
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe impact of dihedral angle in aryl groups on the photocyclization reactivity of inverse-type diarylethenes†
Misato
Suganuma
 a,
Daichi
Kitagawa
*a,
Shota
Hamatani
a,
Hikaru
Sotome
*b,
Cédric
Mittelheisser
c,
Michel
Sliwa
*cd,
Syoji
Ito
b,
Hiroshi
Miyasaka
b and
Seiya
Kobatake
*a
a,
Daichi
Kitagawa
*a,
Shota
Hamatani
a,
Hikaru
Sotome
*b,
Cédric
Mittelheisser
c,
Michel
Sliwa
*cd,
Syoji
Ito
b,
Hiroshi
Miyasaka
b and
Seiya
Kobatake
*a
aDepartment of Chemistry and Bioengineering, Graduate School of Engineering, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: kitagawa@omu.ac.jp; kobatake@omu.ac.jp
bDivision of Frontier Materials Science and Center for Promotion of Advanced Interdisciplinary Research, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan. E-mail: sotome@laser.chem.es.osaka-u.ac.jp
cUniversité de Lille, CNRS, UMR 8516, LASIR, Laboratoire de Spectrochimie Infrarouge et Raman, Lille 59000, France. E-mail: michel.sliwa@univ-lille.fr
dLOB, CNRS, INSERM, École Polytechnique, Institut Polytechnique de Paris, Palaiseau 91120, France
First published on 17th January 2025
Abstract
Photoreactivity in crystals is one of the essential properties for creating photo-functional crystalline materials. This study explores the impact of the dihedral angle in aryl groups on the photocyclization reactivity of inverse-type diarylethenes, both in solution and crystalline phases. By synthesizing various diarylethene derivatives with different dihedral angles, the relationship between structural geometry and photoreactivity is systematically examined. We find that larger dihedral angles between the thiophene and phenyl rings enhance photocyclization reactivity in solution, indicating that destabilized π-conjugation lowers the activation barrier. In fact, ultrafast spectroscopy confirms that the cyclization time constant decreases with larger dihedral angles. In the crystalline phase, X-ray crystallographic analysis shows that all diarylethene derivatives adopt ideally photoreactive anti-parallel conformations, but only crystals with a dihedral angle exceeding approximately 81° exhibit photocyclization. These findings indicate that a certain threshold dihedral angle is essential for photocyclization to occur in crystals. The results of this work provide new insights into the role of molecular geometry in photoreactivity and offer a strategy for designing functional photochromic materials that operate efficiently in the solid state.
Introduction
Stimuli-responsive molecular crystals are being intensively investigated as next-generation functional materials because they exhibit interesting properties such as superelasticity,1,2 plasticity,3,4 self-healing,5–8 fluorescence,9,10 gas adsorption/desorption,11,12 and photomechanical effects.13–15 In particular, photo-responsive molecular crystals have the advantage that changes in their physicochemical properties can be induced in a non-contact and spatially selective manner.16–20 Therefore, photoreactivity in crystals is one of the important molecular properties.21 One interesting feature of photoreactions in crystals is topochemistry, where reactions take place with minimal atomic displacements that can be observed using X-ray crystallography techniques.22–24 Moreover, Schmidt's extensive research into the [2 + 2] photodimerization of olefins and [4 + 4] photodimerization of anthracenes revealed that solid-state photodimerization can only proceed when adjacent molecules are aligned in parallel and the distance between olefins is less than 4.2 Å.25–29 This rule is called “Schmidt's criteria” and most solid-state photoreactions follow this rule.20,30–33 However, there are a few cases that do not follow Schmidt's criteria. For instance, there have been several reports that the [2 + 2] cyclization reaction in coordination polymers or coordination complexes proceeds even when the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond distance is longer than 4.2 Å.34–37 Conversely, there are cases where the reaction does not proceed even though the molecules in crystals geometrically satisfy Schmidt's criteria.38–40 Recently, Lu and coworkers revealed that the intermolecular interactions around the CC double bond affect the [2 + 2] cycloaddition reactivity except for Schmidt's criteria by investigating the photochemistry of chalcone derivatives in crystals.41 Therefore, investigation of the photoreactivity of molecules in crystals is thus required not only for creating new functional materials, but also for gaining insight into the factors that control photoreactivity in crystals.
C double bond distance is longer than 4.2 Å.34–37 Conversely, there are cases where the reaction does not proceed even though the molecules in crystals geometrically satisfy Schmidt's criteria.38–40 Recently, Lu and coworkers revealed that the intermolecular interactions around the CC double bond affect the [2 + 2] cycloaddition reactivity except for Schmidt's criteria by investigating the photochemistry of chalcone derivatives in crystals.41 Therefore, investigation of the photoreactivity of molecules in crystals is thus required not only for creating new functional materials, but also for gaining insight into the factors that control photoreactivity in crystals.
With such a background, we focused on the photoreactivity of diarylethene in crystals.42–46 Diarylethene is known as one of the photochromic molecules which reversibly isomerizes between open and closed-ring forms based on a 6π electrocyclic reaction.47 Most of the diarylethene derivatives that have been studied so far have thiophene rings as the aryl groups48–52 and can be classified into two types, normal and inverse, with respect to the orientation of the thiophene ring.53–55 Regarding the photoreactivity of diarylethene in crystals, there is an empirical rule that the normal-type diarylethene derivatives can undergo a reversible photochromic reaction in the crystalline state when the molecules adopt an antiparallel conformation in the crystals and the distance between reactive carbons is less than 4.2 Å, similar to Schmidt's criteria.56,57 In contrast, however, we previously found that most inverse-type diarylethene derivatives cannot undergo photocyclization reactions from the open-ring form to the closed-ring form in crystals even though the molecules satisfy the above reaction conditions in the crystalline state,40,58–61 which are singular cases that do not follow the empirical rule. It has also been confirmed that photoreactivity, i.e. the photocyclization quantum yield, of an inverse-type diarylethene decreases when the viscosity of the solvent is high (from 0.2 in n-hexane to 0.1 in cis-decalin), suggesting that the loss of the photoreactivity in the crystalline state would be due to the property of the molecule itself, rather than being influenced by intermolecular interactions or the size of voids in the crystal.54 It remains an open question as to what controls the photoreactivity of inverse-type diarylethenes in crystals. To gain insight into the answer to this question, we recently focused on the host–guest chemistry of cyclodextrins (CDs), which restricts the molecular geometrical change like in the crystalline phase, and investigated the effect of inclusion into CDs on the photochromic behavior of an inverse-type diarylethene derivative.62 As a result, we found that inclusion of the inverse-type diarylethene into βCD reduced the photocyclization quantum yield from 0.11 to 0.056. A detailed study of the interaction between diarylethene and βCD suggests that the reduced photocyclization reactivity is ascribed to a restriction of the rotational motion of the bond between the thiophene and phenyl rings. Based on this result, we hypothesized that the photoreactivity of inverse-type diarylethenes in crystals depends on the dihedral angle between the thiophene and the phenyl rings.
