ceilley, Adv. Ther., 2017, 34, 599–610 CrossRef PubMed.
- Y. Dong and Z. Wang, Front. Bioeng. Biotechnol., 2023, 11, 1304835 CrossRef PubMed.
- H. Zhao, J. Huang, Y. Li, X. Lv, H. Zhou, H. Wang, Y. Xu, C. Wang, J. Wang and Z. Liu, Biomaterials, 2020, 258, 120286 CrossRef CAS PubMed.
- L. Deng, C. Du, P. Song, T. Chen, S. Rui, D. G. Armstrong and W. Deng, Oxid. Med. Cell. Longevity, 2021, 2021, 8852759 CrossRef PubMed.
- F. He, P. Xu, Z. Zhu, Y. Zhang, C. Cai, Y. Zhang, J. Shao, F. Jin, Q. Li and J. You, Adv. Healthcare Mater., 2024, 2400150 Search PubMed.
- Y. Kuninaka, Y. Ishida, A. Ishigami, M. Nosaka, J. Matsuki, H. Yasuda, A. Kofuna, A. Kimura, F. Furukawa and T. Kondo, Sci. Rep., 2022, 12, 20327 CrossRef CAS PubMed.
- L. A. DiPietro, T. A. Wilgus and T. J. Koh, Int. J. Mol. Sci., 2021, 22, 950 Search PubMed.
- A. Hassanshahi, M. Moradzad, S. Ghalamkari, M. Fadaei, A. J. Cowin and M. Hassanshahi, Cells, 2022, 11, 2953 CrossRef CAS PubMed.
- Y. J. Fu, Y. F. Shi, L. Y. Wang, Y. F. Zhao, R. K. Wang, K. Li, S. T. Zhang, X. J. Zha, W. Wang, X. Zhao and W. Yang, Adv. Sci., 2023, 10, e2206771 CrossRef PubMed.
- S. Lin, Q. Wang, X. Huang, J. Feng, Y. Wang, T. Shao, X. Deng, Y. Cao, X. Chen, M. Zhou and C. Zhao, Biomed. Pharmacother., 2023, 157, 114052 Search PubMed.
- S. J. Wolf, W. J. Melvin and K. Gallagher, Semin. Cell Dev. Biol., 2021, 119, 111–118 CrossRef CAS PubMed.
- M. He, X. Gao, Y. Fan, L. Xie, M. Yang and W. Tian, J. Mater. Chem. B, 2021, 9, 1096–1106 Search PubMed.
- N. Xu, Y. Gao, Z. Li, Y. Chen, M. Liu, J. Jia, R. Zeng, G. Luo, J. Li and Y. Yu, Chem. Eng. J., 2023, 466, 143173 CrossRef CAS.
- W. Jia, L. Zhou, L. Li, P. Zhou and Z. Shen, Pharmaceuticals, 2023, 16, 101 CrossRef CAS PubMed.
- İ. Gülçin, Z. Huyut, M. Elmastaş and H. Y. Aboul-Enein, Arabian J. Chem., 2010, 3, 43–53 CrossRef.
- L. Alechinsky, F. Favreau, P. Cechova, S. Inal, P. A. Faye, C. Ory, R. Thuillier, B. Barrou, P. Trouillas, J. Guillard and T. Hauet, Biomolecules, 2020, 10, 439 CrossRef CAS PubMed.
- Y. Shen, T. Jia, J. Zeng, J. Wang, Z. Zhao, Y. Liu, Y. Jing, J. Pan, M. Ma, Y. Fu, S. Wei, J. Li, D. Wang, C. Wang and G. Chen, ACS Appl. Mater. Interfaces, 2024, 16, 63241–63254 CrossRef CAS PubMed.
- Y. Liu, M. Zhu, J. Ou, K. Li, X. Ju, Y. Tian and Z. Niu, Int. J. Biol. Macromol., 2024, 278, 134896 CrossRef CAS PubMed.
- J. G. Hodge, D. S. Zamierowski, J. L. Robinson and A. J. Mellott, Biomater. Res., 2022, 26, 50 CrossRef PubMed.
- J. W. Park, S. R. Hwang and I. S. Yoon, Molecules, 2017, 22, 1259 CrossRef PubMed.
- S.-Y. Zheng, X.-X. Wan, P. A. Kambey, Y. Luo, X.-M. Hu, Y.-F. Liu, J.-Q. Shan, Y.-W. Chen and K. Xiong, World J. Diabetes, 2023, 14, 364 CrossRef PubMed.
- M. J. White, P. S. Briquez, D. A. White and J. A. Hubbell, npj Regener. Med., 2021, 6, 76 CrossRef CAS PubMed.
- Y. Chu, D. Yu, P. Wang, J. Xu, D. Li and M. Ding, Wound Repair Regen., 2010, 18, 499–505 Search PubMed.
- X. Liu, Y. Liu, J. Zhou, X. Yu, J. Wan, J. Wang, S. Lei, Z. Zhang, L. Zhang and S. Wang, ACS Biomater. Sci. Eng., 2024, 10, 975–986 CrossRef CAS PubMed.
