
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancement of intersystem crossing in asymmetrically substituted BODIPY photosensitizers†
Mikhail A. Filatov *a,
Tatsiana Mikulchykb,
Maxime Hodéec,
Metodej Dvoraceka,
Venkata N. K. Mamillapallia,
Aimee Sheehana,
Craig Newmana,
Sergey M. Borisovd,
Daniel Escudero*c and
Izabela Naydenovab
*a,
Tatsiana Mikulchykb,
Maxime Hodéec,
Metodej Dvoraceka,
Venkata N. K. Mamillapallia,
Aimee Sheehana,
Craig Newmana,
Sergey M. Borisovd,
Daniel Escudero*c and
Izabela Naydenovab
aSchool of Chemical and Biopharmaceutical Sciences, Technological University Dublin, City Campus, Grangegorman, D07 ADY7 Dublin, Ireland. E-mail: mikhail.filatov@tudublin.ie
bCentre for Industrial & Engineering Optics, School of Physics, Clinical and Optometric Sciences, Technological University Dublin, D07 ADY7 Dublin, Ireland
cQuantum Chemistry and Physical Chemistry, Department of Chemistry KU Leuven, Celestijnenlaan 200F, Leuven 3001, Belgium. E-mail: daniel.escudero@kuleuven.be
dInstitute of Analytical Chemistry and Food Chemistry, Graz University of Technology, Stremayrgasse 9, Graz 8010, Austria
First published on 18th February 2025
Abstract
We present a novel method to promote intersystem crossing (ISC) and triplet state formation in boron dipyrromethenes (BODIPYs) through the asymmetrical introduction of functional groups within the chromophore. This approach enables the development of new BODIPY photosensitizers without relying on the incorporation of heavy atoms or large electron-donating aromatic groups. Demonstrated on a series of 14 synthesized asymmetrical BODIPY (aBDP) compounds, it significantly enhances photosensitization efficiency compared to the reference symmetrical BODIPYs. In particular, the asymmetrical introduction of ethoxycarbonyl groups into pyrrolic rings of the BODIPY core lead to efficient ISC and singlet oxygen generation, with quantum yields reaching 0.76 in non-polar solvents. Quantum chemical calculations elucidated the ISC mechanism, revealing an S1 → T2 pathway facilitated by a reduced singlet–triplet energy gap (ΔES–T). The new photosensitizers were successfully applied in holographic recording through photopolymerization of acrylamide monomers in a cellulose acetate–based photopolymer, using irradiation with a 532 nm laser. The material containing an asymmetrical BODIPY–anthracene dyad exhibited the highest exposure sensitivity, achieving diffraction efficiencies up to 71% for volume transmission gratings at an exposure energy of 2.3 J cm−2. The practical potential of these dyes was demonstrated by fabricating a holographic optical element – an off-axis lens – with a uniform diffraction efficiency of 38 ± 3% across the lens.
Introduction
Photosensitizers capable of forming triplet excited states via the intersystem crossing (ISC) process are critically important for various technologies, including photocatalysis, solar energy conversion, and photomedicine.1 In these applications, formation of long-lived triplet excited states is used to harvest light energy and drive various photochemical processes. Traditionally, the design of photosensitizers has focused on incorporating heavy atoms, such as transition metals (e.g., Ru, Pd, Pt) and halogens (Br, I), into organic chromophores.2 Heavy atoms facilitate spin–orbit coupling, significantly enhancing ISC rates and leading to high triplet state yields (ΦT).3 However, this strategy introduces significant challenges, such as shortened triplet lifetimes, elevated costs, and increased toxicity,4 which hinder the broader practical applications of these dyes.Recently, there has been growing interest in exploring novel approaches to enhance ISC in organic dyes that do not contain heavy atoms, referred to as heavy-atom-free photosensitizers. Spin–orbit charge-transfer intersystem crossing (SOCT-ISC)5 has emerged as a versatile strategy applicable to various classes of dyes.6 SOCT-ISC occurs in donor–acceptor dyads composed of orthogonal aromatic fragments, e.g. meso-aryl BODIPYs (Fig. 1a). In these systems, triplet excited states are generated from a singlet charge-transfer (1CT) state formed as a result of an electron transfer between the BODIPY unit (acceptor) and the aryl group (donor). This approach offers significant advantages over the introduction of heavy atoms, e.g., allowing on–off switching of the electron transfer and ISC processes.7 However, achieving efficient triplet state formation through the SOCT-ISC mechanism is a complex task, as it requires precise tuning of the molecular structures to ensure (i) orthogonal geometry and (ii) optimal driving force for electron transfer (ΔGET) between the subunits.8 Recently, a new method utilizing through-space charge transfer (TSCT) in face-to-face electron donor–acceptor dyads has been introduced, providing greater flexibility in molecular design but also demanding more synthetic efforts to access such structures.9
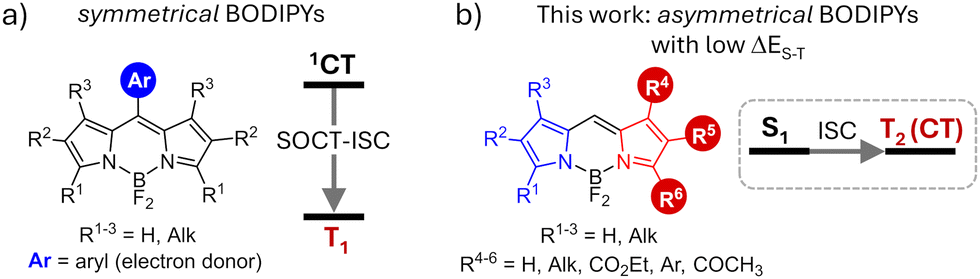 | ||
| Fig. 1 General structures of (a) symmetrical BODIPY donor–acceptor dyads undergoing SOCT-ISC process and (b) asymmetrical BODIPY dyes investigated in this work. | ||
A promising strategy for designing photosensitizers relies on reducing the energy gap between the singlet and triplet excited states (ΔES–T). A key advantage is that it removes the need for heavy atom incorporation or specific solvent conditions to achieve high triplet state yields. In such molecules, ISC can occur through two pathways: (1) direct spin–orbit coupling between S1 and T1 or (2) coupling between S1 and a higher triplet state (Tn), followed by internal conversion from Tn to T1.10 According to the energy gap law,11 ISC is more efficient when the singlet and triplet states have similar energy levels. This approach has been successfully applied in some recent works. For example, thionation of carbonyl groups in organic dyes has been shown to reduce the energy gap between Sn and Tn states, leading to efficient ISC.12 Aggregation of donor–π–acceptor (D–π–A) dyes has also been proven effective for enhancing ISC via this mechanism.13 Börjesson and co-workers demonstrated that in oligomers containing two to four dye units, exciton coupling shifts the singlet state energy levels, thereby decreasing ΔES–T.14 Olivier et al. showed that charge transfer (CT) delocalization allows to decrease the singlet–triplet energy gap without deteriorating the oscillator strength.15 However, a significant challenge remains: there is no versatile strategy and finding structures with small ΔES–T values is still based on a trial and error approach. The lack of guidelines limits the applications of this method, making it challenging to tune ISC efficiency by modulating ΔES–T values.
In this work, we demonstrate that asymmetrical substitution of the two pyrrolic rings in the BODIPY chromophore effectively reduces the singlet–triplet energy gap, enabling efficient ISC via the S1 → T2 pathway. A comprehensive screening of asymmetrical BODIPYs (aBDPs, Fig. 2) showed that this approach is applicable to a wide range of structures with different substitution patterns. Specifically, we explored BODIPYs featuring an electron-rich alkyl-substituted pyrrolic unit (highlighted in blue in Fig. 2) and an electron-deficient pyrrolic unit (highlighted in red), which may contain a carbonyl and/or aryl group, or may be simply unsubstituted. The resulting compounds exhibited singlet oxygen generation quantum yields (ΦΔ) of up to 0.76. The photosensitization efficiency of aBDPs was found to be solvent-dependent, with the highest ΦΔ values observed in non-polar solvents, which are particularly favourable for photocatalysis applications.
