
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and phase purity of the negative thermal expansion material ZrV2O7†
Aistė
Miliūtė
 ab,
Joana
Bustamante
a,
Stephanos
Karafiludis
ab,
Moritz
Zöllner
ab,
Mustapha
Eddah
a,
Franziska
Emmerling
ab,
Björn
Mieller
*a,
Janine
George
*ac and
Tomasz M.
Stawski
*a
ab,
Joana
Bustamante
a,
Stephanos
Karafiludis
ab,
Moritz
Zöllner
ab,
Mustapha
Eddah
a,
Franziska
Emmerling
ab,
Björn
Mieller
*a,
Janine
George
*ac and
Tomasz M.
Stawski
*a
aFederal Institute for Materials Research and Testing (BAM), Berlin, Germany. E-mail: bjoern.mieller@bam.de; janine.george@bam.de; tomasz.stawski@bam.de
bHumboldt Universität zu Berlin, Department of Chemistry, Berlin, Germany
cFriedrich Schiller University of Jena, Institute of Solid-State Theory and Optics, Jena, Germany
First published on 10th December 2024
Abstract
Synthesis of pure, homogeneous, and reproducible materials is key for the comprehensive understanding, design, and tailoring of material properties. In this study, we focus on the synthesis of ZrV2O7, a material known for its negative thermal expansion properties. We investigate the influence of solid-state and wet chemistry synthesis methods on the purity and homogeneity of ZrV2O7 samples. Our findings indicate that different synthesis methods significantly impact the material's characteristics. The solid-state reaction provided high-purity material through extended milling time and repeated calcination cycles, while the sol–gel reaction enabled a “near-atomic” level of mixing, and therefore, homogenous phase-pure ZrV2O7. We confirmed purity via X-ray diffraction and Raman spectroscopy, highlighting differences between phase-pure and multiphase ceramics. These analytical techniques allowed us to distinguish subtle differences in the structure of the material. Based on ab initio simulated phonon data, we were able to interpret the Raman spectra and visualise Raman active atom vibrations. We show that phase purity enables the unbiased characterisation of material properties such as negative thermal expansion.
 Janine George | Janine George is a junior research group leader and a Professor of Materials Informatics at the Federal Institute for Materials Research and Testing (BAM) in Berlin and at Friedrich Schiller University in Jena (FSU Jena), Germany. She completed her MSc in Chemistry in 2013 and, in 2017, her doctorate in computational solid-state chemistry at RWTH Aachen University. In 2018, she started her postdoc and, later, Marie-Skłodowska-Curie fellowship at UCLouvain in Belgium. Since 2021 is a junior group leader at BAM and FSU Jena. In 2023, she became a Professor of Materials Informatics in Jena. She was a Falling Walls Finalist for “Breakthrough of the Year” in the “Physical Sciences” in 2021. She received a junior scientist award from the Stiftung Werner-von-Siemens-Ring in 2023. In 2024, she received the ERC Starting Grant. She is using atomistic simulations and machine learning to design material properties. To do so, she is linking quantum-chemical bonding information and material properties. |
Introduction
Ceramic materials with unique thermal expansion properties have attracted considerable interest in recent years due to their potential applications in various fields.1,2 Among these, zirconium vanadate (ZrV2O7) stands out for its remarkable negative thermal expansion (NTE) behaviour over a wide temperature range.3 This unusual property, in which the material contracts when heated, opens up exciting possibilities for creating composites with tailored thermal expansion properties.In zirconium vanadate (ZrV2O7), a unit cell contracts isotropically with increasing temperature over a range of 130 °C to 800 °C.3,4 The synthesis of zirconium vanadate ceramics, exhibiting NTE, enables the fabrication of composites where the overall expansion coefficient can be tailored to a specific negative, positive, or neutral value.5 Consequently, such composite materials are attractive for many device applications because they can compensate for damage caused by thermal expansion. NTE composites are relevant to optical systems, electronic and biomedical applications.5 Furthermore, ZrV2O7 has attracted further interest due to its photocatalytic activity6 and specific discharge capacity.7 The photocatalytic activity of ZrV2O7 and its substituted analogues has been considered for the degradation of various dyes,8 in sensors and inorganic ion exchangers.6 Also, Yb3+/Er3+ co-doped ZrV2O7 may have potential application in the display technology.9 The NTE property, which prevents damage from volume expansion, combined with the specific discharge capacity of ZrV2O7, makes it a promising cathode material for secondary lithium batteries.7 However, while ZrV2O7 exhibits high specific capacity as a cathode material, its capacity retention could be improved. In this context, this promising electrochemical application and the associated challenges highlight complex dependencies between the intrinsic properties of crystal structures and the microstructure of the material. Namely, there is a correlation between crystallite sizes, porosity, and measured capacity retention,7 demonstrating the need to tune the purity, size, and homogeneity of synthesised particles. The potential applications in which the material must be tuned simultaneously in terms of NTE and other physicochemical properties exemplify the fact that the microstructural qualities of the material are fundamental.
An important part of NTE materials research is related to lowering the offset temperature of linear negative thermal expansion to room temperature. This can be achieved, for example, by partially substituting V with P,4 Mo8,10 or W.11 Or lowering thermal expansion via double substitution of Fe/Mo,12 Cu/P13 or Nb, Y/P14 for Zr/V sites. The goal is to tune the phase transition temperature and extend the NTE property to manage the thermal expansion of materials more effectively, especially in devices where dimensional stability is essential at lower temperatures.
Despite the growing number of studies investigating the potential applications of ZrV2O7, the existing literature on the synthesis and resulting quality of ZrV2O7 ceramics remains inconclusive. Many synthesis methods have been reported throughout the 80 years of research, including single crystal, various solid-state synthesis attempts, and wet-chemistry methods. Currently, our understanding of the physicochemical properties of the material is mainly based on extensive single-crystal studies by Evans et al.15 In this regard, single crystals enable us to evaluate the fundamental crystallographic principles of NTE. However, material characteristics will inevitably differ in the case of bulk polycrystalline powder samples and sintered ceramics. The structural comparison among powders derived through different methods and strategies would be an important milestone towards any larger-scale production attempt.
ZrV2O7 is derived by reacting stoichiometric amounts of ZrO2 and V2O5. Thus, at its core, the synthesis challenge is related to the extent of mixing of zirconium and vanadium precursors. The phase diagram suggests that if zirconium and vanadium precursors are not well mixed and remain separated over a more considerable local distance, final local stoichiometry can diverge and lead to the formation of competing phases such as Zr3V3Ox (x = 1.0 or 0.6)16 or residual ZrO2 and V2O5. Analysis of a multi-phase material might lead to misleading results, in which the overall elemental composition would match that of ZrV2O7, but other non-NTE crystalline phases would be present. Although it may appear to be a trivial matter, as we discuss further, there is commercially available “zirconium vanadate”, which contains, in fact, very little actual ZrV2O7. Here, we systematically investigate various synthesis methods, including solid-state, sol–gel, and solvothermal, to determine reliable routes for producing phase-pure ZrV2O7.
Experimental section
Synthesis strategy
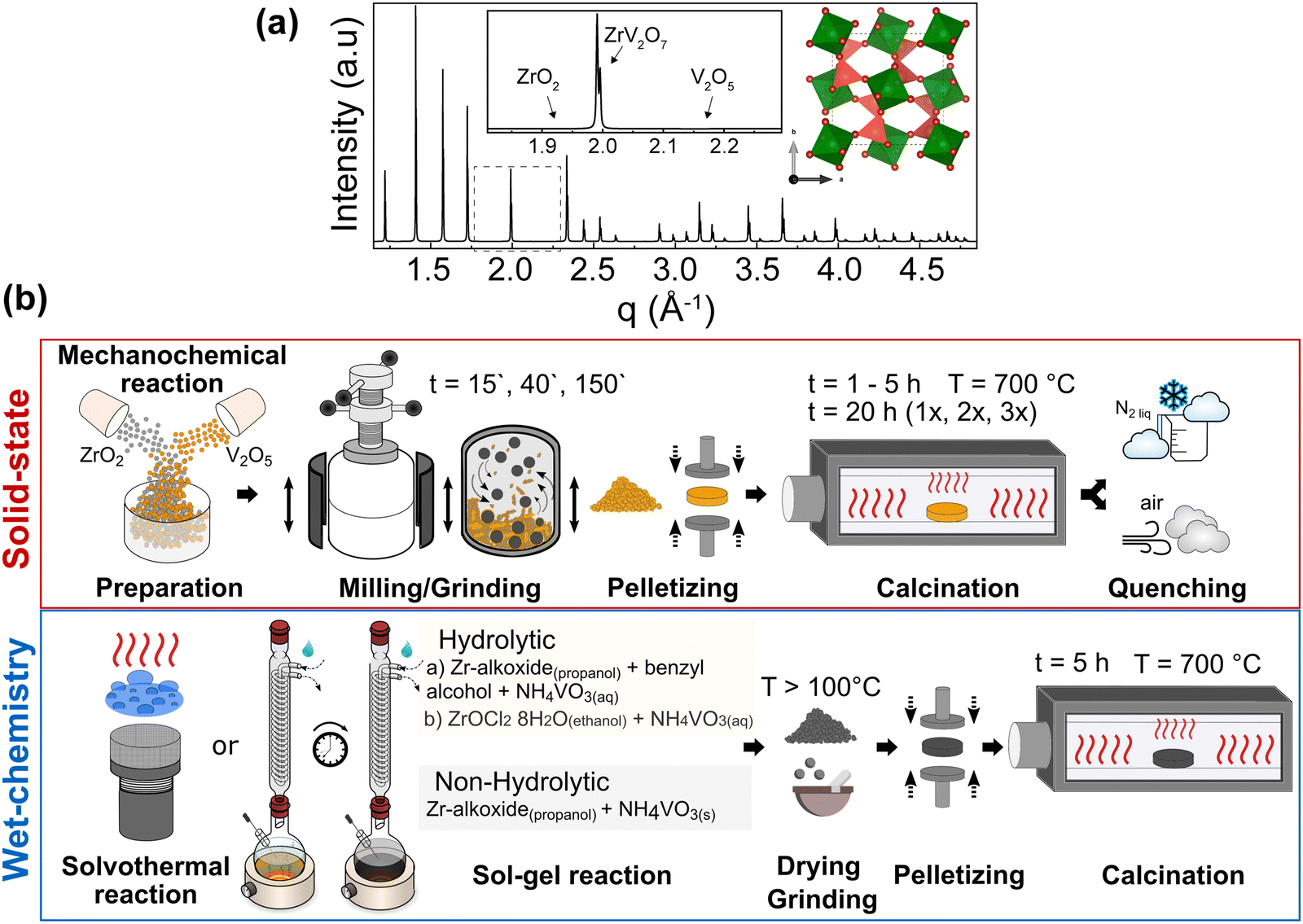 | ||
| Fig. 1 (a) A simulated X-ray diffraction pattern from ZrV2O7 crystal structure. Green polyhedra correspond to ZrO6 octahedra and dark pink tetrahedra to VO4. The selected representative area from a broader range is shown as an inset. (b) Overview of solid-state and wet-chemistry reactions. Solid-state method variations include milling times (15′, 40′, and 180′) and calcination for 5 h and 20 h cycles repeated 1, 2, or 3 times at 700 °C. Selected samples were heated for 5 h and quenched in air or liquid nitrogen (b top). Wet-chemistry methods include solvothermal, nonhydrolytic hot injection, and hydrolytic sol–gel reactions (b bottom). Further 2–7 figures include symbols used here to associate results with corresponding synthesis methods and synthesis steps. | ||
It is clear that uniform elemental mixing is crucial to obtaining high-quality products and needs to be improved. Reaction and processing parameters such as milling time and heating cycles with intermediate grinding steps could improve the purity of the final product. We refer to purity as the amounts of reagents remaining in the mixture and other side products limited to the minimum or removed altogether. Milling techniques can reduce particle size, leading to better homogeneity of the reactants and improved reactivity due to a higher surface–volume ratio. Additionally, repeated calcination cycles with intermediate grinding can promote a more thorough mixing of elements, ensuring a more uniform reaction throughout the material. Furthermore, quenching the product after synthesis can improve its purity by preventing undesired low-temperature processes.