In this study, we focus on the relationship between the photoreactivity of the inverse-type diarylethene and the dihedral angle. We synthesize various inverse-type diarylethenes (1–6) (Scheme 1) to tune the dihedral angle and examine the photoreactivity in solution and in crystals. We find that the photocyclization quantum yield in solution increases with an increase in the dihedral angle. Ultrafast spectroscopy reveals that the time constant of cyclization tends to decrease with an increase in the dihedral angle, which is interpretable from the viewpoint of the destabilization due to breaking of the π-conjugation. Moreover, we find that the photoreactivity in crystals strongly depends on the dihedral angle and that there is a dihedral angle threshold determining photoreactivity. These results provide new insights into not only the reaction dynamics between hexatriene and cyclohexadiene skeletons but also the molecular design strategy to achieve photoreactivity in the crystalline state.
 | ||
| Scheme 1 Inverse-type diarylethenes investigated in this work. | ||
Results and discussion
Molecular design by quantum chemical calculations
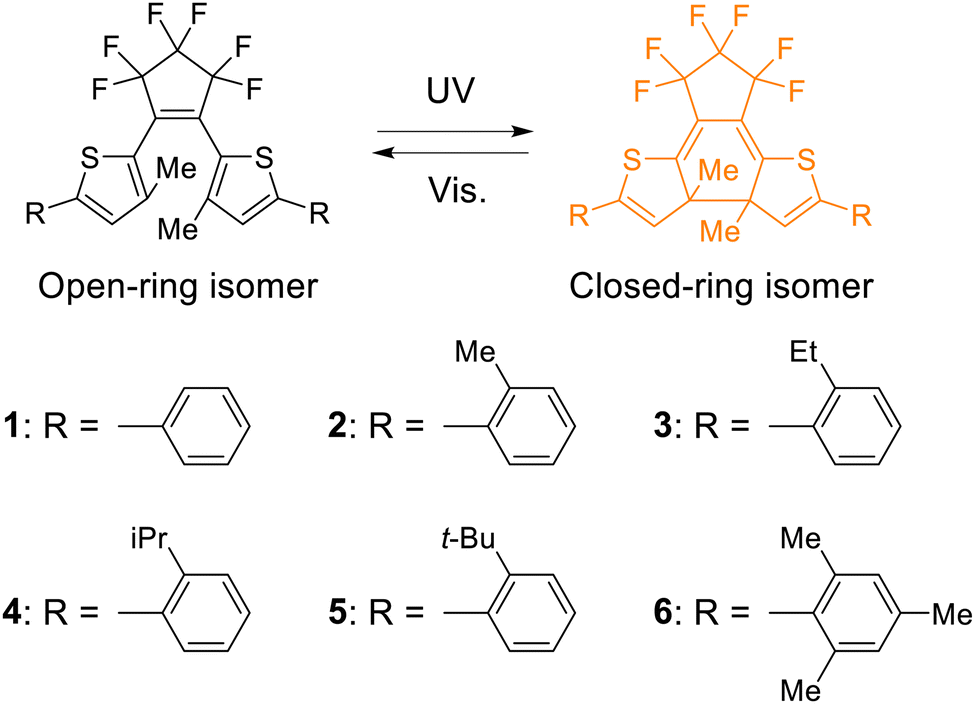
To prepare the inverse-type diarylethenes having different dihedral angles between the thiophene and the phenyl rings (hereafter, the dihedral angles in aryl groups), we newly designed compounds 2–6 that have various alkyl groups at the ortho position of the phenyl rings focusing on the steric hindrance. To estimate the dihedral angles in aryl groups in solution in the ground state (φsoln) for compounds 1–6, density functional theory (DFT) calculations were conducted. The details of the DFT calculations are described in the ESI† and the results are summarized in Fig. 1a and Fig. S1 (ESI†) and Table 1. The φsoln values for the open-ring isomers 1o–6o were estimated to be 27, 41, 46, 50, 72, and 86°, respectively. Moreover, the φsoln values for the closed-ring isomers 1c–6c were estimated to be 31, 45, 49, 51, 82, and 86°, respectively, as shown in Fig. S2 (ESI†) and Table 1. Thus, φsoln increased in the order of 1–6 for both open- and closed-ring isomers. These results indicate that by introducing different alkyl groups such as methyl, ethyl, isopropyl, and tert-butyl groups into the ortho position of the lateral phenyl ring, it is possible to prepare inverse-type diarylethenes with various dihedral angles in aryl groups. Therefore, we synthesized 2o–6o using a procedure similar to that described in the literature.63 The details are described in the ESI.† The chemical structures were confirmed by 1H and 13C NMR spectroscopy and high-resolution mass spectrometry.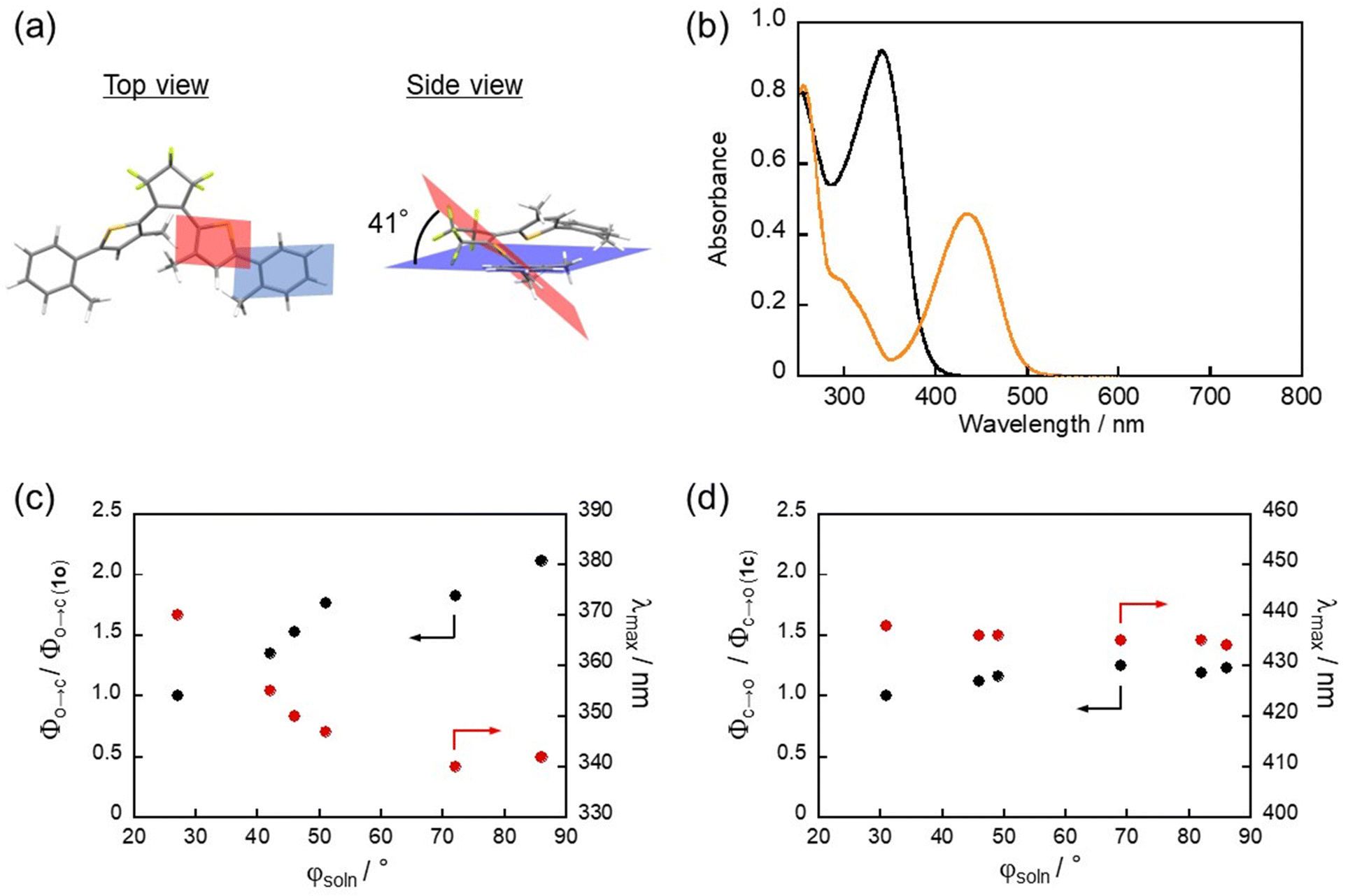 | ||
| Fig. 1 (a) Molecular structure of 2o optimized in the ground state using DFT calculations, (b) absorption spectra of 2o and 2c in n-hexane (3.2 × 10−5 mol L−1), and the relationship between the quantum yields of (c) the cyclization reaction and (d) the cycloreversion reaction and φsoln when compared to the quantum yields of 1. The dependence of the λmax of the open-ring isomer and the closed-ring isomer on φsoln is also shown in (c) and (d), respectively. | ||
| Open-ring isomer | Closed-ring isomer | Quantum yield | ||||||
|---|---|---|---|---|---|---|---|---|
| λ max/nm | ε/M−1 cm−1 | φ soln/° | λ max/nm | ε/M−1 cm−1 | φ soln/° | Φ o→c | Φ c→o | |
| a Ref. 63. | ||||||||
| 1 | 370 | 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 800 |
27 | 438 | 5250 | 31 | 0.17 | 0.48 |
| 2 | 355 | 19000 |
41 | 436 | 6820 | 45 | 0.23 | 0.54 |
| 3 | 350 | 17100 |
46 | 436 | 6670 | 49 | 0.26 | 0.56 |
| 4 | 347 | 15000 |
50 | 435 | 6530 | 51 | 0.30 | 0.60 |
| 5 | 342 | 14900 |
72 | 435 | 7440 | 82 | 0.31 | 0.59 |
| 6 | 340 | 16000 |
86 | 434 | 7310 | 86 | 0.36 | 0.57 |
Photochromic reactivity in solution
Photochromic behaviors of 2o–6o in n-hexane were investigated. Fig. 1b shows the absorption spectral change of 2o. Diarylethene 2o has an absorption maximum (λmax) at 355 nm in n-hexane. Upon irradiation with 365 nm light, the colorless solution turned yellow, in which an absorption maximum was observed at 436 nm. This spectral change is ascribed to the photoisomerization from the open-ring isomer to the closed-ring isomer. The yellow color disappeared by irradiation with visible light (>470 nm) and the absorption spectrum returned to that of 2o. Similarly, 3o–6o exhibited reversible photochromism upon alternating irradiation with UV and visible light as shown in Fig. S3 (ESI†). Table 1 shows the steady-state photophysical