
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimizing the short-wavelength infrared photoluminescence quantum yield and brightness of Er3+, Yb3+ co-doped yttrium orthophosphate phosphors for tracer-based sorting†
Nisrin Mohamed
Bhiri‡
a,
Herman
Duim
 b,
Eduard
Madirov
a,
Justine
Nyarige
a,
Bryce S.
Richards
*ac and
Andrey
Turshatov‡
*a
b,
Eduard
Madirov
a,
Justine
Nyarige
a,
Bryce S.
Richards
*ac and
Andrey
Turshatov‡
*a
aInstitute of Microstructure Technology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: bryce.richards@kit.edu; andrey.turshatov@kit.edu
bDelmic BV, Oostsingel 209, 2612 HL Delft, The Netherlands
cLight Technology Institute, Karlsruhe Institute of Technology, Engesserstrasse 13, 76131 Karlsruhe, Germany
First published on 7th January 2025
Abstract
Development of efficient inorganic luminescent tracers is essential for various applications, including plastic sorting. In this study, yttrium orthophosphate (YPO4) phosphors co-doped with Er3+ and Yb3+ lanthanide ions across a broad range of concentrations were synthesised via a solid-state reaction (SSR). The optimisation of SSR, and the structural and photoluminescence properties – including photoluminescence quantum yield (PLQY) and brightness (B) – of these new tracers are reported. The PLQY for the short-wave infrared (SWIR) emission of Er3+ under excitation with a 976 nm laser reaches a maximum of 67% for the sample with a nominal composition of 3 at% Er3+ and 3 at% Yb3+. The maximum brightness was achieved for a sample with a nominal composition of 3 at% Er3+ and 24 at% Yb3+. The brightest sample was printed using an industry-relevant process on a polyethylene pouch. Hyperspectral SWIR imaging under simultaneous irradiation of the pouch using a halogen lamp and 978 nm laser resulted in a strong luminescence signal peaking at 1540 nm. which was easily distinguishable against the reflectance background spectrum of polyethylene. This demonstration paves the way for future exploration of SWIR luminescence tracers in the next generation of plastic sorting approaches – tracer-based sorting.
1. Introduction
Over the past few decades, lanthanide activated materials have attracted the attention of many researchers around the world. Many authors have focused on their synthesis protocol, and structural, morphological and luminescence properties. This growing interest is due to the vast amount of their industrial applications as phosphors for light-emitting diodes (LEDs),1–3 laser,4,5 optical amplifiers,6 display devices,7 scintillation materials8 and luminescent tracers in medicine8,9 and recently in plastic recycling.10In 2021 in the European Union (EU), 84.3 million tons (mt) of packaging waste was collected, including 16 mt (19%) of plastic packaging waste.11 This 16 mt is roughly 26% of all plastic materials produced in the EU in 2021.12 While 64% of the total volume of 84.3 mt is currently being recycled, a significant challenge persists, with 60% (9.6 mt) of plastic packaging waste remaining unrecycled.
Polymer packaging materials have found extensive use across various sectors such as food, cosmetics, personal and health care, and medical industries. An impediment to achieving a higher recycling rate in the food industry is the stringent safety regulations. Even a small fraction of non-food grade packaging can cause migration of the chemical of concern to food grade plastic and can completely preclude the use of recyclate in food contact products.13 In addition, food-grade plastic packaging often involves a special multilayer design with a barrier function and the use of ink printing for consumer information. This, in turn, requires special processes for delamination (in the case of multilayer structures) and cleaning (including deinking and deodorisation) to integrate the recyclate into a closed-loop material production process for food-grade packaging.14–16
Extracting these specialised food grade materials from the overall plastics stream therefore requires a process that maximises yield, purity and efficiency. Achieving this goal is challenging with the current state-of-the-art near infrared (NIR) reflectance-based sorting technology, which relies on the chemical bonds in different polymers to provide a detectable reflectance spectrum. However, the advent of innovative sorting techniques such as tracer-based sorting (TBS) promises to make this goal achievable in the future.17
In the field of TBS applications, the shortwave infrared (SWIR) luminescence in the range of 1000 to 2000 nm emitted by various trivalent lanthanide ions (Ln3+) – such as Yb3+, Er3+, Pr3+, Tm3+ and Ho3+ – exhibits significant promise. The SWIR luminescence is detected as a reflection signal by the InGaAs detectors found in conventional NIR sorters. Implementing TBS in an NIR sorter requires only minor modifications – in particular, in addition to the conventional broadband light source (halogen lamp), a laser with an excitation wavelength of around 940–980 nm is required (as shown in Fig. 1). The choice of 940–980 nm is strategic because of the strong absorption of Yb3+, which can either emit with a peak at 1014 nm or sensitize the luminescence of Er3+, Pr3+, Tm3+ and Ho3+ at longer wavelengths. Thus, the TBS approach enhances the sorting capabilities of plastic recycling machines with minimal adjustments.17 In this regime, the reflectance of plastics and luminescence of tracers are measured simultaneously. While reflectance is inversely proportional to absorbance and is always a number less than one, the luminescence appears in reflectance spectra as a peak with apparent reflectance significantly greater than one (Fig. 1). If the luminescence signal is detected, the plastic object can be sorted out using compressed air, a tilt tray sorter or a robotic sorting system.
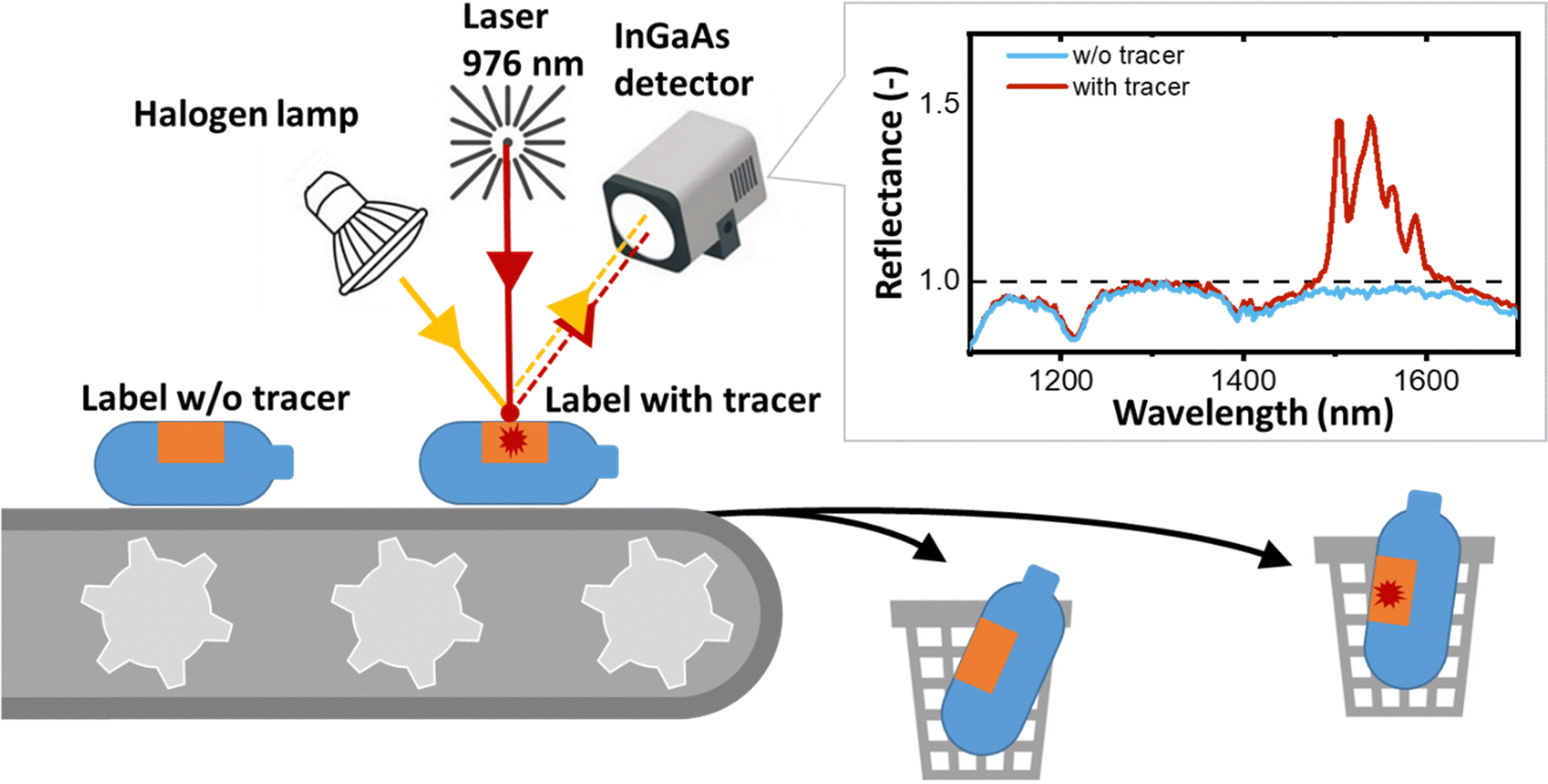 | ||
| Fig. 1 Schematic of a conventional NIR sorter upgraded with a 976 nm laser to detect SWIR luminescence from a plastic object labelled using a printed tracer, thus now achieving tracer-based sorting (TBS). | ||
For a given tracer concentration (typically μg per cm2 of the printed label), the amplitude of this luminescence peak should increase with increasing intrinsic photoluminescence quantum yield (PLQY), which is defined as the ratio of the number of photons emitted to the number of photons absorbed.18 PLQY is a fundamental parameter of the luminescence process that provides information about the competing rates of both radiative and non-radiative transitions, but does not take into account the percentage of incident radiation absorbed by a tracer, i.e. absorptance or absorption efficiency (A, units: dimensionless). Absorptance can be increased either by developing materials with a higher absorption cross-section (a, units: cm2) or by simply increasing the concentration of Yb3+ ions. In turn, the concentration of Yb3+ ions can be increased either via the increase of the tracer concentration in a printed label (C, expressed as mass of tracer per area of the label [g cm−2]) or via the increase of the doping concentration of Yb3+ in a tracer (NYb3+, units at%). If the absorptance is also taken into account, a second figure-of-merit (FOM) parameter appears – the brightness (B = A × PLQY, units: dimensionless), as defined by Wong et al.18 As absorption efficiency is directly proportional to a and NYb3+, brightness (still dimensionless) can be expressed as B ∼ a × NYb3+ × C × PLQY. The definition of brightness given above should not be confused with the definition used for liquids (B [M−1 cm−1] = α × PLQY, where α is the extinction coefficient) or the definition of brightness for LEDs, persistent luminescence, astronomical objects and microscopic objects as described by Wong et al. in an attempt to standardise the definition of brightness.18 It should also be noted that when it comes to scattering, the use of micro-sized tracers is more advantageous than nano-sized ones. When micro-sized materials are dispersed in a polymer or ink, they scatter the excitation light more strongly. This increases the excitation path length, resulting in increased absorption and luminescence brightness.19
