
Deciphering carbon–sulfur rotational distribution in a crystalline host for enhanced red persistent organic phosphorescence†
Sakuya
Ueda
,
Kazuki
Fujita
,
Bahadur
Sk
 and
Shuzo
Hirata
*
and
Shuzo
Hirata
*
Department of Engineering Science, The University of Electro-Communications, 1-5-1 Chofugaoka, Chofushi, 1828585, Tokyo, Japan. E-mail: shuzohirata@uec.ac.jp; Tel: +81 (0)42 4435264
First published on 12th December 2024
Abstract
Persistent room-temperature phosphorescence (pRTP) from organic host–guest solids has been used for autofluorescence-free high-resolution imaging. However, from the lowest triplet excited state, the radiative transition rate (kp) decreases and the non-radiative transition rate (knr) increases with increasing wavelength. Therefore, the pRTP yield (Φp) at long wavelengths is still low. Here we report an enhanced red pRTP from dibenzo[g,p]chrysene substituted with a phenylthio group (PhSDBC) as a guest in a crystalline benzophenone host. Host–guest crystals composed of deuterated PhSDBC and benzophenone showed red pRTP with Φp = 28.8% and a lifetime of 1.47 s. Investigation of the correlation between experimental and calculated values of kp and knr revealed that the distribution of C–S rotational conformations of PhSDBC was constrained to a narrow range in the crystalline host. In the constrained distribution, the phenylthio substituents enhanced the cooperation of spin–orbit couplings and the transition dipole moment associated with the higher singlet excited state, which resulted in the selective increase of kp and in turn the enhanced Φp.
Introduction
Persistent room-temperature phosphorescence (pRTP), which can be observed from organic matter within 100 ms after the excitation light irradiation is stopped, is beginning to be used as a technique to obtain two-dimensional (2D) information without observing autofluorescence with a 2D photodetector.1–3 Many works have been reported, focusing on anti-counterfeiting4–7 and bioimaging.8–11 Because pRTP can produce a higher instantaneous brightness than the long persistent luminescence caused by recombination after excited charge separation, attempts are being made to use pRTP for high-resolution imaging.12,13To fully use pRTP and extract information about emission color and lifetime, preparation of materials with highly efficient pRTP in the blue-green-red and near-infrared regions is desirable. For blue and green pRTP, host–guest molecular crystals with a pRTP yield (Φp) of nearly 100% have been reported.14,15 This molecular structure displaying efficient blue and green pRTP is a motif already known in other fields, and there has been much discussion of the crystal structure of guest chromophores in crystalline hosts. Φp is expressed by the phosphorescence rate constant (kp), the non-radiative deactivation rate (knr) from the lowest triplet excited state (T1) of guest chromophores, and the triplet quenching rate caused by intermolecular electron transfer and exchange between the guest and host (kq). Because knr increases as the square of the wavelength and kp is inversely proportional to the cube of the wavelength, shorter wavelengths generally tend to produce higher Φp16 when kq is well suppressed. Therefore, for short-wavelength colors, the use of existing chemical structures may be sufficient to obtain large Φp. However, in the long-wavelength region, knr increases while kp does not. Therefore, it is predicted that new π structures may be necessary to control kp and knr using the molecular framework in the long-wavelength region. pRTP at long wavelengths such as in the red and near-infrared regions is important for bioimaging applications because such long-wavelength light penetrates living organisms. Although materials with a pRTP peak wavelength (λp) exceeding 600 nm and an average pRTP lifetime (τp) longer than 100 ms have been reported, their Φp is still below 21%.11,17–24
Molecular structures that show high fluorescence quantum yields in the red and near-infrared regions have been developed.25,26 Therefore, a major scientific question is why is it still difficult to increase kp above knr in the long-wavelength region in organic phosphorescence? In an attempt to address this question, we have pioneered computational techniques to estimate kp and knr for a wide range of molecules. It has been confirmed that to statistically discuss kp and knr of organic guest chromophores, it is necessary to consider the structural displacement caused by molecular vibration in the T1 state of the chromophores.24,27–30 An approach correlating experimental and calculation data considering the thermal distribution of molecular geometry may clarify molecular designs that can realize efficient pRTP at long wavelengths.
Here we report a phenylthio (PhS)-substituted chromophore that exhibits enhanced red pRTP and demonstrate that the structural distribution of the PhS bridge site is confined in a solid host. A dibenzo[g,p]chrysene (DBC) substituted with a PhS group is synthesized as a guest chromophore. In a benzophenone crystalline host, this chromophore shows higher Φp in the red region than DBC substituted with a benzoyl group, even though benzoyl substituents are often reported to enhance Φp in the blue and blue-green regions.14 A deuterated form of the PhS-substituted DBC shows red pRTP with Φp = 28.8%, τp = 1.47 s, and λp = 611 nm. The C–S rotation of the guest exhibiting the red pRTP is calculated to be free in a vacuum, but the analysis of the correlation between the experimental and calculated values of kp as well as knr reveals that the distribution of such free C–S rotation at room temperature (RT) is greatly suppressed in the benzophenone host. With its limited distribution of C–S rotations, PhS substitution allows cooperative enhancement of the spin–orbit coupling (SOC) between the nth-order singlet excited state (Sn) and T1 (n ≥ 1) and the transition dipole moment between Sn and the ground state (S0). This cooperation leads to selective enhancement of kp relative to knr, which in turn results in the increase of Φp.