properties of 1–6. The λmax for the open-ring isomers 1o–6o are 370, 355, 350, 347, 342, and 340 nm, respectively, are blue-shifted as φsoln increased (Fig. 1c). This is because the twisting of the phenyl ring reduces the effective π-conjugation. On the other hand, the λmax for the closed-ring isomers 1c–6c are 438, 436, 436, 435, 435, and 434 nm, respectively, and are independent of φsoln (Fig. 1d). This is because the π-conjugation of the closed-ring isomer for the inverse-type diarylethenes is localized within the central thiophene rings.53 To quantitatively discuss the relationship between photochromic reactivity and φsoln, the cyclization quantum yields (Φo→c) and the cycloreversion quantum yields (Φc→o) were determined. The Φo→c for 1–6 is 0.17, 0.23, 0.26, 0.30, 0.31, and 0.36, respectively, while the Φc→o for 1–6 is 0.48. 0.54, 0.56, 0.60, 0.59, and 0.57, respectively. Fig. 1c and d show the plots of the quantum yields relative to φsoln when compared to quantum yields of 1. The Φo→c values of 1–4 increase with an increase of φsoln compared to those of 5 and 6 adopting a value similar to that of 4, whereas Φc→o is almost constant relative to the value of φsoln. Namely, the cyclization reactivity is largely affected by the dihedral angle in the aryl group, while that of the cycloreversion is independent of the angle. These relation behaviors would be ascribed to the difference in the two reactions: the photocyclization reaction of the inverse-type diarylethene with aryl groups like 1–6 has an activation barrier in the excited state before reaching the reactive conical intersection, while the cycloreversion reaction has no one.53It should be noted that the open-ring isomer of diarylethene derivatives is present as reactive and unreactive conformers such as the anti-parallel (AP) and parallel (P) forms, and their relative population affects the apparent cyclization reaction yield. In our previous work, three conformers (AP1, P1, AP2) were demonstrated to be dominant in the ground state of 1o. AP1 is the only reactive conformer, and P1 and AP2 are unreactive.54 We evaluated the relative energies of each conformer using quantum chemical calculation and quantified the relative population of AP1, P1 and AP2 by assuming the thermal equilibrium under the Boltzmann distribution (Fig. S4 and Table S1, ESI†). The relative portion of AP1, P1 and AP2 are respectively 60–65%, 30–35% and ∼5%, which is almost constant in a series of the compounds although 5o shows a slight deviation. This result indicates that the change in Φo→c in 1–6 is less affected by the relative population of the conformers and predominantly originates from the intrinsic cyclization reactivity of the reactive AP form (AP1).
As 6o shows the highest photocyclization quantum yield, we firstly investigated the photocyclization reaction dynamics of 6o using transient absorption spectroscopy with an instrument allowing the recording of transient spectra until 1000 nm as the excited state absorption (ESA) of 1o was reported to extend to the near-infrared region in our precedent work.54Fig. 2 shows transient absorption spectra of 6o in n-hexane in the femtosecond and later timescales. At the time zero, a positive absorption band was observed at 610 nm concomitantly with a small negative band at around 435 nm. This negative one is ascribable to stimulated emission and is dynamically shifted toward the longer wavelength region within 0.2 ps. It reaches a maximum at 490 nm similar to the steady-state emission maximum shown in Fig. S5 (ESI†), indicating that the molecules undergo structural relaxation on the excited state potential energy surface. The resultant stimulated emission at around 490 nm and ESA at around 635 nm decayed on a picosecond timescale. The transient spectrum at 100 ps is characterized by two positive bands centered at 430 and 610 nm. The first one corresponds to the absorption band of the newly produced closed-ring isomer (strictly speaking, it is a differential spectrum between the open- and closed-ring isomers), and the second one is assigned to the long-lived species of the unreactive conformer such as P1 and AP2. A dip at around 520 nm is attributable to the stimulated emission of these conformers from the spectral agreement with the fluorescence maximum wavelength (Fig. S5, ESI†) and persists from several hundreds of picoseconds to a few nanoseconds. The overall spectral features of 6o are similar to those of 1o with phenyl groups reported in the previous work54 although its timescales become shorter.
