
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccessing mixed cluster rare-earth MOFs with reduced connectivity via linker expansion and desymmetrization: co-assembly of 6-c and 10-c hexanuclear clusters in RE-stc-MOF-1†
Edward
Loukopoulos
a,
Constantinos
Tsangarakis
a,
Konstantinos G.
Froudas
a,
Maria
Vassaki
a,
Giasemi K.
Angeli
 ab and
Pantelis N.
Trikalitis
*a
ab and
Pantelis N.
Trikalitis
*a
aDepartment of Chemistry, University of Crete, Voutes, 71003 Heraklion, Greece. E-mail: ptrikal@uoc.gr
bTheoretical and Physical Chemistry Institute, National Hellenic Research Foundation, 48 Vassileos Constantinou Avenue, Athens, 11635, Greece
First published on 3rd December 2024
Abstract
MOFs based on hexanuclear rare-earth (RE) clusters with reduced connectivity (<12) are rare. The co-assembly of RE clusters with distinct reduced connectivity provides access to novel structures. We report a new family of MOFs denoted as RE-stc-MOF-1, displaying a unique (3,3,3,3,6,10)-connected network, based on 6-c and 10-c RE hexanuclear clusters.
As a distinct class of porous materials, metal–organic frameworks (MOFs) have been rapidly expanding in the last few decades. The versatile and tunable chemistry of these hybrid inorganic/organic materials offers unparalleled control and design opportunities, leading to structures with specific features, topologies, pore characteristics and associated properties.1 In this context, the choice of suitable building blocks is clearly crucial in rational MOF design and synthesis.2 Recently, there has been increased interest in synthetic approaches that employ uncommon or seemingly incompatible building units.3 In terms of ligand selection and engineering, particular emphasis has been given in the utilization of linkers with reduced symmetry.4,5 Conceptually, such linkers can be designed by modifying selected features of perfectly symmetric ligands, including their shape6,7 (partially extending or shortening specific parts of the ligand) or connectivity8–10 (insertion, removal or displacement of selected coordination groups). While these strategies can introduce structural complexity and unpredictability, they have also provided a platform towards novel frameworks and topologies that cannot be obtained otherwise.11,12
The potential of low-symmetry linkers in MOF synthesis can be even more appealing when combined with an appropriate inorganic secondary building unit (SBU). Rare-earth (RE) metals have recently gained significant traction as an exceptional choice for generating unique frameworks. Their rich coordination chemistry (in terms of coordination number and geometry) can dictate the formation of diverse, highly connected SBUs, such as polymeric chains or clusters with varying nuclearity.13 At the same time, the resulting materials can have exciting and tailorable application potential, owed to the distinct properties of the RE metals.13 A combination of such an inorganic building block with non-symmetric ligand systems could therefore offer a unique opportunity to develop truly novel materials with advanced properties. Initial research on this concept has already shown that the resulting RE-MOFs can display traditionally unattainable networks with metal nodes of lower connectivity (e.g., lxl,14jun,14pek15 and thc16), as well as peculiar gas storage/trapping capabilities.17,18 Generally, the existence of unsaturated metal sites in RE-MOFs remains a challenge due to associated stability issues, however it makes them much desired for various applications or further functionalization.19,20 Therefore, more efforts are necessary to fully explore and harness the capabilities of low symmetry RE-MOF systems.21
Herein, we report the discovery of a new series of RE-MOFs using the tetratopic carboxylate-based linker H4L, a ligand with two benzoic and one isophthalic acid groups, designed by lowering the symmetry of the analogous C3 linkers to C2 (Fig. 1). The resulting frameworks are based on two different RE6 clusters of 10- and 6-connectivity. The latter inorganic building block, as well as the resulting network, denoted as stc (six and ten connected) are reported for the first time. The results, as detailed below, validate this synthetic approach as a powerful method to gain new insights in MOF design and particularly in frameworks based on RE elements.