Ultimately, the increase of PLQY (as a part of brightness) is an important process in the development of luminescence tracers for TBS. Recently, we investigated the effect of Yb3+ and Er3+ concentrations on the SWIR luminescence of the molybdate host Li3Ba2La(3−x−y)(MoO4)8 and were able to demonstrate a very high PLQY of 70% (measured using an integrating sphere).20 Furthermore, we have used this material as a luminescent tracer to print a unique luminescent label (onto a polyethylene terephthalate bottle) that can be identified using a SWIR hyperspectral camera. To continue the investigation of suitable tracers with high PLQY for the TBS process, we turned our attention to another potential host, yttrium-orthophosphate (YPO4). In our work, we have deliberately chosen to focus on phosphate-based tracers rather than fluoride-based tracers, which are also known for their efficient Ln3+ luminescence. This decision is based on previous concerns regarding the potential degradation and dissolution of Ln3+-doped fluorides, particularly in the context of bio-applications.21
In general, the incorporation of Ln3+ ions into inorganic materials (hosts) results in various luminescence emission peaks whose intensities, line widths, and intensity ratios are largely influenced by the nature of the host compound. Among many possible inorganic crystalline hosts, YPO4 is known for its high thermal and chemical stability; low symmetry tetragonal crystal lattice and high refractive index (that enhance partially forbidden radiative transitions in Ln3+); its relatively high phonon energy of 1060 cm−1 (similar to phonon energy of the efficient molybdate host20); and its ability to accept high doping concentrations of Ln3+ ions.22–27 It has been reported that the Yb3+,Er3+-activated YPO4 host exhibits potentially strong SWIR luminescence corresponding to the Er3+:4I13/2 → 4I15/2 transition, after sensitization via the Yb3+:2F7/2 → 2F5/2 transition.28–30 However, there is a notable lack of PLQY measurements for this particular process in the existing literature. To address this research gap, our study involved the meticulous fine-tuning of Yb3+ and Er3+ doping concentrations in a YPO4 host synthesized by a high-temperature solid-state process with the aim of identifying a composition with the highest PLQY and B in the SWIR region. Furthermore, we investigated the correlation between PLQY values and the level of intermixing of Yb3+ and Er3+ ions using the cathodoluminescence (CL) technique. Although this technique has previously been used to study the distribution of luminescent ions such as Tb3+ or Eu2+ in single or dual phase hosts,31–34 this is the first time it has been used to study the mixing of two lanthanide ions. Additionally, we demonstrated that YPO4:Yb3+,Er3+ tracers can be printed onto PE pouches using a research-scale implementation of an industry-relevant printing process. This new approach may be more feasible than adding tracers to bulk polymers, as previously reported.35–38 An SWIR hyperspectral camera was then used to visualise this labelled pouch with the aim of differentiating between two classes of PE-based packaging materials, specifically food grade and non-food grade plastics.
2. Experimental section
2.1. Synthesis procedure
Undoped YPO4 micro-phosphors as well as hosts co-doped with Ln3+ ions with a general composition of x at% Er3+, y at% Yb3+ were prepared following a solid-state reaction method (and labelled as YPO4:xEr,yYb throughout the manuscript). The starting raw materials from Sigma Aldrich: Y2O3 (99.99%), Er2O3 (99.9%) and Yb2O3 (99.9%) were weighed in stoichiometric proportions except for the (NH4)2HPO4 phosphate that was used with an excess of 10%. The mixtures were manually ground and preheated at 200 °C, then ground again and heated at 1200 °C for variable times (from 12 to 60 hours, following the protocol explained in Fig. S1, ESI†) in a muffle furnace (KLS 05/13, Thermconcept) under ambient atmosphere. Finally, the obtained powders (after 12, 24, 36, 49 and 60 hours of heating at 1200 °C) were ground again for subsequent structural analyses and optical measurements.2.2. Structural characterization
The crystal structure of the YPO4 materials was identified using a powder X-ray diffractometer (pXRD, Bruker D8 Discover) equipped with a Cu Kα radiation source. The measurements were performed over a 2θ angle range of 10–70° with a step width of 0.01° s−1. Scanning electron microscopy (SEM) images of the as-prepared powders were taken using a Zeiss Supra 60 VP, and the particle size was estimated using Image J software. Samples for CL measurements were prepared by sprinkling a small amount of phosphor powder on an adhesive carbon tab that was then placed in the SEM. CL measurements were performed on a FEI Helios Nanolab 600 SFEG SEM equipped with a Delmic SPARC Spectral CL detector. The SPARC Spectral CL detector is equipped with a PMT for intensity mapping, and a sensitive CCD for spectroscopy. A grating of 300 lines per mm blazed at 760 nm was used at different angles, corresponding to center wavelengths of 600 nm and 900 nm, to cover the full spectral range of interest (385–1100 nm). The samples were measured using the following beam conditions: 5 kV, 43 pA and dwell time of 15 μs for the CL intensity maps, and 5 and 10 kV, 43 pA and dwell times of 15 ms for the spectroscopic measurements.2.3. Optical characterization
Room temperature absorption spectra in the UV-Vis-NIR range were recorded using a Lambda 950 spectrophotometer (PerkinElmer) equipped with an integrating sphere in the absorbance mode. The photoluminescence excitation (PLE) spectra of YPO4 materials were obtained using a spectrofluorometer (Edinburgh Instruments FS5) equipped with a built-in xenon lamp as the excitation source.The thermal imaging of samples under laser excitation was performed using a FLIR ONE PRO (Teledyne FLIR) thermal camera.
The PLQY was measured in a custom-built optical system. The sample was placed in an integrating sphere (Labsphere, 6 in. ∅, 3 P-LPM-060-SL) and excited with a continuous-wave laser diode operating either at 940 nm and 180 mW (Thorlabs, M9-940-0200), or 976 nm and 190 mW (Roithner, RLT980-200GS), or 1510 nm and 15 mW (Roithner, RLT1510-15MGS). The excitation source was mounted on a temperature-controlled mount (Thorlabs, TCLDM9) and driven by a diode laser controller (Thorlabs, ITC4001). Detection of the SWIR emission spectra, excitation laser intensity, and PLQY of the Er3+:4I13/2–4I15/2 emission was performed using a NIR spectrometer (NIRQuest, Ocean Insight). To eliminate the scattered excitation radiation, a sharp turn-on 980 nm long-pass filter (Semrock, LP02-980RE-25) was placed between the output of the integrating sphere and the collecting fiber (Thorlabs, BFY200MS02).