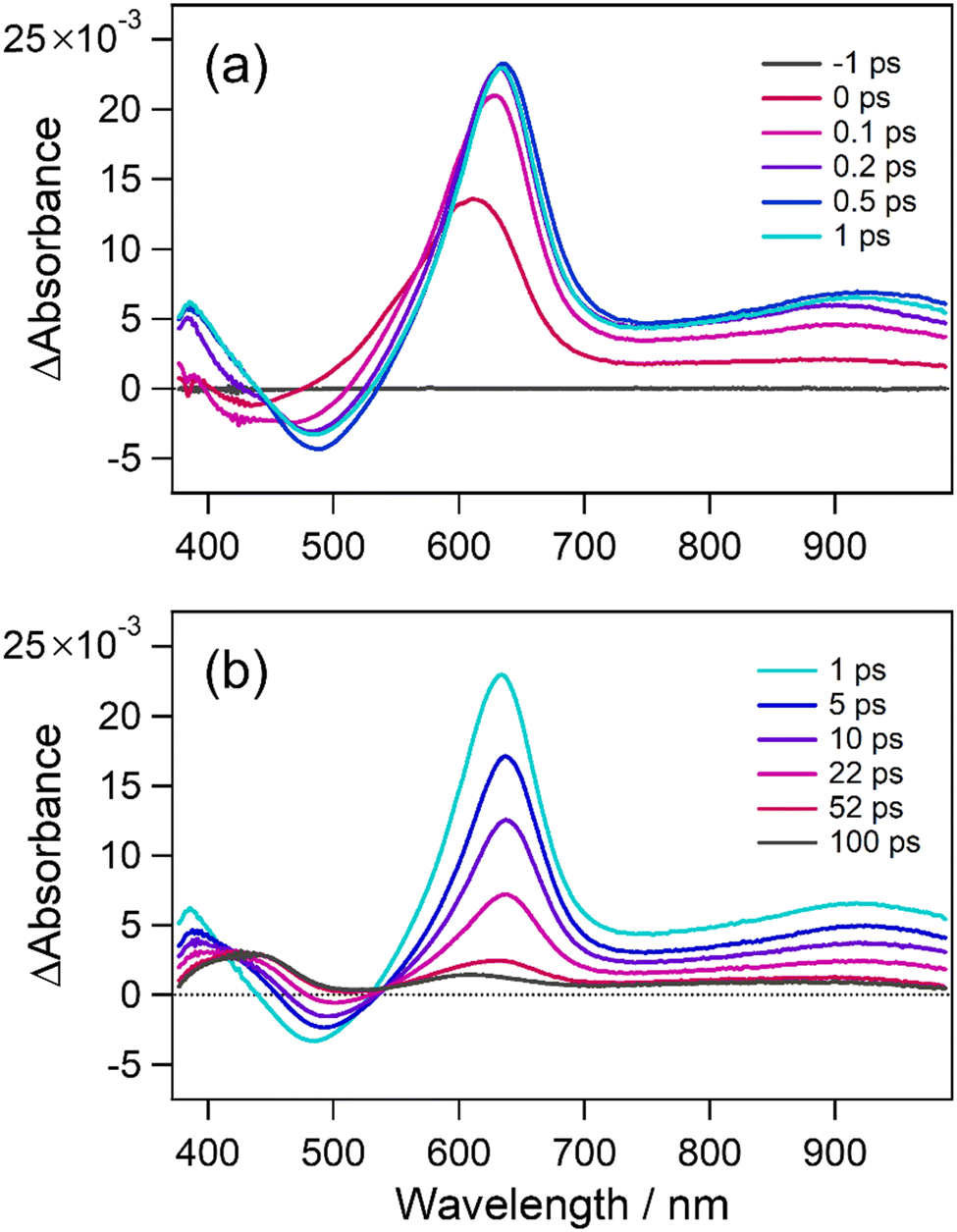 | ||
| Fig. 2 Transient absorption spectra of 6o in n-hexane solution (1.0 × 10−3 mol L−1) in (a) early and (b) late time regions. The sample was excited with a femtosecond laser pulse at 350 nm. The excitation power was 120 nJ per pulse. | ||
As well as for the cyclization dynamics of 1o, increase of transient absorbance corresponding to the formation of the closed-ring isomer was unfortunately elusive due to the spectral overlap with the absorption and stimulated emission of the precursor. Thus, we applied global fitting analysis to the transient absorption data in Fig. 2 to obtain the formation time of the closed-ring isomer, which is essential information for discussion on the topology of the excited state potential. The time evolution of the transient absorption was well reproduced with four components in the 100 ps time window, as shown in Fig. 3a. Similarly to 1o, an oscillatory feature in the first 2 ps is ascribable to coherent vibrations triggered by the pulsed excitation, and detailed analysis with assignment is shown in Fig. S6 and S7 (ESI†). Four decay-associated spectra (DAS) thus yielded are shown in Fig. 3b with the corresponding time constants of 0.12, 2.9, 17 ps and infinity (offset component).
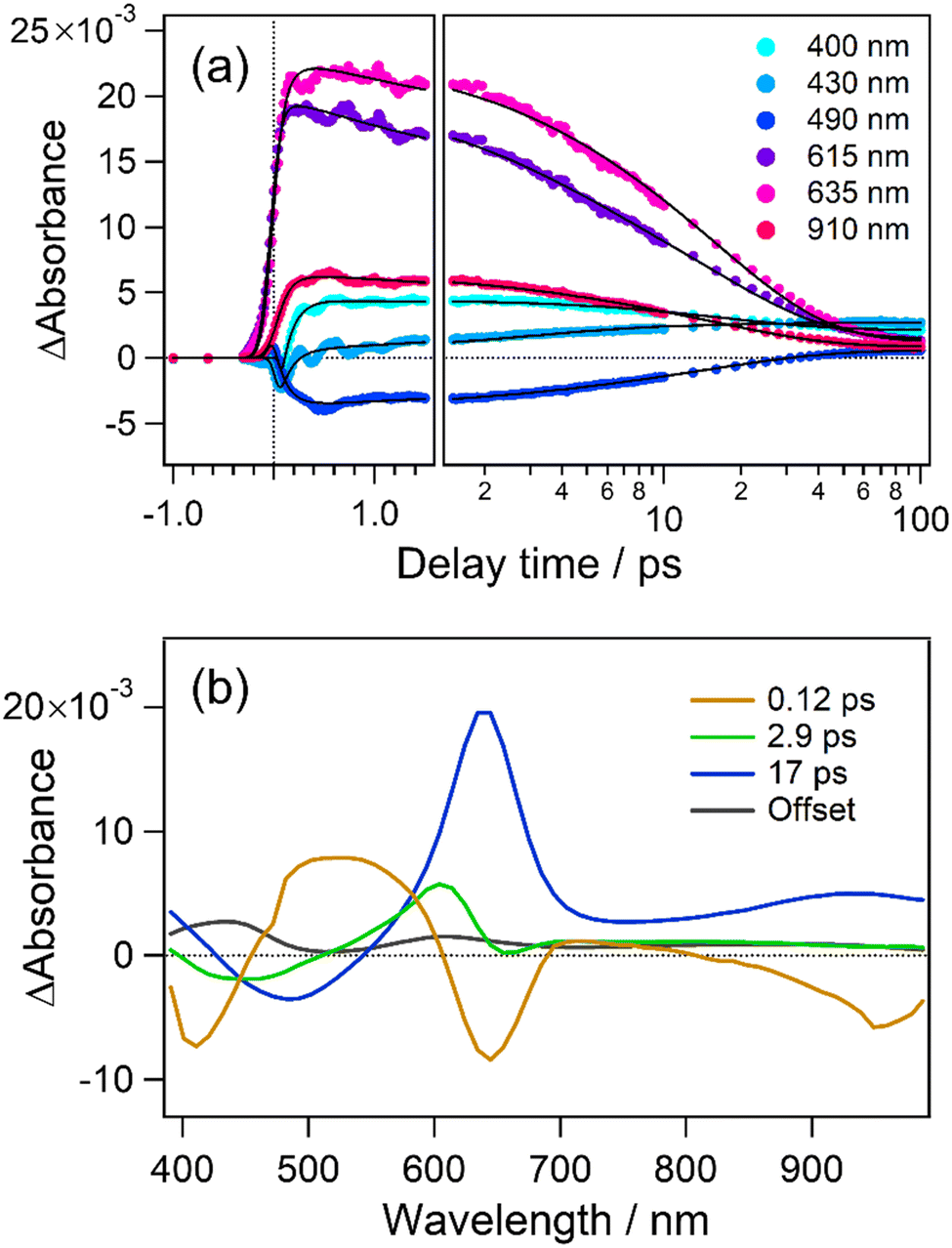 | ||
| Fig. 3 (a) Time profiles of transient absorbance of 6o in n-hexane monitored at selected wavelengths. (b) Decay-associated spectra obtained from global fitting analysis. | ||
The first DAS (0.12 ps) shows negative (<450 nm) and positive components (450–600 nm), which are attributable to the red shift of stimulated emission. A negative signal at 640 nm is also related to the initial geometrical relaxation in the ultrafast timescale. For the second DAS (2.9 ps), a negative component lies at around 440 nm and indicates the formation of a new species and/or the recovery of the stimulated emission. In the present case, this negative feature is well correlated with a positive component at 440 nm in the fourth DAS (offset) showing the absorption of the closed-ring isomer. This spectral agreement shows that the closed-ring isomer is produced with a time constant of 2.9 ps. On the other hand, the third DAS (17 ps) is assignable to the decay of the unreactive conformer in the excited state from the spectral similarity to 1o observed in the previous work.54 The offset component is the superposition of absorption bands due to the closed-ring isomer and long-lived species of the unreactive conformer. In our previous report, time constants were 36 ps (cyclization), 130 and 390 ps for two unreactive (recovery of SE band) conformers. To check if we do not have any extra long time species, we used commercial apparatus (Ultrafast Systems, HELIOS) that allows the measurement of transient absorption until 8 ns (see the ESI†) and found time constant for 6o of 0.11, 2.1, 18 ps and 890 ps, a minor second unreactive conformer (Fig. S9e, ESI†).