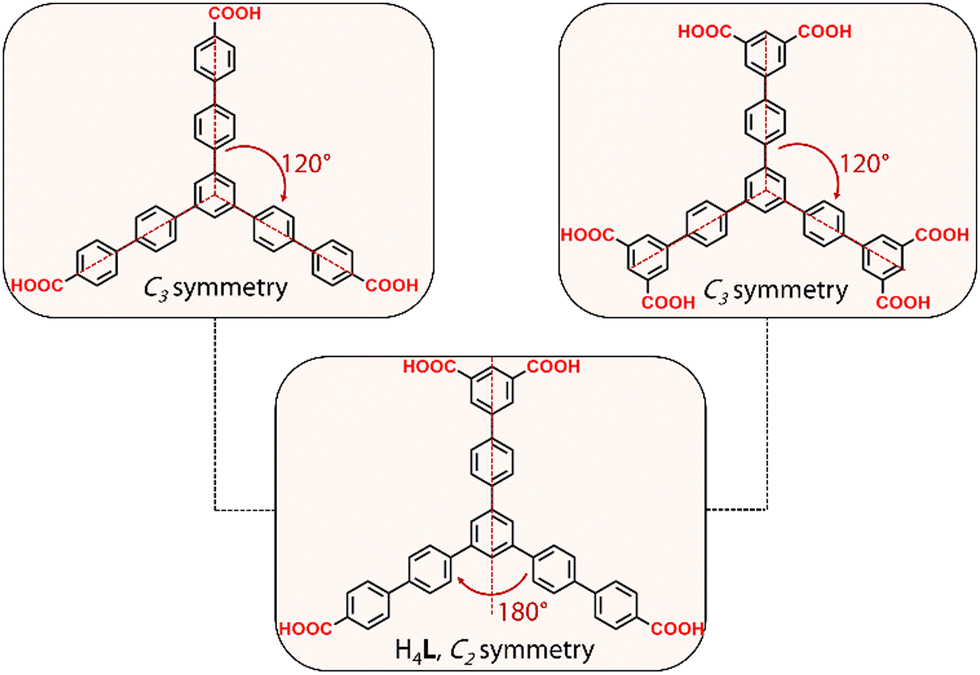 | ||
| Fig. 1 Conceptual design of the reduced symmetry H4L linker used in this work. | ||
Colourless polyhedral crystals of the RE-stc-MOF-1 family were afforded after an extensive synthetic screening of several parameters, also detailed in Table S1 (ESI†). The optimized protocol required a solvothermal reaction of H4L with rare earth nitrates at 120 °C, in the presence of N,N′-dimethylformamide (DMF), H2O and the organic modulator 2-fluorobenzoic acid (HFBA) (Pages S8 and S9, ESI†). This method led to the synthesis of the Y3+, Sm3+, Eu3+, Tb3+ and Dy3+ isostructural analogues (Fig. 2 and Fig. S12, ESI†). On the other hand, amorphous material was formed when other types of co-modulating ligands (e.g., formic, acetic or benzoic acid) were used instead. The result confirms the multiple function of fluorinated modulators towards the formation of polynuclear RE-cluster SBUs. As established in other studies, these hydrophobic carboxylic acids delay the crystallization process, prohibiting fast polymerization into extended RE chain SBUs.13 At the same time, the presence of fluorine in the modulator also affects the chemical composition of the bridging anions within the inorganic node itself, resulting in the formation of fluorinated RE-clusters.22 Utilization of a DMF/H2O solvent mixture was also found to be critical for the formation of phase-pure material. Several RE-cluster based frameworks have been reported to be anionic, with the charge balance completed by dimethylammonium cations derived from the hydrolytic decomposition of DMF at high temperatures.17 In accordance, a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of DMF to H2O was found to provide optimal conditions for this effect, while small changes in the ratio led to mixtures also containing non-crystalline solid.
:1 ratio of DMF to H2O was found to provide optimal conditions for this effect, while small changes in the ratio led to mixtures also containing non-crystalline solid.
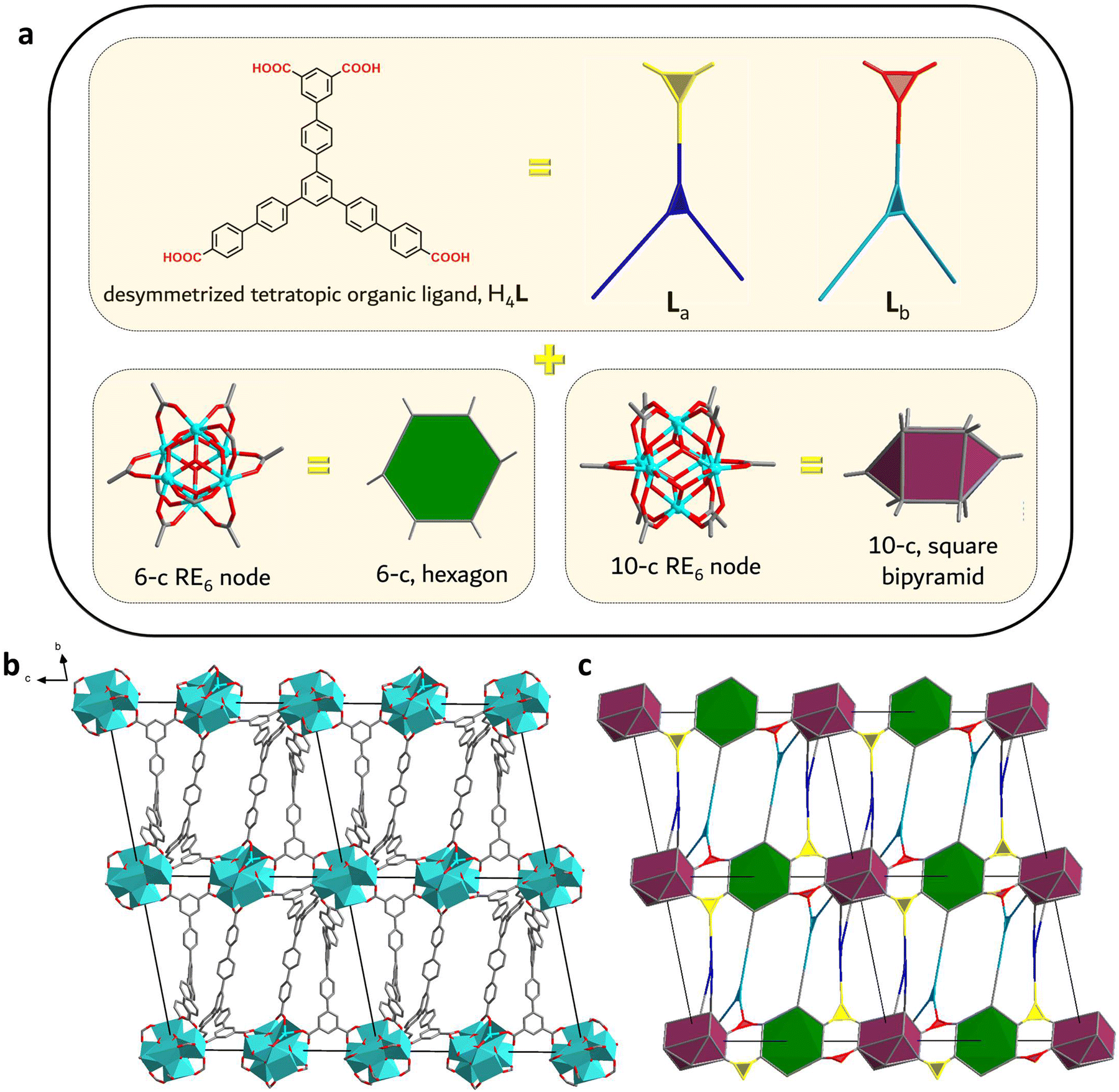 | ||
| Fig. 2 (a) The combination of the desymmetrized, tetratopic H4L organic linker with 6-c and 10-c RE6-clusters resulted in the formation of RE-stc-MOF-1 series of MOFs. Their geometric representation used for the topological analysis is shown where crystallographically distinct components have different colors. (b) The structure of Eu-stc-MOF-1 looking down the a-axis. (c) The unique six nodal, (3,3,3,3,6,10)-c, augmented stc-a net derived from the topological analysis of Eu-stc-MOF-1. | ||