Results and discussion
To demonstrate the enhanced red pRTP performance, the five molecules shown in Fig. 1a were examined as guest compounds. Pyrene and DBC (1h) were obtained commercially and purified by sublimation before use. Chromophore 2h was synthesized from 1h by Friedel–Crafts acylation. Chromophore 3h was synthesized from 1hvia monobromination followed by reaction with diphenyl disulfide using a palladium catalyst. The structures of 2h and 3h were confirmed by nuclear magnetic resonance and high-resolution mass spectral analyses, and the purity of 2h and 3h was confirmed by elemental analysis (Fig. S1 and S2, ESI†). Chromophore 3d was synthesized by deuteration of 1h followed by monobromination and subsequent reaction with diphenyl disulfide using a palladium catalyst (Fig. S3 and S4, ESI†). Pyrene, 1h, 2h, 3h, and 3d were each doped in crystalline benzophenone at a concentration of 0.3 wt% to prepare films denoted as pyrene, 1h, 2h, 3h, and 3d, respectively (Fig. 1b). For these films, Φp was measured by comparing the luminescence quantum yield upon irradiation with excitation light with the emission spectrum 20–40 ms after the excitation light irradiation was stopped (Fig. S5–S7, ESI†). Φp of the pyrene, 1h, 2h, 3h, and 3d films were 1.5%, 7.6%, 8.8%, 12.4%, and 28.8%, respectively, after excitation at 310 nm (Fig. 1c and Movie S1, ESI†). Φp of film 3d (28.8%) is the highest achieved to date among materials with τp ≥ 100 ms and λp ≥ 600 nm (Fig. 1d and Table S1, ESI†). For pyrene doped in crystalline benzophenone, Φp was reported to be close to 9.2%.11 However, we investigated many films with different amounts of pyrene doped in benzophenone and their crystallization conditions, and found that similar phosphorescence lifetimes were observed but with much lower Φp (1.5%). In fact, bright red pRTP under ultraviolet (UV) light and immediately after UV light exposure was not observed in the pyrene film; a much brighter red pRTP was observed in the 1h, 2h, 3h, and 3d films (Fig. 1c and Movie S1, ESI†). It should also be noted that the absorption spectra of these thin films were comparable from 300 to 400 nm (Fig. S8a, ESI†). Observation of red pRTP soon after ceasing the UV excitation under white indoor light was reasonable considering the excitation power of the UV light and intensity of the indoor light (Fig. S8b–e, ESI†). Previous reports have shown that direct substitution of polyaromatic hydrocarbons with a benzoyl group can markedly improve Φp of blue to green emission.14,31,32 However, benzoyl substitution of DBC (2h) did not enhance Φp compared with that of unsubstituted DBC (1h). Comparison of Φp of films 1h–3h confirmed that direct substitution of DBC with a PhS group enhanced red pRTP to a greater extent than benzoyl substitution.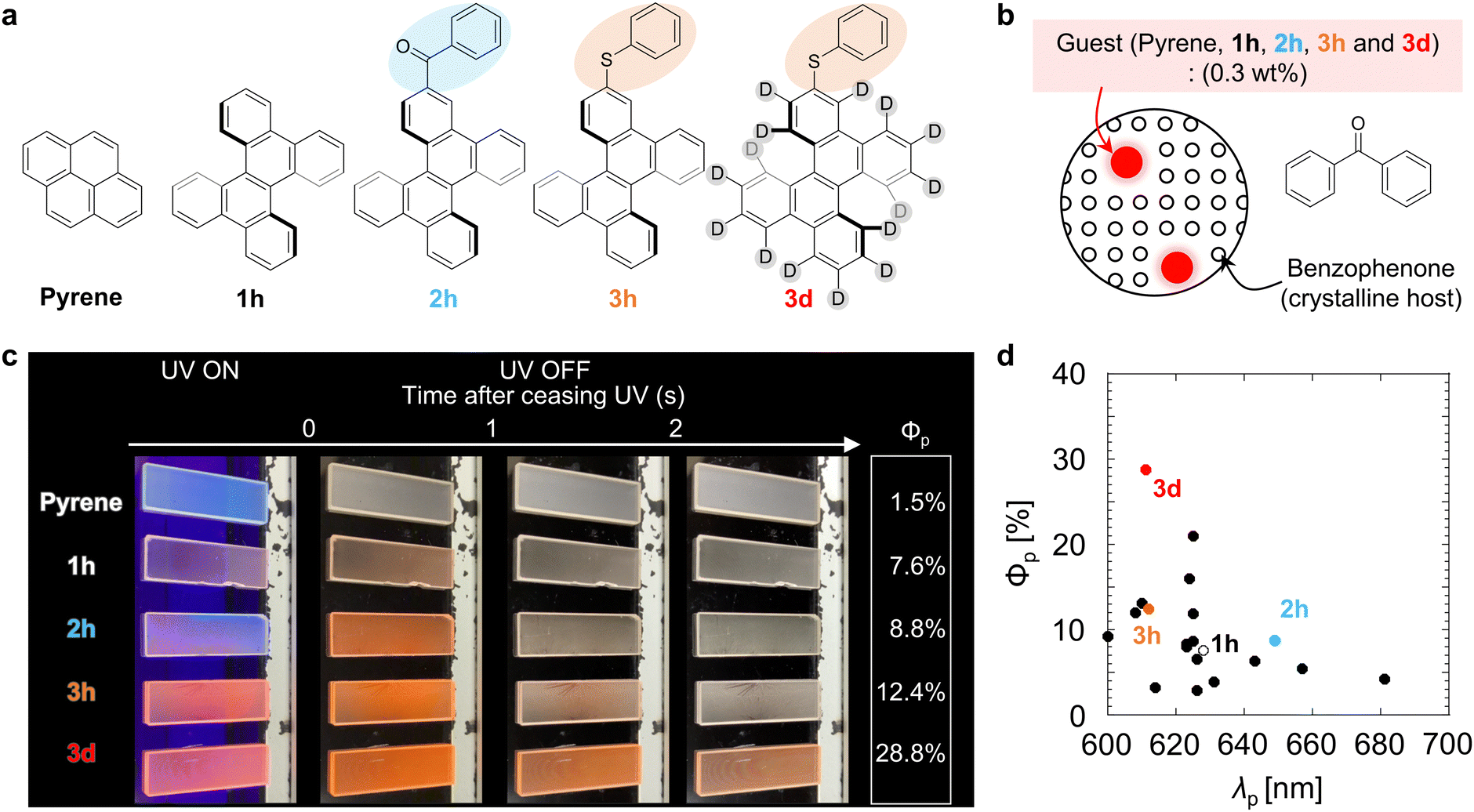 | ||
| Fig. 1 (a) Chemical structures of guest chromophores. (b) Schematic diagram of the host–guest crystal. (c) Luminescence from host–guest crystalline thin films during and after UV light irradiation under indoor lighting (0.12 mW cm−2). When the films were photographed separately, the emission intensity of each film appeared to vary depending on the automatic change of camera settings. To mitigate this problem, five films were photographed simultaneously using one UV light source (0.22 mW cm−2). (d) The performance of the films in terms of Φpvs. λp relative to previously reported materials with τp ≥ 100 ms. | ||
Next, quantitative optical properties were measured to explore the origin of the enhancement of Φp from 1h to 3h. λp of the 1h, 2h, 3h, and 3d films were 628, 649, 612, and 611 nm, respectively (Fig. 2a). τp of the 1h, 2h, 3h, and 3d films were 0.49, 0.34, 0.48, and 1.47 s, respectively (Fig. 2a, inset). Afterglow based on thermally activated delayed fluorescence has a large initial decay by 100 ms. In contrast, pRTP with τp > 100 ms does not have such a large decrease in the initial stage of the afterglow decay (Fig. S9, ESI†). Therefore, bright 2D afterglow emissions can be observed using 2D photodetectors, which makes it difficult to increase the frame rate.24Φp is commonly denoted as,
| Φp = Φtkpτp, | (1) |
| τp = 1/(kp + knr + kq), | (2) |
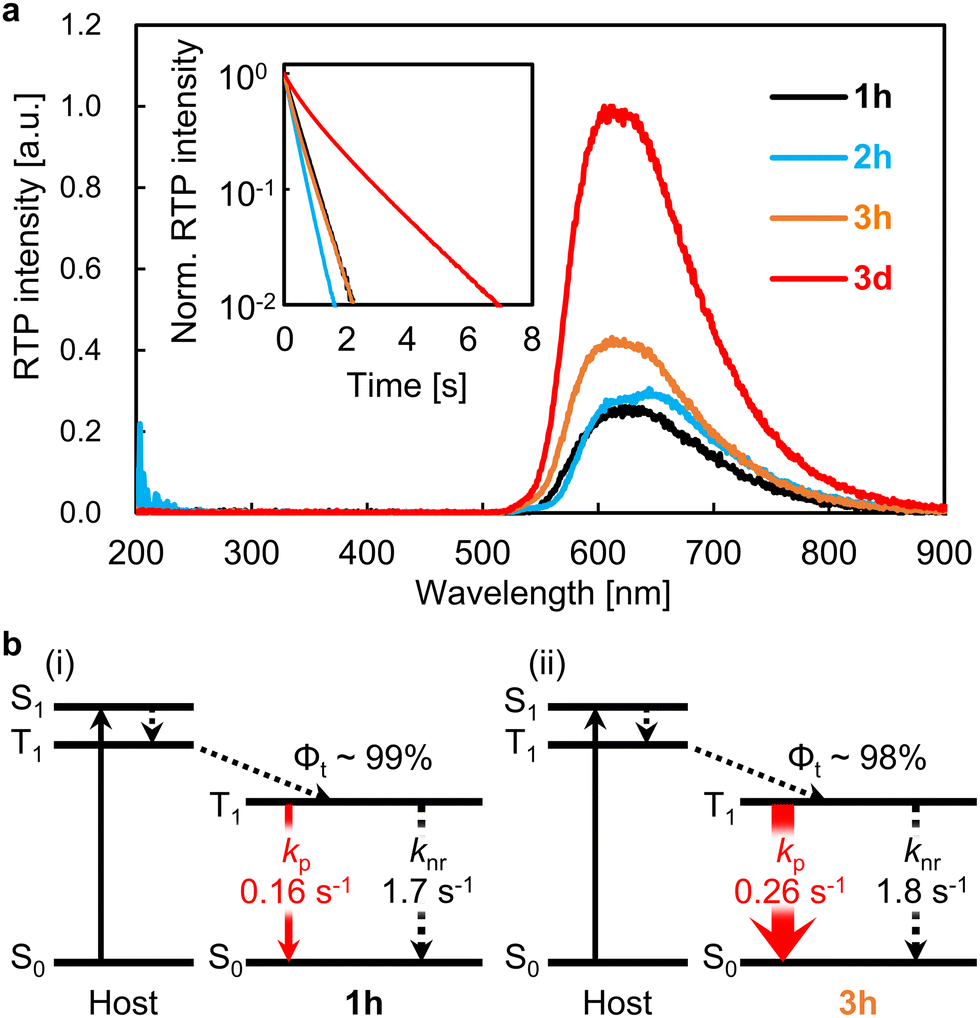 | ||
| Fig. 2 (a) pRTP spectra and decay characteristics (inset) of the 1h, 2h, 3h, and 3d films soon after ceasing excitation at 310 nm. The spectral height of the RTP spectra was set so that the area of the phosphorescence spectrum was proportional to Φp. (b) Jablonsky diagrams showing pRTP of (i) film 1h and (ii) film 3h. | ||