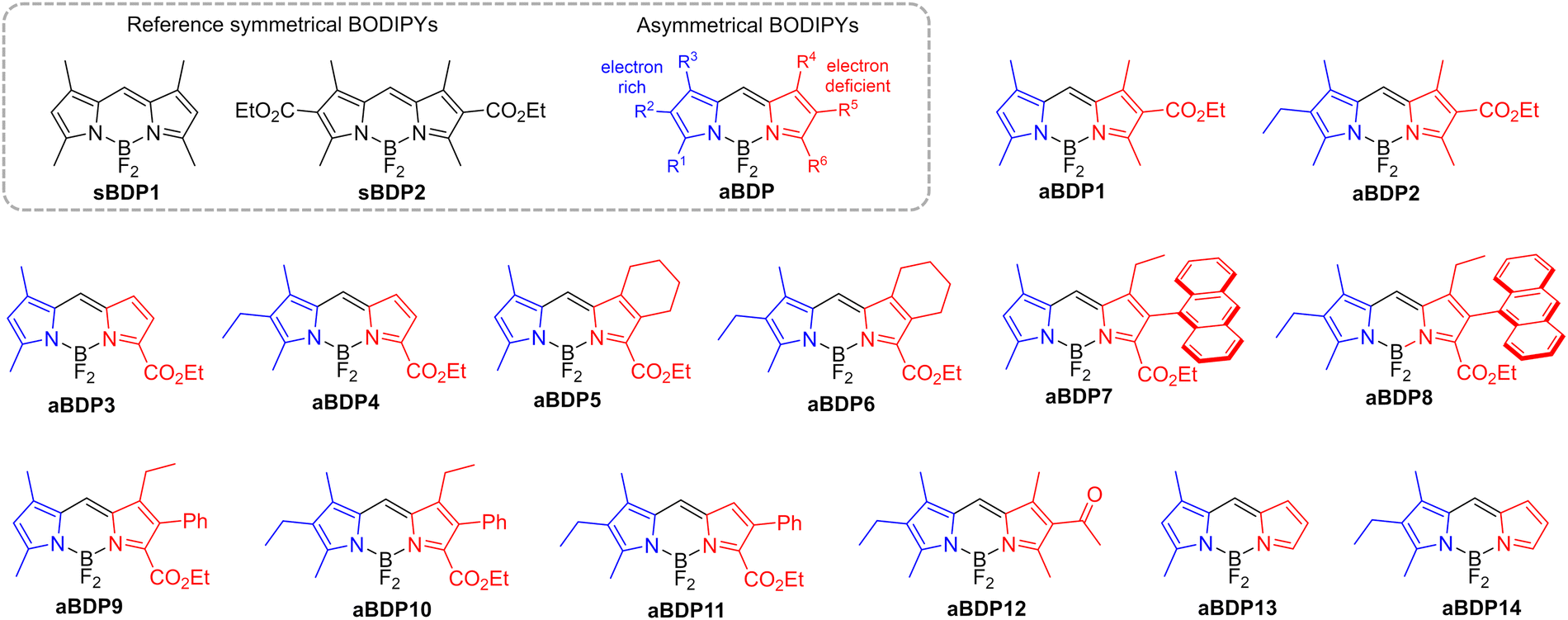 | ||
| Fig. 2 Chemical structures of asymmetrical and reference symmetrical BODIPY (sBDP) compounds investigated in this work. | ||
Manipulating molecular symmetry to enhance ISC represents a completely new and highly promising strategy for accessing triplet states in BODIPY dyes. aBDPs can be synthesized from readily available pyrrolic precursors with a wide range of substituents and thus enabling fine-tuning of the molecular properties for specific applications. To further explore the potential of these novel dyes, we employed them as light-harvesting components in a cellulose acetate-based photopolymer for holographic recording of diffractive structures. The lead compound, aBDP8, demonstrated exceptional photopolymerization efficiency, providing a diffraction efficiency of up to 71% in holographic transmission gratings. Additionally, the resulting material was used to fabricate a holographic optical element – an off-axis lens with a uniform diffraction efficiency of 38 ± 3% across the element.
Results and discussion
Molecular design
Introduction of substituents at the pyrrolic positions of the BODIPY core is known to significantly affect its electronic structure and has been used to optimize its optical properties.16 For example, introduction of alkyl groups results in increased photostability, molar absorption coefficients and fluorescence quantum yields (ΦFl). Therefore, derivatives of the symmetrical 1,3,5,7-tetramethylBODIPY (sBDP1, Fig. 2a) are widely used as fluorophores.17 In our recent work, we explored symmetrical 2,6-diethoxycarbonyl BODIPY (sBDP2, Fig. 2a) as efficient electron acceptors for the creation of donor–acceptor dyads.18The incorporation of ethoxycarbonyl groups attenuates the reduction potentials of BODIPY by 0.2–0.3 eV,19 while preserving high fluorescence quantum yields (>90%). Interesting optoelectronic properties of the diethoxycarbonyl BODIPY scaffold prompted us to examine its derivatives in which the ethoxycarbonyl group is introduced asymmetrically, i.e., into only one pyrrolic ring. Condensation of corresponding pyrrole aldehyde precursors with 2,4-dimethylpyrrole or 3-ethyl-2,4-dimethylpyrrole, followed by boron insertion (see ESI† for synthetic details), yielded the asymmetrical compounds aBDP1 and 2, respectively (Fig. 2). Fluorescence studies revealed that asymmetrical substitution leads to substantial quenching of fluorescence in polar solvents. For example, the emission quantum yield of aBDP2 in acetonitrile (ΦFl = 0.69) is significantly lower than that of the symmetrical reference compound sBDP2 in the same solvent (ΦFl = 0.96), highlighting the impact of asymmetrical substitution.
Given the unexpected reduction in fluorescence quantum yields, we hypothesized that enhanced intersystem crossing in aBDPs competes with fluorescence emission, leading to triplet state formation. Indeed, the formation of triplets was confirmed by the enhanced singlet oxygen phosphorescence at 1276 nm in an air-saturated solution of aBDP2 (Fig. 3b), which gave ΦΔ values of up to 24% in acetonitrile (Table 1).
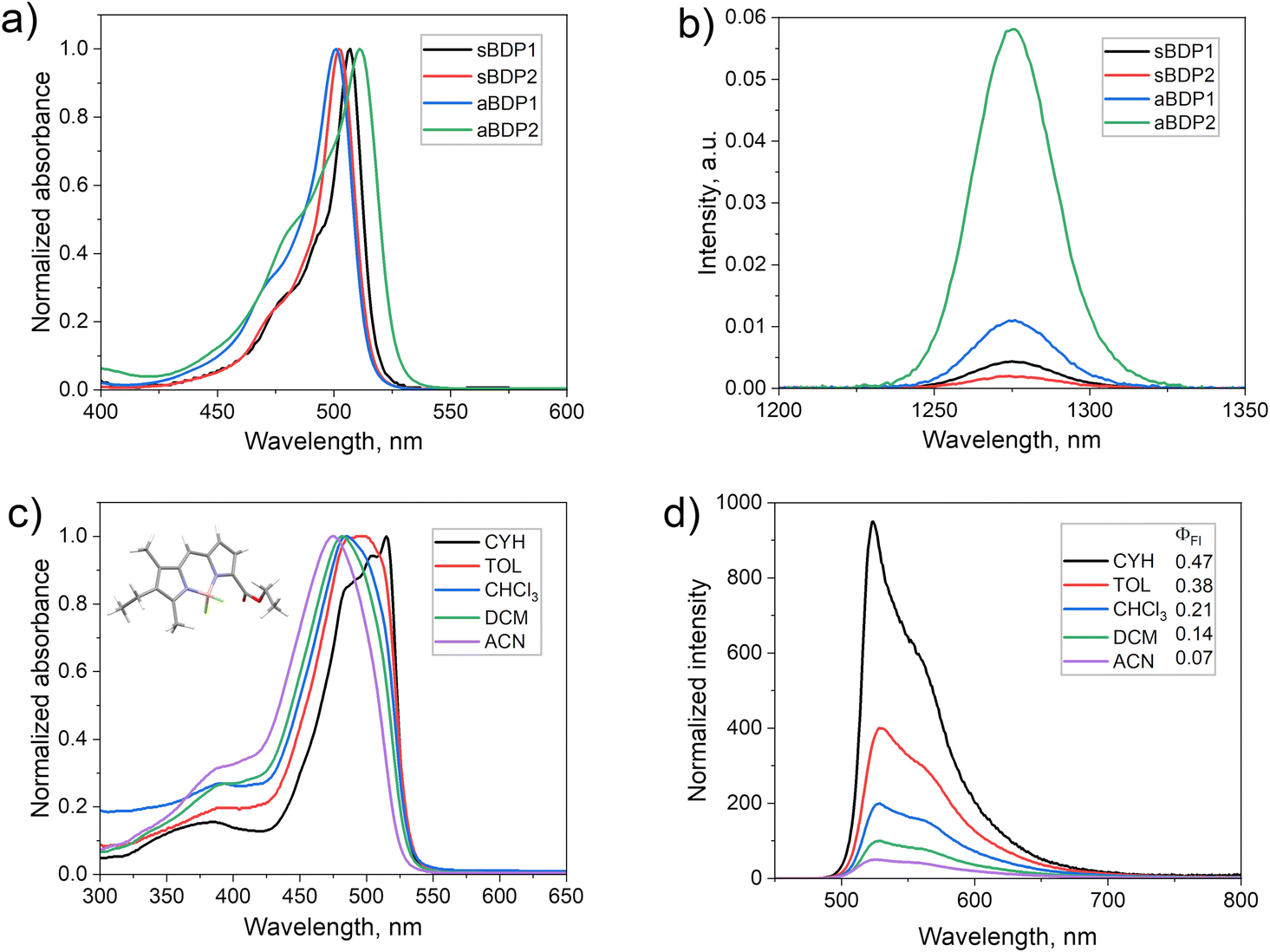 | ||
| Fig. 3 (a) UV-vis absorption of compounds sBDPs 1–2 and aBDPs 1–2 dissolved in DCM and (b) NIR phosphorescence spectra recorded for air-saturated solutions of these dyes in carbon disulfide. The samples’ concentrations were adjusted to have equal absorption at the excitation wavelength (520 nm). (c) UV-vis absorption spectra of aBDP4 in different solvents. Inset: Computed molecular geometry in the ground state. (d) Fluorescence spectra of aBDP4 in different solvents. | ||
| Compound | Solventa (εr)b | λabs (nm) | λemc (nm) | ΦFld | ΦΔe | Compound | Solvent | λabs (nm) | λem (nm) | ΦFl | ΦΔ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a ACN – acetonitrile, DCM – dichloromethane, TOL – toluene, CYH – cyclohexane.b Dielectric constant.c Fluorescence was recorded by exciting the samples at the vibrational shoulder of the BODIPY absorption (450–470 nm).d Fluorescence quantum yields were measured using Rhodamine 6G as a standard (ΦFl = 0.95 in EtOH).20e Measured using 1,9-dimethylanthracene as a singlet oxygen sensor and 2,6-diiodo-8-phenylBODIPY as a reference photosensitizer (ΦΔ = 0.85 in toluene).21 | |||||||||||
| sBDP1 | ACN(37.5) | 501 | 507 | 0.875 | 0.09 | sBDP2 | ACN(37.5) | 496 | 505 | 0.958 | 0.02 |
| DCM(8.9) | 506 | 510 | 0.911 | 0.06 | DCM(8.9) | 502 | 511 | 0.919 | 0.03 | ||
| TOL(2.4) | 509 | 517 | 0.938 | 0.06 | TOL(2.4) | 507 | 517 | 0.966 | 0.03 | ||
| CYH(2.0) | 508 | 511 | 0.945 | 0.05 | CYH(2.0) | 504 | 511 | 0.96 | 0.03 | ||
| aBDP1 | ACN(37.5) | 493 | 502 | 0.901 | 0.12 | aBDP2 | ACN(37.5) | 504 | 516 | 0.69 | 0.24 |
| DCM(8.9) | 501 | 508 | 0.939 | 0.06 | DCM(8.9) | 510 | 520 | 0.809 | 0.19 | ||
| TOL(2.4) | 504 | 513 | 0.943 | 0.06 | TOL(2.4) | 515 | 524 | 0.889 | 0.13 | ||
| CYH(2.0) | 504 | 508 | 0.944 | 0.05 | CYH(2.0) | 514 | 520 | 0.915 | 0.08 | ||
| aBDP3 | ACN(37.5) | 483 | 508 | 0.383 | 0.11 | aBDP4 | ACN(37.5) | 475 | 527 | 0.071 | 0.07 |
| DCM(8.9) | 494 | 511 | 0.431 | 0.26 | DCM(8.9) | 483 | 529 | 0.14 | 0.14 | ||
| TOL(2.4) | 502 | 516 | 0.52 | 0.23 | TOL(2.4) | 494 | 527 | 0.313 | 0.38 | ||
| CYH(2.0) | 505 | 509 | 0.695 | 0.24 | CYH(2.0) | 514 | 523 | 0.472 | 0.47 | ||
| aBDP5 | ACN(37.5) | 510 | 527 | 0.662 | 0.21 | aBDP6 | ACN(37.5) | 512 | 540 | 0.291 | 0.25 |
| DCM(8.9) | 518 | 533 | 0.683 | 0.13 | DCM(8.9) | 527 | 546 | 0.493 | 0.28 | ||
| TOL(2.4) | 525 | 535 | 0.756 | 0.06 | TOL(2.4) | 535 | 547 | 0.654 | 0.18 | ||
| CYH(2.0) | 526 | 530 | 0.854 | 0.08 | CYH(2.0) | 535 | 542 | 0.529 | 0.27 | ||
| aBDP7 | ACN(37.5) | 500 | 520 | 0.005 | 0.02 | aBDP8 | ACN(37.5) | 495 | 547 | 0.003 | 0.04 |
| DCM(8.9) | 509 | 517 | 0.01 | 0.33 | DCM(8.9) | 508 | 551 | 0.007 | 0.49 | ||
| TOL(2.4) | 515 | 525 | 0.016 | 0.76 | TOL(2.4) | 524 | 552 | 0.014 | 0.76 | ||
| CYH(2.0) | 515 | 589 | 0.033 | 0.73 | CYH(2.0) | 524 | 581 | 0.104 | 0.67 | ||
| aBDP9 | ACN(37.5) | 501 | 544 | 0.539 | 0.21 | aBDP10 | ACN(37.5) | 504 | 544 | 0.429 | 0.21 |
| DCM(8.9) | 509 | 546 | 0.625 | 0.18 | DCM(8.9) | 517 | 546 | 0.571 | 0.27 | ||
| TOL(2.4) | 501 | 517 | 0.803 | 0.09 | TOL(2.4) | 499 | 527 | 0.673 | 0.15 | ||
| CYH(2.0) | 516 | 542 | 0.797 | 0.11 | CYH(2.0) | 527 | 549 | 0.761 | 0.14 | ||
| aBDP11 | ACN(37.5) | 500 | 571 | 0.554 | 0.17 | aBDP12 | ACN(37.5) | 508 | 516 | 0.675 | 0.17 |
| DCM(8.9) | 510 | 570 | 0.56 | 0.17 | DCM(8.9) | 512 | 523 | 0.808 | 0.15 | ||
| TOL(2.4) | 524 | 571 | 0.815 | 0.17 | TOL(2.4) | 518 | 527 | 0.811 | 0.06 | ||
| CYH(2.0) | 529 | 566 | 0.734 | 0.1 | CYH(2.0) | 516 | 523 | 0.892 | 0.08 | ||
| aBDP13 | ACN(37.5) | 490 | 501 | 0.806 | 0.09 | aBDP14 | ACN(37.5) | 499 | 517 | 0.591 | 0.38 |
| DCM(8.9) | 497 | 507 | 0.846 | 0.08 | DCM(8.9) | 507 | 519 | 0.623 | 0.35 | ||
| TOL(2.4) | 501 | 509 | 0.821 | 0.07 | TOL(2.4) | 512 | 524 | 0.705 | 0.21 | ||
| CYH(2.0) | 500 | 504 | 0.882 | 0.06 | CYH(2.0) | 510 | 517 | 0.734 | 0.25 | ||
Effects of asymmetrical substitution of the BODIPY core in pyrrolic positions on its fluorescence quantum yields and lifetimes was reported by the groups of Peña-Cabrera22 and Ortiz;23 however, the impact of such structural modifications on the ISC process has not been studied to date.
To investigate the impact of molecular asymmetry on triplet state generation, we extended the aBDP series by introducing alkyl and other functional groups at various positions on the pyrrolic rings to modulate their electronic properties. This included variations in the position of the ethoxycarbonyl group and the nature of other substituents (hydrogen, alkyl, or aryl) in the pyrrole core, resulting in a series of compounds aBDP 3–11 (Fig. 2). By systematically varying these substituents, we aimed to understand how these subtle structural changes influence the triplet state generation. To explore the potential scope of the approach, an asymmetrical BODIPY containing an acetyl group (aBDP12), instead of an ethoxycarbonyl group, and alkyl-substituted derivatives aBDP13 and 14 were studied. The synthesized compounds were characterized using NMR, high resolution mass-spectroscopy and optical spectroscopic techniques. Their ability to generate triplet states in different solvents was assessed by measuring singlet oxygen generation quantum yields (ΦΔ).
Photophysical properties
The steady-state optical properties of aBDP compounds 1–14 in solvents of different polarity (Table 1) revealed a notable solvatochromic behaviour correlating with the substitution pattern on the pyrrolic rings. While the reference symmetrical compounds sBDP1 and sBDP2 exhibit sharp intense absorption bands in all the tested solvents, aBDPs exhibit hypsochromic shifts with increasing solvent polarity (Fig. 3c and Fig. S22–S25, ESI†). This contrasts with symmetrical electron donor–acceptor BODIPY dyads (Fig. 1a), whose absorption bands remain virtually unaffected by changes in solvent polarity.7 Across all solvents, absorption spectra of aBDPs display a broad S0 → S1 transition band, with fullwidth at half-maximum absorption values significantly larger than those observed for sBDPs.The effect is more pronounced for derivatives containing the 3-ethyl-2,4-dimethylpyrrole unit. For example, compound aBDP4 displays a larger blue shift (39 nm) in the absorption maximum when transitioning from cyclohexane to acetonitrile (Fig. 3c), compared to aBDP3 (22 nm). Asymmetry has a notable impact on the molar absorption coefficient, with values progressively decreasing as the substitution becomes more asymmetric. Among the series, aBDP4 exhibits the lowest molar extinction coefficient of 1.44 × 104 (Table S1, ESI†). At the same time, although the position of the emission maxima is almost unaffected by the solvent, the emission is strongly quenched as solvent polarity increases (Fig. 3d). The blue shift in the emission spectra of aBDPs with increasing solvent polarity indicates that the excited state has a lower dipole moment than that of the ground state (GS). This behaviour is similar to that observed in merocyanine dyes, where the formation of a stable zwitterionic species stabilizes the ground state, leading to blue shifts in the spectra.24
Asymmetrical introduction of alkyl substituents in one of the pyrrolic rings, leads to a decrease in fluorescence quantum yields (Table 1) and lifetimes (Table S4, ESI†). This trend is also observed for compounds lacking the ester group, such as aBDPs 13 and 14, and is in line with previous reports.22,23 The introduction of an ethoxycarbonyl group on one of the rings further enhances this effect, which can be attributed to an asymmetric charge distribution between the pyrrolic rings, reducing the aromaticity and increasing non-radiative decays.
The effect is most pronounced for compounds aBDP7 and aBDP8, which contain an anthracenyl group on the pyrrole ring and exhibit low ΦFl values even in non-polar cyclohexane (0.033 and 0.104, respectively). The emission spectra of both compounds showed a broad, red-shifted band, centered at 580–590 nm, particularly prominent in non-polar solvents (Fig. S22–S25, ESI†). This emission exhibited biexponential decay, with lifetimes of 0.41 and 3.49 ns for aBDP7, and 0.4 and 3.66 ns for aBDP8 (Table S4, ESI†). The observed behaviour suggests the presence of two distinct emissive states: a locally excited (LE) state, centered on the BODIPY chromophore, and a charge-transfer (CT) state. The CT emission and biexponential decays are indicative of intramolecular electron transfer from the anthracene group to the BODIPY core, forming the CT state. Similar CT state signatures have been observed in other BODIPY-anthracene dyads studied by us25 and others.26 The biexponential decay corresponds to the emission from both the LE and CT states. It should be noted that the CT emission is weak, and the overall fluorescence quantum yields are low (<1%, as shown in Table 1), consistent with the known poor emissivity of CT states,27 which is also strongly solvent-dependent.28 In ACN and DCM, this emission feature was not observed for aBDP7, likely due to the extremely low emissivity of the CT state in these solvents. For aBDP8, we were able to measure the CT emission lifetimes only in toluene, as in other solvents, the decay components could not be reliably resolved due to low intensity.
BODIPY triplet states are known to be poorly emissive at room temperature,29 therefore we attempted to detect phosphorescence of aBDPs at 77 K in a frozen glass matrix (toluene - THF, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v). For the reference 2,6-diiodo-8-phenylBODIPY (PhBDPI2), the emission decays (Fig. S26, ESI†) confirmed the phosphorescent nature of the emission, displaying a monoexponential decay profile with a calculated lifetime of 1.48 ms, consistent with the value reported by Dick et al.30 However, attempts to detect phosphorescence for aBDPs were unsuccessful (Fig. S27, ESI†), indicating that their triplet states are subject to rapid non-radiative decay under these conditions.
:3 v/v). For the reference 2,6-diiodo-8-phenylBODIPY (PhBDPI2), the emission decays (Fig. S26, ESI†) confirmed the phosphorescent nature of the emission, displaying a monoexponential decay profile with a calculated lifetime of 1.48 ms, consistent with the value reported by Dick et al.30 However, attempts to detect phosphorescence for aBDPs were unsuccessful (Fig. S27, ESI†), indicating that their triplet states are subject to rapid non-radiative decay under these conditions.
To assess the efficiency of triplet state formation across different solvents, singlet oxygen quantum yields were determined using a combination of two complementary methods: (1) chemical trapping of singlet oxygen by 9,10-dimethylanthracene probe31 in various solvents, and (2) direct detection of singlet oxygen phosphorescence at 1276 nm in carbon disulfide (CS2). CS2 was selected as the medium for 1O2 phosphorescence measurements due to its previously demonstrated ability to provide excellent resolution, attributed to the long lifetimes of singlet oxygen in this solvent.32 Notably, the dielectric constant of CS2 (2.6) closely matches that of toluene (2.4), allowing for meaningful comparison of ΦΔ values obtained by both methods and thereby ensuring consistency in the results for both experimental approaches.
The symmetrical reference compound, sBPD1 and aBDP2, exhibited low ΦΔ values in the range of 5–9% across all solvents tested, which aligns with their high fluorescence quantum yields. The aBDP derivatives exhibited moderate variation in ΦΔ values depending on the solvent, with the highest values observed in non-polar solvents, indicating their ability to facilitate efficient triplet state formation. For instance, aBDP4 displayed a ΦΔ value of 47% in cyclohexane. Compounds aBDP7 and aBDP8, which incorporate an anthracene subunit, showed the most efficient triplet state formation, with ΦΔ values reaching as high as 76% in toluene. This is significantly higher compared to previously reported BODIPY–anthracene compounds shown in Table S2 (ESI†) for comparison. For example, Zhao et al. reported a structurally similar BODIPY compound with an anthracenyl group at position 2, which yielded a maximum singlet oxygen generation of only 24% in DCM.33 The ΦΔ values measured in toluene using the chemical trapping method were consistent with those measured in CS2 using singlet oxygen phosphorescence detection (Table S3, ESI†), further confirming the accuracy of the results.
A possible explanation for the higher ΦΔ values observed for aBDPs 7 and 8, compared to other aBDPs, is the involvement of multiple ISC pathways, leading to triplet states localized either on the BODIPY or anthracene subunits. The co-existence of multiple ISC pathways have been reported for other BODIPY donor–acceptor dyads.34 This explanation is consistent with the results obtained for structurally similar compounds aBDPs 9–11, containing a phenyl group in place of the anthracene group. The highest ΦΔ value, obtained for aBDP10 in DCM (27%), is more than two-fold lower than for the anthracene-containing structure. Thus, it seems likely that (1) SOCT-ISC occurs in aBDPs 7 and 8 due to the feasibility of electron transfer from the anthracene to the BODIPY subunit and (2) another ISC pathway is occurring in parallel in these molecules.
For aBDP7 and aBDP8, ΦΔ values decreased in more polar solvents, which has been observed for other donor–acceptor dyads.35 In highly polar solvents, the overstabilization of the CT state may leads to reduced triplet yields due the enhancement of the ground state recombination (1CT → S0) rate.36 Other studied compounds, particularly aBDPs 12–14 showed higher ΦΔ values in polar solvents. For example, aBDP14 showed ΦΔ of 38% in DCM, further demonstrating the influence of solvent on the triplet states formation. Overall, these results illustrate the complex interplay between molecular structure, solvent environment and triplet state formation efficiency. To shed light on the effect of asymmetrical substitution on ISC, quantum chemical calculations have been performed.
Quantum chemistry calculations
Structurally related pairs of compounds were chosen for computational investigations and analyzed in comparison: (1) derivatives aBDP3 and aBDP4, which represent the simplest structures across the series containing ethoxycarbonyl group yet showing efficient ISC as indicated by their high ΦΔ yields; (2) alkyl-substituted derivatives aBDP13 and aBDP14, which lack ethoxycarbonyl group but still exhibit triplet state formation; (3) compounds aBDP7 and aBDP8, to evaluate the effect of substitution with anthracene; and (4) sBDP1 and sBDP2 as they are the two symmetrical reference structures. Finally, aBDP1 was also selected for the computational study to assess the effect of substituting the ethoxycarbonyl group at different positions. Details on the computational protocol are provided in the ESI.†All dyes possess predominantly rigid structures, but for some of them there is certain degree of conformational flexibility, where the ester group can adopt different configurations. For instance, in Fig. S32 (ESI†) it is shown a rigid scan of the dihedral angle between the ester group and the BODIPY molecular plane for aBDP3 and aBDP4, revealing minima at an orthogonal orientation relative to the molecule, while a completely linear conformation leads to high-energies due to significant steric hindrance. This near-perpendicular arrangement, consistent across all examined electronic states, may limit the charge transfer (CT) character, reducing the electron-accepting capacity of the ester and the overall asymmetry. In contrast, structures lacking flexible substituents maintain similar geometries across all the studied electronic states, with the exception of aBDP7 and aBDP8, which exhibits at the S1 state a decrease of the dihedral angle between the BODIPY backbone and the anthracene moiety.
Vertical excitation and emission energies, along with oscillator strengths computed at the SCS-ADC(2) level, are presented in Table S14 (ESI†). For all dyes considered, small Stokes shifts were calculated, indicating minimal geometrical reorganization between S0 and S1, in agreement with the experimental features. A qualitative agreement is found between the experimental and theoretical transition energies. All compounds show S1 vertical excitation energies between 2.67 and 2.72 eV, with vertical emission energies ranging between 2.42 eV (aBDP7 and aBDP8) and 2.64 eV (aBDP12). Large oscillator strengths (ca. 0.5 at the S1 optimized geometries) were calculated, consistent with the strong emissive properties of these dyes in non-polar solvents. The differences of emissive properties, albeit small, are likely linked in the case of aBDP7 and 8 to their longer π-conjugated backbones which expand up to the anthracene moiety. For the other systems, the variations in both energies and oscillator strengths are small and may be linked to the slight differences of geometries brought on by the different moieties, or the small addition of polarization brought on by one of the side groups (for instance when comparing aBDP13 and aBDP14).
The computed vibrationally resolved absorption and emission spectra, accounting for vibronic coupling between the ground and excited singlet states, are presented in Fig. S33 (ESI†). The spectra were calculated for aBDPs 3, 4, 13 and 14 as representative compounds across the series. The vibrational profiles of the computed spectra closely match the experimental ones for the investigated molecules. The influence of donor and acceptor groups on the BODIPY core is evident: aBDP13, which contains an unsubstituted pyrrole unit, shows the most structured spectra, while the presence of electron-accepting ester or electron-donating ethyl groups leads to broader spectra, indicating increased vibrational freedom. Fluorescence rate constants (kr) were calculated by integrating the emission spectra and are shown in Table S15 (ESI†) for the four dyes. In all cases, the computed rate constants closely align with the experimental values, validating the accuracy of the vibronic calculations.
Next, we discuss the character of the involved excited states. In Table 2 are presented the results of the charge transfer (CT) analysis conducted following the methodology established by Le Bahers et al.37 Notably, for aBDPs 1, 3, 4, 12 and 13, a decrease in both the DCT (charge transfer distance) and the permanent static dipole moment (μ) is observed when moving from the ground state (GS) situation to S1. Thus, in these molecules, their GS is more polar than their emissive state, which aligns with the experimental observations of negative solvatochromism. This solvatochromic effect is particularly pronounced for aBDP3 and aBDP4, for which the larger differences between GS and S1 dipole moments are calculated. The two anthracene substituted dyes, aBDP7 and aBDP8, show experimentally only a very small hypsochromic shift. This is correlated with the small differences in the computed μ is values between the GS and S1, especially for aBDP7. Moreover, a notable hybrid localized charge-transfer (HLCT) character can be seen for those two derivatives, as observed on Fig. 4.38
| GS | S1 | T1 | T2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| QCT | DCT | μ | QCT | DCT | μ | QCT | DCT | μ | QCT | DCT | μ | |
| sBDP1 | 0.51 | 0.6 | 4.23 | 0.5 | 0.58 | 4.31 | 0.43 | 0.56 | 4.08 | 0.44 | 0.74 | 6.65 |
| sBDP2 | 0.52 | 0.82 | 0.61 | 0.52 | 0.59 | 0.7 | 0.42 | 0.66 | 1.27 | 0.44 | 0.84 | 7.8 |
| aBDP1 | 0.52 | 1.01 | 2 | 0.51 | 0.78 | 2.19 | 0.42 | 0.61 | 2.44 | 0.63 | 2.47 | 7.89 |
| aBDP3 | 0.55 | 1.46 | 9.6 | 0.52 | 0.93 | 7.98 | 0.43 | 0.53 | 8.45 | 0.62 | 2.3 | 12.4 |
| aBDP4 | 0.55 | 1.27 | 10.07 | 0.54 | 0.82 | 8.4 | 0.44 | 0.51 | 8.78 | 0.6 | 2.48 | 13.96 |
| aBDP7 | 0.53 | 1.35 | 6.55 | 0.59 | 3.29 | 6.44 | 0.42 | 0.39 | 6.51 | 0.46 | 0.92 | 6.5 |
| aBDP8 | 0.53 | 1.32 | 6.81 | 0.56 | 2.11 | 6.44 | 0.43 | 0.37 | 6.5 | 0.28 | 0.03 | 6.5 |
| aBDP13 | 0.53 | 1.1 | 5.5 | 0.53 | 0.85 | 4.76 | 0.43 | 0.54 | 4.6 | 0.62 | 2.34 | 7.95 |
| aBDP14 | 0.54 | 0.97 | 5.86 | 0.53 | 0.7 | 5.01 | 0.44 | 0.52 | 4.84 | 0.6 | 2.55 | 9.58 |
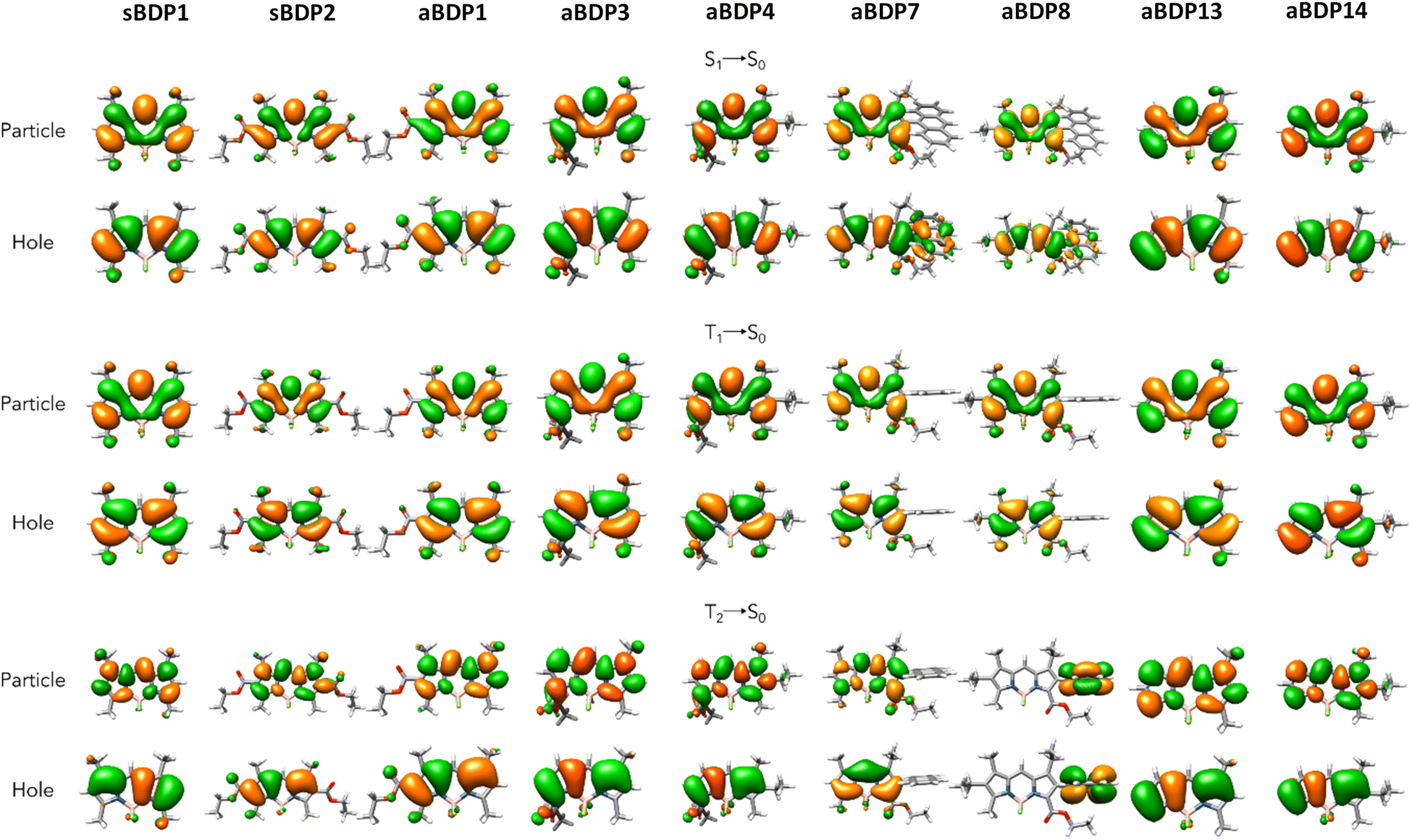 | ||
| Fig. 4 Comparison of NTOs for sBDPs 1 and 2, aBDPs 1, 3, 4, 7, 8, 13 and 14 at the S1, T1 and T2 excited states (isovalue is equal to 0.02 e− Å−3). | ||
Nevertheless, all the asymmetric structures discussed here demonstrate a notable evolution in their charge transfer capacities when comparing their ground and first singlet excited states. Given the solvatochromic effect, which is observed experimentally and is present at various degrees for all of the derivatives, these calculations demonstrate that the solvation effects are indeed caused by their asymmetric nature. In contrast, the symmetrical reference compounds do not present such effects at all.
Regarding the triplet states, the second triplet state (T2) exhibits generally stronger CT character than the other states. This is especially evident from the DCT values for all compounds and is calculated at different degrees. The difference in transition character is clearly reflected in the orbitals involved in the transitions, as shown in Fig. 4 through natural transition orbitals (NTOs). All states exhibit a π → π* character, consistent with the non-zero oscillator strengths and the large calculated radiative rate constants. The discrepancy between aBDP7 and aBDP8 stands out, with the localization of the exciton being on the BODIPY core for the former, and on the anthracene for the latter. The first triplet state, however, do not present such a strong CT character, as well as the two symmetric derivatives sBDP1 and sBDP 2 which present an expected less marked CT behaviour.
Intersystem crossing mechanisms
Table 3 presents the calculated ΔEST values along with the corresponding spin–orbit coupling matrix elements (SOCMEs), to evaluate the feasibility of ISC between S1 and the first two excited triplet states (T1 and T2). According to Fermi-Golden's rule, the ΔEST values are the main factor determining the speed of the ISC process. The magnitude of the SOCMEs also play an important role in accelerating the ISC processes. For ISC between the S1 and T1 states, all compounds show large ΔEST values, ranging between 0.67 and 0.84 eV. While these values are accompanied by non-zero SOCMEs, with the notable exception of sBDP1, the relatively large splitting may hinder efficient ISC between these states. In contrast, the ΔEST values between S1 and T2 are significantly smaller, approaching near-degeneracy in several compounds, and with SOCMEs ranging between 0.21 cm−1 (aBDP7) and 1.10 cm−1 (aBDP8). aBDPs 3, 4, 8 and 14 show the smallest ΔES1–T2 values. We conclude that the S1 → T2 ISC process should be the main ISC process for these compounds. As mentioned above, T2 possesses a more pronounced charge transfer (CT) character in many of these compounds. In contrast, and apart from the T2 of aBDP8, S1 displays a more localized excitation character, thus leading to non-zero SOCs between the two states and thereto to enhanced ISC processes. This is in accordance with the “El-Sayed rule”39–41 and/or the SOCT-ISC mechanisms,5 but a particularly enhanced ISC process is attained in this case because of the relatively small ΔEST value between S1 and T2. We note that slightly thermodynamically uphill processes are calculated in some cases, but in view of the expected accuracy of the SCS-ADC(2)/aug-cc-pVTZ calculations, we believe the two states are isoenergetic for compounds aBDP3, 4, 8, 13 and 14.| ΔES1–T1 | ΔES1–T2 | SOCMEs S1 – T1 | SOCMEs S1 – T2 | |
|---|---|---|---|---|
| sBDP1 | 0.84 | −0.25 | 0 | 0.24 |
| sBDP2 | 0.72 | −0.34 | 0.04 | 0.51 |
| aBDP1 | 0.79 | −0.2 | 0.04 | 0.35 |
| aBDP3 | 0.8 | −0.09 | 0.7 | 0.56 |
| aBDP4 | 0.77 | 0.03 | 0.12 | 0.46 |
| aBDP7 | 0.7 | 0.22 | 0.09 | 0.21 |
| aBDP8 | 0.7 | −0.08 | 0.09 | 1.1 |
| aBDP13 | 0.67 | −0.12 | 0.29 | 0.46 |
| aBDP14 | 0.79 | −0.01 | 0.04 | 0.37 |
We now examine the trends observed between the experimental singlet oxygen quantum yields and the computational results. It is important to note that these trends are qualitative, as singlet oxygen quantum yield values are determined not only by the triplet formation quantum yields but also by other factors beyond ISC efficiency. These include non-radiative relaxation from both singlet and triplet states, as well as bimolecular triplet–triplet annihilation processes involving triplet oxygen.32 Nevertheless, useful insights can be drawn by analyzing the expected ISC efficiency based on the data in Table 3.
The very low ΦΔ values of approximately 5% measured for the symmetrical sBDP1 and sBDP2 are consistent with the thermodynamically uphill S1 → T2 ISC process (T2 being 0.25 eV above S1, see Table 3). The S1 → T1 ISC process is similarly disfavoured in these compounds due to negligible SOCME values and a very large ΔES1–T1 value. aBDP1, which exhibits similar computed magnitudes to sBDP1, also shows a similar low ΦΔ value of approximately 5%. In contrast, compounds with nearly degenerate S1 and T2 levels (i.e., ΔES1–T2 values of ca. ± 0.1 eV, such as aBDP3, aBDP4, and aBDP14) should enhance the S1 → T2 ISC process. As a result, intermediate ΦΔ values ranging from 21% to 47% are observed for these compounds in apolar solvents. Finally, aBDP7 and aBDP8 emerge as the most efficient compounds for generating singlet oxygen (Table 1).
In the case of aBDP7, this correlates with a thermodynamically downhill S1 → T2 ISC process across the series (T2 being 0.22 eV below S1, which likely falls in the optimal Marcus region, see Table 3). However, aBDP8 behaves as an outlier. Given its computed magnitudes, one would expect a similar behaviour to that of, for example, aBDP3, aBDP4, and aBDP14, but a similar ΦΔ value to that of aBDP7 is obtained. The different character of T2, i.e., enhanced CT character and correspondingly larger SOCME values (see Table 3), along with the feasibility of another ISC pathway (such as SOCT-ISC), are likely behind the increased ISC efficacy in aBDP8.
Holographic structures recording
Triplet excited states formation via SOCT-ISC is highly dependent on solvent polarity, with higher yields typically observed in polar solvents.42 This limits its use in photocatalysis and photopolymerization, where the polarity of the media is dictated by the substrate or monomer. The aBDPs studied demonstrated efficient triplet formation in non-polar solvents, which is advantageous for photopolymerization, as monomers and their formulations are generally non-polar.43 Additionally, the overall polarity decreases during the polymerization process as polar carbon–carbon double bonds are converted into less polar single bonds.44 To assess their potential as photosensitizers, we used aBDP dyes to fabricate diffractive structures in photopolymer layers via holographic patterning. Such materials are crucial for technologies requiring precise light control, such as solar energy harvesting,45,46 sensing,47 LED lighting,48 and augmented reality displays.49A photopolymer based on cellulose acetate–polyethylene glycol (CA–PEG) matrix, with acrylamide as a monomer and N,N′-methylenebisacrylamide as a cross-linker was used in this study. CA–PEG offers low cost, easy synthesis, mechanical stability, and compatibility with BODIPY dyes.18 It is also self-developing, eliminating the need for post-recording treatment, and ensures hologram stability in humid environments.50
aBDP dyes were employed in a two-component photoinitiating system containing N-phenylglycine (NPG) as a co-initiator. NPG is known react with triplet excited states of dyes, forming a radical cation (NPG˙+) which undergoes deprotonation/decarboxylation to give an anilinomethyl radical (PhNHCH2˙) that initiates polymerization of methacrylate and acrylamide monomers.51
The performance of the aBDP-NPG photoinitiating system for photopolymerization of acrylamide in the cellulose-PEG matrix was evaluated by recording volume transmission gratings with a spatial frequency of 800 lines per mm. Holographic recording was carried out in photopolymer layers sensitized with aBDPs 3, 4, 7, and 8, which were selected based on their high singlet oxygen generation quantum yields, the highest in the investigated series of compounds. The recording was performed using the setup shown in Fig. S35 (ESI†). Details of the recording conditions are provided in Section 10.1 of the ESI.† A transmission grating forms when two collimated laser beams overlap in the photosensitive layer (Fig. 5b), creating an interference pattern that results in refractive index modulation. Photopolymerization occurs in the illuminated regions, and its kinetics are monitored through changes in diffraction efficiency, measured as the fraction of light diffracted by the grating (Fig. 5b). The evolution of diffraction efficiency during grating formation is shown in Fig. 5c and d.
 | ||
| Fig. 5 Formation of the transmission grating under holographic exposure and its characterization. (a) Photograph of the photopolymer layer with recorded gratings (dashed line) containing aBDP8. (b) Holographic recording using two collimated beams forming the transmission grating and refractive index variation. Green curve marks bright fringes, and lines denote the areas illuminated with maximum light intensity. (c) Real-time growth curves of diffraction efficiency in CA–PEG layers with aBDP4 (130 μm), aBDP7 (140 μm), and aBDP8 (164 μm). (d) Evolution of refractive index modulation (RIM) during grating recording. (e) Bragg selectivity curves of recorded transmission gratings. Inset: Photograph of the diffraction pattern under white light illumination. | ||
The composition containing aBDP8 exhibited the highest photosensitivity (1.15 J−1 cm2), reaching a diffraction efficiency of 71% with an exposure energy of about 2.3 J cm−2. Photosensitivity was estimated from the linear region of the growth curves (Section 10.4, ESI†). In comparison, aBDP7 achieved 32% diffraction efficiency, while aBDP4 reached 62% after exposure to 7.5 J cm−2 and 33 J cm−2, respectively. Attempts to record transmission gratings with aBDP3-sensitized photopolymer were unsuccessful, as no diffracted beam was observed after exposure to intensities of 50–100 mW cm−2 for 70 seconds. This indicates that aBDP3 is inactive in initiating photopolymerization in this composition, likely due to its insufficient triplet state yield.
The angular selectivity curves show a sinusoidal shape with symmetrical side lobes, indicating uniform refractive index modulation across the grating thickness (Fig. 5e). Refractive index modulation, induced by photopolymerization, was calculated using eqn (S6) (ESI†), yielding values of 0.86 × 10−3, 1.22 × 10−3, and 1.39 × 10−3 for aBDP7 (140 μm), aBDP8 (164 μm), and aBDP4 (130 μm), respectively. The modulation achieved in aBDP7 and aBDP8 is higher than the 1.1 × 10−3 obtained in CA–PEG layers sensitized with Erythrosine B (100 μm), as reported in our previous work.52 These results confirm that aBDP dyes are effective as photosensitizers in holographic recording materials.
The composition based on aBDP8, identified as having the highest exposure sensitivity, was selected for further investigation by recording an off-axis holographic lens. This lens was recorded by employing interference of a diverging laser beam with a collimated laser beam (Fig. 6a) using a transmission geometry setup (Fig. S36, ESI†). According to the modelling results (Section 10.5 of the ESI†), the off-axis holographic lens forms a complex diffractive structure within the volume of the photosensitive layer (Fig. 6b), characterized by a spatial frequency range from 215 to 590 lines per mm (Fig. S38a, ESI†) and a slant angle variation from 6.0° to 2.2° (Fig. S38b, ESI†) across the element.
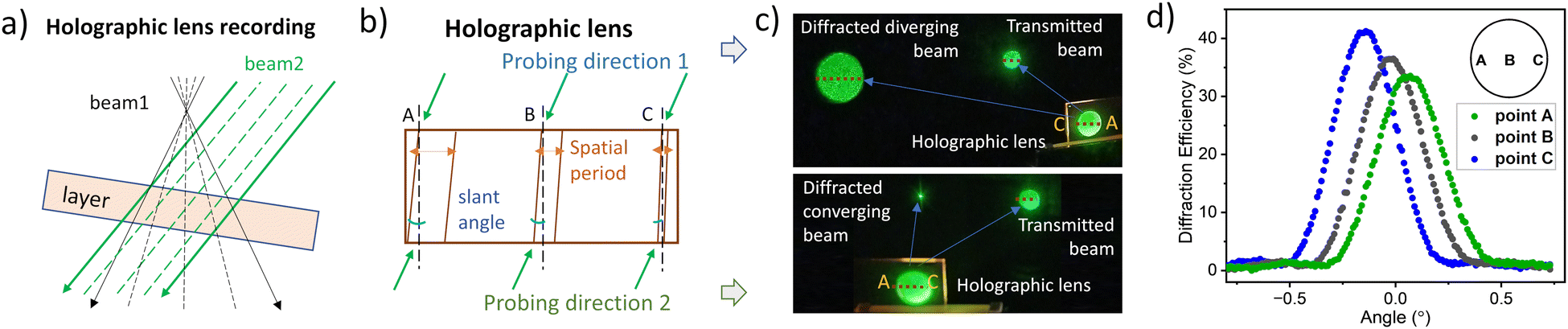 | ||
| Fig. 6 (a) Recording of the holographic lens by overlapping a divergent laser beam (beam1) and a collimated laser beam (beam2). (b) Schematic representation of the spatial period and the slant angle across the holographic lens. (c) Photographs of the diffraction produced by the holographic lens when probing from the same direction as recording (probing direction 1) and the opposite one (probing direction 2), respectively. (d) Diffraction efficiency along the horizontal diameter. | ||
The intricate fringe pattern enables the holographic lens to diverge or converge light, depending on the probing direction. Fig. 6c shows the lens performance under illumination with a collimated beam of a 532 nm wavelength. When probed from the recording direction (original reference beam), it produces a diverging beam. In contrast, a focused beam is achieved when probed from the opposite direction (conjugate reference beam).
To ensure sustainable performance, uniform diffraction efficiency across the holographic lens is crucial. The uniformity was assessed by probing along the horizontal diameter. Fig. 6d shows the Bragg angular selectivity curves measured along the horizontal diameter (points A, B, and C). A diffraction efficiency of 38% was achieved at the centre, with a variation of ±3% across the horizontal diameter.
Each section of the holographic lens, comparable in size to the probing beam, acts as a local grating. This makes the lens a collection of local holographic gratings with varying spatial frequencies and slant angles, resulting in in both redirection of the light beam and its shaping. Such light-manipulating technology can be used in applications as smart, sustainable and energy-saving LED lighting (to get light efficiency to target area, reducing waste and light pollution),47 solar energy collection (re-direction and concentration to solar cells)45 and a wide range of other photonics applications.
Conclusions
Symmetrical BODIPY compounds are widely used in the design of functional dyes for optical applications, thanks to their simple synthesis and favourable photophysical properties. In this work, we demonstrate that asymmetrical substitution in the BODIPY core opens new avenues for enhancing triplet state formation by reducing the energy gap between the S1 and T2 states. This approach eliminates the need for heavy atoms or bulky aromatic groups to promote intersystem crossing (ISC), enabling efficient ISC even in compact BODIPY molecules, such as aBDP3 and aBDP4, which feature different substitution patterns on the pyrrolic rings. We propose that this asymmetrical substitution strategy can be combined with other ISC enhancement methods, such as SOCT-ISC, to create photosensitizers with multiple ISC pathways contributing to triplet state formation, leading to improved overall performance.The practical potential of aBDP dyes for free-radical polymerization in a cellulose–PEG matrix was demonstrated through the holographic recording of high-efficiency transmission gratings. Specifically, aBDP8-sensitized CA–PEG has shown promise in developing holographic optical elements with complex diffractive structures. An off-axis holographic lens was successfully recorded, achieving a diffraction efficiency of up to 38%, with uniformity within ±3%, highlighting its potential for advanced light manipulation applications.
These findings not only introduce a new direction in the design of heavy-atom-free BODIPY photosensitizers but also have broad implications for a wide range of applications that rely on the generation of triplet states and/or fluorescence. By precisely modulating the ISC crossing through asymmetrical substitution, this approach facilitates the design of dyes with a well-defined ratio between fluorescence and triplet state formation. Such dyes are highly sought after for applications like triplet–triplet annihilation, where they could pave the way for novel upconversion systems with enhanced performance and functionality. Moreover, these dyes hold significant promise for bioimaging and theranostics, where a delicate balance between emissivity and ISC generation is critical. We believe that this strategy for ISC enhancement can be applied to a wide range of chromophores, making it a promising area for future investigation.
Data availability
The data supporting this article have been included as a part the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. A. F. acknowledges Research Ireland award 21/FFP-A/9214 (DyeSICPhoto) for support of this work. T. M. acknowledges Research Ireland award 22/PATH-S/10837 (PolyGlass4AR). M. A. F. and A. S. acknowledge the TU Dublin Research Scholarship programme.Notes and references
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- C. Imberti, P. Zhang, H. Huang and P. J. Sadler, Angew. Chem., Int. Ed., 2020, 59, 61–73 CrossRef CAS PubMed.
- C. M. Marian, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2012, 2, 187–203 CAS.
- A. Turksoy, D. Yildiz and E. U. Akkaya, Coord. Chem. Rev., 2019, 379, 47–64 CrossRef CAS.
- (a) J. W. Verhoeven, J. Photochem. Photobiol., C, 2006, 7, 40–60 CrossRef CAS; (b) Z. E. Dance, S. M. Mickley, T. M. Wilson, A. B. Ricks, A. M. Scott, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 4194–4201 CrossRef CAS PubMed; (c) J. W. Verhoeven, H. J. van Ramesdonk, M. M. Groeneveld, A. C. Benniston and A. Harriman, ChemPhysChem, 2005, 6, 2251–2260 CrossRef CAS PubMed.
- Y. Hou, X. Zhang, K. Chen, D. Liu, Z. Wang, Q. Liu, J. Zhao and A. Barbon, J. Mater. Chem. C, 2019, 7, 12048–12074 RSC.
- M. A. Filatov, Org. Biomol. Chem., 2020, 18, 10–27 RSC.
- (a) P. Spenst, R. M. Young, M. R. Wasielewski and F. Würthner, Chem. Sci., 2016, 7, 5428 RSC; (b) N. Epelde-Elezcano, E. Palao, H. Manzano, A. Prieto-Castañeda, A. R. Agarrabeitia, A. Tabero, A. Villanueva, S. de la Moya, Í. López-Arbeloa, V. Martínez-Martínez and M. J. Ortiz, Chem. – Eur. J., 2017, 23, 4837 CrossRef CAS PubMed; (c) E. Bassan, A. Gualandi, P. G. Cozzi and P. Ceroni, Chem. Sci., 2021, 12, 6607 RSC.
- H. Liang, X. Zhang, M. Lu, X. Chen, W. Li, S. Li, M.-D. Li, J. Zhao, Y. Huo and S. Ji, Angew. Chem., Int. Ed., 2024, 63, e202402774 CrossRef CAS PubMed.
- S. Xu, Y. Yuan, X. Cai, C.-J. Zhang, F. Hu, J. Liang, G. Zhang, D. Zhang and B. Liu, Chem. Sci., 2015, 6, 5824 RSC.
- C. M. Marian, Annu. Rev. Phys. Chem., 2021, 72, 617 CrossRef CAS PubMed.
- V.-N. Nguyen, S. Qi, S. Kim, N. Kwon, G. Kim, Y. Yim, S. Park and J. Yoon, J. Am. Chem. Soc., 2019, 141, 16243–16248 CrossRef CAS PubMed.
- (a) W. Wu, D. Mao, F. Hu, S. Xu, C. Chen, C.-J. Zhang, X. Cheng, Y. Yuan, D. Ding, D. Kong and B. Liu, Adv. Mater., 2017, 29, 1700548 CrossRef PubMed; (b) D. Wang, M. M. S. Lee, G. Shan, R. T. K. Kwok, J. W. Y. Lam, H. Su, Y. Cai and B. Z. Tang, Adv. Mater., 2018, 30, 1802105 CrossRef PubMed.
- C. Schäfer, R. Ringström, J. Hanrieder, M. Rahm, B. Albinsson and K. Börjesson, Nat. Commun., 2024, 15, 8705 CrossRef PubMed.
- A. Pershin, D. Hall, V. Lemaur, J.-C. Sancho-Garcia, L. Muccioli, E. Zysman-Colman, D. Beljonne and Y. Olivier, Nat. Commun., 2019, 10, 597 CrossRef CAS PubMed.
- N. Boens, B. Verbelen, M. J. Ortiz, L. Jiao and W. Dehaen, Coord. Chem. Rev., 2019, 399, 213024 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- A. Sheehan, T. Mikulchyk, C. S. P. De Castro, S. Karuthedath, W. Althobaiti, M. Dvoracek, Sabad-e-Gul, H. J. Byrne, F. Laquai, I. Naydenova and M. A. Filatov, J. Mater. Chem. C, 2023, 11, 15084–15096 RSC.
- A. Sheehan, I. A. Okkelman, G. Groslambert, C. Bucher, R. I. Dmitriev and M. A. Filatov, Chem. – Eur. J., 2025, e202404188 CrossRef CAS PubMed.
- R. F. Kubin and A. N. Fletcher, J. Lumin., 1982, 27, 455–462 CrossRef.
- Y. Zhao, R. Duan, J. Zhao and C. Li, Chem. Commun., 2018, 54, 12329–12332 RSC.
- A. de J. Gómez-Infante, J. Bañuelos, I. Valois-Escamilla, D. Cruz-Cruz, R. Prieto-Montero, I. López-Arbeloa, T. Arbeloa and E. Peña-Cabrera, Eur. J. Org. Chem., 2016, 5009–5023 CrossRef.
- J. Bañuelos-Prieto, A. R. Agarrabeitia, I. Garcia-Moreno, I. Lopez-Arbeloa, A. Costela, L. Infantes, M. E. Perez-Ojeda, M. Palacios-Cuesta and M. J. Ortiz, Chem. – Eur. J., 2010, 16, 14094–14105 CrossRef PubMed.
- J. O. Morley, R. M. Morley, R. Docherty and M. H. Charlton, J. Am. Chem. Soc., 1997, 119, 10192–10202 CrossRef CAS.
- (a) M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, M. Telitchko, T. Wiesner, F. Laquai and M. O. Senge, Phys. Chem. Chem. Phys., 2018, 20, 8016–8031 RSC; (b) N. Kiseleva, M. A. Filatov, M. Oldenburg, D. Busko, M. Jakoby, I. A. Howard, B. S. Richards, M. O. Senge, S. M. Borisov and A. Turshatov, Chem. Commun., 2018, 54, 1607–1610 RSC.
- (a) Z. Wang, A. A. Sukhanov, A. Toffoletti, F. Sadiq, J. Zhao, A. Barbon, V. K. Voronkova and B. Dick, J. Phys. Chem. C, 2019, 123, 265–274 CrossRef CAS; (b) K. Chen, W. Yang, Z. Wang, A. Iagatti, L. Bussotti, P. Foggi, W. Ji, J. Zhao and M. Di Donato, J. Phys. Chem. A, 2017, 121, 7550–7564 CrossRef CAS PubMed.
- C. Chen and C. Fang, Chemosensors, 2023, 11, 87 CrossRef CAS.
- N. Kiseleva, D. Busko, B. S. Richards, M. A. Filatov and A. Turshatov, J. Phys. Chem. Lett., 2020, 11, 6560–6566 CrossRef CAS PubMed.
- X.-F. Zhang, X. Yang, K. Niu and H. Geng, J. Photochem. Photobiol., A, 2014, 285, 16–20 CrossRef CAS.
- Z. Wang, A. Toffoletti, Y. Hou, J. Zhao, A. Barbon and B. Dick, Chem. Sci., 2021, 12, 2829 RSC.
- S. Mondal, R. B. Jethwa, B. Pant, R. Hauschild and S. A. Freunberger, Faraday Discuss., 2024, 248, 175–189 RSC.
- M. Bregnhøj, F. Thorning and P. R. Ogilby, Chem. Rev., 2024, 124, 9949–10051 CrossRef PubMed.
- Z. Wang and J. Zhao, Org. Lett., 2017, 19, 4492–4495 CrossRef CAS PubMed.
- Y. E. Kandrashkin, Z. Wang, A. A. Sukhanov, Y. Hou, X. Zhang, Y. Liu, V. K. Voronkova and J. Zhao, J. Phys. Chem. Lett., 2019, 10, 4157–4163 CrossRef CAS PubMed.
- N. Kiseleva, M. A. Filatov, J. C. Fischer, M. Kaiser, M. Jakoby, D. Busko, I. A. Howard, B. S. Richards and A. Turshatov, Phys. Chem. Chem. Phys., 2022, 24, 3568 RSC.
- J. T. Buck, A. M. Boudreau, A. DeCarmine, R. W. Wilson, J. Hampsey and T. Mani, Chem, 2018, 5, 1 Search PubMed.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS PubMed.
- J. Li, M. Zhang, T. Li, D. Guo, T. Tian and H. Zhang, J. Mater. Chem. C, 2022, 10, 13124–13136 RSC.
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- K. Stavrou, L. G. Franca, T. Böhmer, L. M. Duben, C. M. Marian and A. P. Monkman, Adv. Funct. Mater., 2023, 2300910 CrossRef CAS.
- (a) A. A. Buglak, A. Charisiadis, A. Sheehan, C. J. Kingsbury, M. O. Senge and M. A. Filatov, Chem. – Eur. J., 2021, 27, 9934 CrossRef CAS PubMed; (b) P. P. Chebotaev, A. A. Buglak, A. Sheehan and M. A. Filatov, Phys. Chem. Chem. Phys., 2024, 26, 25131 RSC.
- A. Uddin, S. R. Allen, A. K. Rylski, C. J. O’Dea, J. T. Ly, T. A. Grusenmeyer, S. T. Roberts and Z. A. Page, Angew. Chem., Int. Ed., 2023, 62, e202219140 CrossRef CAS PubMed.
- M. Topa, F. Petko, M. Galek, K. Machowski, M. Pilch, P. Szymaszek and J. Ortyl, Polymers, 2019, 11, 1756 CrossRef CAS PubMed.
- E. Alfaro, T. Lloret, J. M. Vilardy, M. Bastidas, M. Morales-Vidal and I. Pascual, Polymers, 2024, 16, 732 CrossRef CAS PubMed.
- N. Jiang, S. Davies, Y. Jiao, J. Blyth, H. Butt, Y. Montelongo and A. K. Yetisen, ACS Sens., 2021, 6, 915–924 CrossRef CAS PubMed.
- M. Murray, I. Naydenova and S. Martin, Opt. Mater. Express, 2023, 13, 3481–3501 CrossRef CAS.
- C. Bigler, P. Blanche and K. Sarma, Appl. Opt., 2018, 57, 2007–2013 CrossRef PubMed.
- (a) Y. Liu, T. Shen, B. Kang, S. Li, Z. Zheng, H. Lv and J. Zheng, Opt. Laser Technol., 2022, 156, 108580 CrossRef CAS; (b) H. Berneth, F. K. Bruder, T. Fäcke, S. Hansen, K. Kawamura, L. Pitzer, S. Kern, B. Wewer and T. A. Rölle, Polymers, 2021, 13, 3517 CrossRef CAS PubMed; (c) A. Narita, J. Oshima, Y. Iso, S. Hasegawa and Y. Tomita, Opt. Mater. Express, 2021, 11, 614–628 CrossRef; (d) R. Castagna, A. Di Donato, O. Francescangeli and D. E. Lucchetta, Photonics, 2022, 9, 751 CrossRef CAS; (e) B. Guo, M. Wang, D. Zhang, M. Sun, H. Gao, Y. Bi and Y. Zhao, ACS Appl. Polym. Mater., 2024, 6, 2964–2971 CrossRef.
- Sabad-e-Gul, J. Cassidy and I. Naydenova, Photonics, 2021, 8, 329 CrossRef.
- S. Ikeda, S. Murata, K. Ishii and H. Hamaguchi, Chem. Lett., 1999, 1009 CrossRef CAS.
- T. Mikulchyk, S. Karuthedath, C. S. P. De Castro, A. A. Buglak, A. Sheehan, A. Wieder, F. Laquai, I. Naydenova and M. A. Filatov, J. Mater. Chem. C, 2022, 10, 11588–11597 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, optical spectroscopy and computational data. See DOI: https://doi.org/10.1039/d4tc04850d |
| This journal is © The Royal Society of Chemistry 2025 |