We further performed transient absorption measurements of 2o, 3o, 4o and 5o (Fig. S8, ESI†) and extracted the cyclization time constant for investigation of dihedral angle dependence. For 5o, three time constants were needed to model unreactive conformers (7 ps, 26 ps, and 1 ns) and can be rationalized with a tert-butyl group that increases the existence of different conformers. The full datasets of transient absorption data and obtained DAS are described in Fig. S8 and S9 (ESI†). The obtained cyclization timescales of 1o–6o are shown in Table 2, in which the maximum wavelengths of ESA are also shown for comparison. The cyclization time constant decreases with an increase of the dihedral angle reaching a plateau for 5o and 6o. A similar trend can be seen in the dihedral angle dependence of the absorption maximum wavelength of the positive band in 600–700 nm in the transient absorption spectra. These results can be interpreted as follows: approaching the planar geometry with less bulky substituents extends the π-conjugation and leads to stabilization of the energy minimum of the excited state. This is verified by a relaxation time constant of a few ps for 2o and 3o concomitant with the red-shift of the ESA maximum although it is also affected by the excited state of the unreactive conformer. Thus, the activation energy substantially becomes large along the cyclization coordinate toward the energetic barrier, which results in the deceleration of the cyclization process. The comparable time constants in 4o–6o are probably due to sufficiently small activation energy by almost complete breaking of π-conjugation.
Taken together with the above results, we built the cyclization reaction scheme of inverse-type diarylethenes with different dihedral angles. As shown in Fig. 4, photoexcitation first populates the molecule into the Franck–Condon region of the excited state potential energy surface. This excited molecule undergoes slight geometrical relaxation such as the decrease of the dihedral angle, which extends π-conjugation and stabilizes the S1 energy minimum. The calculated molecular geometries theoretically verify that the dihedral angle changes of 1o from the initial 27° to 12° at this minimum.54 From the transient absorption data the timescale of this structural evolution was estimated to be 120 fs. Subsequently, the molecule in the S1 minimum evolves on the excited state potential and overcomes the activation barrier therein. However, this barrier is sufficiently small and the S1 molecule can easily overcome this within a few picoseconds in 6o where the energy stabilization is lower due to shorter π-conjugation. On the other hand, the activation energy of 1o is larger than that of 6o because the S1 minimum of 1o is more stabilized, which leads to the deceleration of the cyclization process (36 ps) and the existence of vibrational relaxation time constant for 2o and 3o. The slower time constant for the cyclization increases the probability of internal conversion, reducing the cyclization quantum yield. Actually, the cyclization reaction of an inverse-type derivative without any aromatic substituents proceeds with a time constant of 0.8 ps according to a previous work64 and this barrierless reaction behavior supports the above consideration. In this way, the dihedral angle of the aromatic groups at the molecular terminals modulates the topology of the excited state potential through the stabilization due to the π-conjugation and plays a crucial role in the evolution of the cyclization process in the solution phase. The slight increase of cyclization quantum yield for 6o in comparison to 5o can be assigned to a lower concentration of unreactive conformer and an increase of internal conversion without the cyclization for 5o due to the tert-butyl group.
 | ||
| Fig. 4 Schematic representation of the photocyclization reaction dynamics of inverse-type diarylethenes with different dihedral angles. | ||
Photochromic reactivity in the crystalline state
To obtain more detailed information on the relationship between the photocyclization reactivity and the dihedral angle in aryl groups, we investigated the photochromic behaviors in the crystalline phase where the molecular motion and alignment are fixed, and the dihedral angles can be precisely determined by X-ray crystallographic analysis. Fortunately, single crystals of all compounds could be obtained by a slow solvent evaporation method, and X-ray crystallographic analysis was successful for all crystals (Table S2, ESI†). As reported previously, 1o has polymorphic forms of 1o-α and 1o-β.40 In addition, 6o was found to crystallize into polymorphs of 6o-α and 6o-β as well. From the results of single-crystal X-ray structural analysis, all 1o–6o molecules are fixed in anti-parallel conformations, and the distances between the reactive carbons are within 4.2 Å, indicating that 1o–6o can potentially undergo the photocyclization reaction in the crystalline phase. Moreover, the dihedral angles of aryl groups in the crystalline phase (φcry) were investigated. The results are shown in Fig. 5a and Fig. S10 (ESI†) and summarized in Table 3. Note that dihedral angle pairs of both left and right aryl groups are listed. Also in solution, the φcry increases in the order of 1o–6o.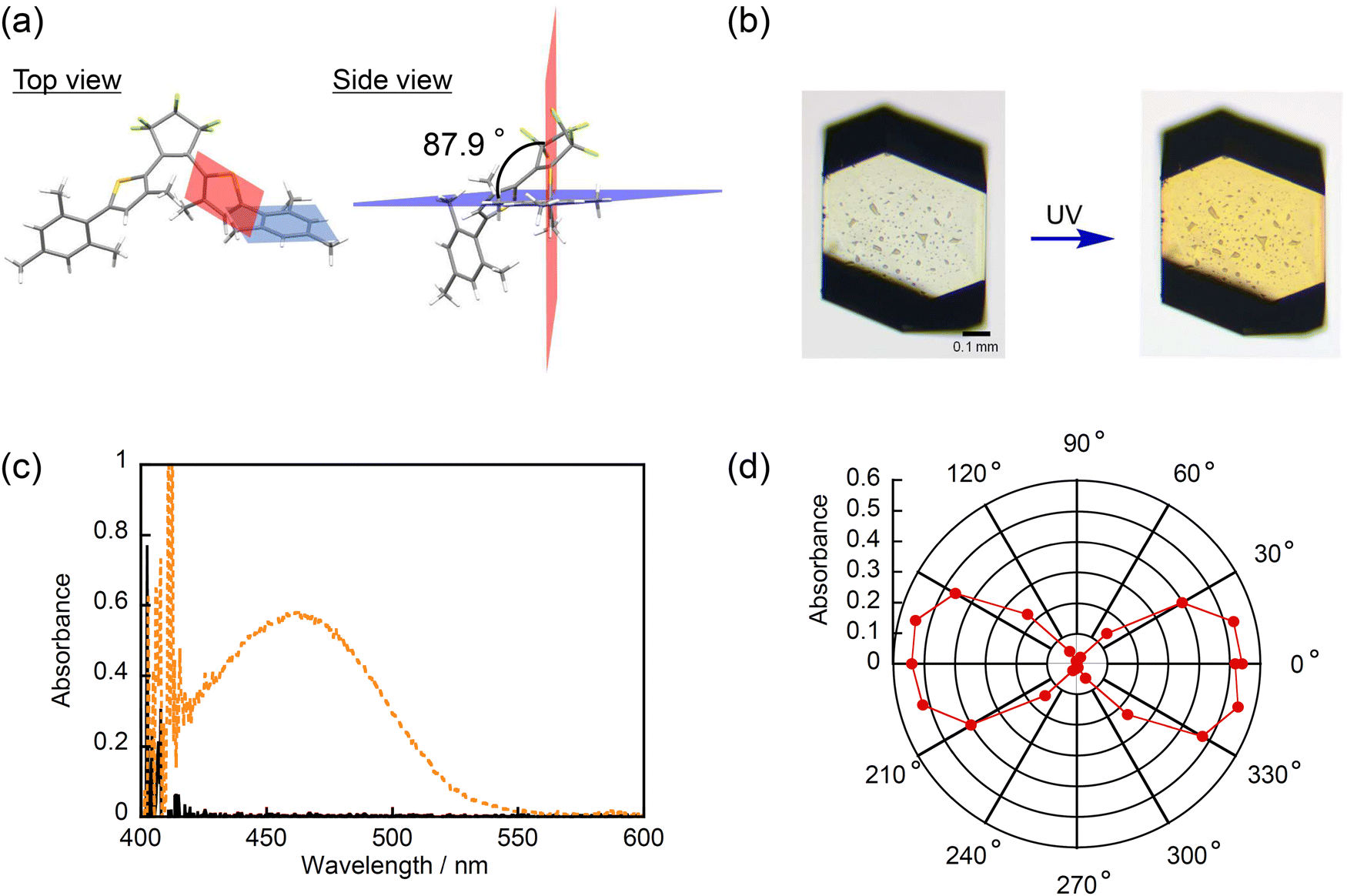 | ||