It should be noted that in many cases the PLQY of a powder sample can be accurately measured using the integrating sphere, but the result obtained ideally describes the PLQY of the sample, not the intrinsic PLQY of the phosphors. One of the main reasons for the discrepancy between the measured value and the intrinsic PLQY is the strong re-absorption of the luminescence by the sample due to the very small Stokes shift for Ln3+ luminescence. Unfortunately, simple dilution of the sample to reduce re-absorption also reduces the absorptance inside the integrating sphere (for a reliable estimate of PLQY, the absorptance should be greater than ∼5%) and prevents accurate measurement of the intrinsic PLQY of the phosphor. Nevertheless, the dilution experiment can help to perform the PLQY correction to obtain the intrinsic PLQY of the phosphor from the experimental PLQY of the sample, as described in our previous work.20 Briefly, in the present work luminescence spectra were measured for a powder sample inside the integrating sphere and for a diluted (using BaSO4 powder) powder sample without applying the integrating sphere. The correction of the PLQY taking reabsorption into account was performed following the method described by Wilson and Richards.39 The long-wavelength tail of the Er3+:4I13/2–4I15/2 luminescence peak was adjusted for spectra obtained for two measurements, and the PLQY for the diluted powder sample (assumed to be equal to the PLQY of the phosphor) was calculated using eqn (1):
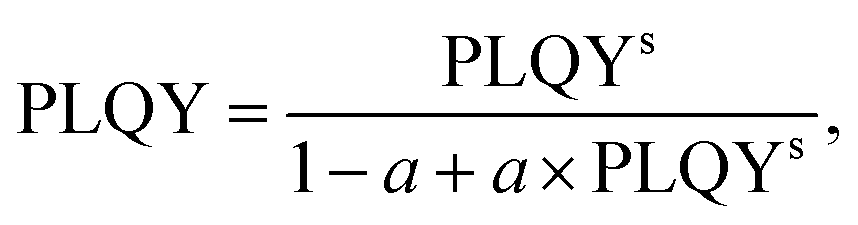 | (1) |
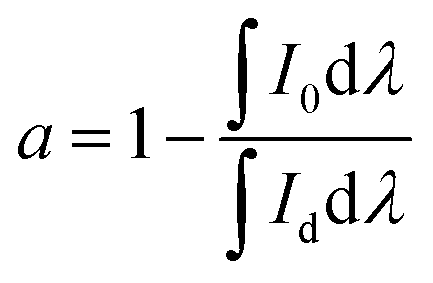 with I0 being the normalised emission intensity of a sample and Id being the normalised emission intensity of a 1
with I0 being the normalised emission intensity of a sample and Id being the normalised emission intensity of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 diluted powder sample. The chosen integration ranges are 1430–1700 nm to cover the 4I13/2–4I15/2 emission band of Er3+.
:9 diluted powder sample. The chosen integration ranges are 1430–1700 nm to cover the 4I13/2–4I15/2 emission band of Er3+.
To determine the intensity-dependent PLQY efficiency, the power of the incident laser radiation is varied with a neutral density (ND) filter wheel (Thorlabs, NDC-100C-2) attached to a stepper motor. The entire system is automated and controlled using custom software developed using the LabView environment.
2.4. Printing of labelled samples
The luminescent tracers on the surface of the PE pouch were printed by Siegwerk (Siegwerk Druckfarben AG & Co. KGaA) using Printing Proofer Equipment (Erichsen GmbH & Co. KG). Briefly, an ink with luminescent tracer fills the engraved cells in the printing plate. The doctor blade forces the ink into the engraving and wipes the overrun away. The ink is transferred to a substrate, by the rolling movement of the engraved plate. Subsequently, the solvent is evaporated by an air stream. The transfer efficiency is defined by the depth of the engraving, the pressure of the drum and the printing speed.2.5. SWIR imaging
The SWIR image of the PE pouch was obtained as follows: the pouch was placed on a moving platform and data were acquired using a SWIR hyperspectral camera (Specim, FX17). The sample was illuminated with two light sources: a 150 W halogen lamp (Elro, HL150) with an intensity of about 100 W cm−2 (in the whole emission range) on a sample placed 30 cm from the light source, and a 978 nm fibre laser (CNILaser, FC-W-980H) with an intensity of ∼3 W cm−2. The sample was scanned at a rate of 2 cm s−1 to simulate the motion of plastic packaging on a conveyor belt. The integration time for a frame was set to 20 ms. The process was controlled using in-house-developed software. Reflectance spectra were obtained by dividing the spectra in each pixel by the reference spectrum of a white calibration tile (Specim, OPRS200.25.10).3. Results and discussion
3.1. Absorption spectra and crystal structure of Er3+, Yb3+ activated YPO4 microcrystals
The success of the TBS process depends on the careful design and performance of the luminescent tracers. The sensitiser ions strongly absorb the 976 nm laser excitation, resulting in the emission of photons within the optimal detection range of an InGaAs camera (1000–1650 nm). Fig. 2(a) displays absorption spectra of YPO4:3Er, YPO4:3Er,3Yb and YPO4:3Er,24Yb phosphors obtained using an integrating sphere. Several absorption peaks were observed in the analyzed samples, the most prominent ones being located at 380, 490, 524 and 657 nm. These are attributed to the corresponding Er3+:4I15/2–4G11/2, 4I15/2–4F7/2, 4I15/2–2H11/2 and 4I15/2–4F9/2 electronic transitions, respectively. In addition, a broad absorption band was observed at about 976 nm, which is assigned to the Er3+:4I15/2–4I11/2 and Yb3+:2F7/2–2F5/2 overlapping bands. The absorption peak in the SWIR range (1504 nm) corresponds to the Er3+:4I15/2–4I13/2 transition. There are three notable findings from the data: (i) the absorptance of Er3+ at 976 nm is of a similar order of magnitude to that of Yb3+. Consequently, when using a 976 nm laser for excitation, one can assume a comparable probability of excitation of Er3+ and Yb3+ ions in the YPO4:3Er,3Yb sample; (ii) the absorption peak of Er3+ is much narrower than that of Yb3+. This means that when using a laser with a wavelength approximately 2–3 nm longer or shorter than 976 nm, the predominant excitation will be of Yb3+ ions, however, here dual sensitisation is leveraged; (iii) in samples heavily doped with Yb3+ (e.g. YPO4:3Er,24Yb), excitation in the range of 940–980 nm will predominantly result in the excitation of Yb3+ ions. The fraction of light from