After a series of single-crystal X-ray diffraction (SCXRD) studies on the Eu and Sm analogues, it was determined that RE-stc-MOF-1 crystallize in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (unit cell parameters for the Eu analogue: a = 24.354 Å, b = 25.255 Å, c = 26.050 Å, α = 79.367°, β = 88.674°, γ = 62.912°, V = 13985.9 Å3). Evidently, desymmetrization of the linker also translates to a low symmetry space group, in contrast to the vast majority of reported RE-MOFs based on polynuclear clusters, which crystallize in crystal systems such as orthorhombic, tetragonal or cubic.13 The framework features a unique 3D network that contains two crystallographically independent L4− organic linkers, denoted as La and Lb, and two different types of hexanuclear RE6 clusters (Fig. 2) as the inorganic building units. Both clusters are based on a main [RE6(μ3-X)8]10+ core, where X = OH− or F−. The presence of either or both of these bridging anions in MOFs based on polynuclear RE clusters has been already detailed in previous reports by us and others.22–24 In agreement with these data, the existence of μ3-F species was verified through 19F-NMR experiments on representative analogues (Fig. S4 and S5, ESI†). The corresponding spectra showed a singlet peak at approximately −165 ppm, consistent with the presence of inorganic F− anions.
space group (unit cell parameters for the Eu analogue: a = 24.354 Å, b = 25.255 Å, c = 26.050 Å, α = 79.367°, β = 88.674°, γ = 62.912°, V = 13985.9 Å3). Evidently, desymmetrization of the linker also translates to a low symmetry space group, in contrast to the vast majority of reported RE-MOFs based on polynuclear clusters, which crystallize in crystal systems such as orthorhombic, tetragonal or cubic.13 The framework features a unique 3D network that contains two crystallographically independent L4− organic linkers, denoted as La and Lb, and two different types of hexanuclear RE6 clusters (Fig. 2) as the inorganic building units. Both clusters are based on a main [RE6(μ3-X)8]10+ core, where X = OH− or F−. The presence of either or both of these bridging anions in MOFs based on polynuclear RE clusters has been already detailed in previous reports by us and others.22–24 In agreement with these data, the existence of μ3-F species was verified through 19F-NMR experiments on representative analogues (Fig. S4 and S5, ESI†). The corresponding spectra showed a singlet peak at approximately −165 ppm, consistent with the presence of inorganic F− anions.
More importantly, both types of RE6 clusters display reduced connectivity to carboxylate groups of bridging ligands, acting as 6-c and 10-c SBUs respectively. These structural features are highly uncommon in MOF chemistry: to the best of our knowledge, the RE-stc-MOF-1 series represents the first ever example of such a combination of inorganic SBUs. Using the more optimal crystallographic data of the Eu analogue, the MOF structure was further studied in order to completely understand the unique features and coordination environment of these building blocks. Viewing the framework along the ab plane, a sheet-like architecture is observed, formed by the benzoate groups of L4-. The network also extends along the c axis as the isophthalate groups link neighbouring clusters of alternating 6- and 10-connectivity (Fig. 2b and c). Furthermore, looking at the particular connectivity of the crystallographically distinct organic linkers, La is bridging three 10-c and one 6-c clusters while Lb is connected to two 10-c and two 6-c SBUs (Fig. S10, ESI†). As shown in Fig. 2c, the linkers are alternated in an upside-down fashion, in a set of two (La–La, Lb–Lb) along the c-axis. Considering the 4-c organic linker as two interconnected 3-c building units, the topological analysis using the software ToposPro25 revealed the formation of a unique (3,3,3,3,6,10)-c 6-nodal net (Fig. 2a, c and Fig. S10, ESI†).
The 6-c SBU in Eu-stc-MOF-1 is formulated as [Eu6(μ3-X)8(COO)6(FBA)4(DMF)4(H2O)2], connecting to 4 isophthalate (2 La and 2 Lb) and 2 benzoate (2 Lb) groups from six different ligands. Considering the position of the C atoms of these coordinated –COO− groups as its points of extension, the cluster is best interpreted as a 6-c hexagonal SBU. This particular type of SBU has not been observed in RE-MOF chemistry: the only reported instances of 6-c clusters in RE-MOFs are RE-CU-4526,27 and RE-urx-MOF28 where the building block is best described as a trigonal antiprism or an octahedron respectively. Even in the entire MOF family of materials, there are limited examples of 6-c hexagonal SBUs as in the case of Zr-based MOFs, PCN-22429 and pbz-MOF-1.30 Notably, in these particular cases, the Zr6 clusters connect to highly symmetrical linkers in an almost perpendicular fashion (dihedral angles between the coordinated carboxylate groups and the Zr2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O2 planes calculated to be in the 177–179° range). On the other hand, the employment of a reduced symmetry linker in Eu-stc-MOF-1 leads to a larger range in respective angles, also accommodated by the broader coordination capabilities of rare-earth metals compared to zirconium. This is mostly evident in the case of the isophthalates, which connect to the 6-c cluster in a notably bent fashion (respective coordination angles ranging from 134.4 to 162.7°).
O2 planes calculated to be in the 177–179° range). On the other hand, the employment of a reduced symmetry linker in Eu-stc-MOF-1 leads to a larger range in respective angles, also accommodated by the broader coordination capabilities of rare-earth metals compared to zirconium. This is mostly evident in the case of the isophthalates, which connect to the 6-c cluster in a notably bent fashion (respective coordination angles ranging from 134.4 to 162.7°).
The other distinct inorganic SBU found in the stc framework is a 10-c cluster formulated as [Eu6(μ3-X)8(COO)10(DMF)2(OH)2]. In this case, the inorganic building block can be best viewed as an elongated square bipyramid in which 4 isophthalate (2 La and 2 Lb) and 6 benzoate (4 La and 2 Lb) groups from ten different L4- ligands contribute to the total connectivity. Up to now, such an SBU has only been observed in NTUniv-5718 and PCN-991,21 both RE-MOFs employing respective 2-c and 3-c linkers with decreased symmetry. Again, certain isophthalate moieties display highly bent coordination angles that reach down to 135.1°. While these unusual isophthalate torsions in both clusters may imply an increased framework energy state, the structure is further stabilized by several intermolecular C–H⋯π interactions involving the remaining benzoate moieties (Table S4, ESI†), which provide the necessary energy to overcome these deformations.
The combination of these inorganic and organic building blocks leads to a three-dimensional framework with large elongated cavities of 16 × 16 × 30 Å in size (Fig. S11, ESI†). The key role of the low symmetry of H4L in the formation of this unique network is evident, considering the above structural analysis. In contrast, utilization of the archetypal higher C3 symmetry linker with 3 benzoate groups (Fig. 1, top left) is known to guide the formation of a highly connected (14-c) frt network when combined with RE elements, published recently by our group.23 From a chemical point of view, the stc framework has an anionic nature and charge balance is provided by dimethylammonium counter cations, verified by 1H-NMR measurements which revealed signals at 2.48 and 8.94 ppm (see the NMR section in the ESI†), in line with previous studies.31 These experiments also revealed a 1:1 ratio of ligand to FBA moieties as observed in SCXRD experiments, further confirming the complete chemical formula of the structure as [(CH3)2NH2]2[RE6(μ3-X)8(L)1.5(FBA)4(DMF)4(H2O)2][RE6(μ3-X)8(L)2.5(DMF)2(OH)2]. This formula is also supported by TGA analysis as we describe below.