| Fig. 5 (a) Molecular structure of 6o in crystal 6o-α determined by X-ray crystallographic analysis, (b) optical microphotographs of crystal 6o-α before and after UV irradiation, (c) absorption spectra of crystal 6o-α before and after UV irradiation, and (d) polar plots of the polarized absorption spectra of crystal 6o-α at 462 nm on the (001) face. | ||
| Conformation | C–C distance | φ cry/°° | Photoreactivity | |
|---|---|---|---|---|
| a Ref. 40. | ||||
| 1o-α | Antiparallel | 3.530/3.441 | 6.42, 23.8/7.18, 8.98 | Inert |
| 1o-β | Antiparallel | 3.561 | 10.2, 28.8 | Inert |
| 2o | Antiparallel | 3.537 | 28.4, 40.79 | Inert |
| 3o | Antiparallel | 3.536 | 38.09, 41.8 | Inert |
| 4o | Antiparallel | 3.582 | 47.1, 53.2 | Inert |
| 5o | Antiparallel | 4.023 | 74.7, 74.7 | Inert |
| 6o-α | Antiparallel | 3.554 | 87.9, 87.9 | Reactive |
| 6o-β | Antiparallel | 3.536 | 80.5, 89.8 | Reactive |
| 7o | Antiparallel | 3.518/3.512 | 85.2, 87.8, 89.3 | Reactive |
To experimentally examine the photochromic reactivity in the crystalline state, the absorption spectral changes of crystals 2o–6o upon UV irradiation were measured. Note that crystals 1o-α and 1o-β are photo-inert as reported previously. The optical microphotographs and the absorption spectra for crystals 2o–6o before and after UV irradiation are shown in Fig. 5b, c and Fig. S11, S12 (ESI†). In the case of crystals 2o–5o, there were no changes in the color of the crystals and the absorption spectra, indicating that the photocyclization reaction did not take place even though they were satisfying the required conditions for the photochromic reaction in the crystalline phase. On the other hand, interestingly, the changes in the color of the crystals and the absorption spectra upon UV irradiation were confirmed for crystals 6o-α and 6o-β as shown in Fig. 5b, c and Fig. S11e, S12e (ESI†). Fig. 5d and Fig. S13 (ESI†) show the polar plots of the polarized absorption spectra at 462 nm on the (001) face for crystal 6o-α and at 455 nm on the (001) face 6o-β, respectively. Note that the face indices were determined by powder X-ray diffraction pattern as shown in Fig. S14 (ESI†). The absorption intensity was dependent on the rotation angle, and there is clear dichroism of the absorption, indicating that the photochromic reaction takes place in the single crystalline phase. Table 3 also summarizes the relationship between the photoreactivity and the φcry. As can be seen, crystals with the φcry less than 74.7° were photo-inert, but crystal 6o-β with the φcry of 80.5° was photo-reactive, indicating that the dihedral angle threshold that determines photoreactivity exists between 74.7° and 80.5°. These results rationalize the previous works that inverse-type diarylethenes bearing hydrogen or methyl groups in place of the aryl ring can undergo photocyclization in solution with a time constant of 0.8 ps and in the crystalline phase, since twisting of the aryl ring is not required.9,64,65
To further verify the effect of the dihedral angle on the photoreactivity in the crystalline state, we synthesized an additional compound 7 shown in Fig. 6a that would have twisted aryl groups. From X-ray crystallographic analysis, it was found that 7o also satisfies the reaction conditions in the crystalline state (the antiparallel conformation in the crystalline state and the distance between the reactive carbons less than 4.2 Å), and the smallest φcry of 7o is 85.2° (Fig. S15 and Table 3, Table S2, ESI†). Upon irradiation with UV light, the crystal color changed from colorless to yellow as shown in Fig. 6b. In addition, the clear dichroism of the absorption was also observed from the polar plots of the polarized absorption spectra shown in Fig. S16 and S17 (ESI†), indicating 7o is photoreactive in the crystalline state. This result follows the relationship between the photoreactivity and the dihedral angle described above, which supports the validity of our explanation of the reaction mechanism of inverse-type diarylethenes.
 | ||
| Fig. 6 (a) Molecular structure of compound 7 and (b) optical microphotographs of crystal 7o before and after UV irradiation. | ||
Summing up the results described above, the twisting of the phenyl groups is necessary for inverse-type diarylethenes to overcome the activation barrier in the excited state, resulting in the photocyclization reaction. In particular, it was found that the dihedral angle should be larger than at least 75° to undergo photocyclization as revealed by the investigation of the photoreactivity in the crystalline phase. This characteristic molecular property is the reason why the photoreactivity of inverse-type diarylethene derivatives in crystals does not follow the empirical rule reported previously.