different NIR sources absorbed by the samples is estimated in Table S2 and Fig. S2 (ESI†). Upon selective excitation of only Yb3+ using a 940 nm laser, all YPO4:Er,Yb samples exhibit two distinct emission peaks (Fig. 2(b)). The first peak, reaching its maximum at 1004 nm, signifies the combined emission from Yb3+:2F5/2–2F7/2 and Er3+:4I13/2–4I15/2 states. Meanwhile, the second peak, peaking at 1540 nm, corresponds to the 4I13/2–4I15/2 transition in Er3+. It can be postulated that upon Yb3+ sensitization with a 940 nm laser, the energy is efficiently transferred from the excited Yb3+ ion in the 2F5/2 state to the corresponding excited Er3+ ion in the 4I11/2 state. Subsequently, the Er3+ ion in the 4I11/2 state undergoes relaxation through either radiative or non-radiative transitions, ultimately reaching the Er3+ ion in the 4I13/2 state. As a consequence of this transition, a photon with a wavelength of approximately 1540 nm is emitted from the 4I13/2 level, falling within the SWIR region. The efficient energy transfer from Yb3+ to Er3+ is also confirmed in Fig. 2(c), where the decays of two representative samples, YPO4:3Er,24Yb and YPO4:24Yb, are shown. Without Er3+ co-doping, the Yb3+ luminescence excited at 940 nm and measured at 1000 nm shows a decay time of 1.2 ms. However, this becomes significantly shorter (0.53 ms) after additional co-doping with Er3+, demonstrating the presence of energy transfer. The normalized PLE spectrum, with luminescence monitored at 1540 nm, as depicted in Fig. S3 (ESI†), reveals multiple narrow excitation lines within the longer wavelength range of 350–830 nm. These lines closely align with the absorption spectra of the YPO4:Er,Yb samples. Simultaneously, a broad PLE peak emerges at 940–980 nm, also signifying the occurrence of energy transfer from Yb3+:2F5/2 to Er3+:4I13/2. Considering the practical application of YPO4:Er,Yb tracers, the use of a laser with a precise wavelength of 976 nm in a sorting machine offers the potential to improve the PLQY and B of Er3+ SWIR luminescence. This improvement is due to the increase of the contribution of directly excited Er3+ ions alongside those predominantly excited by energy transfer from Yb3+ ions. In the process of solid-state synthesis of YPO4 tracers from oxide precursors (Y2O3, Yb2O3 and Er2O3), it is crucial to find the optimum temperature and time for ion intermixing. Effective intermixing is essential for two main reasons: (i) increasing the probability of energy transfer from Yb3+ to Er3+, and (ii) reducing self-quenching in Yb3+ and Er3+ ions. Although an optimum temperature of 1200 °C and a calcination time of 12 hours has been quoted in the literature,40 the clarity of effective ion intermixing under these conditions is uncertain.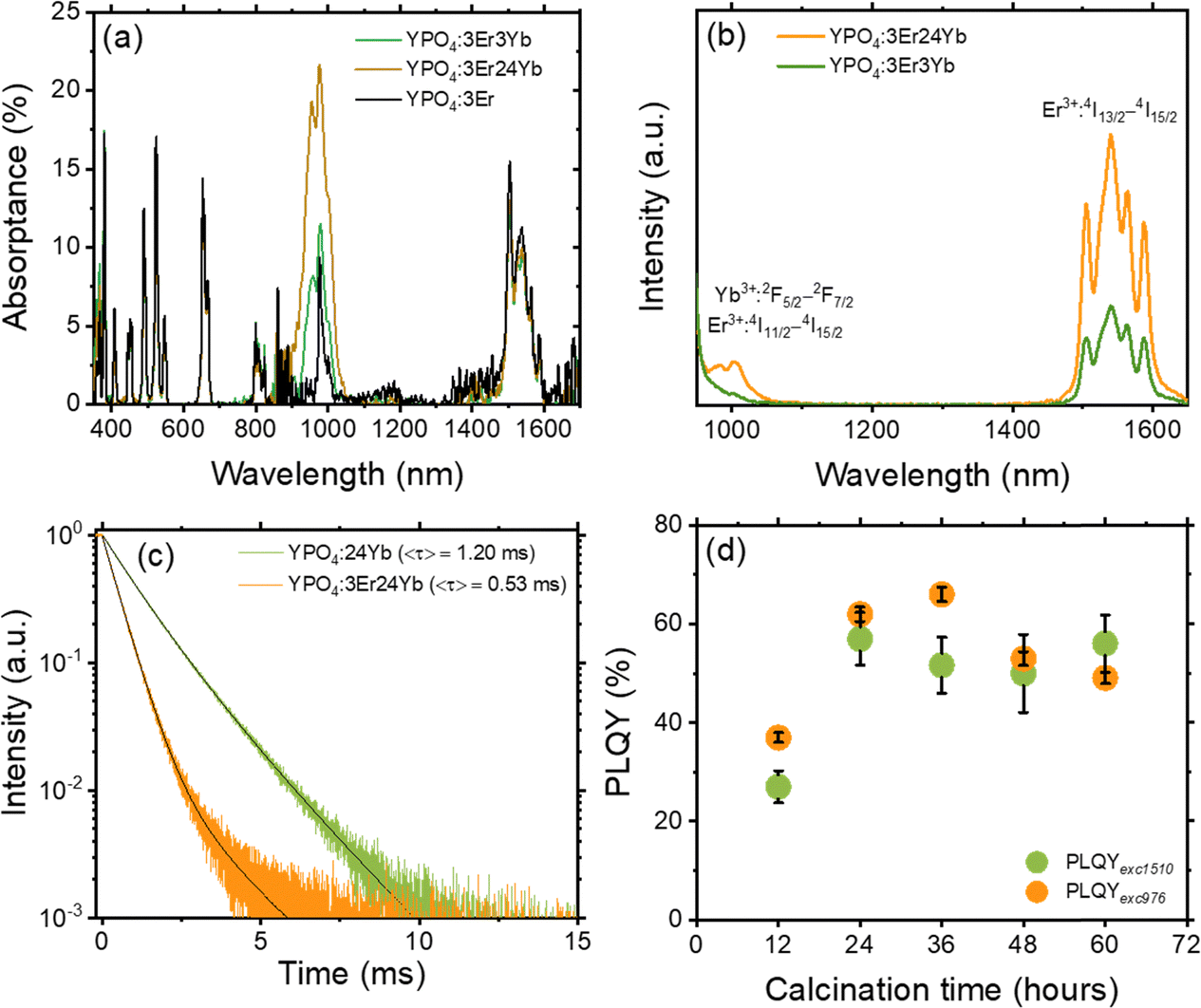 | ||
| Fig. 2 (a) Absorption spectra of samples: YPO4:3Er, YPO4:3Er3Yb and YPO4:3Er24Yb; (b) luminescence of YPO4:3Er3Yb and YPO4:3Er24Yb samples under 940 nm excitation. Excitation intensity of 2 W cm−2; (c) decay of Yb3+ luminescence in samples YPO4:24Yb and YPO4:3Er24Yb; and (d) dependence of PLQY of 1540 nm emission on calcination time (12–60 hours). Two excitation wavelengths were used: 1510 nm (PLQYexc1510) and 976 nm (PLQYexc976). Excitation intensity of 0.1 W cm−2. The calculation of the error bars is described in the Tables S4–S10 (ESI†). | ||
To address this issue, we subjected the YPO4:3Er24Yb sample to a calcination process for a total duration of 60 hours and measured the absolute PLQY after every 12 hours of calcination (Fig. 2(d)). It is noteworthy that an increase in calcination temperature and time can lead to the growth of larger particles. To mitigate the size effect, we maintained a constant temperature of 1200 °C and introduced grinding steps after every 12 hours of calcination. Under these synthetic conditions, we observed no changes in both the crystal structure (Fig. 3(a)) and particle size after a total heating time of 60 hours at 1200 °C (Fig. 3(b)–(d) and Fig. S4, S5, ESI†).
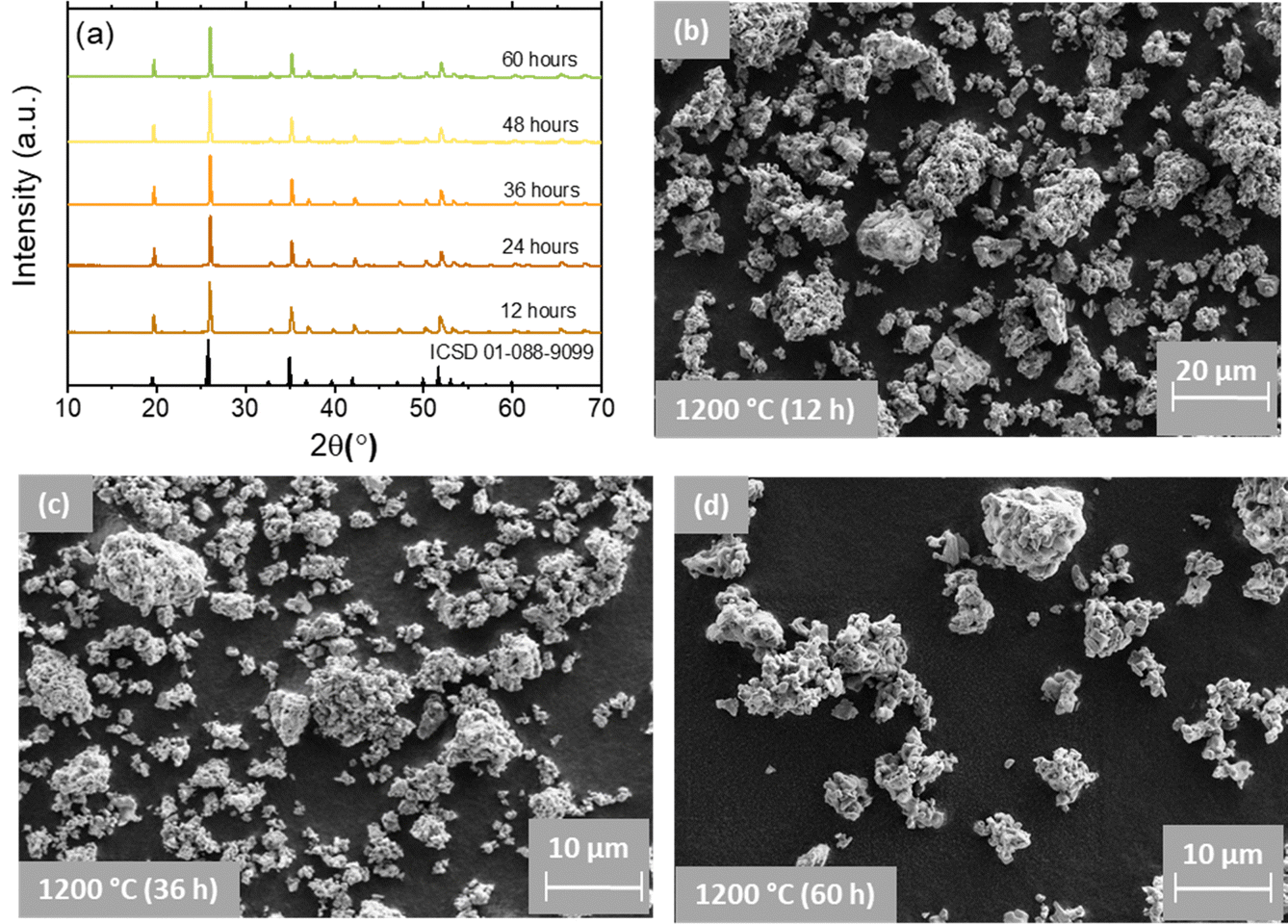 | ||
| Fig. 3 (a) pXRD of the representative YPO4:3Er24Yb sample synthesized using different calcination time (12–60 hours). (b)–(d) SEM images of YPO4:3Er24Yb microcrystals obtained using different calcination time. | ||
Regardless of the calcination time, SEM images exhibit numerous irregularly agglomerated grains with oval- and spherical-like morphologies, forming condensed clusters. In some regions, the observed particles take on longitudinal or square cube shapes, and the grain boundary between them is clearly visible. The SEM images also illustrate the sintering of small crystals (with size ∼0.5–1 μm) into larger particles with sizes in the range of 10 to 20 μm. Simultaneously, the pXRD results (Fig. 3(a)) indicate the presence of the pure YPO4 phase.41 All the observed patterns are very consistent with the standard data of the tetragonal phase of YPO4 with space group I41/amd referenced in the ICSD 01-088-9099.42 This confirms well that a single pure phase of the obtained samples has been formed and the substituting elements have no effect on phase purity of the host lattice. The observed narrow and intense diffraction peaks indicate the as-prepared samples are well crystallized. The position of the pXRD peaks shows an independence from the calcination time. In contrast, the pXRD peaks become narrower as the calcination time increases. Calculation of the crystallite size (Table S3, ESI†) shows that it increases from 38.6 to 52.1 nm as the calcination time increases from 12 to 36 hours. At longer calcination times (48 and 60 hours) the crystallite size does not increase anymore, giving 49.2 and 48.7 nm respectively.