To identify the factor that causes the selective increase of kp of 3h compared with that of 1h, the conformational distribution of 3h was considered. It is possible to estimate kp and knr for many chromophores using elements from the second row of the periodic table onwards by performing quantum chemical calculations considering vibrational displacements at RT.24,27–30 In 3h, the two dihedral angles (θ1 and θ2) defined in Fig. 3a vary easily at low thermal energy because of the weak C–S bonds (Fig. 3a). In this paper, θ1 and θ2 at RT were calculated using density functional theory (DFT) and time-dependent DFT techniques (Section S11, ESI†). Initially, the optimized structure of 3h at T1 in a vacuum was calculated. For that structure, the potential curves in the T1 state were calculated when θ1 and θ2 were changed. Using the activation energy (ΔE) of the potential curve of T1 (Fig. 3b), the possibility of the geometry of 3h with θ1 and θ2 [p(θ1, θ2)] was calculated using the following equation (Fig. 3c),
| p(θ1, θ2) = exp[−ΔE(θ1, θ2)/kT]/∑exp[−ΔE(θ1, θ2)/kT] | (3) |
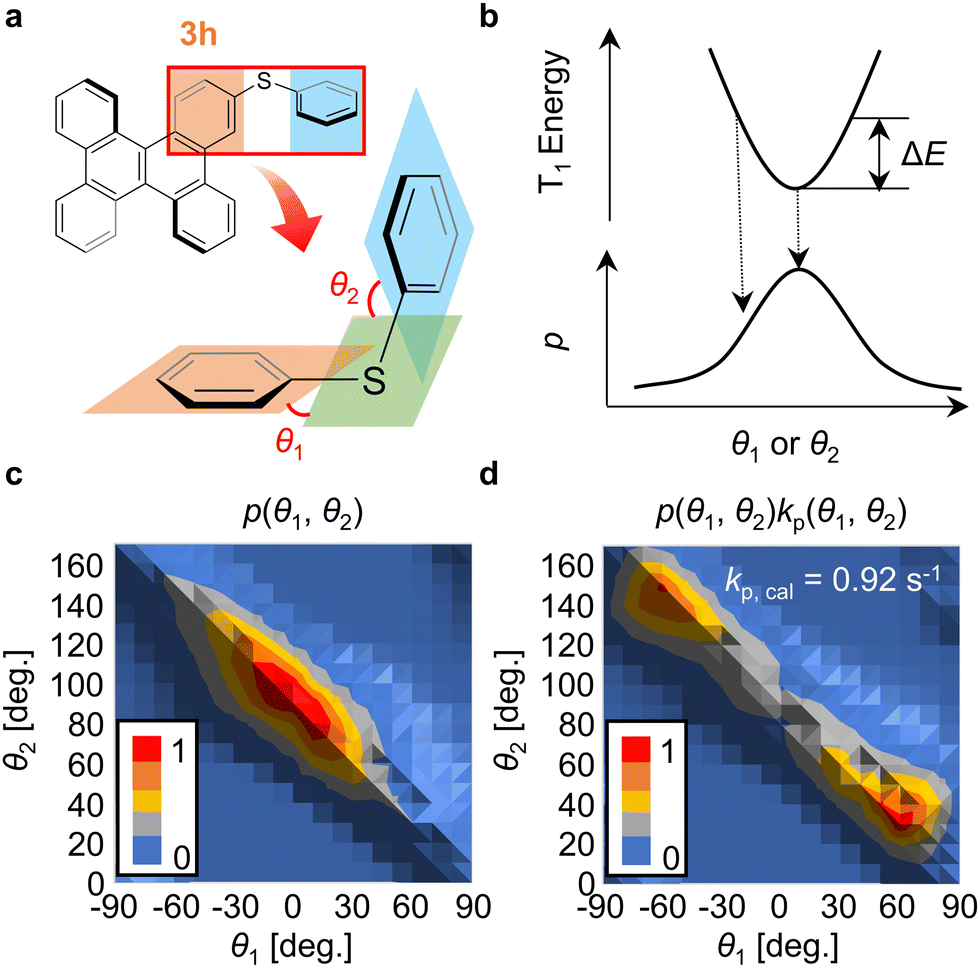 | ||
| Fig. 3 (a) Depiction of the definition of θ1 and θ2 in 3h. (b) Relationship between the energy change (ΔE) and state probability (p) associated with the change of θ1 and θ2 from the optimized structure of T1. (c) Distribution of p calculated in a vacuum for 3h with respect to θ1 and θ2. (d) Distribution of the product of kp and p calculated in a vacuum for 3h with respect to θ1 and θ2. | ||
To confirm the confinement of the distributions of θ1 and θ2 of 3h in the crystalline film, we theoretically narrowed the distributions of θ1 and θ2 to confirm whether the correlation between experimental and calculation results could be improved in terms of both kp and knr. A model was proposed in which when C–S rotation occurs in benzophenone crystals, the steric hindrance between the host and guest increases ΔE of the potential curve of T1 for the same θ1 and θ2 displacement of 3h (Fig. 4a and 4b). For example, when ΔE increases by a factor of eight (Fig. 4c), p(θ1, θ2) in a vacuum converges to a narrow range based on eqn (3) (Fig. S19, ESI†). When p(θ1, θ2) narrows, the histogram of kp(θ1, θ2)p(θ1, θ2) changes from that in Fig. 4d to that in Fig. 4e. From the integration of the histogram in Fig. 4e, kp,cal was estimated to be 0.28 s−1. Therefore, when considering the distributions of θ1 and θ2 where the host increases the guest ΔE by a factor of eight, kp,exp and kp,cal correlate well (Fig. 4f). The correlation of experimental and calculated values of knr provides further evidence that this estimate is reasonable.
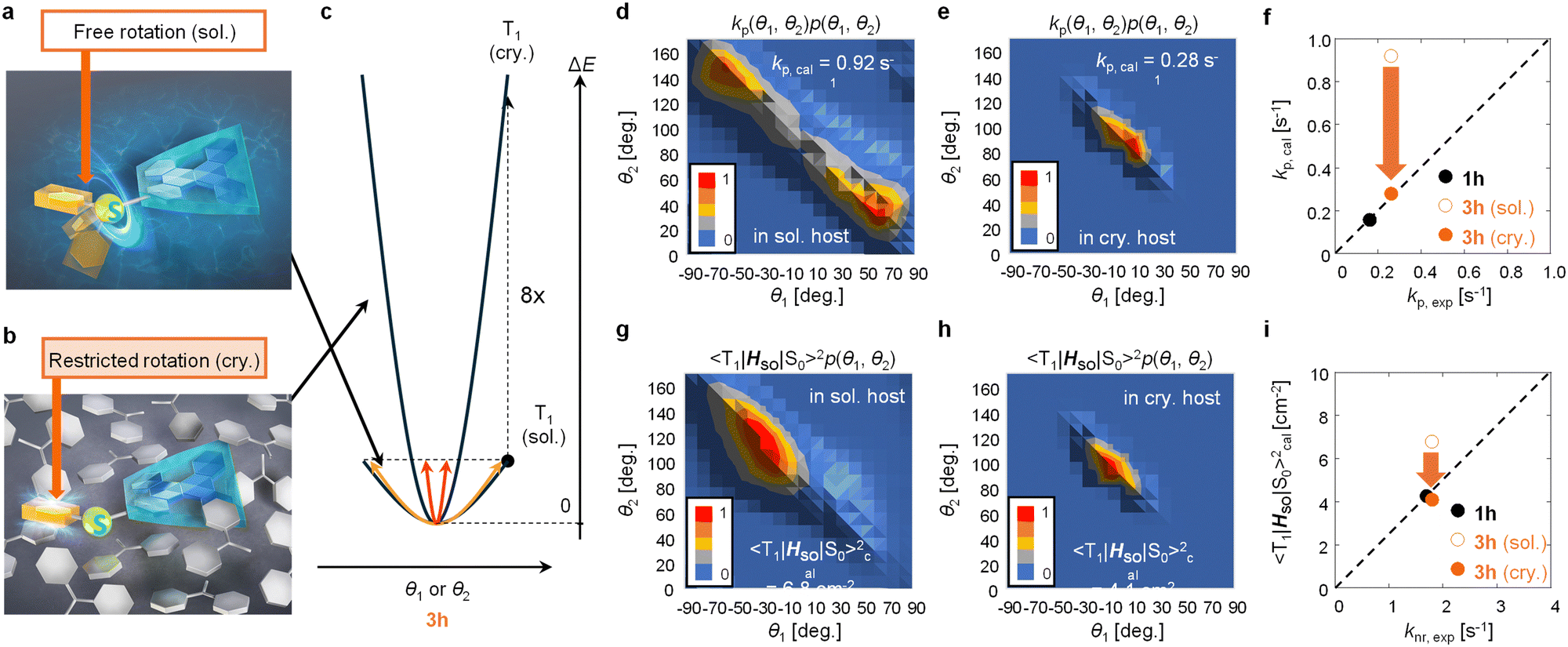 | ||
| Fig. 4 Estimated molecular motions of 3h in (a) solution and (b) a solid host. (c) Estimated potential surface of 3h depending on the change of θ1 and θ2 in solution and solid media. Histograms of normalized kp(θ1, θ2)p(θ1, θ2) in (d) solution and (e) a crystalline host. (f) The relationship between kp,cal and kp,exp for 1h and 3h. Histograms of normalized 〈T1|HSO|S0〉(θ1, θ2)2p(θ1, θ2) in (g) solution and (h) a crystalline host. (i) The relationship between 〈T1|HSO|S0〉cal2 and knr,exp for 1h and 3h. | ||
In a model in which the distributions of θ1 and θ2 of the guest were confined by steric hindrance with the host, the experimental and calculated values of knr showed a strong correlation. knr is proportional to the square of the SOC between T1 and S0 (〈T1|HSO|S0〉) and also depends on the phosphorescence energy. Therefore, the difference of knr among guest chromophores could be approximated by 〈T1|HSO|S0〉2 when the phosphorescence energy does not change much between guests.16,29 When ΔE increases by a factor of eight, the histogram of 〈T1|HSO|S0〉 (θ1, θ2)2p(θ1, θ2) changes from that in Fig. 4g to that in Fig. 4h. The calculated average values of 〈T1|HSO|S0〉2 (〈T1|HSO|S0〉cal2) were determined to be 6.8 and 4.1 cm−2 from the integration of Fig. 4g and h, respectively. For 1h, it was calculated that 〈T1|HSO|S0〉cal2 = 4.3 cm−2 using the optimized T1 structure. For the distributions of θ1 and θ2 in a vacuum at 3h, the correlation between optically determined knr (knr,exp) and 〈T1|HSO|S0〉cal2 was poor (Fig. 4i). Conversely, when considering the distributions of θ1 and θ2 where the host increases the ΔE of the guest by a factor of eight, a much stronger correlation between knr,exp and 〈T1|HSO|S0〉cal2 was observed (Fig. 4i).
When considering kp and knr, it is essential to include the conformational displacement of vibrations.24,27–30 Generally, compounds up to the second row of the periodic table form strong bonds, so the changes of displacement at RT are limited.19,24 Therefore, kp may not change that much between soft and rigid environments. Conversely, in compounds containing elements from the third row of the periodic table or later, the bonds between the elements and carbon are generally weak, and many of the compounds display free rotation. This study demonstrated from both experimental and computational aspects of kp and knr that such large free conformational changes are strongly suppressed in solid hosts, resulting in a narrow structural distribution. When the enhancement of ΔE induced by the crystalline host is much less than a factor of eight, kp,cal is still overestimated compared with kp,exp. The best correlation between the experimental and calculated kp and knr values could be observed when considering the distribution of θ1 and θ2 where the host increases ΔE of the guest by a factor of eight. Not only was there a strong correlation between the experimental and calculated kp and knr values of the confined 3h geometry and 1h, but also the equivalent phosphorescence energies of 1h and 3h observed experimentally were corroborated by the calculations (Fig. S17, ESI†).
The strong correlation between the experimental and calculation results obtained by considering the state distribution makes it possible to clarify the factor causing the increase of kp of 3h relative to that of 1h. kp is logically related to the product of the SOC between Sn and T1 (〈Sn|HSO|T1〉) and the transition dipole moment of Sn and S0 (μSn–S0).16,27,35,36 More specifically, because kp is approximately proportional to 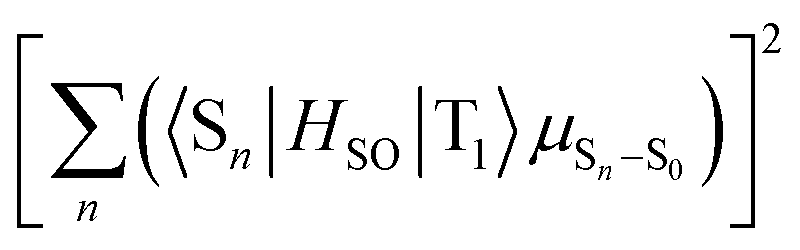 , kp increases as the product of 〈Sn|HSO|T1〉 and μSn–S0 increases for each n. In the conformation where θ1 approaches 0° and θ2 approaches 90° in Fig. 4e and h, which is close to the T1 optimized geometry of 3h. The introduction of PhS-substituents at the confined angles increases the cooperativity of 〈Sn|HSO|T1〉 and μSn–S0 for a wide range of n (Fig. 5). On the other hand, the introduction of phenylthio-substituents in the geometry does not increase knr because 〈T1|HSO|S0〉2 of 3h (3.6 cm−2) is comparable to that of 1h (4.3 cm−2), where 〈T1|HSO|S0〉2 is logically proportional to knr. Therefore, the cooperativity of 〈Sn|HSO|T1〉 and μSn–S0 for a wide range of n in 3h compared with the case for 1h allows the selective enhancement of kp, which in turn leads to the increase of Φp of the 3h film compared with that of the 1h film. The comparable 〈T1|HSO|S0〉 values of 1h and 3h contain important information on the heavy atom effect of S. When S is directly bonded to the phosphorescent chromophore, the heavy atom effect might be in play. However, similar 〈T1|HSO|S0〉 values were calculated at 1h and 3h in benzophenone crystals, and the knr values determined by optical measurements were also comparable between them. Therefore, it can be seen that the increase in the molecular weight of S does not have the effect of significantly increasing at least 〈T1|HSO|S0〉.
, kp increases as the product of 〈Sn|HSO|T1〉 and μSn–S0 increases for each n. In the conformation where θ1 approaches 0° and θ2 approaches 90° in Fig. 4e and h, which is close to the T1 optimized geometry of 3h. The introduction of PhS-substituents at the confined angles increases the cooperativity of 〈Sn|HSO|T1〉 and μSn–S0 for a wide range of n (Fig. 5). On the other hand, the introduction of phenylthio-substituents in the geometry does not increase knr because 〈T1|HSO|S0〉2 of 3h (3.6 cm−2) is comparable to that of 1h (4.3 cm−2), where 〈T1|HSO|S0〉2 is logically proportional to knr. Therefore, the cooperativity of 〈Sn|HSO|T1〉 and μSn–S0 for a wide range of n in 3h compared with the case for 1h allows the selective enhancement of kp, which in turn leads to the increase of Φp of the 3h film compared with that of the 1h film. The comparable 〈T1|HSO|S0〉 values of 1h and 3h contain important information on the heavy atom effect of S. When S is directly bonded to the phosphorescent chromophore, the heavy atom effect might be in play. However, similar 〈T1|HSO|S0〉 values were calculated at 1h and 3h in benzophenone crystals, and the knr values determined by optical measurements were also comparable between them. Therefore, it can be seen that the increase in the molecular weight of S does not have the effect of significantly increasing at least 〈T1|HSO|S0〉.
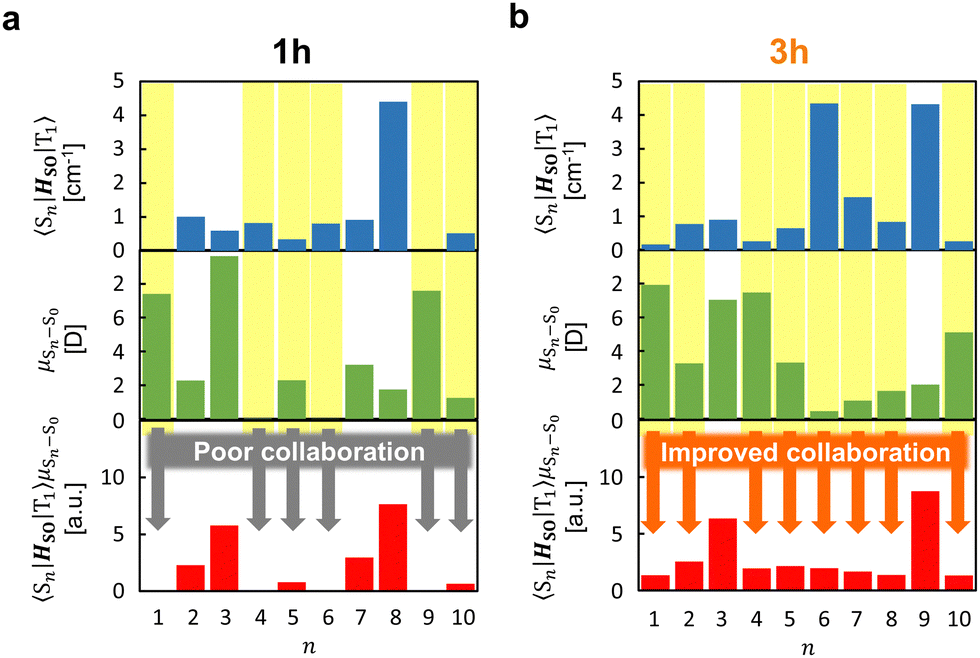 | ||
| Fig. 5 Relationship between 〈Sn|HSO|T1〉, μSn–S0, and 〈Sn|HSO|T1〉μSn–S0 from n = 1 to n = 10 in 1h (a) and 3h (b). | ||
Green pRTP properties have often been achieved using motifs in which two benzene groups are linked by a six-membered ring containing at least one group-16 element, such as phenothiazine, 9H-thioxanthene, and thianthrene.37–42 In terms of the group-16 element, it has been reported that the effect of sulfur on green pRTP is considerably smaller than that of selenium.39,40 In this paper, we showed that direct substitution of sulfur into the red pRTP backbone, even without using heavy atoms such as selenium, enhances Φp considerably in the long-wavelength range and maintains a large τp compared with other substitutions. By clarifying the confined conformational distribution of the guest dye in the crystal host, we also elucidated that the Φp enhancement is caused by the cooperative strengthening of SOCs and the transition dipole moment associated with Sn.
Conclusions
Direct substitution of DBC with PhS produced 3h, which showed enhanced red pRTP over that of DBCs with other substituents in a crystalline benzophenone host. Deuterated DBC with a PhS substituent in a crystalline benzophenone host achieved values of τp = 1.47 s and Φp = 28.8% with λp = 611 nm. Correlation of experimental and calculated values of kp and knr showed that the free rotation of C–S of 3h considered to occur in a vacuum is largely suppressed in the crystalline benzophenone host. For the geometry with a limited distribution of C–S rotations, the PhS group induces cooperative enhancement of the Sn–T1 SOC and the Sn–S0 transition dipole moment for a variety of states with n ≥ 1, which leads to the selective enhancement of kp over knr. The selective enhancement of kp results in the increase of Φp. In host–guest solid materials, quantum chemical calculations taking vibrations into account are necessary to elucidate the relationship between kp, knr, and molecular structure. Although guest chromophores in solution show large fluctuations because of the absence of surrounding intermolecular forces at RT, thermally activated displacements are also present within the crystalline host, but their distribution is greatly constrained. There are few reports of one-to-one correspondence between kp, knr, and molecular structure determined through a combination of experiments and calculations among the many recent studies on pRTP. Recently, it has been reported that the RTP from molecular crystals that are not host–guest is not due to the molecular structure, but rather to impurities present in the crystals, which has become clearer.43–45 A thorough collaborative analysis of experiments and theories as described in this paper will contribute to identifying chromophores that emit phosphorescence and their conformations and distributions.Author contributions
S. U. and S. H. wrote the manuscript. S. U. performed the organic synthesis with advice from B. K. S. U. performed transient absorption analysis with K. F. S. U. performed the quantum chemical calculations with the help of S. H. S. H. supervised this project.Data availability
Detailed information on organic synthesis, optical property measurements, and quantum chemical calculations is provided in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the assistance with high-resolution mass spectrometry measurements received from the Coordinated Center for UEC Research Facilities of the University of Electro-Communications. This work was supported by Fusion Oriented Research for disruptive Science and Technology (FOREST) of the Japan Science and Technology Agency (Grant Number JPMJFR201T) and the Japan Society for the Promotion of Science (Grant Number 24K01567).Notes and references
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS.
- A. S. Mathew, C. A. DeRosa, J. N. Demas and C. L. Fraser, Anal. Methods, 2016, 8, 3109–3114 RSC.
- Y. Deng, D. Zhao, X. Chen, F. Wang, H. Song and D. Shen, Chem. Commun., 2013, 49, 5751–5753 RSC.
- P. Long, Y. Feng, C. Cao, Y. Li, J. Han, S. Li, C. Peng, Z. Li and W. Feng, Adv. Funct. Mater., 2018, 28, 1800791 CrossRef.
- M. Louis, H. Thomas, M. Gmelch, A. Haft, F. Fries and S. Reineke, Adv. Mater., 2019, 31, 1807887 CrossRef.
- B. Zhou, G. Xiao and D. Yan, Adv. Mater., 2021, 33, 2007571 CrossRef CAS.
- X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef.
- S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160–12164 CrossRef CAS PubMed.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- F. Xiao, H. Gao, Y. Lei, W. Dai, M. Liu, X. Zheng, Z. Cai, X. Huang, H. Wu and D. Ding, Nat. Commun., 2022, 13, 186 CrossRef CAS PubMed.
- I. Bhattacharjee and S. Hirata, Adv. Mater., 2020, 32, 2001348 CrossRef CAS.
- E. H. Badriyah, K. Hayashi, B. Sk, R. Takano, T. Ishida and S. Hirata, Adv. Sci., 2023, 10, 2304374 CrossRef CAS PubMed.
- W. Ye, H. Ma, H. Shi, H. Wang, A. Lv, L. Bian, M. Zhang, C. Ma, K. Ling, M. Gu, Y. Mao, X. Yao, C. Gao, K. Shen, W. Jia, J. Zhi, S. Cai, Z. Song, J. Li, Y. Zhang, S. Lu, K. Liu, C. Dong, Q. Wang, Y. Zhou, W. Yao, Y. Zhang, H. Zhang, Z. Zhang, X. Hang, Z. An, X. Liu and W. Huang, Nat. Mater., 2021, 20, 1539–1544 CrossRef CAS PubMed.
- H. Ma, L. Fu, X. Yao, X. Jiang, K. Lv, Q. Ma, H. Shi, Z. An and W. Huang, Nat. Commun., 2024, 15, 3660 CrossRef CAS.
- S. Hirata, Appl. Phys. Rev., 2022, 9, 011304 CAS.
- Y. Katsurada, S. Hirata, K. Totani, T. Watanabe and M. Vacha, Adv. Opt. Mater., 2015, 3, 1726 CrossRef CAS.
- B. Chen, W. Huang, X. Nie, F. Liao, H. Miao, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16970–16973 CrossRef CAS PubMed.
- K. Fukasawa, Y. Sugawara, R. Tsuru, T. Yamashita and S. Hirata, J. Phys. Chem. Lett., 2022, 13, 7788 CrossRef CAS PubMed.
- D. Li, J. Yang, M. Fang, B. Z. Tang and Z. Li, Sci. Adv., 2022, 8, eabl8392 CrossRef CAS PubMed.
- J. Jovaišaitė, S. Kirschner, S. Raišys, G. Kreiza, P. Baronas, S. Juršėnas and M. Wagner, Angew. Chem., Int. Ed., 2023, 62, e202215071 CrossRef.
- X. Shen, Y. Luo, K. Chen, C. Fu, R. Liu, Y. Huang, K. Wu, J. Luoa and Z. Lu, Chem. Commun., 2023, 59, 7036–7039 RSC.
- X. Zheng, Q. Han, Q. Lin, C. Li, J. Jiang, Q. Guo, X. Ye, W. Z. Yuan, Y. Liu and X. Tao, Mater. Horiz., 2023, 10, 197–208 RSC.
- B. Sk and S. Hirata, Adv. Sci., 2024, 11, 2308897 CrossRef CAS.
- C. Qiang, T. Stefan, S. Sven, S. Dieter, M. Klaus, N. Akimitsu and B. Thomas, J. Am. Chem. Soc., 2019, 141, 16439–16449 CrossRef.
- I. Hattori, M. Hagai, M. Ito, M. Sakai, H. Narita, K. J. Fujimoto, T. Yanai and S. Yamaguchi, Angew. Chem., Int. Ed., 2024, 63, e202403829 CrossRef CAS PubMed.
- S. Hirata, J. Phys. Chem. Lett., 2018, 9, 4251–4259 CrossRef CAS.
- S. Hirata and I. Bhattacharjee, J. Phys. Chem. A, 2021, 125, 885–894 CrossRef CAS.
- I. Bhattacharjee, K. Hayashi and S. Hirata, JACS Au, 2021, 1, 945–954 CrossRef CAS PubMed.
- B. Sk, R. Tsuru, K. Hayashi and S. Hirata, Adv. Funct. Mater., 2023, 33, 2211604 CrossRef CAS.
- M. Shimizu, R. Shigitani, M. Nakatani, K. Kuwabara, Y. Miyake, K. Tajima, H. Sakai and T. Hasobe, J. Phys. Chem. C, 2016, 120, 11631–11639 CrossRef CAS.
- Y. Zhang, Z. Wang, Y. Su, Y. Zheng, W. Tang, C. Yang, H. Tang, L. Qu, Y. Li and Y. Zhao, Research, 2021, 8096263 CAS.
- J. Cao, M. Zhang, M. Singh, Z. An, L. Ji, H. Shi and Y. Jiang, Front. Chem., 2021, 9, 781294 CrossRef CAS PubMed.
- G. W. Robinson and R. P. Frosch, J. Chem. Phys., 1963, 38, 1187 CrossRef CAS.
- S. K. Lower and M. A. El-Sayed, Chem. Rev., 1966, 66, 199 CrossRef CAS.
- P. N. Lai, C. H. Brysacz, M. K. Alam, N. A. Ayoub, T. G. Gray, J. M. Bao and T. S. Teets, J. Am. Chem. Soc., 2018, 140, 10198 CrossRef CAS.
- P. Pander, A. Swist, R. Turczyn, S. Pouget, D. Djurado, A. Lazauskas, R. Pashazadeh, J. V. Grazulevicius, R. Motyka, A. Klimash, P. J. Skabara, P. Data, J. Soloducho and F. B. Dias, J. Phys. Chem. C, 2018, 122, 24958–24966 CrossRef CAS.
- N. A. Kukhta, R. Huang, A. S. Batsanov, M. R. Bryce and F. B. Dias, J. Phys. Chem. C, 2019, 123, 26536–26546 CrossRef CAS.
- D. R. Lee, K. H. Lee, W. Shao, C. L. Kim, J. Kim and J. Y. Lee, Chem. Mater., 2020, 32, 2583–2592 CrossRef CAS.
- W. Shao, H. Jiang, R. Ansari, P. M. Zimmerman and J. Kim, Chem. Sci., 2022, 13, 789–797 RSC.
- W. Shao and J. Kim, Acc. Chem. Res., 2022, 55, 1573–1585 CrossRef CAS PubMed.
- M. Schmiedtchen, I. Maisuls, H. Siera, J. Balszuweit, C. Wölper, M. Giese, G. Haberhauer, C. A. Strassert and J. Voskuhl, Angew. Chem., Int. Ed., 2024, e202414326 Search PubMed.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2021, 20, 175–180 CrossRef CAS.
- Z. Wu, J. C. Roldao, F. Rauch, A. Friedrich, M. Ferger, F. Würthner, J. Gierschner and T. B. Marder, Angew. Chem., Int. Ed., 2022, 61, e202200599 CrossRef CAS PubMed.
- Z. Wu, C. Herok, A. Friedrich, B. Engels, T. B. Marder and Z. M. Hudson, J. Am. Chem. Soc., 2024, 146, 31507–31517 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc04829f |
| This journal is © The Royal Society of Chemistry 2025 |