Conclusions
In this study, we investigated the relationship between the dihedral angle in aryl groups and the photocyclization reactivity of inverse-type diarylethenes both in solution and in the crystalline state to address a long-standing question about why most inverse-type diarylethene derivatives do not undergo photocyclization in crystals, despite fulfilling the spatial requirements known as an empirical rule. Through quantum chemical calculations, the dihedral angles of various diarylethenes were modified by introducing different substituents. It was found that the photocyclization quantum yield in solution increases as the dihedral angle between the thiophene and phenyl rings increases. This was attributed to the destabilization of π-conjugation, which reduces the activation barrier for cyclization and some changes at the conical intersection. Ultrafast spectroscopy further supported this result, showing that the time scale for cyclization decreases with larger dihedral angles. In the crystalline state, X-ray crystallography was used to confirm the anti-parallel conformation and appropriate distances for photoreactivity in all tested compounds. However, only compounds with a dihedral angle of approximately 81° displayed photocyclization reactivity. This finding suggests that overcoming the activation barrier in the excited state is largely dependent on achieving sufficient phenyl group twisting. The study reveals that the dihedral angle is a crucial determinant of photoreactivity for inverse-type diarylethenes, both in solution and crystals. This understanding provides insight into designing photoresponsive molecular systems.Author contributions
Misato Suganuma: investigation, formal analysis, visualization, and writing – original draft; Daichi Kitagawa: conceptualization, methodology, formal analysis, project administration, resources, supervision, writing – original draft, and writing – review & editing; Shota Hamatani: investigation, formal analysis, and visualization; Hikaru Sotome: methodology, formal analysis, resources, writing – original draft, and writing – review & editing; Cédric Mittelheisser: investigation, formal analysis, and visualization; Michel Sliwa: methodology, formal analysis, resources, and writing – review & editing; Syoji Ito: formal analysis, resources, and writing – review & editing; Hiroshi Miyasaka: formal analysis, resources, and writing – review & editing; Seiya Kobatake: resources, project administration, supervision, and writing – review & editing.Notes
All authors have given approval to the final version of the manuscript.Data availability
All data that support the findings of this study are available in the published article and/or the ESI† for this article.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was partly supported by the CNSR IRP Nanosynergetics2, JSPS KAKENHI Grant Numbers JP23K26619 and JP24K01458 (D. K.), JP23H03956, JP23H04877, and JP24K21794 (H. S.), JP22J21941 (S. H.), and JP21KK0092, JP23K26616, JP22K19007, JP21H04640, and JP21H04964 (S. I.), and the JST-Mirai Program (Grant Number JPMJMI21G1) (S. I.). We thank Thomas Roland for laser alignment and access to the Ultrafast Spectroscopy Instrument of the advanced characterization platform at the Chevreul Institute.References
- T. Mutai, T. Sasaki, S. Sakamoto, I. Yoshikawa, H. Houjou and S. Takamizawa, Nat. Commun., 2020, 11, 1824 CrossRef CAS PubMed.
- S. Sakamoto, T. Sasaki, A. Sato-Tomita and S. Takamizawa, Angew. Chem., Int. Ed., 2019, 58, 13722–13726 CrossRef CAS PubMed.
- J. Ravi, T. Feiler, A. Mondal, A. A. L. Michalchuk, C. M. Reddy, B. Bhattacharya, F. Emmerling and R. Chandrasekar, Adv. Opt. Mater., 2023, 11, 2201518 CrossRef CAS.
- G. R. Krishna, R. Devarapalli, G. Lal and C. M. Reddy, J. Am. Chem. Soc., 2016, 138, 13561–13567 CrossRef CAS PubMed.
- P. Commins, H. Hara and P. Naumov, Angew. Chem., Int. Ed., 2016, 55, 13028–13032 CrossRef CAS PubMed.
- S. Mondal, P. Tanari, S. Roy, S. Bhunia, R. Chowdhury, A. K. Pal, A. Datta, B. Pal and C. M. Reddy, Nat. Commun., 2023, 14, 6589 CrossRef CAS PubMed.
- M. B. Al-Handawi, P. Commins, A. S. Dalaq, P. A. Santos-Florez, S. Polavaram, P. Didier, D. P. Karothu, Q. Zhu, M. Daqaq, L. Li and P. Naumov, Nat. Commun., 2024, 15, 8095 CrossRef CAS PubMed.
- J. R. Pathan, H. Balan, P. Commins, A. Ravi, M. B. Al-Handawi, I. C.-Y. Hou, P. Naumov and K. M. Sureshan, J. Am. Chem. Soc., 2024, 146, 27100–27108 CrossRef CAS PubMed.
- T. Fukaminato, T. Kawai, S. Kobatake and M. Irie, J. Phys. Chem. B, 2003, 107, 8372–8377 CrossRef CAS.
- S. Ishida, D. Kitagawa, S. Kobatake, S. Kim, S. Kurihara and T. Fukaminato, Chem. Commun., 2019, 55, 5681–5684 RSC.
- K. J. Msayib, D. Book, P. M. Budd, N. Chaukura, K. D. M. Harris, M. Helliwell, S. Tedds, A. Walton, J. E. Warren, M. Xu and N. B. McKeown, Angew. Chem., Int. Ed., 2009, 48, 3273–3277 CrossRef CAS PubMed.
- Z. Wang, N. Sikdar, S.-Q. Wang, X. Li, M. Yu, X.-H. Bu, Z. Chang, X. Zou, Y. Chen, P. Cheng, K. Yu, M. J. Zaworotko and Z. Zhang, J. Am. Chem. Soc., 2019, 141, 9408–9414 CrossRef CAS PubMed.
- I. Tahir, E. Ahmed, D. P. Karothu, F. Fsehaye, J. Mahmoud Halabi and P. Naumov, J. Am. Chem. Soc., 2024, 146, 30174–30182 CrossRef CAS PubMed.
- R. Samanta, D. Kitagawa, A. Mondal, M. Bhattacharya, M. Annadhasan, S. Mondal, R. Chandrasekar, S. Kobatake and C. M. Reddy, ACS Appl. Mater. Interfaces, 2020, 12, 16856–16863 CrossRef CAS PubMed.
- K. Morimoto, D. Kitagawa, H. Sotome, S. Ito, H. Miyasaka and S. Kobatake, Angew. Chem., Int. Ed., 2022, 61, e202212290 CrossRef CAS PubMed.
- H. Wang, P. Chen, Z. Wu, J. Zhao, J. Sun and R. Lu, Angew. Chem., Int. Ed., 2017, 56, 9463–9467 CrossRef CAS PubMed.
- C. J. Barrett, J. Mamiya, K. G. Yager and T. Ikeda, Soft Matter, 2007, 3, 1249–1261 RSC.
- D. Kitagawa and S. Kobatake, Chem. Commun., 2015, 51, 4421–4424 RSC.
- P. Naumov, D. P. Karothu, E. Ahmed, L. Catalano, P. Commins, J. Mahmoud Halabi, M. B. Al-Handawi and L. Li, J. Am. Chem. Soc., 2020, 142, 13256–13272 CrossRef CAS PubMed.
- D. A. Sherman, R. Murase, S. G. Duyker, Q. Gu, W. Lewis, T. Lu, Y. Liu and D. M. D’Alessandro, Nat. Commun., 2020, 11, 2808 CrossRef CAS PubMed.
- L. R. MacGillivray, J. L. Reid and J. A. Ripmeester, J. Am. Chem. Soc., 2000, 122, 7817–7818 CrossRef CAS.
- M. D. Cohen, Angew. Chem., Int. Ed. Engl., 1975, 14, 386–393 CrossRef.
- K. Biradha and R. Santra, Chem. Soc. Rev., 2013, 42, 950–967 RSC.
- B. B. Rath and J. J. Vittal, Acc. Chem. Res., 2022, 55, 1445–1455 CrossRef CAS PubMed.
- G. M. J. Schmidt, J. Chem. Soc., 1964, 2014–2021 RSC.
- J. Bregman, K. Osaki, G. M. J. Schmidt and F. I. Sonntag, J. Chem. Soc., 1964, 2021–2030 RSC.
- M. D. Cohen, G. M. J. Schmidt and F. I. Sonntag, J. Chem. Soc., 1964, 2000–2013 RSC.
- G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647–678 CrossRef CAS.
- E. Heller and G. M. J. Schmidt, Israel J. Chem., 1971, 9, 449–462 CrossRef CAS.
- A. Briceño, D. Leal, R. Atencio and G. Díaz De Delgado, Chem. Commun., 2006, 3534–3536 RSC.
- J. Liu, K. Ye, Y. Shen, J. Peng, J. Sun and R. Lu, J. Mater. Chem. C, 2020, 8, 3165–3175 RSC.
- L.-F. Wang, W.-M. Zhuang, G.-Z. Huang, Y.-C. Chen, J.-Z. Qiu, Z.-P. Ni and M.-L. Tong, Chem. Sci., 2019, 10, 7496–7502 RSC.
- M. Irie, S. Kobatake and M. Horichi, Science, 2001, 291, 1769–1772 CrossRef CAS PubMed.
- M. Nakagawa, S. Kusaka, A. Kiyose, T. Nakajo, H. Iguchi, M. Mizuno and R. Matsuda, J. Am. Chem. Soc., 2023, 145, 12059–12065 CrossRef CAS PubMed.
- A. M. P. Peedikakkal, J. Chem. Sci., 2017, 129, 733–739 CrossRef CAS.
- K. Yadava and J. J. Vittal, Cryst. Growth Des., 2019, 19, 2542–2547 CrossRef CAS.
- Y. Sonoda, M. Goto, S. Tsuzuki, H. Akiyama and N. Tamaoki, J. Fluorine Chem., 2009, 130, 151–157 CrossRef CAS.
- S. Ariel and J. Trotter, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 2084–2086 CrossRef.
- D. Kanagapushpam, V. Ramamurthy and K. Venkatesan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1987, 43, 1128–1131 CrossRef.
- D. Kitagawa, T. Nakahama, K. Mutoh, Y. Kobayashi, J. Abe, H. Sotome, S. Ito, H. Miyasaka and S. Kobatake, Dyes Pigm., 2017, 139, 233–238 CrossRef CAS.
- Y. Yue, J. Dai, L. Jin, C. Liu, J. Sun, K. Ye and R. Lu, Chem. – Eur. J., 2023, 29, e202301525 CrossRef CAS PubMed.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- M. Morimoto and M. Irie, J. Am. Chem. Soc., 2010, 132, 14172–14178 CrossRef CAS PubMed.
- T. Kodani, K. Matsuda, T. Yamada, S. Kobatake and M. Irie, J. Am. Chem. Soc., 2000, 122, 9631–9637 CrossRef CAS.
- M. Irie and K. Uchida, Bull. Chem. Soc. Jpn., 1998, 71, 985–996 CrossRef CAS.
- S. Kobatake and M. Irie, Bull. Chem. Soc. Jpn., 2004, 77, 195–210 CrossRef CAS.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- A. Bianco, C. Bertarelli, J. F. Rabolt and G. Zerbi, Chem. Mater., 2005, 17, 869–874 CrossRef CAS.
- M. Herder, B. M. Schmidt, L. Grubert, M. Pätzel, J. Schwarz and S. Hecht, J. Am. Chem. Soc., 2015, 137, 2738–2747 CrossRef CAS PubMed.
- T. Fukaminato, T. Sasaki, T. Kawai, N. Tamai and M. Irie, J. Am. Chem. Soc., 2004, 126, 14843–14849 CrossRef CAS PubMed.
- C. Zhang, S. Pu, Z. Sun, C. Fan and G. Liu, J. Phys. Chem. B, 2015, 119, 4673–4682 CrossRef CAS PubMed.
- X. Dong, T. Guo, D. Kitagawa, S. Kobatake, P. Palffy-Muhoray and C. J. Bardeen, ACS Appl. Mater. Interfaces, 2022, 14, 27149–27156 CrossRef CAS PubMed.
- T. Kudernac, T. Kobayashi, A. Uyama, K. Uchida, S. Nakamura and B. L. Feringa, J. Phys. Chem. A, 2013, 117, 8222–8229 CrossRef CAS PubMed.
- H. Sotome, D. Kitagawa, T. Nakahama, S. Ito, S. Kobatake, M. Irie and H. Miyasaka, Phys. Chem. Chem. Phys., 2019, 21, 8623–8632 RSC.
- Y. Tatsumi, J. Kitai, W. Uchida, K. Ogata, S. Nakamura and K. Uchida, J. Phys. Chem. A, 2012, 116, 10973–10979 CrossRef CAS PubMed.
- M. Irie, T. Lifka, S. Kobatake and N. Kato, J. Am. Chem. Soc., 2000, 122, 4871–4876 CrossRef CAS.
- S. Kobatake, K. Uchida, E. Tsuchida and M. Irie, Chem. Commun., 2002, 2804–2805 RSC.
- T. Yamaguchi, K. Uchida and M. Irie, Bull. Chem. Soc. Jpn., 2008, 81, 644–652 CrossRef CAS.
- D. Kitagawa, Y. Seto, M. Suganuma, T. Nakahama, H. Sotome, S. Ito, H. Miyasaka and S. Kobatake, ChemPhotoChem, 2024, 8, e202400081 CrossRef CAS.
- T. Nakahama, D. Kitagawa, H. Sotome, S. Ito, H. Miyasaka and S. Kobatake, Dyes Pigm., 2019, 160, 450–456 CrossRef CAS.
- D. Kitagawa, Y. Seto, M. Suganuma, T. Nakahama, H. Sotome, S. Ito, H. Miyasaka and S. Kobatake, ChemPhotoChem, 2024, 8, e202400081 CrossRef CAS.
- M. Suganuma, D. Kitagawa, S. Hamatani, H. Sotome, S. Ito, H. Miyasaka and S. Kobatake, ChemPhotoChem, 2024, 8, e202300244 CrossRef CAS.
- K. Uchida, T. Matsuoka, S. Kobatake, T. Yamaguchi and M. Irie, Tetrahedron, 2001, 57, 4559–4565 CrossRef CAS.
- S. Aloïse, R. Yibin, I. Hamdi, G. Buntinx, A. Perrier, F. Maurel, D. Jacquemin and M. Takeshita, Phys. Chem. Chem. Phys., 2014, 16, 26762–26768 RSC.
- M. Morimoto, S. Kobatake and M. Irie, J. Am. Chem. Soc., 2003, 125, 11080–11087 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized structures obtained by DFT calculations, absorption spectra of 3–6 in n-hexane, the full datasets of transient absorption data and the obtained DASs for 2–5, the detailed data for the investigation of the photoreactivity in the crystalline phase, and the detailed procedures of synthesis (PDF), X-ray crystallographic data (CIF), and cartesian coordinates of the optimized structures (EXCEL). CCDC 2401652–2401657 and 2416015. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc04853a |
| This journal is © The Royal Society of Chemistry 2025 |