The resolving of the PLQY for the 1540 nm emission (Fig. 2(d)) involved employing two distinct excitation wavelengths: a 1510 nm laser for the direct excitation of the Er3+:4I13/2 state (referred to as PLQYexc1510) and a 976 nm laser for the direct excitation of the Yb3+:2F5/2 state (referred to as PLQYexc976). In the latter case, the Er3+:4I13/2 state is populated through the energy transfer process Yb3+:2F5/2 → Er3+:4I11/2, followed by subsequent relaxation to the Er3+:4I13/2 state. It is important to note that the solid-state reaction begins with non-luminescent Yb2O3 and Er2O3 raw materials, where Yb3+ and Er3+ luminescence is completely quenched. During the formation of the YPO4:Er,Yb solid solution, the local concentration of Er3+ and Yb3+ decreases, mitigating concentration quenching. Therefore, when directly exciting the Er3+:4I13/2 state, PLQYexc1510 serves as a measure of concentration quenching. In contrast, the PLQYexc976 for the 1540 nm emission under 976 nm excitation serves as an indicator of energy transfer between Yb3+ and Er3+.
Fig. 2(d) illustrates that after 12 hours of calcination, PLQYexc1510 is only 27%, while PLQYexc976 is 37%. In a normal scenario it can be expected that PLQYexc1510 ≥ PLQYexc976 since the additional energy transfer step Yb3+:2F5/2 → Er3+:4I11/2 can only lower the overall PLQYexc976. In our experiments, the unusual scenario with PLQYexc1510 < PLQYexc976 highlights the inhomogeneous distribution of co-dopants in the YPO4:Er,Yb solid solution. This is realised by the fact that the use of a 1510 nm laser results in the excitation of all Er3+ ions, with varying degrees of concentration quenching. In contrast, excitation with a 976 nm laser (and subsequent energy transfer) selectively activates well-intermixed Er3+ ions with minimised effect of concentration quenching resulting in PLQYexc1510 < PLQYexc976.
With an extended calcination time of 24 hours, both PLQYexc1510 and PLQYexc976 increase to 57% and 62%, respectively. Further prolongation of the calcination time does not demonstrate a clear trend in PLQYexc1510 changes and the value fluctuates near 54 ± 3%. In turn, PLQYexc976 reaches the highest value of 65% (after 36 hours calcination) and decreases down to 53% and 49% for calcination time of 49 and 60 hours, respectively. In an additional experiment with a sample featuring a significantly lower Yb3+ concentration (3Er,3Yb), the PLQYexc1510 and PLQYexc976 demonstrate a consistently rise after 24 and 36 hours of calcination (Fig. S6, ESI†). The PLQYexc1510 reaches a maximum value of 87% after 48 hours calcination and then decreases. For the same sample, the PLQYexc976 decreases after 36 hours calcination with a maximum value of 67%. It is interesting to note that for the 3Er,3Yb sample, the normal scenario with PLQYexc1510 > PLQYexc976 is observed starting from calcination time of 24 hours, suggesting easier ion intermixing in sample with low doping concentrations. Based on this data, a calcination time of 36 hours was chosen as optimal for subsequent variations in Er3+ and Yb3+ doping concentrations to identify the best-performing sample.
To confirm the hypothesis of limited co-dopant intermixing, we examined two samples (after 12 and 36 hours of calcination) using CL measurements.
Under an electron beam, the co-doped YPO4 samples show luminescence as depicted in Fig. 4(a). Fig. 4(c) shows an overlay of images of scattered electrons (Fig. 4(b) – typical SEM image) and CL (the luminescence has been detected panchromatically over the spectral range 350–870 nm). It is evident that the majority of the microparticles are luminescent. However, there are several examples (highlighted with white arrows in Fig. 4(c)) that are not luminescent. These non-luminescent particles can be undoped YPO4 or particles with a very high concentration of Yb3+ and Er3+. In the latter case the luminescence can be very weak due to concentration quenching.
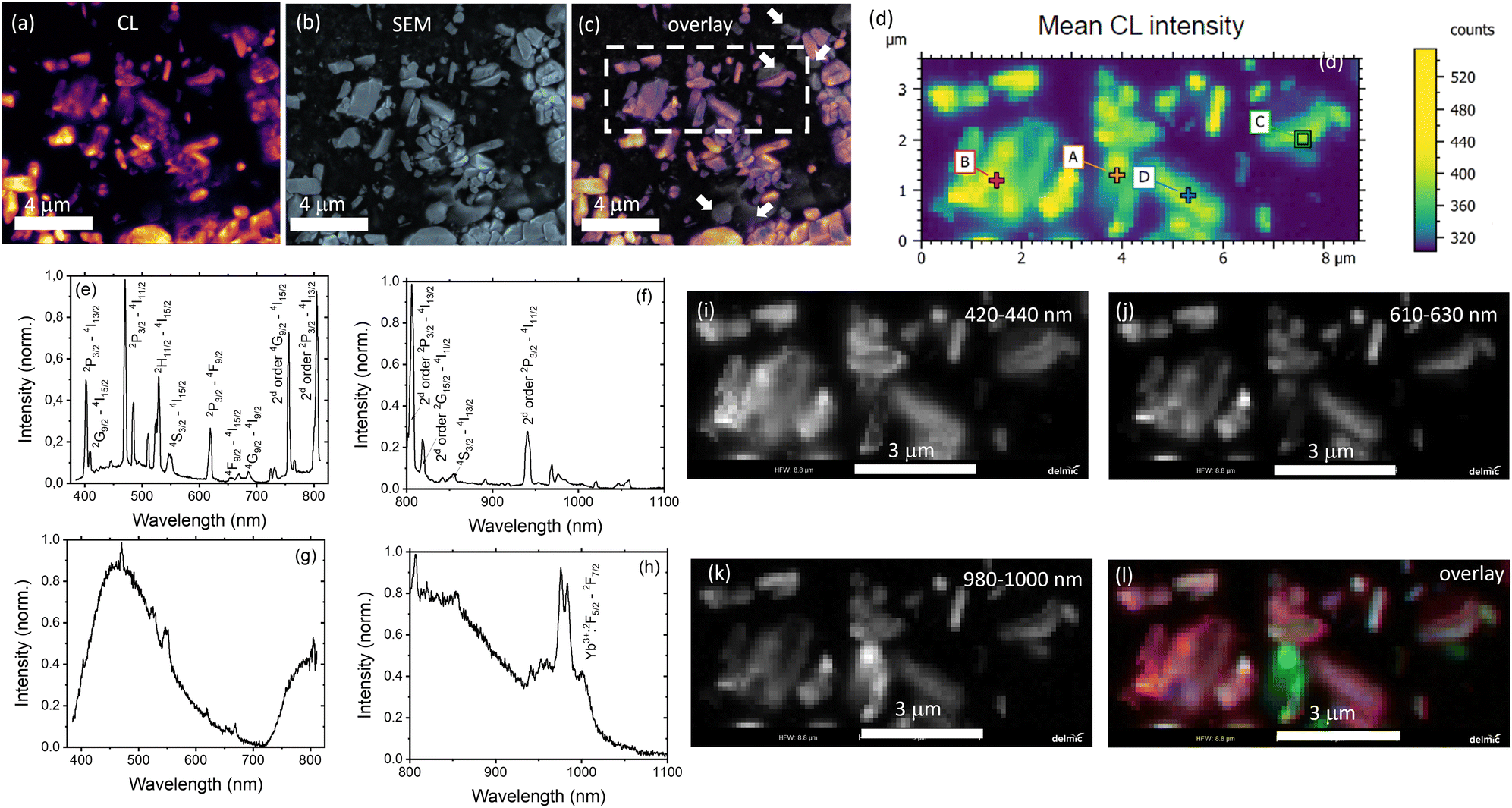 | ||
| Fig. 4 (a) Cathodoluminescence image (CL is integrated from 385 to 1100 nm); (b) SEM image; (c) overlay of (a) and (b); (d) hyperspectral CL intensity map for the area highlighted by the dashed line in (c) (where each pixel contains also spectral information); (e) and (f) spectrum for the point A in (d) taken at two different center wavelengths of the grating; and (g) and (h) spectrum for the point C in (d); image (d) was broken in three channels 420–440 nm (i) – Yb2+ luminescence; 610–630 nm (j) – Er3+ luminescence; 980–1000 nm (k) – Yb3+ luminescence; (l) is a pseudocolor overlay of (i), (j) and (k) where arbitrary 420–440 nm (blue), 610–630 nm (green) and 980–1000 nm (red). | ||
An arbitrarily chosen area is presented in Fig. 4(d) with each pixel providing additional spectral information. For instance, luminescence spectra measured in points A and C (Fig. 4(d)) are shown in Fig. 4(e)–(h). The luminescence spectrum contains a number of sharp peaks corresponding to the luminescence of Er3+ and Yb3+. It should be noted that the measurements in the broad spectral range were carried out without any additional optical filters, so that a second order diffraction in the spectrometer grating gives additional peaks (noticed in Fig. 4(d) and (f)). The origin of the broad peak at 400–600 nm (and an additional broad peak at 700–1100 nm due to second order diffraction) is debatable. We suggest that it is the luminescence of Yb2+ formed by the reduction of Yb3+ under the electron beam during the CL measurement. It has been reported that Yb2+ in the phosphate host exhibits luminescence peaking at 450 nm.43 The possible reduction of Yb3+ to Yb2+ under electron irradiation was also recently discovered by Arnold et al.44
The region of interest (Fig. 4(d)) was then presented as a pseudocolour overlay (Fig. 4(l)), where blue colour corresponds to the 420–440 nm range (emission of Yb2+, Fig. 4(i)), green colour – 610–630 nm range (emission of Er3+. Fig. 4(j)) and red colour – 980–1000 nm range (emission of Yb3+, Fig. 4(k)). Fig. 4(l) shows the excellent co-location between blue and red channels. It means the simultaneous presence of Yb2+ and Yb3+ in microcrystals. In contrast, there are several areas where the Er3+ luminescence is stronger than average, indicating that the Er3+ distribution is far from homogeneous.