We considered all these approaches within the ZrV2O7 system to assess the possible limits and relevant attempts of the solid-state reaction for purity and homogeneity. However, even with perfectly tuned parameters of solid-state synthesis, the extent of mixing within a bulk volume remains limited, and at best, Zr and V are separated from each other over distances of a few tens of nm or more. The presence of the remaining unreacted materials can be established when compared with the phase pure simulated X-ray diffraction (XRD) pattern from the optimised ZrV2O7 crystal structure (Fig. 1a). For clarity, a selected representative q range from a broader diffraction pattern will be displayed in the figures, which includes peak positions corresponding to ZrV2O7, V2O5 and ZrO2. (See solid state synthesis).
We investigated the influence of external reaction parameters on sample purity, including a variation of reagents, solvents, and reaction temperature (Fig. 1b). We followed solvothermal,29 nonhydrolytic30 and hydrolytic31 sol–gel methods for the analysis (Fig. 1b). The main difference between these methods is that the hydrolytic sol–gel method uses water. The hydrolysis and condensation reactions lead to the formation of a sol, which then undergoes further condensation to form a gel. In the nonhydrolytic and solvothermal methods, organic oxygen donors, such as ethers, alcohols, or alkoxides, replace water to supply the oxygen needed for the oxide network formation.31 In addition, the solvothermal method involves high-pressure and temperature conditions to improve chemical diffusion and reactivity, among others. In principle, the goal of all these wet-chemical approaches is to mix and stabilise Zr and V in the form of “dissolved” species such as ions, clusters, small macromolecules, etc., thus enabling the higher diffusivity and near-atomic level of elemental mixing better than that of solid-state methods.
Nonhydrolytic sol–gel hot injection synthesis was selected to provide a controlled environment for direct elemental reactions through mixing precursors at elevated temperatures. In contrast, hydrolytic sol–gel reactions allow a comparison of how the hydrolysis rate influences the purity and homogeneity of ZrV2O7. Previous methods and solvothermal reactions increase the sample size and, therefore, the reliability of the results. We also used an already-reported synthesis method (sol–gel_Cl)26 that allows better insight into already published data and reproducibility attempts. (See solution-based synthesis methods).
Furthermore, the comparison or validation of results with theoretical methods requires high-quality reference data, which can only be obtained on highly pure and homogeneous samples. For example, predicted (simulated) Raman, IR spectra, or XRD patterns can only be compared with high-quality measured data. Otherwise, excessive peak overlap, data fitting challenges, and difficult-to-resolve structures prevent the reliable comparison and validation of computational results. In particular, the prediction of temperature effects on material properties (e.g., lattice parameter changes with temperature, temperature-dependent vibrational spectra) still poses difficulties for ab initio simulations, and methods dealing with anharmonic effects are still under development, so good reference data are particularly important.32–34 (See Simulated and experimental Raman spectra comparison and Thermal expansion of multi-phase materials).
Materials
Commercially available ZrV2O7 was purchased from Onyxmet (CAS-Nr 113981-20-9). The reagents used for the solid-state reactions were zirconium oxide ZrO2 (≥99%, Carl Roth) and vanadium oxide V2O5 (99.9%, Chempur). Analytical grade ZrO2 and V2O5 were additionally dried for 15 h at 100 °C to remove any moisture and to weigh correct stoichiometric amounts. Powders were kept in a desiccator for further use. Particle size distribution of ZrO2 and V2O5 were measured as described in the methods section. Median particle size was d50.3(ZrO2) = 1.808 μm and d50.3(V2O5) = 1.412 μm.Wet-chemical syntheses were performed using ammonium vanadate NH4VO3 (≥99% Sigma-Aldrich) and Zr-alkoxide 70% solution in 1-propanol (Thermo Scientific Chemicals) with benzyl alcohol (≥99.8% Chemsolute). We selected benzyl alcohol as an effective solvent because it can provide monodisperse nanoparticles without adding surfactant35 and stabilise Zr-alkoxide, which is otherwise sensitive to moisture in the air. Zr-alkoxide also serves as a chloride- and nitrate-free reagent to avoid additional contaminations that are complex to remove during the calcination step. Further information is summarised in the corresponding synthesis sections.
Solid state synthesis
8.08 g of ZrO2 and 11.9 g of V2O5 reagents were mixed at a Zr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V = 1:2 molar ratio and ground in a planetary ball mill using plastic containers with 3 mm and 5 mm zirconia balls in ethanol media for 15 min, 40 min, or 3 h (15′, 40′, 180′). The homogenised powder was prepressed at 20 MPa for a few seconds, then pressed at 60 MPa, and dwelt for 1 min. The obtained pellets were 2 cm in diameter and 3 mm in height. Pellets were fired in a tubular furnace at a 5 °C min−1 heating rate, held at 700 °C for 1–5 h, 10 h, or 20 h in an ATV PEO 601 annealing furnace, and then cooled naturally to room temperature (RT). Further, calcination for 20 h was repeated two to three times with intermediate grinding in an agate mortar to support a homogenous reaction. Additional air and liquid nitrogen-quenched samples were prepared after 5 h calcination at 700 °C.
:V = 1:2 molar ratio and ground in a planetary ball mill using plastic containers with 3 mm and 5 mm zirconia balls in ethanol media for 15 min, 40 min, or 3 h (15′, 40′, 180′). The homogenised powder was prepressed at 20 MPa for a few seconds, then pressed at 60 MPa, and dwelt for 1 min. The obtained pellets were 2 cm in diameter and 3 mm in height. Pellets were fired in a tubular furnace at a 5 °C min−1 heating rate, held at 700 °C for 1–5 h, 10 h, or 20 h in an ATV PEO 601 annealing furnace, and then cooled naturally to room temperature (RT). Further, calcination for 20 h was repeated two to three times with intermediate grinding in an agate mortar to support a homogenous reaction. Additional air and liquid nitrogen-quenched samples were prepared after 5 h calcination at 700 °C.
Wet chemistry synthesis methods
(b) For sol–gel_Cl reaction: the samples were prepared as reported by Yanase et al.26 A 0.05 M aqueous NH4VO3 solution and a 0.05 M ethanol solution of ZrOCl2·8H2O (molar ratio Zr:V = 1:2) were stirred over 20 h at 60 °C, then dried in the air at 100 °C. Dried samples were ground in an agate mortar to obtain powder samples.
Heat treatment step
All products of the solution-based synthesis methods exhibit amorphous or partially amorphous XRD patterns. Further heating steps that lead to nucleation, growth, and calcination of the zirconium vanadate phase were required. Pellets of 0.5 cm × 0.2 cm (diameter × height) were pressed and heated at 700 °C for 5 h (5 °C min−1 heating rate).Methods
kV and a working distance of 5702.2 μm. The powder was conductive enough to prevent any discharge effects on the carbon tape. Additional images (Fig. 2b) were collected with an SEM/FIB FEI Quanta 3D electron microscope in the secondary electron detection mode, with a 10kV acceleration voltage and a working distance of 10.0 mm.
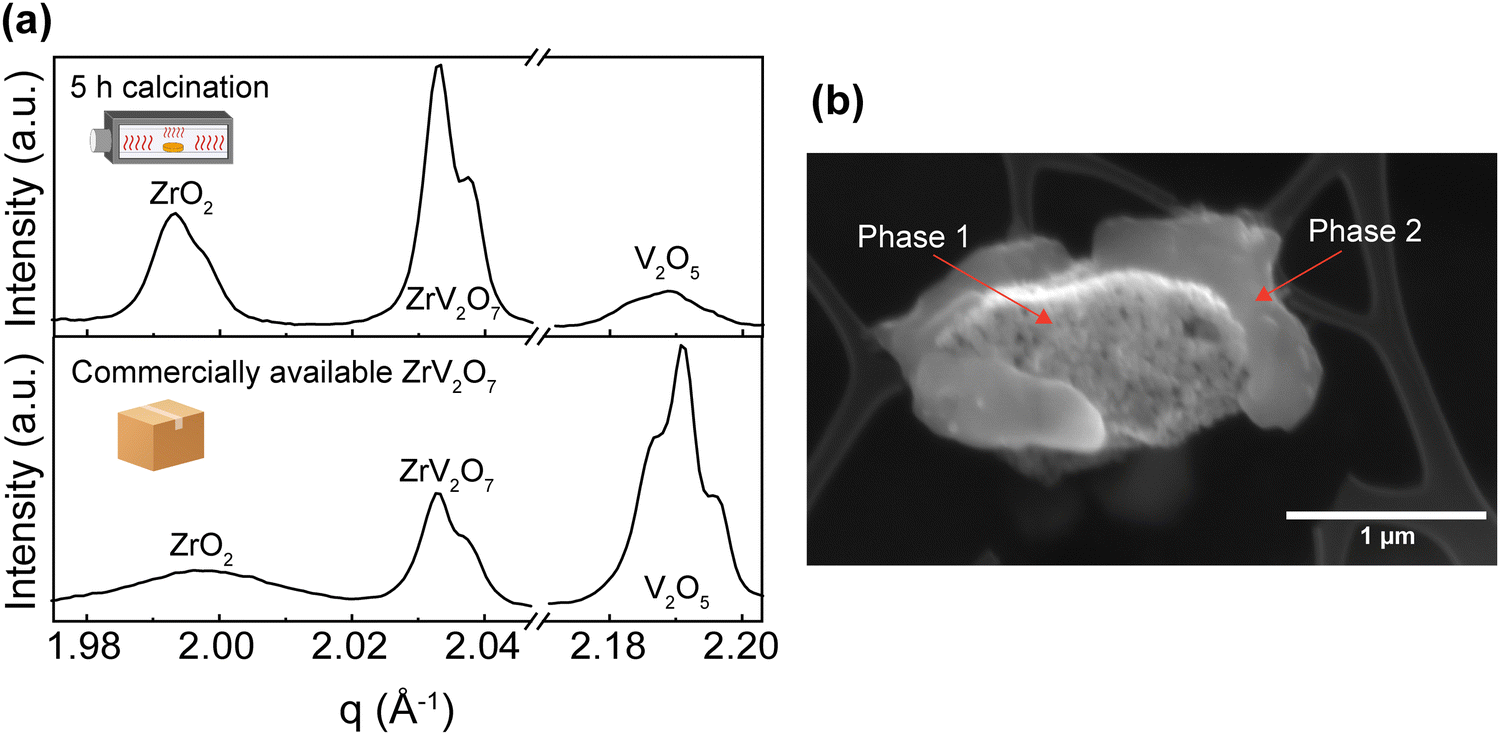 | ||
| Fig. 2 (a) XRD of a solid-state 40′ 5 h multi-phase material and commercially available ZrV2O7 material. (b) SEM image (20 kV, SE) corresponds to the multi-phase solid-state 40′ 5 h material. | ||
:V = 1:2 molar ratio and ground in a planetary ball for 10, 40, 90, 120 and 180 minutes. Further information on sample preparation and measurement procedure can be found in ESI.†
Computational simulations
In addition to experimental work, we performed ab initio computations. Structure optimisation and vibrational properties were computed using Density Functional Theory (DFT) with the Perdew–Burke–Ernzerhof (PBEsol) exchange–correlation functional36 for solids as implemented in the Vienna ab initio simulation package (VASP).37–40 Our calculations used the projected augmented-wave method40,41 with a kinetic energy cut-off of 520 eV. We computed Raman-active modes with Phonopy42,43 at the Gamma point by analysing the irreducible representations. Computational data was compared with experimental results. The ab initio spectra of phase-pure materials are based on ideal crystal structures and are therefore, unaffected by additional phases or instrumental errors. The simulated spectra can be used as a reference and to interpret the experimental spectra. More details on structure optimisation, vibrational properties computations and extraction of Raman-active modes are provided in the ESI,† computational simulations section.Results and discussion
Solid state synthesis
These experiments confirmed previous publications17,44 in which short firing time was reported to affect purity and failed to provide a single-phase product. Our results highlight that a short, up to 10-hour calcination step at 700 °C failed to produce phase-pure ZrV2O7 from micrometre-sized starting powder. This is most likely caused by insufficient diffusion of elements, leading to a heterogeneous reaction throughout the sample. In contrast, some authors reported such an approach as successful and analysed electrical properties23 and up-conversion luminescence9 of the 2–8 h calcined material. In such cases, unreacted materials were not detected, however, no quantitative analysis was reported to confirm phase purity. It is worth noting that part of the reported XRD experimental data on ZrV2O7 is provided as JCPDS (Joint Committee on Powder Diffraction Standards) cards. This data, possibly, has been included as a part of modern databases (for example, Inorganic Crystal Structure Database (ICSD)), but the labels were changed in the process. Therefore, it is difficult to access said data and compare, determine or clarify phase purity.
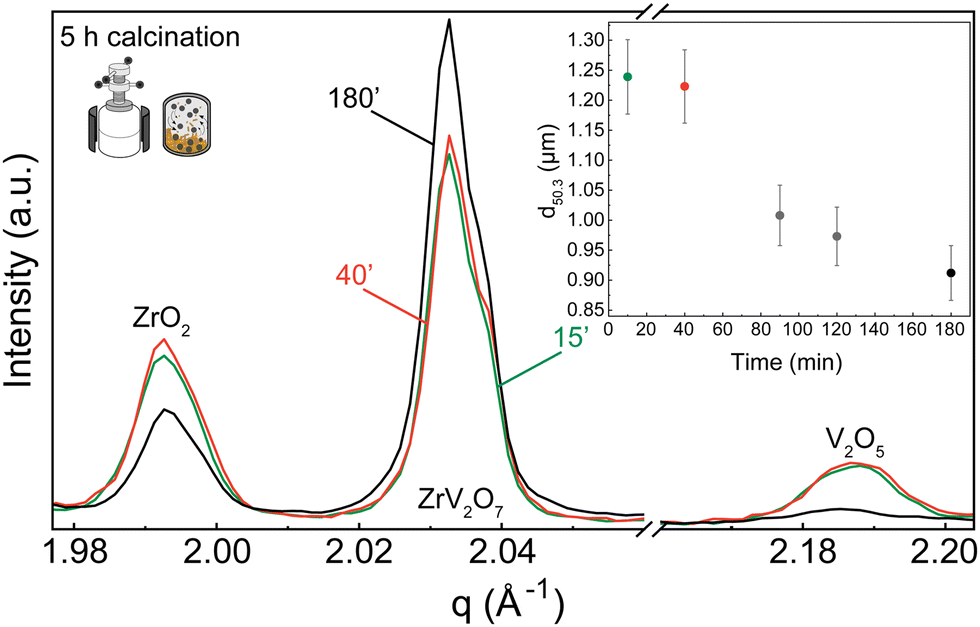 | ||
| Fig. 3 XRD patterns of 15′, 40′, or 180′ milled precursors that were calcined for 5 h. The figure shows selected reflections corresponding to ZrO2 (COD ID 9007485) and V2O5 (COD ID 9012221) phases diminishing with increasing milling time. Samples were heat treated for 5 h at 700 °C. Median particle size distribution after milling the ZrO2 and V2O5 mixture in a planetary ball mill for 15′ (green), 40′ (red), 90′ (grey), 120′ (grey) and 180′ (black) (SD = 0.312 μm) are shown in the inset. Before the experiment, the ZrO2 median particle size distribution was 1.808 μm and 1.412 μm for V2O5. | ||
XRD patterns were collected for 15 min, 40 min, and 180 min milling experiments after 5 h calcination at 700 °C. All three samples contain residual phases of ZrO2 and V2O5 because a shorter thermal treatment time was deliberately selected to demonstrate the importance of milling. The results correlate with the reduction in median particle size at around 1.5 h of milling. For 15 min and 40 min milled samples, the recorded XRD patterns are equivalent, and there is no clear improvement. However, increasing the milling time to 3 hours resulted in a decrease in unreacted oxides (Fig. 3). Following XRD patterns and particle size distribution, we can conclude that longer milling times reduce the particle size of reactants, improve reactivity, and create a more homogenous mixture, thus improving the quality of the final product.
In contrast to our approach, many published solid-state synthesis methods of ZrV2O7 and its substituted variations included combining ZrO2 and V2O5 in an agate mortar.4,9,10,13,20,22,23 Only Kul’bakin et al.21 implemented a planetary mill to pre-disperse oxides – a more efficient tool than manual grinding. We report that increasing the milling time in the planetary ball mill to 180′ improved the purity of solid-state reaction products. However, it was insufficient to produce phase-pure materials, and further steps need to be taken. For further improvement, we considered two approaches: introducing calcination cycles by maintaining 40 or 180-minute milling of the reactants or implementing quenching techniques.
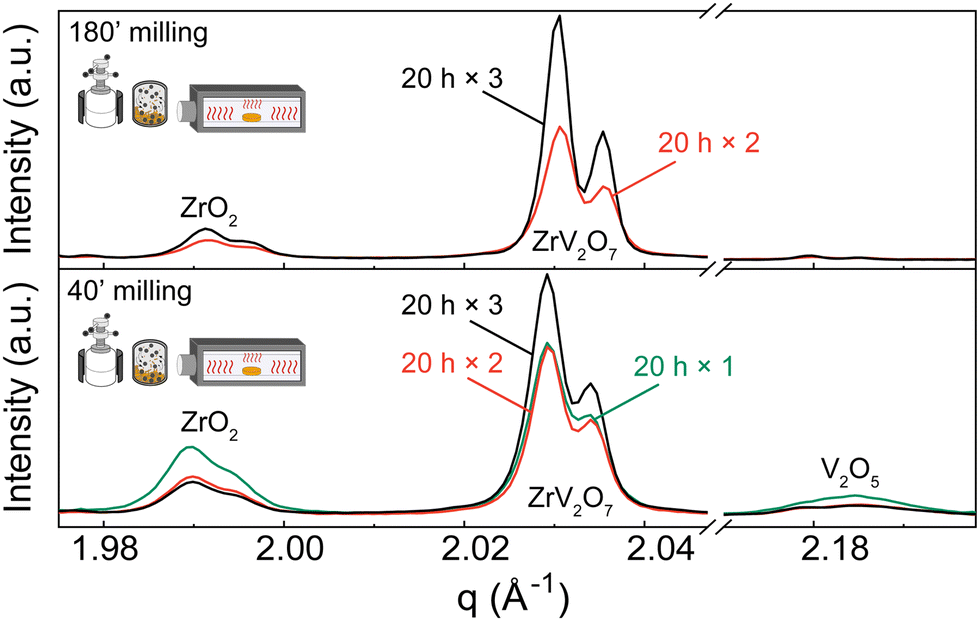 | ||
| Fig. 4 XRD patterns of (a) 180′ and (b) 40′ milled samples calcined for 20 h one, two or three times with intermediate grindings in an agate mortar. Patterns show ZrO2 and V2O5 phases diminishing with additional calcination cycles. Q shift occurs due to height misalignment during measurements. Systematic error is accounted for in Rietveld refinement. Refinement data for samples of interest is included in the ESI.† Experimental XRD patterns can be found at DOI: https://doi.org/10.5281/zenodo.12688634 (ref. 45) or upon request. | ||
We observed that during heat treatments V2O5 content was diminishing faster than ZrO2, with only later remaining in the sample after three calcination cycles. Based on the thermogravimetric analysis (TGA) experiments described in the ESI† (Fig. S12), only a negligible amount of V2O5 could be lost during heat treatments due to a higher volatilization ratio of V2O5. Therefore, we hypothesize that different residual ratio is related to a more complex reaction mechanism that is beyond the scope of this work. For instance, as amorphous V2O5 can be obtained by ultrafast cooling from the melt,46 such a process may occur in small amounts under slower cooling conditions. V2O5 remains present as an amorphous content that is untraceable in the XRD analysis, creating an apparent ZrO2 excess. This is further discussed in the Quenching Experiments section. Alternatively, due to phase equilibrium conditions, there is a shift towards ZrO2 phase formation.
While we cannot draw definite conclusions about residual ZrO2, nonetheless, extended milling and repeated calcination with intermediate grinding provide sufficient elemental mixing and can produce a high-quality pure sample comparable with the ZrV2O7 synthesised via wet-chemical methods. With 95.5% zirconium vanadate purity and high crystallinity, we consider this sample suitable for further analysis and processing. We confirm that ZrO2 and V2O5 solid-state reactions are very slow and thus require long processing times.17
A 5 h calcined sample was air-quenched from 700 °C to room temperature, and another 5 h calcined material was immersed in liquid nitrogen to achieve an extremely fast cooling effect. Quenching in liquid nitrogen showed an insignificant 3.5% reduction of unreacted materials compared to a slowly cooled-down sample. Subsequent phase quantification using Rietveld refinement revealed the composition to be 76.4% ZrV2O7, 13.4% V2O5, and 10.2% ZrO2 (Fig. 5 top). Collected XRD patterns of air-quenched samples matched the ones of the corresponding slowly cooled-down counterparts. Thus, air-quenching proved insufficient to produce any observable difference (Fig. 5 bottom). According to our findings, quenching 5 h calcined ZrV2O7 in air or liquid nitrogen does not sufficiently improve sample purity and cannot replace repeated heating cycles and extended milling time. The synthesis of phase pure ZrV2O7 renders rigorous quenching protocols irrelevant.
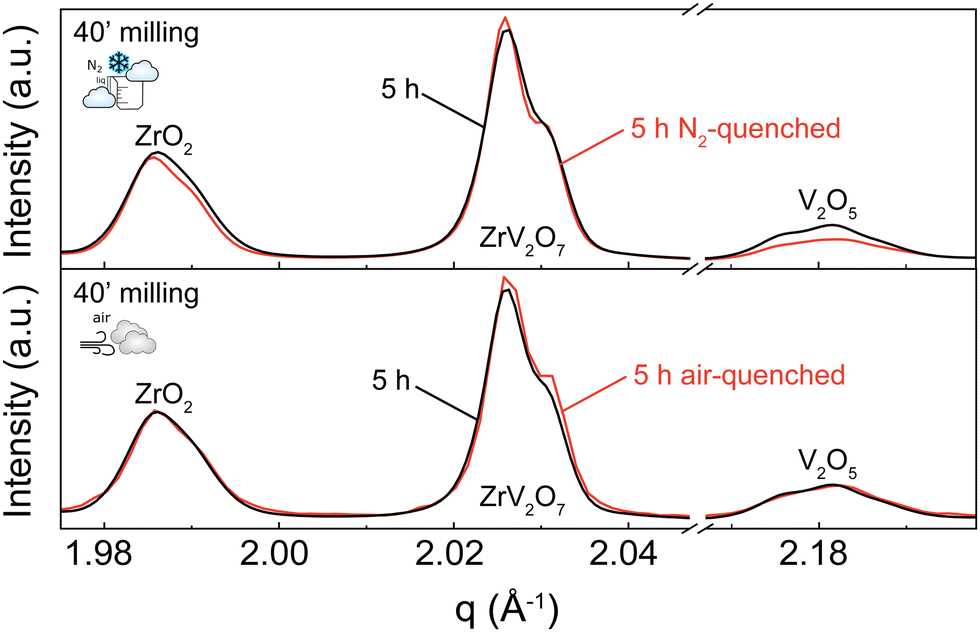 | ||
| Fig. 5 XRD patterns comparing a 40′ 5 h sample quenched in liquid nitrogen versus a gradually cooled down 40′ 5 h sample (top) and an air-quenched 40′ 5 h sample versus the same 40′ 5 h sample (bottom). | ||
While we consider the 3.5% total reduction of unreacted material insignificant, we observed different diminishing rates of V2O5 and ZrO2. Quenching experiments in liquid nitrogen showed a reduction of V2O5 of 2.2% and only 1.3% of ZrO2 compared with the 40′ 5 h sample. This information can further support the hypothesis that depending on processing conditions, V2O5, in addition to reaction with ZrO2, can partially transition to its amorphous state, thus remaining present though untraceable in the XRD patterns.
The synthesis steps followed here were reported by Zhang et al.22 and Wei et al.10 They described 4 h calcination and quenching to obtain Zr0.70V1.33Mo0.67O6.73 and ZrV2−xMoxO7+δ materials. Zhang et al.22 did not report any unreacted materials remaining in the sample. The investigation relied on energy dispersive spectroscopy (EDS), X-ray photoelectron spectroscopy (XPS), and XRD data to confirm the sample composition. The selected quenchant was not specified. Wei et al.10 used cold water as a quenchant. They analysed the purity of the samples by refining powder XRD and synchrotron-radiation X-ray diffraction (SXRD), indicating that the samples were pure. Possibly, the quenching step is a crucial requirement for compositions involving molybdenum (Mo) substitution.
In the case of elemental substitution with Mo, quenching techniques can be employed to enhance purity by preventing the exsolution and stabilising a specific desired phase. Additional elements, for example, Mo may lead to the formation and even stabilisation of undesired secondary phases; therefore, synthesis requires better control of phase formation. It is achieved by quenching a desired, possibly non-equilibrium state while preventing undesired, low-temperature processes, such as phase transformations, by slowing down the reaction kinetics. However, before considering substitution, it is crucial to know if quenching experiments can produce phase-pure material or if the problem of residual ZrO2 and V2O5 phases would remain, creating a complex multi-phase system. If an additional quenching step does not improve the purity and produce multiphase material within the ZrV2O7 system, it remains true for a substituted material.
Solution-based synthesis methods
Hydrolytic and nonhydrolytic reactions were considered for our investigation of solution-based synthesis methods. We selected solvothermal, nonhydrolytic sol–gel hot injection methods (yielding sol–gel_160 °C and sol–gel_100 °C samples), “classical” hydrolytic sol- implementing Zr-alkoxide as a starting material (sol–gel_RO−) and compared them with the already reported sol–gel method using NH4VO3 and ZrOCl2·8H2O as precursors (sol–gel_Cl).26Phase quantification of all sol–gel synthesis products, except the hydrolytic (sol–gel_RO−) reaction, pointed directly to a single-phase ZrV2O7. Sol–gel_RO− showed a high purity of 97.1%, with only 2.9% of ZrO2 detected. Phase quantification of the solvothermally prepared sample showed a detectable amount of ZrO2 phase of 9.6% (Fig. 6a).
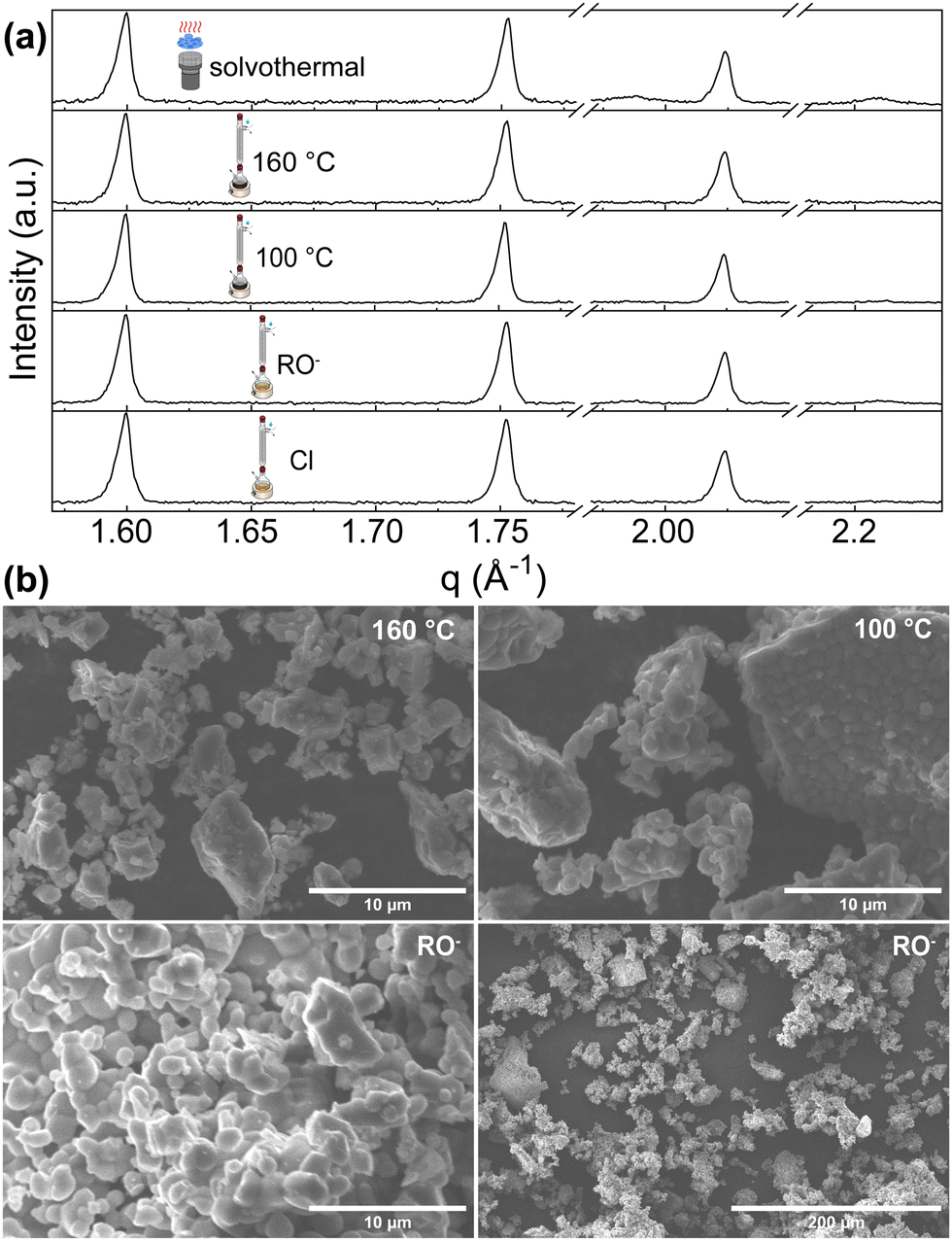 | ||
| Fig. 6 (a) XRD patterns of ZrV2O7 from solution-based synthesis methods. Solvothermal synthesis results in a small amount of ZrO2 phase. (b) SEM images of sol–gel_100 °C, sol–gel_160 °C and sol–gel_RO− samples. The sol–gel_100 °C (b top right) and sol–gel_RO− (b bottom) show smaller, more homogenous particles. The sol–gel_160 °C reaction image (top left) shows powder morphology, which is also similar to solvothermal and sol–gel_Cl reactions. SEM images of solvothermal and sol–gel_Cl reactions are provided in the ESI† Fig. S11. | ||
The morphology of the synthesis products mentioned above is shown in Fig. 6b. SEM images exhibit structures composed of dense particles that vary in size and form larger agglomerates. For sol–gel_160 °C, sol–gel_RO−, sol–gel_Cl, and solvothermal reactions, the particles’ size varies from <1 μm up to 10 μm and forms larger agglomerates reaching 60 μm in size (Fig. 6b, sol–gel_160 °C). The formation of smaller, more homogenous particles was observed for the nonhydrolytic (sol–gel_100 °C) and the hydrolytic (sol–gel_RO−) reactions (Fig. 6b). Dominating ∼1–2 μm sized sphere-like shaped particles were observed. While both reactions yield smaller crystallites than other discussed methods, the sol–gel_RO− reaction visibly provides the largest surface area.
The simple goal of improving Zr and V precursors mixing is driven by the sophisticated chemistry and physics of colloids. In the realm of hydrolytic synthesis, a common challenge arises from varying hydrolysis and condensation rates among different precursors (i.e., those containing different metal centres).31 They can affect elements' mixing in hydrolytic synthesis, potentially leading to inhomogeneous materials.47,48 A phenomenon of phase separation may be effectively induced.48,49 The addition of water can worsen the mixing effect, particularly in highly porous materials where the high surface tension of the water tends to cause the pores to shrink upon drying.50 In contrast, operating under non-aqueous conditions offers improved control over solvolysis and polycondensation kinetics, thus enabling better homogeneity in the elemental mixing.47,50 While the mechanistic details of solvolysis and condensation are beyond the scope of this work, they correlate with the phase composition of the final ZrV2O7.
According to our findings, both the non-hydrolytic sol–gel benzyl alcohol route and classical sol–gel approaches with varying reaction parameters were sufficient to produce high-purity samples. Hydrolysis rate control did not promote a more uniform mixing of elements. In addition to this, there was no significant difference in the phase composition (Fig. 6) when changing stirring time, reaction temperature, time, or reagents while implementing different synthesis routes. This may be due to an arbitrary threshold level of sufficient elemental mixing that ensures optimal results. All abovementioned solution-based reactions pass this threshold and modification of parameters does not show further improvement.
On the contrary, the solvothermal reaction showed the presence of an additional ZrO2 phase. In the case of solvothermal reactions, we consider that the degree of phase separation depends not only on factors like the difference in precursor reactivities, their relative concentrations, and the solvent used but also on reaction conditions such as temperature and pressure. Even though solvolysis is significantly slower than hydrolysis, phase separation remains possible. When precursors show different reactivities towards the non-hydrolytic condensation reactions, the more reactive species can form a complex metal–organic cluster and phase separate before the less reactive one has a chance to condense into the network.48,49 Such a mechanism could also be present in the case of the sol–gel reaction with alkoxide (sample, sol–gel_RO−); small local alkoxide-based clusters can form and lead to the formation of a small amount of ZrO2 during calcination.
An alternative explanation for the remaining ZrO2 after the solvothermal reaction could indicate that pressure conditions and the elevated temperature led to partial dissolution and recrystallisation of zirconium compounds instead of hydrolysis and condensation reactions, leading to the formation of ZrV2O7.29 Another possible consideration is that the reaction conditions could result in the initially formed ZrV2O7 decomposing to ZrO2. Sakuntala et al.51 reported pressure-induced decomposition at ambient temperatures. In situ, high-pressure Raman spectroscopic studies were carried out on ZrV2O7 samples synthesised via solid-state reaction. A potential shift in vanadium coordination was observed with applied pressure, transitioning from a fourfold coordination in ZrV2O7 to a fivefold in V2O5. In addition, Raman peaks corresponding to ZrO2 were indicated to appear. These observations were assigned to ZrV2O7 decomposition. As increased temperatures can enhance such a process, we hypothesise that a lower pressure but elevated temperature of the solvothermal reaction could lead to partial decomposition and the presence of ZrO2 (Fig. 6, solvothermal synthesis).
We indicate that sol–gel synthesis is a reliable method for obtaining high-purity samples. It also proved suitable for obtaining products with a desirable microstructure. However, elevated solvothermal reaction temperature and pressure conditions might lead to the formation of additional phases and provide lower-purity material.
Simulated and experimental Raman spectra comparison
As emphasized before, the phase composition and purity are crucial factors that allow for reproducible properties of the material. In this section, we provide a vivid illustration of this statement and show that ∼70% or >30% purity ZrV2O7 powders yield different experimental results than phase-pure material. We compare Raman spectra from a solid-state reaction with 5 h calcination time (40' 5 h), commercially available zirconium vanadate and a sol–gel (sol–gel_100 °C) reaction product (Fig. 7). According to the previously discussed XRD data analysis, the commercially available sample mostly contained V2O5, while 40′ 5 h showed a mixture of phases with ∼70% ZrV2O7 (Fig. 3). In contrast, the sol–gel_100 °C sample represents a phase-pure material (Fig. 6). As expected, analysis of Raman spectra revealed the abovementioned differences (Fig. 7). The commercially available sample mostly follows the vanadium oxide spectrum only with traces of ZrV2O7 appearing. Both commercially available and solid-state samples contain significant amounts of ZrO2 and V2O5 oxides, deviating significantly from the desired material. On the contrary, the sol–gel sample shows only five main peaks that do not coincide with the spectra of residual phases. This could indicate and further confirm phase purity.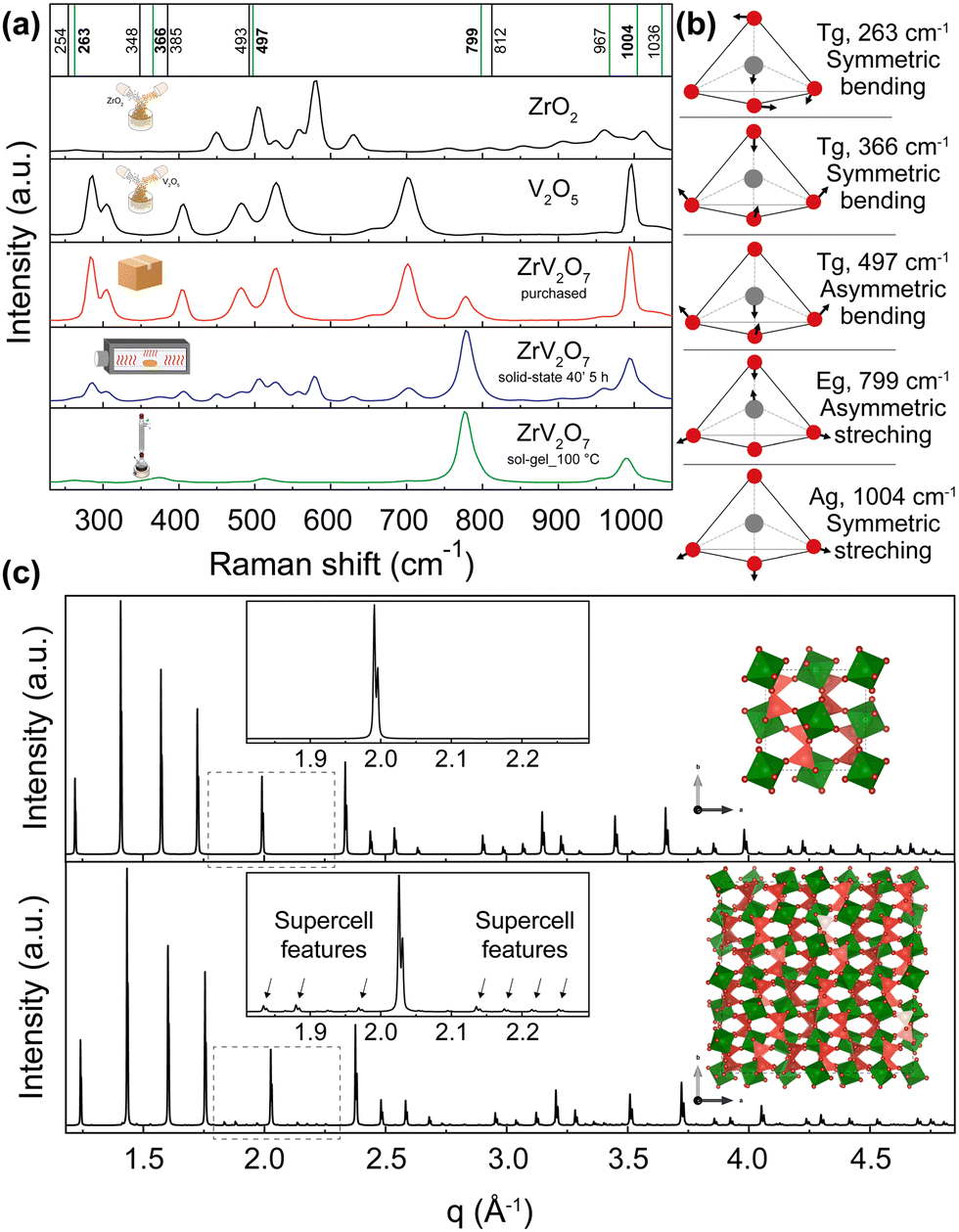 | ||
| Fig. 7 (a) Raman spectra of ZrV2O7 synthesised via sol–gel (sol–gel_100 °C) route (green), 40′ 5 h solid-state reaction (blue) and purchased material (red). These spectra are compared with V2O5 and ZrO2 reagents. The purchased material mostly matches V2O5 spectra, while the solid-state reaction product shows a combination of peaks representing both oxides and ZrV2O7. Vertical lines represent simulated Raman positions. Vibrations marked in green contribute to the Raman spectra and match the sol–gel reaction product within the margin of error. (b) Vibrations that correspond to 263, 366, 497, 799, and 1004 cm−1 frequencies are visualised above in a schematic way. Animations showing these vibrations are provided in the ESI.† (c) The XRD pattern of the ZrV2O7 structure used for computational simulations (c top) compared with the room temperature superstructure used in structure refinements (c bottom). | ||
To interpret the Raman spectra and validate that our product is phase pure, we used ab initio simulated phonon data. The computational results are simulated for a defect-free and phase-pure material. A 40 atoms unit cell was used (Fig. 7c, top) because the actual low-temperature superstructure that includes 1080 atoms in the unit cell makes calculations computationally unfeasible (Fig. 7c, bottom). In the case of the supercell, d spacing between the planes is repeated, and we see all higher-intensity ZrV2O7 peaks present in both structures. Only minor supercell features can be seen due to the slight tilting of ZrO6 octahedra (green) and VO4 tetrahedra (dark pink) (Fig. 7c, bottom). This shows that a smaller, 40 atoms cell, due to its structural similarity to the low-temperature ZrV2O7 phase, is suitable for obtaining computational Raman data.
First, we start with a quantitative comparison of experiment and simulation. We used a Voigt model to fit the experimental spectra. In addition, we carried out VASP and Phonopy ab initio computations to determine Raman active modes and corresponding Raman spectra peak positions. Ab initio values are marked in Fig. 7 as vertical lines. The frequency differences of the simulated and the matched fitted peak positions from the experiment are expressed as Δω = |ωfit − ωcalc|. The comparison was performed for the peak positions that best matched the pattern. Some peaks were neglected as the peak intensities were too low for accurate fitting results. Differences vary from 0.8 cm−1 for the peak at 262 cm−1 and up to a maximum of 21 cm−1 for the peak at 778 cm−1, considering the ab initio frequencies that best fit the experimental pattern. All the fitted peak positions and simulated frequencies are presented in the ESI,† Table S1. The differences were compared with the work of Bagheri and Komsa.52 They simulated Raman spectra based on harmonic phonon computations via a high-throughput computation and compared it with experimental results. The reported examples showed that the most significant average difference for SiO2 was 18.5 cm−1, and the smallest deviation of 6 cm−1. The deviations that occur in our work, therefore, correspond to expectations within the framework of the harmonic approximation and the level of theory (DFT with PBEsol functional). Further improvements could be achieved with the inclusion of additional anharmonic effects (e.g., with temperature-dependent effective potentials32 or other phonon renormalisation methods34 or the quasi-harmonic approximation42,53,54). However, these methods have the disadvantage that they require additional computing time, which makes their implementation much more difficult than the harmonic approximation, especially for a compound such as ZrV2O7 with a rather large 40 atoms unit cell (Fig. 7). For this reason, they are not used here. Overall, the simulated results match the sol–gel_100 °C reaction experimental Raman spectra within the expected error from the level of theory and the harmonic nature of our approximation to the phonons computation. The largest deviation of 21 cm−1 corresponds to a 2.62% error. This discussed example is representative of all discussed sol–gel reactions and solid-state synthesis that can provide a high-purity ZrV2O7 phase through extended milling and calcination times.
We also further interpret the Raman spectra based on the computed phonons through visualisation of the vibrations. Vibrations corresponding to 263, 366, 497, 799, and 1004 cm−1 frequencies are shown in Fig. 7. Also, all of the animated computed vibrations are provided in the ESI.† Raman active frequencies mostly match the vibrations of vanadium and oxygen atoms within the VO4 tetrahedra, except the 256 cm−1 frequency associated with Zr movements in ZrO6 octahedra. Frequencies at 799 and 1036 cm−1 represent asymmetric, and 967 with 1004 cm−1 show symmetric stretching of VO4 tetrahedra. With 799 and 1004 cm−1 vibrations contributing the most to the Raman spectra. Smaller intensity peaks at 263, 366, and 497 cm−1 correspond to symmetric and asymmetric bending of VO4 tetrahedra, respectively. These simulated optically active modes of phase pure ZrV2O7 structure match the frequencies in the ZrV2O7 experimental Raman spectrum. In the case of multiphase materials, due to peak overlap and complex peak assignment, structural information might be lost. In addition to this, validation with theoretical data would be impossible, thus preventing the detailed analysis of Raman active atom vibrations (e.g. by analysing the vibrational eigenvectors from the phonon computation as done in Fig. 7). Therefore, in this case, the necessity of a phase-pure material is further emphasised.
Hemamala et al.55 also analysed Raman spectra of ZrV2O7 from experimental data and ab initio calculations. The authors implemented Perdew–Burke–Ernzerhof (PBE) functional56 and (4 × 4 × 4) k-point sampling mesh. Force convergence of 0.001 eV Å−1 for geometry relaxation was used, and 0.001 meV convergence for self-consistent force calculations. In contrast, we used PBEsol functional, a Γ-centre grid of (6 × 6 × 6) k-points; ΔE < 10−8 and ΔE < 10−6 eV for the electronic and structural optimisation, respectively, thus increasing computational accuracy. Reported Raman-active modes centred at 263, 280, 367, 380, 404, 511, 774, 957, 984, and 1023 cm−1 confirm our results. Similar to our study, authors assigned lower frequency modes to the symmetric VO4 tetrahedra bending, 404 and 511 cm−1 to the asymmetric bending, and 774 cm−1 to the asymmetric stretching. High-frequency modes were assigned to the symmetric stretching of VO4 tetrahedra.
Thermal expansion of multi-phase materials
In this section, we discuss how the phase purity correlates with and affects the NTE property of the materials. Zirconium vanadate garnered interest due to its anomalous isotropic negative thermal expansion behaviour. Negative thermal expansion is a very rare property. Thus, it is very unlikely that secondary phases besides ZrV2O7 in a non-phase-pure product would share such an attribute. In the case of ZrV2O7, many synthesised and even commercially available materials contain unreacted precursors – zirconium and vanadium oxides. As temperature changes, we will inevitably see structural differences between zirconium vanadate and unreacted materials. This directly influences the negative thermal expansion property of a material. For example, in Fig. 8, for the peaks representing ZrO2 and V2O5 phases, when the temperature is increased, we see a decrease in q (where q = 2π/d). This means that d spacing increases, and we observe ordinary unit cell expansion. In the case of ZrV2O7, we see the opposite extraordinary effect of unit cell contraction (Fig. 8). This crucial difference within multiphase materials prevents the evaluation of the fundamental crystallographic principles of negative thermal expansion. Thus, the presence of additional phases would affect any applicational implementation of ZrV2O7 relying on NTE.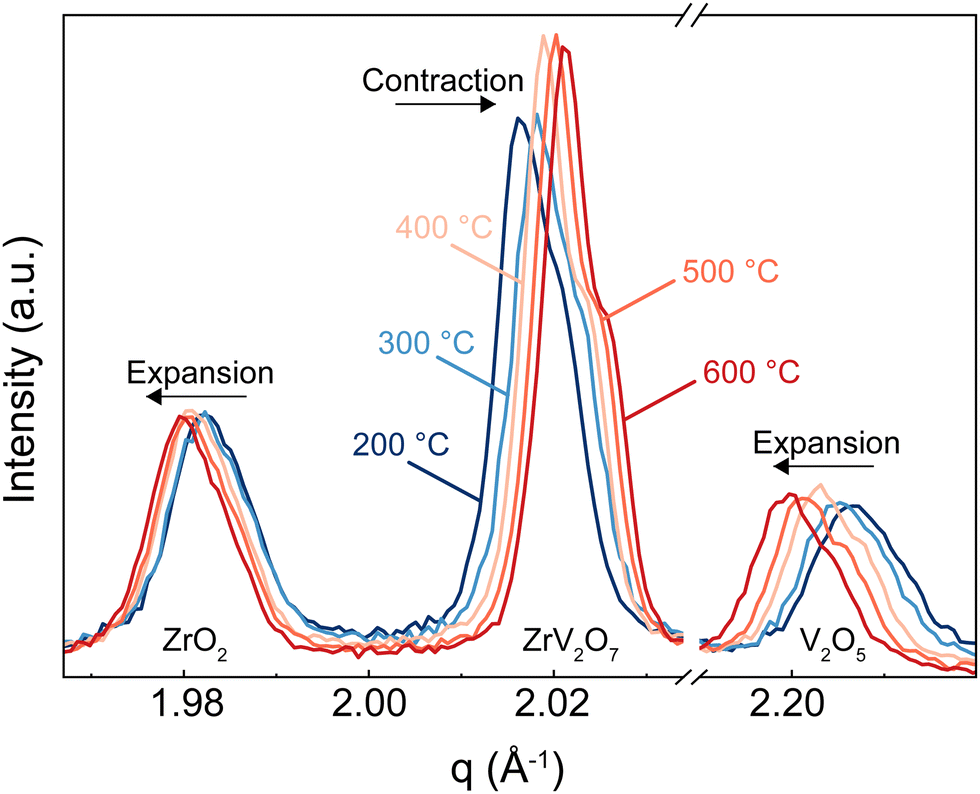 | ||
| Fig. 8 In situ XRD characterisation of ZrV2O7 sample synthesised via solid-state synthesis and calcined for 5 h at 700 °C. The peak corresponding to ZrV2O7 shows unit cell contraction in the 200–500 °C range, while residual ZrO2 and V2O5 phases display opposite unit cell expansion behaviour. | ||
All implemented characterisation methods, such as XRD, SEM and Raman spectroscopy, revealed differences between phase-pure and multiphase materials. Multiphase materials lead to reproducibility issues and difficulty in material characterisation. This is also true if substitution is considered, as it increases the probability of the final local composition diverging and leading to the formation of additional competing phases. Due to the cubic crystal structure, the unit cell of zirconium vanadate contracts isotropically with temperature.4 At the same time, common residual ZrO2 and V2O5 phases show the usual positive thermal expansion. V2O5 adopts an orthorhombic crystal system and ZrO2 a monoclinic one, both showing highly anisotropic behaviour, making it challenging to assess discrepancies in the experimental data. This could create issues in dilatometry measurements. Another commonly applied method is the energy dispersive X-ray (EDX) analysis, but this reveals only the presence of elements and does not allow differentiation between residual ZrO2 and V2O5 phases and ZrV2O7. On the contrary, high-temperature XRD analysis shows clear differences in unit cell expansion between ZrV2O7 and remaining oxides. It is a compelling technique to confirm the purity of negative thermal expansion materials or detect phases showing positive thermal expansion. However, data processing remains more challenging in comparison to phase-pure materials.
Conclusions
The synthesis of pure, homogeneous materials is essential for comprehensive material characterisation. Our study shows that the solid-state synthesis of ZrV2O7 requires careful consideration of various processing parameters to achieve high purity.Initial attempts using a 1 to 5 hours calcination step yielded multiphase materials containing only ∼70% ZrV2O7. However, by optimising the milling time and implementing repeated calcination cycles with intermediate grinding, we successfully produced a 95.5% pure ZrV2O7 sample.
We also investigated alternative wet chemical methods, including sol–gel and solvothermal synthesis. While solvothermal reactions resulted in the formation of an additional ZrO2 phase due to elevated temperature and pressure conditions, sol–gel methods proved highly effective in producing single-phase zirconium vanadate samples.
Raman spectroscopy was used as a powerful tool to assess sample purity. Spectra from multiphase materials showed peaks from unreacted precursors, making meaningful interpretation difficult. In contrast, Raman data from phase-pure samples agreed well with ab initio simulated phonon data, falling within the expected error range of density functional theory calculations within the harmonic approximation. This agreement allowed detailed analysis and visualisation of lattice vibrations, further confirming material purity.
In addition, high-temperature XRD proved to be invaluable in distinguishing between unreacted materials and the desired ZrV2O7 phase, revealing their contrasting thermal expansion behaviour. This technique is critical for verifying sample composition and purity.
In conclusion, our study highlights the importance of meticulous synthesis procedures and comprehensive characterisation techniques in the preparation and confirmation of high-purity ZrV2O7. These results not only advance our understanding of this specific material but also provide valuable insights for the broader field of materials science, highlighting the critical interplay between synthesis methods, characterisation techniques and material properties.
Author contributions
Conceptualization (BM, TMS, JG, AM). Investigation (performing the experiments (AM, BM)), data/evidence collection (MZ collected Raman spectra; AM, TMS, SK, ME conducted synchrotron experiments; JB and AM conducted the theoretical calculations; AM performed XRD and TGA experiments; TMS, AM and SK collected high-temperature XRD). Formal analysis (JB, JG, and AM analysed theoretical computations; AM and TMS performed Raman, TGA, and XRD data analysis; AM analysed SEM and particle size distribution data). Supervision (TMS, BM, JG). Visualization (AM, SK, ME). Writing (original draft (AM); review & editing (all authors contributed to the manuscript)).Data availability
The complete dataset is published on Zenodo (DOI: https://doi.org/10.5281/zenodo.12688634).45 Any other raw data are available from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for providing experimental facilities. Parts of this research were carried out at PETRA III, and we would like to thank Dr Alba San Jose Mendez for assistance in using P02.1 Powder Diffraction and Total Scattering Beamline. Beamtime was allocated for proposal I-20230110. We would also like to thank Franziska Lindemann for particle size distribution measurements and Michaela Lagleder for assistance in SEM imaging performed at the electron microscopy centre, Federal Institute for Materials Research and Testing (Berlin, Germany). AM, JB and JG would like to acknowledge the Gauss Centre for Supercomputing e.V. (https://www.gauss-centre.eu) for funding the DFT-related work by providing generous computing time on the GCS Supercomputer SuperMUC-NG at Leibniz Supercomputing Centre (https://www.lrz.de) (Project pn73da).References
- Q. Li, et al., Chemical Diversity for Tailoring Negative Thermal Expansion, Chem. Rev., 2022, 122, 8438–8486 CrossRef CAS PubMed.
- K. Takenaka, Giant Negative Thermal Expansion Materials: Progress of Research and Future Prospects, Mater. Trans., 2024, 65, 243–252 CrossRef CAS.
- R. L. Withers, J. S. O. Evans, J. Hanson and A. W. Sleight, An in situ temperature-dependent electron and X-ray diffraction study of structural phase transitions in ZrV2O7, J. Solid State Chem., 1998, 137, 161–167 CrossRef CAS.
- V. Korthuis, et al., Negative Thermal Expansion and Phase Transitions in the ZrV2−xPxO7 Series, Chem. Mater., 1995, 7, 412–417 CrossRef CAS.
- J. S. O. Evans, Negative thermal expansion materials, J. Chem. Soc., Dalton Trans., 1999, 0, 3317–3326 RSC.
- Q. Liu, et al., Structural, negative thermal expansion and photocatalytic properties of ZrV2O7: a comparative study between fibers and powders, Mater. Charact., 2014, 96, 63–70 CrossRef CAS.
- L. Shreenivasa, et al., Engineering of highly conductive and mesoporous ZrV2O7: a cathode material for lithium secondary batteries, J. Solid State Electrochem., 2019, 23, 1201–1209 CrossRef CAS.
- P. P. Sahoo, S. Sumithra, G. Madras and T. N. Guru Row, Synthesis, Structure, Negative Thermal Expansion, and Photocatalytic Property of Mo Doped ZrV2O7, Inorg. Chem., 2011, 50, 8774–8781 CrossRef CAS PubMed.
- J. Wang, et al. Temperature-controlled Upconversion Luminescence of Yb3+/Er3+ Co-doped ZrV2O7 Crystals, Asia Communications and Photonics Conference T4A.151 (Optica Publishing Group, Asia Communications and Photonics Conference, 2021) DOI:10.1364/ACPC.2021.T4A.151.
- W. Wei, et al., Realizing isotropic negative thermal expansion covering room temperature by breaking the superstructure of ZrV2O7, Appl. Phys. Lett., 2020, 116, 181902 CrossRef CAS.
- Q. Liu, et al., Influence of W doped ZrV2O7 on structure, negative thermal expansion property and photocatalytic performance, Appl. Surf. Sci., 2014, 313, 41–47 CrossRef CAS.
- B. Yuan, et al., Low thermal expansion over a wide temperature range of Zr1−xFexV2−xMoxO7 (0 ≤ x ≤ 0.9), Mater. Chem. Phys., 2016, 170, 162–167 CrossRef CAS.
- B.-H. Yuan, et al., High Solubility of Hetero-Valence Ion (Cu2+) for Reducing Phase Transition and Thermal Expansion of ZrV1.6P0.4O7, Chin. Phys. Lett., 2014, 31, 076501 CrossRef.
- I. Yanase, T. Kojima and H. Kobayashi, Effects of Nb and Y substitution on negative thermal expansion of ZrV2−XPXO7 (0 ≦ X ≦ 0.8), Solid State Commun., 2011, 151, 595–598 CrossRef CAS.
- J. S. O. Evans, J. C. Hanson and A. W. Sleight, Room-Temperature Superstructure of ZrV2O7, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 705–713 CrossRef.
- O. Yu Khyzhun, V. L. Bekenev, M. V. Karpets and I. Yu Zavaliy, First-principles FP-LAPW calculations and X-ray spectroscopy studies of the electronic structure of Zr3V3O and Zr3V3O0.6 oxides, J. Phys. Chem. Solids, 2012, 73, 1302–1308 CrossRef CAS.
- R. C. Buchanan and G. W. Wolter, Properties of Hot-Pressed Zirconium Pyrovanadate Ceramics, J. Electrochem. Soc., 1983, 130, 1905 CrossRef CAS.
- N. Khosrovani, A. W. Sleight and T. Vogt, Structure of ZrV2O7 from −263 to 470 °C, J. Solid State Chem., 1997, 132, 355–360 CrossRef CAS.
- S. Carlson and A. Andersen, High-pressure properties of TiP2O7, ZrP2O7 and ZrV2O7, J. Appl. Crystallogr., 2001, 34, 7–12 CrossRef CAS.
- T. Hisashige, T. Yamaguchi, T. Tsuji and Y. Yamamura, Phase Transition of Zr1−xHfxV2O7 Solid Solutions Having Negative Thermal Expansion, J. Ceram. Soc. Jpn., 2006, 114, 607–611 CrossRef CAS.
- I. Kul’bakin, S. Fedorov, A. Vorob’ev and V. Belousov, Transport properties of ZrV2O7–V2O5 composites with liquid – channel grain boundary structure, Russ. J. Electrochem., 2013, 49, 878–882 CrossRef.
- M. Zhang, et al., A novel negative thermal expansion material of Zr0.70V1.33Mo0.67O6.73, RSC Adv., 2017, 7, 3934–3940 RSC.
- B. Yuan, et al., Electrical properties and dielectric relaxation behavior of zirconium vanadate, Ceram. Int., 2018, 44, 21621–21625 CrossRef CAS.
- M. F. Elkady, M. A. Alrafaa and N. A. El Essawy, Morphological and electrical properties of zirconium vanadate doped with cesium, Beni-Suef Univ. J. Basic Appl. Sci., 2014, 3, 229–237 Search PubMed.
- Q. Liu, X. Cheng, X. Sun, J.-Y. Yang and H. Li, Synthesis and characterization of sol–gel derived ZrV2O7 fibers with negative thermal expansion property, J. Sol-Gel Sci. Technol., 2014, 72, 502–510 CrossRef CAS.
- I. Yanase, H. Chida and H. Kobayashi, Fabrication and negative thermal expansion properties of P-substituted ZrV2O7 sintered bodies, J. Eur. Ceram. Soc., 2018, 38, 221–226 CrossRef.
- Q. Kuang, Y. Zhao, Y. Dong and Q. Fan, Sol–gel synthesized zirconium pyrovanadate as a high-capacity cathode for rechargeable Li batteries, Electrochim. Acta, 2015, 170, 229–233 CrossRef CAS.
- Q. Li, et al., Superstructure ZrV2O7 nanofibres: thermal expansion, electronic and lithium storage properties, Phys. Chem. Chem. Phys., 2016, 18, 32160–32168 RSC.
- G. Demazeau, Solvothermal reactions: an original route for the synthesis of novel materials, J. Mater. Sci., 2008, 43, 2104–2114 CrossRef CAS.
- A. Vioux and P. Hubert Mutin, Nonhydrolytic Sol–Gel Technology, in L. Klein, M. Aparicio and A. Jitianu (ed.), Handbook of sol–gel Science and Technology, Springer International Publishing, Cham, 2016, pp. 1–27 DOI:10.1007/978-3-319-19454-7_28-1.
- A. E. Danks, S. R. Hall and Z. Schnepp, The evolution of ‘sol–gel’ chemistry as a technique for materials synthesis, Mater. Horiz., 2016, 3, 91–112 RSC.
- O. Hellman, I. A. Abrikosov and S. I. Simak, Lattice dynamics of anharmonic solids from first principles, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 180301 CrossRef.
- Z. Zhu, J. Park, H. Sahasrabuddhe, A. M. Ganose, R. Chang, J. W. Lawson and A. Jain, A high-throughput framework for lattice dynamics, npj Comput. Mater., 2024, 10, 258 CrossRef.
- F. Eriksson, E. Fransson and P. Erhart, The Hiphive Package for the Extraction of High-Order Force Constants by Machine Learning, Adv. Theory Simul., 2019, 2, 1800184 CrossRef.
- M. Niederberger and G. Garnweitner, Organic reaction pathways in the nonaqueous synthesis of metal oxide nanoparticles, Chem. Weinh. Bergstr. Ger, 2006, 12, 7282–7302 CAS.
- J. P. Perdew, et al., Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse, J. Furthmüller and J. Hafner, Theory of the crystal structures of selenium and tellurium: the effect of generalized-gradient corrections to the local-density approximation, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 13181–13185 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- A. Togo, First-principles Phonon Calculations with Phonopy and Phono3py, J. Phys. Soc. Jpn., 2023, 92, 012001 CrossRef.
- A. Togo, L. Chaput, T. Tadano and I. Tanaka, Implementation strategies in phonopy and phono3py, J. Phys.: Condens. Matter, 2023, 35, 353001 CrossRef CAS PubMed.
- V. Cirilli, A. Burdese and C. Brisi, La corrosione di leghe refrattarie da parte di ceneri di petrolio contenenti vanadio, Atti Della R. Accad. Delle Sci. Torino, 1961, 95, 399–424 Search PubMed.
- A. Miliūtė, et al., Synthesis and phase purity of the negative thermal expansion material ZrV2O7, 2024 DOI:10.5281/zenodo.12688634.
- M. Nabavi, C. Sanchez and J. Livage, Structure and properties of amorphous V2O5, Philos. Mag. B, 1991, 63, 941–953 CAS.
- A. Styskalik, D. Skoda, C. E. Barnes and J. Pinkas, The Power of Non-Hydrolytic Sol–Gel Chemistry: A Review, Catalysts, 2017, 7, 168 CrossRef.
- T. M. Stawski, et al., Nanostructure Development in Alkoxide-Carboxylate-Derived Precursor Films of Barium Titanate, J. Phys. Chem. C, 2012, 116, 425–434 CrossRef CAS.
- T. M. Stawski, et al., Development of Nanoscale Inhomogeneities during Drying of Sol–Gel Derived Amorphous Lead Zirconate Titanate Precursor Thin Films, Langmuir, 2011, 27, 11081–11089 CrossRef CAS PubMed.
- V. Smeets, A. Styskalik and D. P. Debecker, Non-hydrolytic sol–gel as a versatile route for the preparation of hybrid heterogeneous catalysts, J. Sol-Gel Sci. Technol., 2021, 97, 505–522 CrossRef CAS.
- T. Sakuntala, et al., Pressure-induced amorphization and decomposition in ZrV2O7: a Raman spectroscopic study, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 174119 CrossRef.
- M. Bagheri and H.-P. Komsa, High-throughput computation of Raman spectra from first principles, Sci. Data, 2023, 10, 80 CrossRef CAS PubMed.
- A. Togo and I. Tanaka, First principles phonon calculations in materials science, Scr. Mater., 2015, 108, 1–5 CrossRef CAS.
- R. P. Stoffel, C. Wessel, M.-W. Lumey and R. Dronskowski, Ab Initio Thermochemistry of Solid-State Materials, Angew. Chem., Int. Ed., 2010, 49, 5242–5266 CrossRef CAS PubMed.
- U. L. C. Hemamala, et al., High-pressure Raman and infrared study of ZrV2O7, Solid State Commun., 2007, 141, 680–684 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed particle size distribution measurement description and summarized results (Table S1). XRD instrumental parameters and analysis description including phase quantification results, plots, and Rietveld refinement parameter list. Phase quantification data covers solid-state 40′ 5 h sample (Fig. S1), commercially available ZrV2O7 (Fig. S2), solid-state 180′ 20 h × 3 (Fig. S3), solid-state 40′ 5 h N2-quenched (Fig. S4), solvothermal reaction product (Fig. S5), sol–gel_160 °C (Fig. S6), sol–gel_100 °C (Fig. S7), sol–gel_RO− (Fig. S8) and sol–gel_Cl reaction product (Fig. S9). Experimental analysis and computational simulations of Raman spectra, including description and plot of room-temperature spectra fitting (Fig. S10) and parameters used to simulate theoretical positions of Raman active modes. A comparison of calculated and fitted experimental frequencies is shown in Table S2. Visualisations of Raman active phonon vibrations are included for the spectral range of 200 to 1100 cm−1 as attached GIF files. Scanning electron microscopy images of solvothermal, sol–gel_Cl and solid state 40′ 20 h × 3 reactions showing the morphology of corresponding samples (Fig. S11). Thermogravimetric analysis (TGA) method description and analysis results. TGA weight loss curves of 40′ milled ZrO2 and V2O5 mixture heated from room temperature to 400 °C and 700 °C, held at that temperature for 20 h and cooled down to room temperature are presented in Fig. S12. See DOI: https://doi.org/10.1039/d4tc04095c |
| This journal is © The Royal Society of Chemistry 2025 |