- O. Eskens and S. Amin, Int. J. Cosmet. Sci., 2021, 43, 123–130 CrossRef PubMed.
- P. Losi, E. Briganti, A. Magera, D. Spiller, C. Ristori, B. Battolla, M. Balderi, S. Kull, A. Balbarini and R. Di Stefano, Biomaterials, 2010, 31, 5336–5344 CrossRef CAS PubMed.
- H. J. Zhang, W. Zhao, S. Venkataraman, M. E. Robbins, G. R. Buettner, K. C. Kregel and L. W. Oberley, J. Biol. Chem., 2002, 277, 20919–20926 CrossRef CAS PubMed.
- Y. Liang, J. He and B. Guo, ACS Nano, 2021, 15, 12687–12722 Search PubMed.
- H. Li, H. Zhang, Y. Peng, X. Liu, J. Du and J. Liao, Adv. Healthcare Mater., 2024, e2401704, DOI:10.1002/adhm.202401704.
- J. Kim, Y. M. Kim and S. C. Song, Adv. Healthcare Mater., 2023, 12, e2202401 CrossRef PubMed.
- Q. Huang, Y. Zou, M. C. Arno, S. Chen, T. Wang, J. Gao, A. P. Dove and J. Du, Chem. Soc. Rev., 2017, 46, 6255–6275 RSC.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed.
- Y. Cai, L. Y. Xin, H. Li, P. Sun, C. Liu and L. Fang, J. Controlled Release, 2024, 365, 161–175 CrossRef CAS PubMed.
- B. E. Jensen, I. Davila and A. N. Zelikin, J. Phys. Chem. B, 2016, 120, 5916–5926 CrossRef CAS PubMed.
- S. Soukasene, D. J. Toft, T. J. Moyer, H. Lu, H.-K. Lee, S. M. Standley, V. L. Cryns and S. I. Stupp, ACS Nano, 2011, 5, 9113–9121 CrossRef CAS PubMed.
- J. Kim, B. B. Seo, K. H. Hong, S. E. Kim, Y. M. Kim and S. C. Song, Acta Biomater., 2022, 144, 183–194 CrossRef CAS PubMed.
- W. Zhang, W. Liu, L. Long, S. He, Z. Wang, Y. Liu, L. Yang, N. Chen, C. Hu and Y. Wang, J. Controlled Release, 2023, 354, 821–834 CrossRef CAS PubMed.
- Y. Wang, L. Chen, D.-Y. Ren, Z.-X. Feng, L.-Y. Zhang, Y.-F. Zhong, M.-Y. Jin, F.-W. Xu, C.-Y. Feng and Y.-Z. Du, Mater. Today Bio, 2022, 15, 100320 CrossRef CAS PubMed.
- N. Annabi, J. W. Nichol, X. Zhong, C. Ji, S. Koshy, A. Khademhosseini and F. Dehghani, Tissue Eng., Part B, 2010, 16, 371–383 CrossRef CAS PubMed.
- S. Naahidi, M. Jafari, M. Logan, Y. Wang, Y. Yuan, H. Bae, B. Dixon and P. Chen, Biotechnol. Adv., 2017, 35, 530–544 CrossRef CAS PubMed.
- G. F. Pierce, T. A. Mustoe, B. W. Altrock, T. F. Deuel and A. Thomason, J. Cell. Biochem., 1991, 45, 319–326 CrossRef CAS PubMed.
- M. S. Ågren, H. H. Steenfos, S. Dabelsteen, J. B. Hansen and E. Dabelsteen, J. Invest. Dermatol., 1999, 112, 463–469 CrossRef PubMed.
- Y. Pei, G. Xu, X. Wu, K. Tang and G. Wang, Polymers, 2019, 11, 548 CrossRef CAS PubMed.
- B. Zhou, X. Hu, J. Zhu, Z. Wang, X. Wang and M. Wang, Int. J. Biol. Macromol., 2016, 91, 68–74 CrossRef CAS PubMed.
- Y. N. Zhao, X. Xu, N. Wen, R. Song, Q. Meng, Y. Guan, S. Cheng, D. Cao, Y. Dong, J. Qie, K. Liu and Y. Zhang, Sci. Rep., 2017, 7, 5524 CrossRef PubMed.
- Y.-N. Chen, C. Jiao, Y. Zhao, J. Zhang and H. Wang, ACS Omega, 2018, 3, 11788–11795 CrossRef CAS PubMed.
- H. N. Antoniades, C. D. Scher and C. D. Stiles, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 1809–1813 CrossRef CAS PubMed.
- S. Perumal, R. Atchudan and W. Lee, Polymers, 2022, 14, 2510 CrossRef CAS PubMed.
- B.-B. Seo, J.-T. Koh and S.-C. Song, Biomaterials, 2017, 122, 91–104 CrossRef CAS PubMed.
- Y.-M. Kim, M.-R. Park and S.-C. Song, ACS Nano, 2012, 6, 5757–5766 CrossRef CAS PubMed.
- A. A. Torrano, R. Herrmann, C. Strobel, M. Rennhak, H. Engelke, A. Reller, I. Hilger, A. Wixforth and C. Bräuchle, Nanoscale, 2016, 8, 13352–13367 RSC.
- S. H. Kim, R. T. Han, H.-S. Han and Y.-M. Kim, Bioact. Mater., 2025, 43, 67–81 CAS.
- H. Byun, Y. Han, E. Kim, I. Jun, J. Lee, H. Jeong, S. J. Huh, J. Joo, S. R. Shin and H. Shin, Bioact. Mater., 2024, 36, 185–202 CAS.
- İ. Gulcin and S. H. Alwasel, Processes, 2023, 11, 2248 CrossRef.
- E. A. Ainsworth and K. M. Gillespie, Nat. Protoc., 2007, 2, 875–877 CrossRef CAS PubMed.
- R. G. Dare, C. V. Nakamura, V. F. Ximenes and S. O. Lautenschlager, Free Radicals Biol. Med., 2020, 160, 342–355 CrossRef CAS PubMed.
- N. Li, Y. Cao, J. Liu, W. Zou, M. Chen, H. Cao, S. Deng, J. Liang, T. Yuan, Q. Wang, Y. Fan and X. Zhang, J. Mater. Chem. B, 2024, 12, 6856–6873 RSC.
- S. Zhan, J. Wang, W. Wang, L. Cui and Q. Zhao, RSC Adv., 2019, 9, 16167–16175 RSC.
- S. Dash, P. N. Murthy, L. Nath and P. Chowdhury, Acta Pol. Pharm., 2010, 67, 217–223 CAS.
- Z. Miao, R. Lv, S. Teng, C. Cao and P. Lu, Int. J. Biol. Macromol., 2022, 222, 403–412 CrossRef CAS PubMed.
- M. Z. Nasrullah, Antioxidants, 2022, 12, 60 CrossRef PubMed.
- W. M. Chou, C. N. Chen, H. T. Hsieh, T. Y. Lo, P. Y. Juan and F. D. Mai, Technol. Health Care, 2015, 24(Suppl 1), S147–S153 Search PubMed.
- Z. Tu, M. Chen, M. Wang, Z. Shao, X. Jiang, K. Wang, Z. Yao, S. Yang, X. Zhang and W. Gao, Adv. Funct. Mater., 2021, 31, 2100924 CrossRef CAS.
- T. A. Wynn and K. M. Vannella, Immunity, 2016, 44, 450–462 CrossRef CAS PubMed.
- S. Das, M. Majid and A. B. Baker, Acta Biomater., 2016, 42, 56–65 CrossRef CAS PubMed.
- R. D. Galiano, O. M. Tepper, C. R. Pelo, K. A. Bhatt, M. Callaghan, N. Bastidas, S. Bunting, H. G. Steinmetz and G. C. Gurtner, Am. J. Pathol., 2004, 164, 1935–1947 CrossRef CAS PubMed.
- E. Mahdipour and A. Sahebkar, J. Diabetes Res., 2020, 2020, 6320514 Search PubMed.
- M. Sadeghi-Ardebili, S. Hasannia, B. Dabirmanesh and R. A. Khavari-Nejad, Sci. Rep., 2024, 14, 1003 CrossRef CAS PubMed.
- F. Cialdai, C. Risaliti and M. Monici, Front. Bioeng. Biotechnol., 2022, 10, 958381 CrossRef PubMed.
- M. Wang, C. Wang, M. Chen, Y. Xi, W. Cheng, C. Mao, T. Xu, X. Zhang, C. Lin, W. Gao, Y. Guo and B. Lei, ACS Nano, 2019, 13, 10279–10293 CrossRef CAS PubMed.
- H. Niu, Y. Guan, T. Zhong, L. Ma, M. Zayed and J. Guan, npj Regener. Med., 2023, 8, 32 CrossRef CAS PubMed.
- S. C. Song, S. B. Lee, J. I. Jin and Y. S. Sohn, Macromolecules, 1999, 32, 2188–2193 CrossRef CAS.
- Z. Sun, H. Xiong, T. Lou, W. Liu, Y. Xu, S. Yu, H. Wang, W. Liu, L. Yang, C. Zhou and C. Fan, Gels, 2023, 9, 381 CrossRef CAS PubMed.
- L. Jiang, X. Yang, Y. Zhang, D. He, Y. Gao, K. Lu, Y. Hao, Y. Gao, D. Lu, X. Jin and C. Li, Biomater. Adv., 2023, 144, 213226 CrossRef CAS PubMed.
- M. Sharifiaghdam, E. Shaabani, R. Faridi-Majidi, S. C. De Smedt, K. Braeckmans and J. C. Fraire, Mol. Ther., 2022, 30, 2891–2908 CrossRef CAS PubMed.
- J. Park, H.-S. Han, Y.-C. Kim, J.-P. Ahn, M.-R. Ok, K. E. Lee, J.-W. Lee, P.-R. Cha, H.-K. Seok and H. Jeon, Sci. Rep., 2015, 5, 18150 CrossRef CAS PubMed.
- J. Kim, H. S. Choi, Y. M. Kim and S. C. Song, Small, 2023, 19, e2203464 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tb00563a |
| This journal is © The Royal Society of Chemistry 2025 |