On the one hand, it is difficult to find particles without CL for the sample after 36 hours of calcination. There are only a few non-luminescent particles in Fig. S7c (ESI†) with the field of view in 25 times larger than in Fig. 4(c). Furthermore, no particles with excessive Er3+ luminescence were found in the examined areas of the sample after 36 h calcination (Fig. S7d and e, ESI†), suggesting that a more homogeneous mixture is achieved after a longer calcination time.
3.2. Optimization of Er3+ and Yb3+ concentrations for SWIR luminescence of Er3+, Yb3+ activated YPO4 microcrystals
The chemical composition of the synthesised samples is detailed in Table S1 (ESI†), which provides an overview of the elemental composition, highlighting any observed deviations between the experimental and nominal compositions. It is noteworthy that these deviations are relatively small, indicating that the synthesised samples are close to the intended chemical compositions. Overall, the experimental values of Er3+ and Yb3+ in the samples, corroborated by the pXRD results (Fig. S8, ESI†), provide compelling evidence for the successful incorporation of Yb3+ and Er3+ ions in place of Y3+ into the YPO4 crystal lattice.Before discussing the optimal concentrations of Yb3+ and Er3+, it should be noted that the PLQY values exhibit a dependence on the excitation intensity, decreasing at higher excitation intensities (Fig. S9, ESI†). The main reason for this effect is the quenching of the luminescence due to local heating induced by the excitation laser beam. Table S4 and Fig. S10–S12 (ESI†) indicate that a temperature of 36.9 °C and 46.1 °C can be achieved within the excitation spot at an excitation intensity of 3 W cm−2 and 300 W cm−2, respectively. For consistency, we discuss the PLQY value at a low excitation intensity of 0.1 W cm−2 throughout the whole manuscript. These PLQY measurements (Fig. 4) provide the most interesting insights. Keeping the Yb3+ concentration constant at 24 at%, increasing the Er3+ content from 1 to 5 at% has minimal effect on the PLQYexc976 values. They are consistently around 51 ± 5%, indicating a negligible influence of concentration quenching on the Er3+:4I13/2 state over the range of Er3+ concentrations studied. Furthermore, if the Er3+ concentration is fixed at 3 at%, while the Yb3+ concentration is varied between 1–30 at%, a clear trend emerges. PLQYexc976 decreases with increasing Yb3+ concentration. This suggests that concentration quenching of the Yb3+:2F5/2 state may be more important within the YPO4 host.
The measured PLQY values are a key parameter when comparing different Yb3+ and Er3+ doped phosphors for practical applications. However, it's important to consider PLQY alongside absorption characteristics to accurately predict sample brightness. To illustrate this, two scenarios with different tracer concentrations are considered (Fig. 6): (1) high tracer concentration with intense absorption and (2) low tracer concentration with weak absorption. In all cases, the luminescence intensity (I) correlates with the B, defined as the product of PLQY and absorptance (A) (eqn (2)):
| I ∼ B = PLQY × A | (2) |
| A = f(α,s,L) = f(a,NYb3+,s,L) | (3) |
| I ∼ B ∼ PLQY × NYb3+ | (4) |
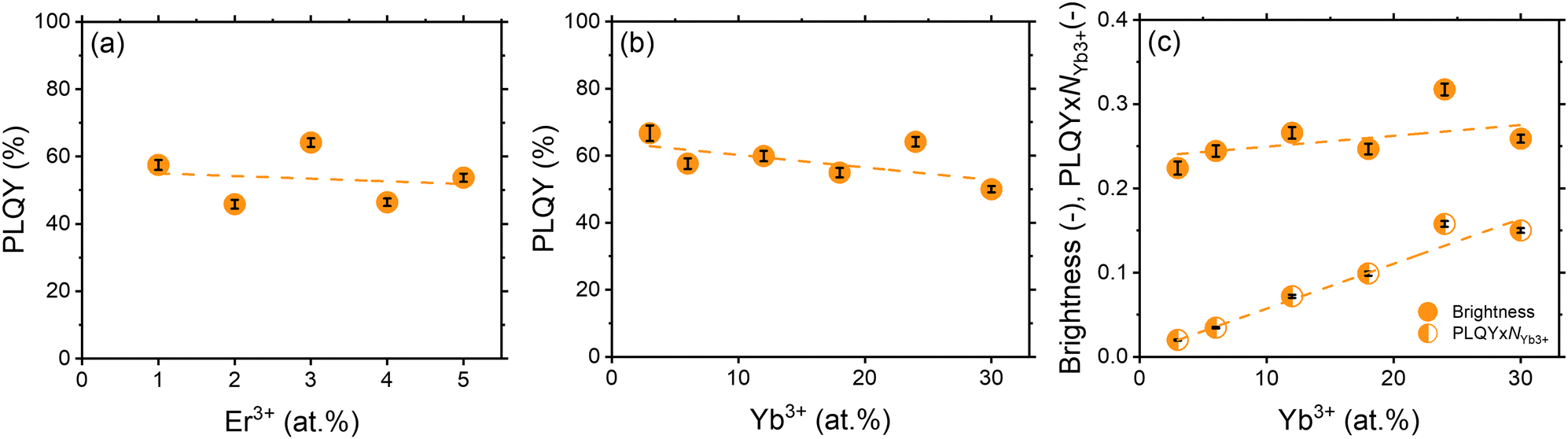 | ||
| Fig. 5 (a) PLQY of YPO4:xEr24Yb samples; (b) PLQY of YPO4:yYb3Er samples; (c) B and PLQY × NYb3+ of YPO4:yYb3Er samples. Excitation wavelength – 976 nm. Excitation intensity – 0.1 W cm−2. Emission intensity was integrated in the range of 1450–1650 nm. Dashed lines are provided to guide the eye. The calculation of the error bars is described in the Tables S4–S10 (ESI†). | ||
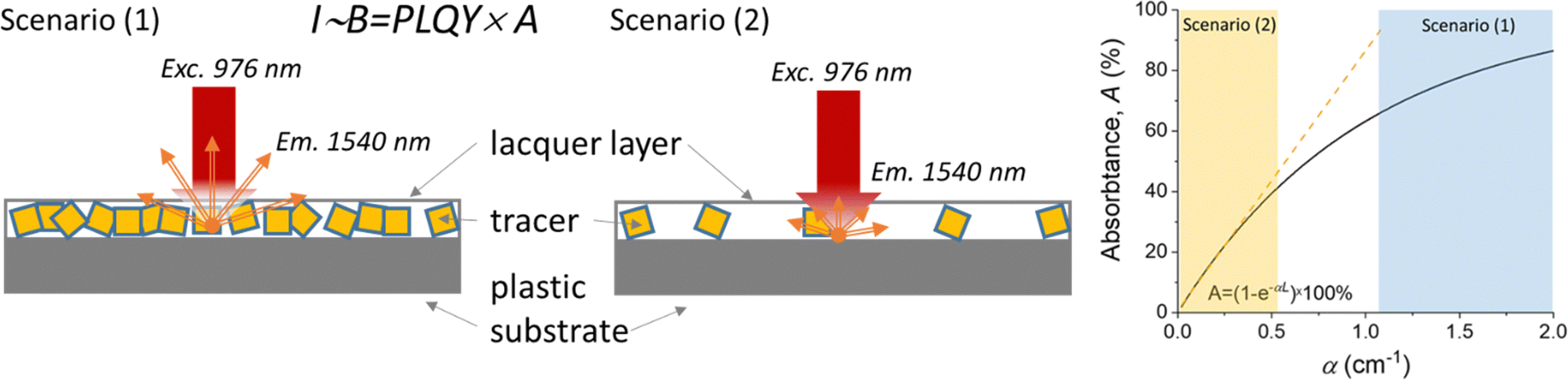 | ||
| Fig. 6 Two typical scenarios with high (1) and low (2) concentration of the luminescent tracer in a printed label. The label is excited by a 976 nm laser and emits 1510 nm luminescence whose intensity is proportional to brightness. The graph shows the linear dependence between absorptance and absorption coefficient in scenario (2) and the saturation of the absorptance with increasing absorption coefficient in scenario (1). | ||
In general, the benefit of a high Yb3+ concentration for brighter YPO4 samples may be even less pronounced, as it has been reported that the Yb3+ absorption cross section may decrease with increasing Yb3+ concentration, as estimated in previous studies on YPO4 single crystals co-doped with different concentrations of Yb3+. For instance, absorption cross-section at 976 nm for σ-polarized light was measured as ∼4.8 × 10−20 cm2 (for 1.2 at% Yb3+),45 ∼2.3 × 10−20 cm2 (for 5 at% Yb3+), ∼3.5 × 10−20 cm2 (for 7.5 at% Yb3+)46 and ∼1.9 × 10−20 cm2 (for 11.5 at% Yb3+).46 However, the results from the present work suggest that a relatively high doping concentration of Yb3+ (24 at%) is still advantageous for achieving the most luminescence from YPO4:Yb3+,Er3+ tracers.