Additional characterization of the crystalline RE-stc-MOF-1 materials was performed through powder X-ray diffraction (PXRD) and scanning electron microscopy (SEM) studies, as detailed in Fig. 3 and Fig. S12–S21 (ESI†). A comparison of the experimental PXRD patterns to the one simulated from the structure of the Eu analogue confirmed phase purity (Fig. 3a and Fig. S12, ESI†). It is worth mentioning that the frameworks remain initially stable in open air despite containing low-connected RE clusters as inorganic nodes. Typically, the presence of such species is destabilizing for the structure, since high connectivity is thermodynamically favoured in RE metals.19 In the case of Eu-stc-MOF-1, a series of successive measurements over time indicated that a partial loss in crystallinity is observed after approximately 2 hours of air exposure (Fig. S13, ESI†). These results were also confirmed for Tb-stc-MOF-1 as shown in Fig. S14 (ESI†). Notably, in this case after 2 hours exposure in air, a significant degree of crystallinity is preserved indicating an enhanced structural stability as compared to Eu-stc-MOF-1. As we describe below, the observed structural stability is also reflected in the porosity of the activated samples.
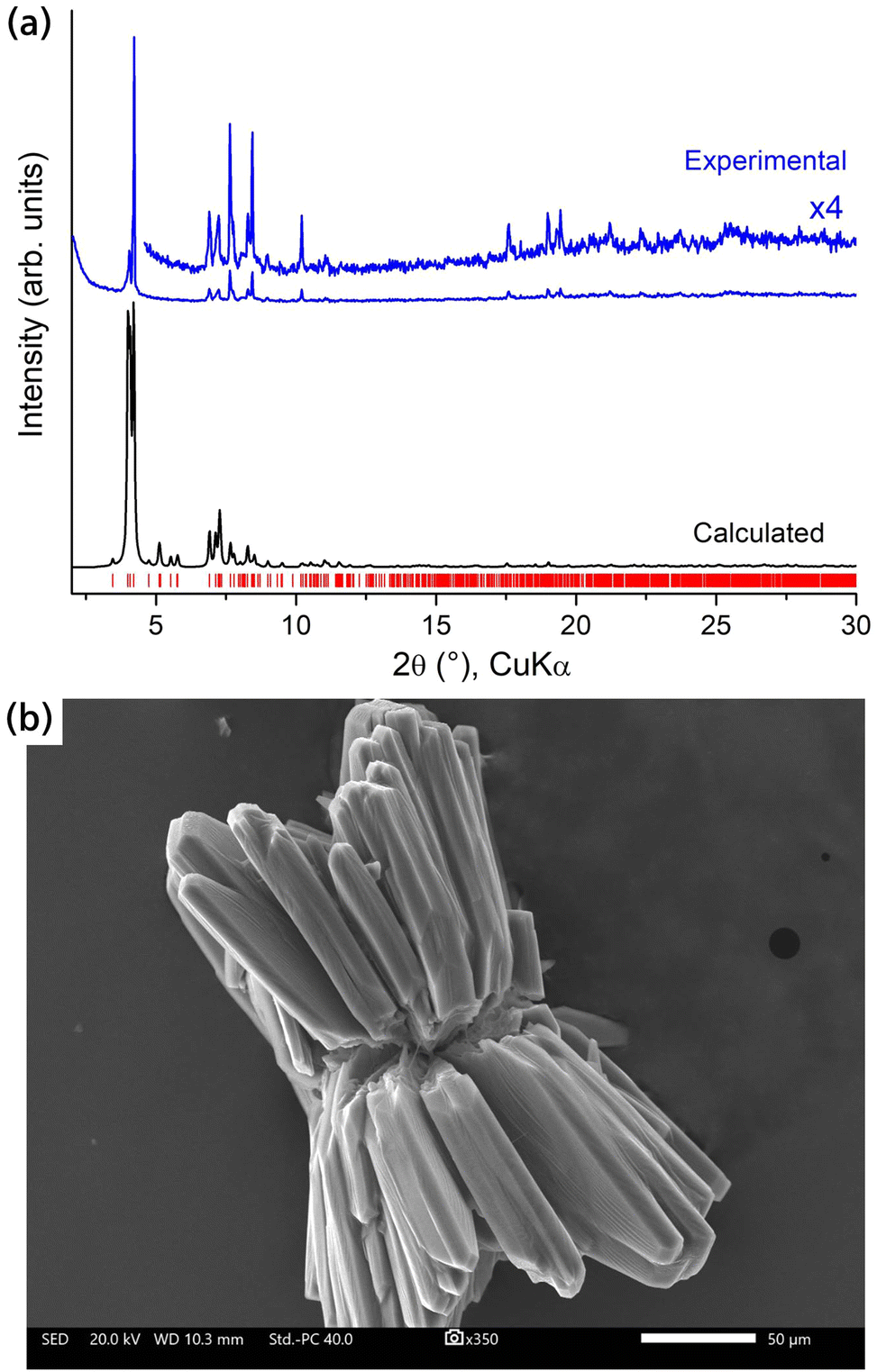 | ||
| Fig. 3 (a) PXRD pattern of as-made Eu-stc-MOF-1 (blue), compared to the pattern calculated from the crystal structure (black). (b) Representative SEM image of the crystals in Eu-stc-MOF-1. | ||
Taking this behaviour into account, we performed initial gas sorption measurements in ethanol-exchanged batches of Tb- and Eu-stc-MOF-1. Complete exchange of DMF to ethanol (EtOH) within the framework was verified by 1H-NMR (Fig. S9, ESI†). PXRD measurements confirmed that both materials are stable in EtOH (Fig. S15 and S16, ESI†). Tb-stc-MOF-1 was then activated under vacuum at various temperatures (25, 40 and 80 °C), followed by experimental N2 isotherm recordings at 77 K. In all cases, the isotherms revealed a type I sorption behaviour, reaching respective uptakes of 191.4, 177.9 and 121.4 cm3 g−1 at 0.99 p/p0 (Fig. S22, ESI†). Corresponding BET areas were calculated at 692, 643 and 438 m2 g−1, respectively (Fig. S23, ESI†). The experimental total pore volume at 0.99 p/p0 for the sample activated at room temperature was calculated at 0.30 cm3 g−1, which is notably lower than the theoretical value determined from the single crystal structure (0.92 cm3 g−1). Given that Tb-stc-MOF-1 is stable in EtOH, it is natural to consider that its partial activation is associated with the evacuation procedure. In particular, the removal of EtOH by vacuum involves a liquid to gas phase transition that could affect the integrity of the MOF framework due to moderate EtOH-framework interactions, including coordination to metal clusters and hydrogen bonds. To eliminate these problems we applied supercritical CO2 (scCO2) activation in EtOH exchanged samples, where first liquid EtOH is exchanged by liquid CO2 and then under supercritical conditions, CO2 escapes the pore structure leaving the structure intact due to very weak CO2-framework interactions. The formation of solvent-free materials is supported by TGA analysis of the activated solids, as we describe below. It is noted that EtOH is the solvent of choice for scCO2 MOF activation because it is miscible with liquid CO2, promoting its facile exchange.
Indeed, argon sorption isotherm of Tb-stc-MOF-1 recorded at 87 K (Fig. 4), revealed a type I isotherm with high uptake at 0.99 p/p0 reaching 556.1 cm3 g−1, corresponding to a total pore volume of 0.71 cm3 g−1, which is significantly improved compared to the value obtained after direct evacuation from EtOH and close to the calculated value of 0.92 cm3 g−1. Taking into account that the calculated value is overestimated because dimethylammonium counterions cannot be located in the single crystal structure, due to their highly disordered nature but occupy pore space, the experimentally obtained total pore volume reflects a successfully activated material. The BET surface area calculated using consistency criteria is 1707.6 m2 g−1 (Fig. S24, ESI†), while the pore size distribution (PSD) curve obtained by a GCMC fitting method available in the software BELMaster™ (Fig. S25, ESI†), shows a main peak centered at 15.8 Å (Fig. 4 inset), which is very close to expected value from the single crystal structure.
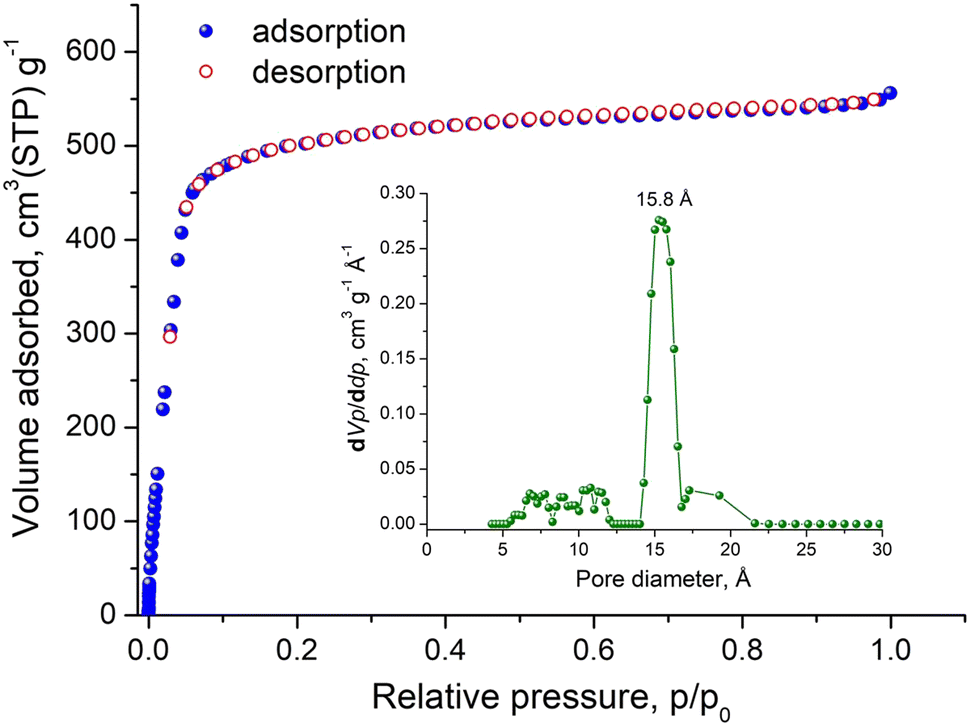 | ||
| Fig. 4 Argon sorption isotherm of activated Tb-stc-MOF-1 recorded at 87 K. Inset shows the corresponding pore size distribution curve obtained by GCMC method. | ||
In the case of scCO2 activated Eu-stc-MOF-1, a very similar Ar isotherm is obtained at 87 K (Fig. S26, ESI†) however, the total uptake at 0.99 p/p0 is 436.9 cm3 g−1, corresponding to a total pore volume of 0.56 cm3 g−1, which is lower compared to that of the Tb-analogue (0.71 cm3 g−1), indicating partial activation. These results are consistent with the proposition based on PXRD data that Tb-stc-MOF-1 is more stable than the Eu-analogue. The BET surface area of Eu-stc-MOF-1 is 1440.5 m2 g−1 and PSD analysis obtained by GCMC (Fig. S27 and S28, ESI†) shows a peak centered at 15.5 Å, which is slightly contracted as compared to the Tb-analogue, indicating partially pore shrinkage, in agreement with the lower total pore volume of this material.
The thermal stability of the scCO2 activated MOFs were studied by thermogravimetric analysis under nitrogen flow (Fig. S29, ESI†). After an initial small weight loss of approximately 5% and 8% for Tb- and Eu-stc-MOF-1 respectively, associated with adsorbed moisture during loading into the TGA instrument, a plateau is observed up to 370 °C followed by a significant weight loss due to the decomposition of the organic part of the framework, which is completed at 575 °C and 610 °C for the Eu- and Tb-stc-MOF-1 respectively (Fig. S29, ESI†). Considering the formation of the corresponding oxides, that is Eu2O3 and Tb2O3, the observed total weight loss is 58.2% and 57.3% respectively, which is very close to the calculated values of 62.31% and 61.40% based on their chemical formula, considering no solvent molecules.
In conclusion, we herein report the discovery of the RE-stc-MOF-1 family using a tetratopic, carboxylate-based linker with reduced symmetry. The structures display high complexity, featuring two distinct types of hexanuclear RE6-clusters with reduced connectivity. This leads to the construction of an unprecedented (3,3,3,3,6,10)-c 6-nodal net, while the stc-family also represents the first example of a 6-c hexagonal node in RE-MOFs. NMR spectroscopy confirms the presence of capping ligands which aid to lower the node connectivity. Tb-stc-MOF-1 and Eu-stc-MOF-1 were selected as representative members to study the porosity in this family of MOFs. Successfully pore activation was achieved by scCO2 drying, revealing high surface areas and pore volumes. Our findings validate the potential of lower symmetry linkers combined with RE elements as a powerful synthetic approach towards novel materials with structural peculiarities that cannot be obtained otherwise. In combination with the already established RE cluster node chemistry, this approach is ideal for the generation of MOFs with accessible unsaturated sites that could be exploited for applications of interest. In this context, we are currently investigating the capability of the RE-stc-MOF-1 materials in sensing and post-synthetic modifications, encouraged by their low connectivity and stability in common organic solvents. Additionally, future efforts will be focused on further tapping the potential of this design approach towards novel frameworks and topologies.
This study was carried out within the framework of the National Recovery and Resilience Plan Greece 2.0 (Award Number TAEDR-0535821), funded by the European Union – NextGenerationEU.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- R. Robson, Chem. Rec., 2024, 24, e202400038 CrossRef CAS.
- V. Guillerm and D. Maspoch, J. Am. Chem. Soc., 2019, 141, 16517–16538 CrossRef CAS PubMed.
- J. Duan, M. Higuchi, J. Zheng, S. I. Noro, I. Y. Chang, K. Hyeon-Deuk, S. Mathew, S. Kusaka, E. Sivaniah, R. Matsuda, S. Sakaki and S. Kitagawa, J. Am. Chem. Soc., 2017, 139, 11576–11583 CrossRef CAS PubMed.
- J. K. Schnobrich, O. Lebel, K. A. Cychosz, A. Dailly, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2010, 132, 13941–13948 CrossRef CAS PubMed.
- R. R. Liang, S. Xu, Z. Han, Y. Yang, K. Y. Wang, Z. Huang, J. Rushlow, P. Cai, P. Samorì and H. C. Zhou, J. Am. Chem. Soc., 2024, 146, 9811–9818 CrossRef CAS PubMed.
- B. Wang, P. Wang, L. H. Xie, R. B. Lin, J. Lv, J. R. Li and B. Chen, Nat. Commun., 2019, 10, 3861 CrossRef PubMed.
- Y. Gao, M. Zhang, C. Chen, Y. Zhang, Y. Gu, Q. Wang, W. Zhang, Y. Pan, J. Ma and J. Bai, Chem. Commun., 2020, 56, 11985–11988 RSC.
- W. Han, X. Ma, J. Wang, F. Leng, C. Xie and H. L. Jiang, J. Am. Chem. Soc., 2023, 145, 9665–9671 CrossRef CAS PubMed.
- S. C. Wang, Q. S. Zhang, Z. Wang, L. Zheng, X. D. Zhang, Y. N. Fan, P. Y. Fu, X. H. Xiong and M. Pan, Angew. Chem., Int. Ed., 2022, 61, e202211356 CrossRef CAS PubMed.
- Y. Wang, L. Feng, K. Zhang, K. Y. Wang, W. Fan, X. Wang, B. Guo, F. Dai, L. Zhang, D. Sun and H. C. Zhou, Adv. Sci., 2019, 6, 1901855 CrossRef CAS.
- Y. Zhu, J. Gu, X. Yu, B. Zhang, G. Li, J. Li and Y. Liu, Inorg. Chem. Front., 2021, 8, 4990–4997 RSC.
- F. Saraci, V. Quezada-Novoa, P. R. Donnarumma and A. J. Howarth, Chem. Soc. Rev., 2020, 49, 7949–7977 RSC.
- X. L. Lv, L. Feng, L. H. Xie, T. He, W. Wu, K. Y. Wang, G. Si, B. Wang, J. R. Li and H. C. Zhou, J. Am. Chem. Soc., 2021, 143, 2784–2791 CrossRef CAS PubMed.
- D. Alezi, A. M. Peedikakkal, L. J. Weselinski, V. Guillerm, Y. Belmabkhout, A. J. Cairns, Z. Chen, L. Wojtas and M. Eddaoudi, J. Am. Chem. Soc., 2015, 137, 5421–5430 CrossRef CAS.
- E. Loukopoulos, G. K. Angeli, C. Tsangarakis, E. Traka, K. G. Froudas and P. N. Trikalitis, Chem. – Eur. J., 2024, 30, e202302709 CrossRef CAS PubMed.
- G. K. Angeli, E. Loukopoulos, K. Kouvidis, A. Bosveli, C. Tsangarakis, E. Tylianakis, G. Froudakis and P. N. Trikalitis, J. Am. Chem. Soc., 2021, 143, 10250–10260 CrossRef CAS PubMed.
- M. Zhang, P. Liu, R. Dang, H. Cui, G. Jiang, J. Wang, M. Wang, T. Sun, M. Wang, G. Qin, S. Wang and Y. Tang, Inorg. Chem., 2022, 61, 18653–18659 CrossRef CAS PubMed.
- Y. Hu, R. S. H. Khoo, A. Pang, S. Yang, C. Fiankor, X. Zhang and J. Zhang, J. Mater. Chem. C, 2024, 12, 2836–2842 RSC.
- J. T. Zhao, G. R. Si, T. He, X. J. Kong and J. R. Li, ACS Mater. Lett., 2024, 6, 1977–1983 CrossRef CAS.
- F. Li, K. Y. Wang, Z. Liu, Z. Han, D. Kuai, W. Fan, L. Feng, Y. Wang, X. Wang, Y. Li, Z. Yang, R. Wang, D. Sun and H. C. Zhou, JACS Au, 2023, 3, 1337–1347 CrossRef CAS PubMed.
- J. P. Vizuet, M. L. Mortensen, A. L. Lewis, M. A. Wunch, H. R. Firouzi, G. T. McCandless and K. J. Balkus, Jr., J. Am. Chem. Soc., 2021, 143, 17995–18000 CrossRef CAS.
- E. Loukopoulos, G. K. Angeli, K. Kouvidis, C. Tsangarakis and P. N. Trikalitis, ACS Appl. Mater. Interfaces, 2022, 14, 22242–22251 CrossRef CAS PubMed.
- A. R. B. J. Lutton-Gething, B. F. Spencer, G. F. S. Whitehead, I. J. Vitorica-Yrezabal, D. Lee and M. P. Attfield, Chem. Mater., 2024, 36, 1957–1965 CrossRef CAS PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CrossRef CAS.
- H. A. Bicalho, F. Saraci, J. J. Velazquez-Garcia, H. M. Titi and A. J. Howarth, Chem. Commun., 2022, 58, 10925–10928 RSC.
- H. A. Bicalho, C. Copeman, H. P. Barbosa, P. R. Donnarumma, Z. Davis, V. Quezada-Novoa, J. J. Velazquez-Garcia, E. Hemmer and A. J. Howarth, Chemrxiv, 2024, preprint DOI:10.26434/chemrxiv-2024-jwscm.
- Z. Chen, Z. Thiam, A. Shkurenko, L. J. Weselinski, K. Adil, H. Jiang, D. Alezi, A. H. Assen, M. O'Keeffe and M. Eddaoudi, J. Am. Chem. Soc., 2019, 141, 20480–20489 CrossRef CAS.
- D. Feng, W. C. Chung, Z. Wei, Z. Y. Gu, H. L. Jiang, Y. P. Chen, D. J. Darensbourg and H. C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105–17110 CrossRef CAS.
- D. Alezi, I. Spanopoulos, C. Tsangarakis, A. Shkurenko, K. Adil, Y. Belmabkhout, M. O’Keeffe, M. Eddaoudi and P. N. Trikalitis, J. Am. Chem. Soc., 2016, 138, 12767–12770 CrossRef CAS PubMed.
- Q. Hu, X. X. Tian, P. Wang, X. Y. Tang, W. H. Zhang and D. J. Young, Inorg. Chem., 2021, 60, 18614–18619 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR, SCXRD, PXRD and sorption data. CCDC 2374028 and 2374029. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc03317e |
| This journal is © The Royal Society of Chemistry 2025 |