3.3. Hyperspectral imaging with the SWIR tracer
To demonstrate the potential of synthesised luminescent tracers in plastics sorting, the tracers were applied to a PE pouch using industrial printing technology provided by Siegwerk and mimicking labelling of multilayer packaging. In Fig. 7(a), a daylight photograph of the pouch is shown. The tracers are printed within the colourless and blue areas. The pouch, mounted on a stand, was moved at a linear speed of 2 cm s−1 through a 978 nm laser with line alignment, and a 2D image was reconstructed as shown in Fig. 7(b).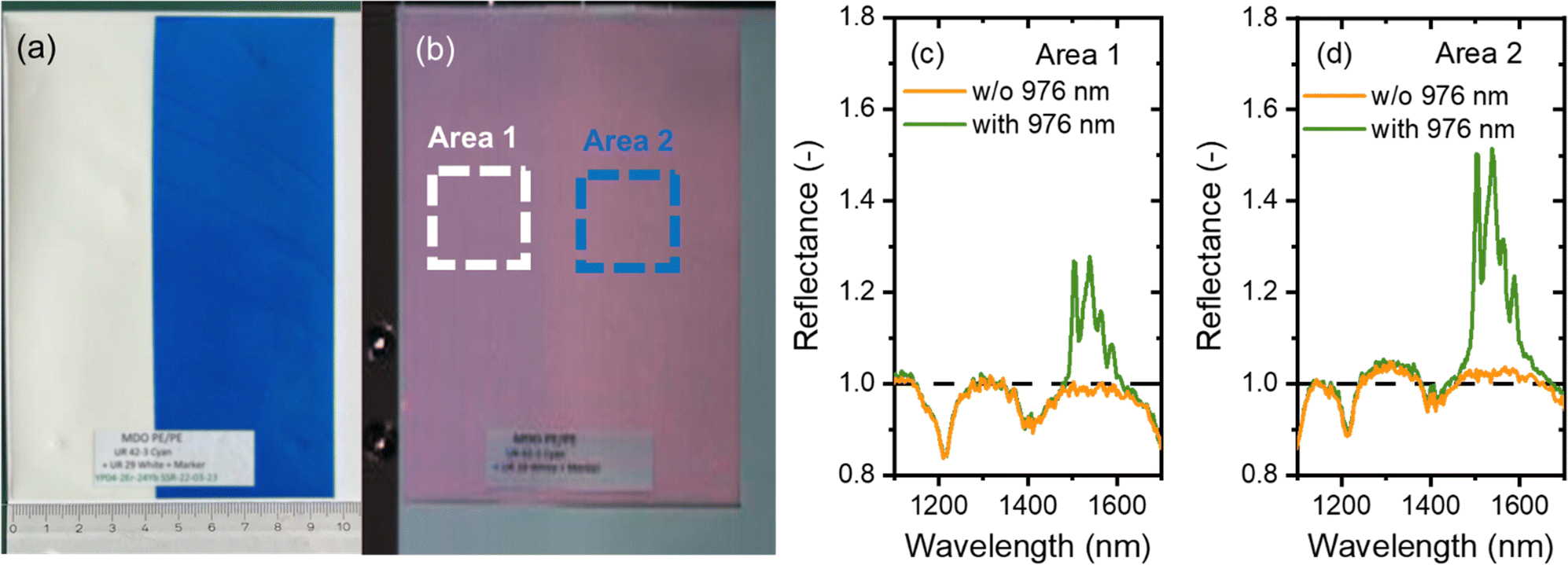 | ||
| Fig. 7 (a) Polyethylene pouch with the printed SWIR tracer. Photo taken with a phone camera (iPhone 13), (b) SWIR hyperspectral image (resolution of 626 × 702 pixels) of the PE pouch obtained in the presence of a halogen lamp and 976 nm laser light, (c) and (d) reflectance spectra obtained with and without (w/o) the 976 nm laser from the PE pouch with the printed tracer. The spectra are averaged over a region highlighted in Fig. 7(b) for area 1 (c) and area 2 (d). | ||
Fig. 7(b) shows a hyperspectral SWIR image where each pixel also provides spectral information in the 1000–1700 nm range. The reflectance spectra from the regions highlighted with white and green frames is plotted in Fig. 7(c) and (d). When illuminated with a halogen lamp, the spectrum shows a reflection signal with intensity <1. Specifically, the spectra show typical reflectance minima at 1085, 1210 and 1390 nm, corresponding to the absorption bands of PE. When the pouch is illuminated simultaneously by both the halogen lamp and the 976 nm laser, a prominent band corresponding to the Er3+ luminescence appears and the luminescence of the tracer becomes easily discernible. It contributes as a signal with intensity >1 to the overall reflectance spectrum (where reflectance signals typically have intensities <1). It is evident that a blue commercial pigment does not reduce the tracer luminescence. As a result, a decision to sort out a product with a printed tracer can be made with confidence.
4. Conclusions
In conclusion, the development of efficient inorganic luminescent tracers is crucial for various applications, particularly in plastic sorting. This study identified a new luminescent YPO4:Er3+,Yb3+ tracer with a high SWIR PLQY of 67% following the optimization of Yb3+ and Er3+ concentrations in the high-temperature solid-state synthesis of the YPO4 host. While the sample co-doped with 3 at% Er3+ and 3 at% Yb3+ exhibited the highest SWIR PLQY, the maximum brightness was observed in the sample co-doped with 3 at% Er3+ and 24 at% Yb3+. The particle size achieved in the synthesis allows the tracer exhibiting maximum brightness to be printed on the PE pouch using a research-scale implementation of an industry-relevant printing process. Hyperspectral SWIR imaging of the foil, conducted under simultaneous irradiation with a halogen lamp and a 978 nm laser, produced a strong luminescence signal peaking at 1540 nm, which was readily distinguishable from the polyethylene reflectance background. Importantly, mixing the luminescent tracer with a commercial blue pigment did not affect the printing performance, and the SWIR luminescence remained unaffected by the blue pigment. Thus, this work demonstrates the potential of SWIR luminescence tracers in advancing plastic recycling technology through tracer-based sorting.Author contributions
Nisrin Mohamed Bhiri conducted a thorough evaluation of synthetic protocols from the literature, developed modifications to the synthetic procedures, synthesized all samples, performed X-ray diffraction and scanning electron microscopy measurements, and proofread the document; Eduard Madirov led the photophysical characterization of all samples, performed the demonstration experiment involving short-wave infrared imaging and participated in proofreading; Herman Duim conducted cathodoluminescence measurements and participated in proofreading; Justin Nyarige performed a range of scanning electron microscopy and X-ray diffraction experiments and contributed to proofreading; Bryce S. Richards secured financial support for the project (personnel and equipment), contributed to the conceptualization of the experiments and manuscript, and performed several rounds of critical proofreading (regarding both structure and wording of the manuscript). Andrey Turshatov contributed to the conception of the experiments and the manuscript, supervised the experiments, wrote large parts of the text for the manuscript, proofread the document and handled the submission process.Data availability
Data are available on request from the corresponding authors.Conflicts of interest
There is no conflict of interest to disclose.Acknowledgements
The financial support provided by the Helmholtz Association is gratefully acknowledged: (i) a Recruitment Initiative Fellowship for B. S. R.; (ii) the funding of chemical synthesis equipment from the Helmholtz Materials Energy Foundry (HEMF); and (iii) Research Field Energy – Program Materials and Technologies for the Energy Transition – Topic 1 Photovoltaics (38.01.05). B. S. R. and A. T. acknowledge funding from the European Union's Horizon 2020 research and innovation project “Circular Foodpack” agreement no: 101003806. The authors acknowledge Dr. Dmitry Busko for fruitful discussion of the experimental results. The authors acknowledge Pascal Wendler (Polysecure GmbH) for their help in conducting hyperspectral imaging. The authors acknowledge Dr. Andreas Kulawig and Dr. Ralf Leineweber (Siegwerk Druckfarben AG & Co. KGaA) for their help in printing the PE pouch.Notes and references
- S. Li, L. Zhou and H. Zhang, Light: Sci. Appl., 2022, 11, 177 CrossRef CAS PubMed.
- E. Erol, N. Vahedigharehchopogh, O. Kıbrıslı, M. Ç. Ersundu and A. E. Ersundu, J. Phys.: Condens. Matter, 2021, 33, 483001 CrossRef CAS.
- Q. Jin, J. Shi, J. Jin, W. Xu, S. Chen and Y. Pan, J. Mater. Chem. C, 2024, 12, 5615–5622 RSC.
- F. Jobin, P. Paradis, Y. O. Aydin, T. Boilard, V. Fortin, J.-C. Gauthier, M. Lemieux-Tanguay, S. Magnan-Saucier, L.-C. Michaud, S. Mondor, L.-P. Pleau, L. Talbot, M. Bernier and R. Vallée, Opt. Express, 2022, 30, 8615–8640 CrossRef CAS PubMed.
- Q. Zhang, Y. Liu, K. Liu and H. Zhang, Nano Res., 2024, 17, 97–111 CrossRef CAS.
- Y. Chigusa, K. Nakazato, S. Hirai, M. Watanabe and S. Suzuki, Optical Amplifiers and Their Applications, Monterey, California, 1990 Search PubMed.
- S. Vaidyanathan, J. Mater. Chem. C, 2023, 11, 8649–8687 RSC.
- Z. Xia and A. Meijerink, Chem. Soc. Rev., 2017, 46, 275–299 RSC.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- I. A. Howard, D. Busko, G. Gao, P. Wendler, E. Madirov, A. Turshatov, J. Moesslein and B. S. Richards, Resour., Conserv. Recycl., 2024, 205, 107557 CrossRef CAS.
- Eurostat, https://ec.europa.eu/eurostat/statistics-explained/index.php?title=Packaging_waste_statistics, (accessed 27.11.2024).
- plasticseurope.org, https://plasticseurope.org/knowledge-hub/plastics-the-fast-facts-2023/, (accessed 27.11.2024).
- B. Geueke, K. Groh and J. Muncke, J. Cleaner Prod., 2018, 193, 491–505 CrossRef CAS.
- O. Horodytska, F. J. Valdés and A. Fullana, Waste Manage., 2018, 77, 413–425 CrossRef CAS.
- S. Ügdüler, T. Van Laere, T. De Somer, S. Gusev, K. M. Van Geem, A. Kulawig, R. Leineweber, M. Defoin, H. Van den Bergen, D. Bontinck and S. De Meester, J. Hazard. Mater., 2023, 452, 131239 CrossRef.
- M. Roosen, T. Van Laere, V. Decottignies, L. Morel, J.-L. Schnitzler, J. Schneider, M. Schlummer, I. S. Lase, A. Dumoulin and S. De Meester, Chemosphere, 2023, 324, 138281 CrossRef CAS.
- Polysecure GmbH, https://www.polysecure.eu/en/sort-reliably, (accessed 27.11.2024).
- K.-L. Wong, J.-C. G. Bünzli and P. A. Tanner, J. Lumin., 2020, 224, 117256 CrossRef CAS.
- T.-H. Yang, B. Glorieux, K.-W. Shiau, Y.-Y. Chang, Y.-W. Yu and C.-C. Sun, Opt. Express, 2022, 30, 47082–47092 CrossRef CAS PubMed.
- K. Rajagopalan, E. Madirov, D. Busko, I. A. Howard, B. S. Richards, H. C. Swart and A. Turshatov, ACS Appl. Mater. Interfaces, 2023, 15, 43985–43993 CrossRef.
- M. F. Torresan and A. Wolosiuk, ACS Appl. Bio Mater., 2021, 4, 1191–1210 CrossRef CAS PubMed.
- I.-S. Cho, G. K. Choi, J.-S. An, J.-R. Kim and K. S. Hong, Mater. Res. Bull., 2009, 44, 173–178 CrossRef CAS.
- J. C. Batista, P. C. de Sousa Filho and O. A. Serra, Dalton Trans., 2012, 41, 6310–6318 RSC.
- A. H. Krumpel, A. J. Bos, A. Bessière, E. van der Kolk and P. Dorenbos, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 085103 CrossRef.
- O. Moune, M. Faucher and N. Edelstein, J. Lumin., 2002, 96, 51–68 CrossRef CAS.
- Z. Wang, J.-G. Li, Q. Zhu, B.-N. Kim and X. Sun, CrystEngComm, 2017, 19, 5230–5243 RSC.
- M. L. Debasu, D. Ananias, J. Rocha, O. L. Malta and L. D. Carlos, Phys. Chem. Chem. Phys., 2013, 15, 15565–15571 RSC.
- O. Lehmann, H. Meyssamy, K. Kömpe, H. Schnablegger and M. Haase, J. Phys. Chem. B, 2003, 107, 7449–7453 CrossRef CAS.
- Y. Che, S. Sun, J. Lu, F. Zheng, G. Yu, Y. Cao, L. Wang, L. Sun, Y. Yin and Z. Wang, J. Lumin., 2022, 248, 118957 CrossRef CAS.
- X. Gao, X. Liu, Q. Wen, X. Yang and S. Xiao, J. Appl. Phys., 2014, 116, 173105 CrossRef.
- L. L. Noto, S. K. K. Shaat, D. Poelman, M. S. Dhlamini, B. M. Mothudi and H. C. Swart, Ceram. Int., 2016, 42, 9779–9784 CrossRef CAS.
- L. L. Noto, S. K. K. Shaat, D. Poelman, P. F. Smet, L. Martin, M. Y. A. Yagoub, S. M. Dhlamini, O. M. Ntwaeaborwa and H. C. Swart, Ceram. Int., 2016, 42, 5497–5503 CrossRef CAS.
- R. L. Nyenge, H. C. Swart, D. Poelman, P. F. Smet, L. I. D. J. Martin, L. L. Noto, S. Som and O. M. Ntwaeaborwa, J. Alloys Compd., 2016, 657, 787–793 CrossRef CAS.
- X. Zhang, J. Zhang, X. Wu, L. Yu, Y. Liu, X. Xu and S. Lian, Chem. Eng. J., 2020, 388, 124289 CrossRef CAS.
- F. Bezati, D. Froelich, V. Massardier and E. Maris, Waste Manage., 2010, 30, 591–596 CrossRef CAS PubMed.
- S. Brunner, P. Fomin and C. Kargel, Waste Manage., 2015, 38, 49–60 CrossRef CAS PubMed.
- A. Arenas-Vivo, F. R. Beltrán, V. Alcázar, M. U. de la Orden and J. Martinez Urreaga, Mater. Today Commun., 2017, 12, 125–132 CrossRef CAS.
- A. Jandric, C. Olscher, C. Zafiu, R. Lielacher, C. Lechner, A. Lassenberger and F. Part, Polymers, 2024, 16, 2591 CrossRef CAS.
- L. R. Wilson and B. S. Richards, Appl. Opt., 2009, 48, 212–220 CrossRef CAS PubMed.
- J. M. Nedelec, D. Avignant and R. Mahiou, Chem. Mater., 2002, 14, 651–655 CrossRef CAS.
- W. Milligan, D. Mullica, G. Beall and L. Boatner, Inorg. Chim. Acta, 1982, 60, 39–43 CrossRef CAS.
- M. M. Zaman and S. M. Antao, Minerals, 2021, 11, 16 CrossRef CAS.
- P. Dorenbos, J. Phys.: Condens. Matter, 2003, 15, 2645 CrossRef CAS.
- M. Arnold, J. Katzmann, A. Naik, A. L. Görne, T. Härtling, J. George and C. Schuster, J. Mater. Chem. C, 2022, 10, 11579–11587 RSC.
- Y. Che, F. Zheng, J. Lu, Y. Yin, Z. Wang, D. Zhong, S. Sun and B. Teng, CrystEngComm, 2021, 23, 8038–8042 RSC.
- Y. Che, S. Sun, J. Lu, F. Zheng, G. Yu, Y. Cao, L. Wang, L. Sun, Y. Yin, Z. Wang, D. Zhong and B. Teng, J. Lumin., 2022, 248, 118957 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc04096a |
| ‡ A. Turshatov and N. M. Bhiri are co-first authors and have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |