
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The intersection of field-limited density of states and matter in radioluminescence: nanophotonic control of energy transfer†
Haley W. Jones ab,
Yuriy Banderaab and
Stephen H. Foulger*abc
ab,
Yuriy Banderaab and
Stephen H. Foulger*abc
aCenter for Optical Materials Science and Engineering Technologies (COMSET), Clemson University, Anderson, SC 29625, USA. E-mail: foulger@clemson.edu
bDepartment of Materials Science and Engineering, Clemson University, Clemson, SC 29634, USA
cDepartment of Bioengineering, Clemson University, Clemson, SC 29634, USA
First published on 13th May 2025
Abstract
The unresolved correlation between a nanostructured environment, like a crystalline colloidal array (CCA), and the Förster resonance energy transfer (FRET) between multiple embedded emitters is a fundamental aspect of quantum light–matter interactions with implications for various high-priority applications, such as telecommunications, energy-efficient lighting, and quantum computing technologies. This highly debated topic was explored in two series (n1 and n2) of organic radioluminescent nanoparticles, containing a copolymerized scintillator and two organic fluorophores, that self-assembled into a liquid ordered structure, or CCA. The three copolymerized emitters exhibited two sequential transfers of energy upon X-ray irradiation, resulting in emission spanning the visible spectrum. Nanophotonic manipulation of the radioluminescence of each nanoparticle series assembled in a CCA and the energy transfer efficiency between the three emitters copolymerized within each nanoparticle series was demonstrated by positioning the partial photonic bandgap of the liquid ordered structure within the spectral regions attributed to each copolymerized emitter. Enhanced and suppressed energy transfer was exhibited in each nanoparticle series, revealing control over FRET in a radioluminescent system through strategic placement of the bandgap.
1 Introduction
The energy transfer process occurring between two quantum emitters when in nanometer proximity, known as Förster resonance energy transfer (FRET), is a vital mechanism exploited in various applications, including biosensors,1–3 fluorescence microscopy techniques,4 and photonic devices.5,6 In FRET, an excited-state donor molecule nonradiatively transfers energy to a nearby ground-state acceptor molecule, typically referred to as a donor/acceptor FRET pair. FRET between quantum emitters can be manipulated through strategic engineering of the spectral properties of the donor and acceptor molecule or by adjusting the orientation of their dipole moments relative to each other.7,8 Given the direct influence on the performance and efficiency of devices such as lasers, light-emitting diodes (LEDs), and quantum computing elements, which capitalize on the unique characteristics of confined structured environments, nanophotonic control over FRET by embedding donor and acceptor molecules within a structured environment remains a highly debated topic within the domain of quantum light–matter interactions.9–21Nanophotonics plays a key role in the ongoing second quantum revolution by providing innovative ways to control and manipulate quantum states of matter at the nanoscale.22 The manipulation of radiative decay, or spontaneous photon emission, of an emitter by a nanostructured environment, such as a photonic crystal, has been well-demonstrated in the literature.23,24 However, studies aimed at characterizing the impact of such environments on the nonradiative interactions between emitters have faced significant challenges, contributing to the unresolved nature of this phenomenon. These challenges include manipulating photonic conditions while preserving the chemical and geometric aspects of the donor and acceptor molecules as well as selecting a precisely comparable reference to quantify photonic effects. Pioneering work by Andrew and Barnes revealed a linear relationship between the local density of optical states (LDOS) experienced by an emitter embedded within a confined structured environment and the rate of energy transfer between donor and acceptor molecules.9 Subsequent theoretical10 and experimental11–13 studies were reported in the literature supporting this relationship. However, contradictory evidence has also been reported, suggesting a quadratic dependence of FRET on the LDOS14 or no dependence at all.17,19–21
Photonic crystals are frequently utilized to manipulate the optical properties of embedded emitters through the photonic bandgap effect.25–27 The inherent photonic bandgap of a photonic crystal corresponds to optical modes which are not allowed to propagate through its highly-ordered structure. Consequently, the LDOS of an embedded emitter is considerably reduced at the photonic bandgap frequency.28 As a result, spontaneous emission at the bandgap frequency is inhibited, an effect characteristic of the weak coupling regime.22 This work exploits colloidal photonic crystals, known as crystalline colloidal arrays, composed of electrostatically-charged nanoparticles that spontaneously self-assemble into a face-centered cubic (FCC) structure.29–31 Due to the low refractive index contrast between the colloidal nanoparticles and liquid medium of a CCA, a CCA exhibits a partial photonic bandgap (i.e., stop-band) in the visible regime.32 CCAs are exploited in this work for their dynamic stop-band position, often referred to as the rejection wavelength (λrw), which can be shifted across the full visible spectrum by diluting the colloid.33 Colloidal photonic crystals have been extensively exploited in various photoluminescent and optoelectronic material applications34,35 and have proven valuable for investigations of light–matter interactions in nanostructured environments.36–41 Importantly, the nanostructured environment of a sterically-packed colloidal crystal has been suggested to enhance nonradiative decay mechanisms, such as intersystem crossing, by the photonic bandgap effect.41 However, investigations of radioluminescent CCAs remain limited.42–44 It is important to note that, while the current work is limited to the weak coupling regime, further engineering of the photonic environment, such as incorporating high-Q cavities, could enable access to the strong coupling regime, where hybrid light–matter states emerge.22 Exploring this transition presents exciting opportunities for future studies in coherent light–matter interactions;22 however, such phenomena are beyond the scope of this work.
In the efforts to explore the unresolved dependence of FRET on the LDOS and expand upon this phenomenon in a radioluminescent system, two nanoparticle series (n1 and n2) were synthesized, where each series contained a copolymerized scintillator and two fluorophores. Notably, n2 was synthesized using 100 times less emitter content than n1. Both n1 and n2 spontaneously self-assemble into a CCA, allowing for dynamic control over the partial photonic bandgap position by increasing the interparticle spacing, achieved by dilution with deionized water, without chemically or geometrically altering the copolymerized emitters.45 In both nanoparticle series, an anthracene derivative (AMMA) serves as the initial donor in the FRET system, resulting in radioluminescent nanoparticles. Anthracene is a well-known organic scintillator (cf. Fig. S1, ESI†) and has a scintillation efficiency comparable to that of bismuth germanium oxide (BGO), a common inorganic scintillator.46 By utilizing an X-ray excitation source, the emitters covalently incorporated within the nanoparticles and nanoparticle building blocks of the colloidal array are collectively excited as opposed to only emitters at the surface of the nanoparticles or nanoparticles at the surface of the crystal structure. Additionally, naphthalimide and rhodamine B derivatives (NMMA and RMMA, respectively) were copolymerized within the nanoparticles, extending the emission to the red region of the visible spectrum through two sequential transfers of energy, where anthracene/naphthalimide and naphthalimide/rhodamine B are known to form FRET pairs with each other.42–45,47 Upon nanoparticle excitation with an X-ray source, the AMMA donor transfers energy to NMMA, acting as an acceptor in the AMMA/NMMA FRET pair. Subsequently, NMMA acts as a donor and transfers energy to RMMA, acting as an acceptor in the NMMA/RMMA FRET pair. Radioluminescence (RL) measurements of n1 and n2 assembled into an ordered structure (OS) as the λrw was shifted across the emission of each nanoparticle series were performed, where the reference systems used to quantify photonic effects were precisely comparable in regard to nanoparticle density and emitter content.
2 Experimental
2.1 Reagents and solvents
All commercial reagents were used without further purification. Deionized (DI) water at a resistivity of 18.2 MΩ cm was acquired from a Nanopure System.2.2 Synthesis
2.3 Optical characterization
Reflectance spectra of the nanoparticles assembled in an ordered structure (OS) was collected normal to the sample surface at the [111] plane of the OS and was obtained using an Ocean Optics USB2000 fiber coupled spectrometer equipped with an Ocean Optics bifurcated fiber optic bundle (cf. Fig. 1E–G and 3E–G). A white light source (Ocean Optics LS-1-CAL) was attached to the input arm of the fiber optic bundle while the output arm was attached to the spectrometer. It is important to note that reflectance peaks in the blue region of the visible spectrum exhibited significant scattering (cf. Fig. 1E and 3E). An Amptek Inc. mini-X X-ray tube equipped with a tungsten (W) target operating at a tube voltage of 158 kV and a tube current of 25 μA was used to irradiate the colloid for RL spectral collections (cf. Fig. 1–3 and Fig. S1, S2, ESI†). A Horiba Jobin-Yvon MicroHR monochromator and a Horiba Jobin-Yvon Synapse cooled CCD detector was used to collect the RL spectra of the colloid. The colloid was irradiated with X-rays normal to the sample surface at the [111] plane of the OS. The RL of the colloid in the forward direction (i.e., along the X-ray propagation axis) was redirected by an aluminum mirror (Thor Labs PF20-03-G01) aligned such that the transmitted light was reflected by 90 degrees into the CCD detector and the signal was collected on a grating with 600 line per mm and a blaze of 500 nm. This geometry allowed for the collection of RL spectra without placing the detector directly behind the sample, minimizing direct X-ray exposure to the detector. The exposure time and detector slit width for all samples was 30 seconds and 1 mm, respectively, unless otherwise noted. RL spectra were collected without correcting for the wavelength-dependent sensitivity of the CCD detector. Photographs of the OS were obtained with a Canon Rebel Ti1 camera (cf. Fig. 2D and E).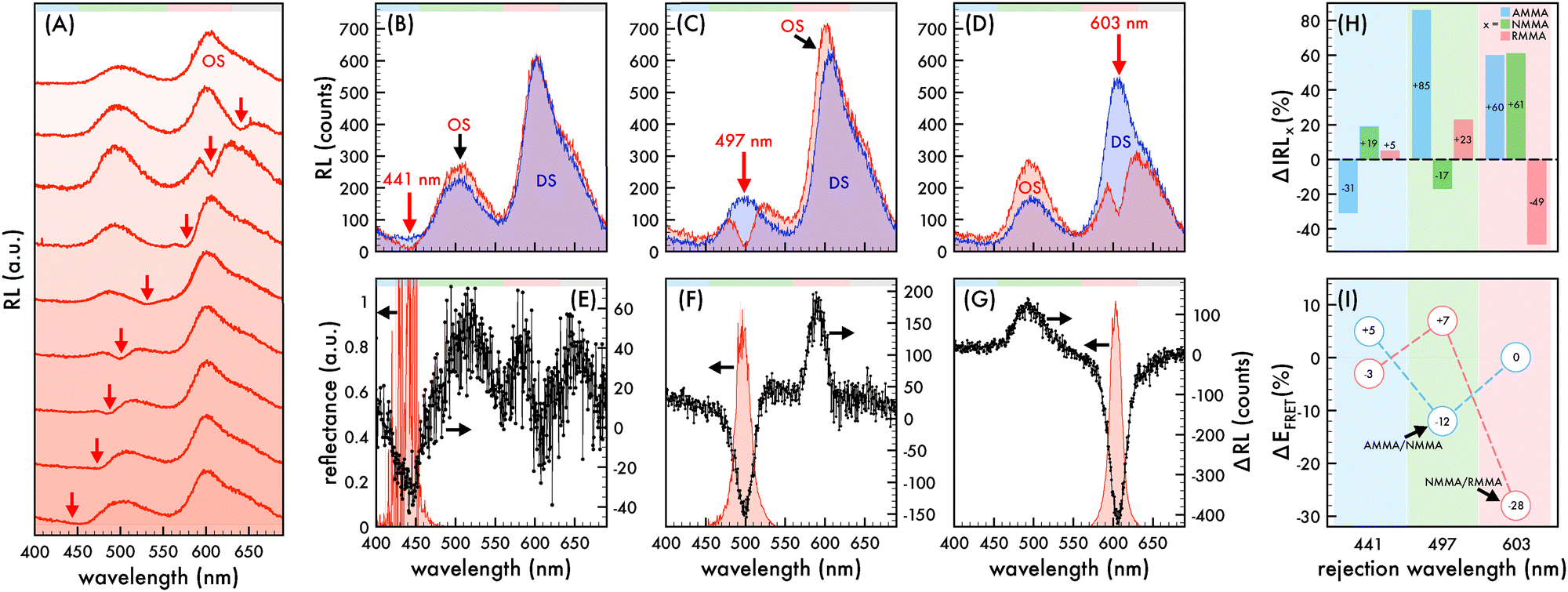 | ||
| Fig. 1 Nanophotonic manipulation of n1 radioluminescence and FRET efficiency. (A) Radioluminescence (RL) spectra of n1 nanoparticles assembled in an ordered structure (OS) as the rejection wavelength (λrw, red arrow) was shifted across the emission spectrum. RL spectra of n1 assembled in an OS (red) in comparison to that of corresponding disordered structures (DS, blue) as the λrw (denoted by a red arrow) was positioned to coincide with AMMA, NMMA, and RMMA emission at (B) 441 nm, (C) 497 nm, and (D) 603 nm, respectively. The difference in RL spectra (ΔRL, black dotted line) of n1 in an OS in comparison to that of the corresponding DS as the λrw was positioned at (E) 441 nm, (F) 497 nm, and (G) 603 nm. The observed reflectance peak of each OS is presented in (E)–(G). Lightly shaded bars at the top of (A)–(G) were included as a visual aid to identify the spectral regions attributed to AMMA (blue), NMMA (green), and RMMA (red) emission. (H) Change (%) in integrated RL (ΔIRL) attributed to AMMA (blue), NMMA (green), and RMMA (red) of n1 assembled in an OS in reference to that of the corresponding DS as the λrw was positioned at 441 nm, 497 nm, and 603 nm. (I) Difference in Förster resonance energy transfer (FRET) efficiency (ΔEFRET) of the AMMA/NMMA (blue circles) and NMMA/RMMA (red circles) pairs copolymerized in n1 when the nanoparticles were assembled in an OS in reference to that of the corresponding DS as the λrw was positioned at 441 nm, 497 nm, and 603 nm. Light shading at each λrw position is included in (H) and (I) as a visual aid to identify the overlapped emitter. | ||
 | ||
| Fig. 2 Comparison of the integrated radioluminescence of n1 assembled in an ordered structure and corresponding disordered structure with dilution. Integrated radioluminescence (IRL) normalized by the concentration of nanoparticles in each dilution of the n1 ordered structure (OS, red circles) and disordered structure (DS, blue circles) emission attributed to (A) RMMA, (B) NMMA, and (C) AMMA as the rejection wavelength (λrw) of the OS was shifted through the visible spectrum. Lightly shaded bars at the top of (A)–(C) were included as a visual aid to identify the spectral regions attributed to AMMA (blue), NMMA (green), and RMMA (red) emission. As an aid to the eye, a trendline for the DS IRL (blue dashed line) is included in (A)–(C). Photographs of the n1 OS under (D) white light and (E) X-ray exposure, where the λrw of the left and right half of the OS droplet is indicated. | ||
 | ||
| Fig. 3 Nanophotonic manipulation of n2 radioluminescence and FRET efficiency. (A) Radioluminescence (RL) spectra of n2 nanoparticles assembled in an ordered structure (OS) as the rejection wavelength (λrw, red arrow) was shifted across the emission spectrum. RL spectra of n2 assembled in an OS (red) in comparison to that of corresponding disordered structures (DS, blue) as the λrw (denoted by a red arrow) was positioned to coincide with AMMA, NMMA, and RMMA emission at (B) 424 nm, (C) 505 nm, and (D) 592 nm, respectively. The difference in RL spectra (ΔRL, black dotted line) of n2 in an OS in comparison to that of the corresponding DS as the λrw was positioned at (E) 424 nm, (F) 505 nm, and (G) 592 nm. The observed reflectance peak of each OS is presented in (E)–(G). Lightly shaded bars at the top of (A)–(G) were included as a visual aid to identify the spectral regions attributed to AMMA (blue), NMMA (green), and RMMA (red) emission. (H) Change (%) in integrated RL (ΔIRL) attributed to AMMA (blue), NMMA (green), and RMMA (red) of n2 assembled in an OS in reference to that of the corresponding DS as the λrw was positioned at 424 nm, 505 nm, and 592 nm. (I) Difference in Förster resonance energy transfer (FRET) efficiency (ΔEFRET) of the AMMA/NMMA (blue circles) and NMMA/RMMA (red circles) pairs copolymerized in n2 when the nanoparticles were assembled in an OS in reference to that of the corresponding DS as the λrw was positioned at 424 nm, 505 nm, and 592 nm. Light shading at each λrw position is included in (H) and (I) as a visual aid to identify the overlapped emitter. | ||
3 Results and discussion
Two series of radioluminescent polystyrene-based nanoparticles (n1 and n2) were synthesized by a general emulsion polymerization procedure with copolymerized derivatives of anthracene, naphthalimide, and rhodamine B (AMMA, NMMA, and RMMA, respectively) described elsewhere.45 The n2 nanoparticles were synthesized with 100× less emitter content than utilized in the synthesis of n1 such that trends observed in the n1 nanoparticles could be corroborated in nanoparticles with less emitter content. The photophysical properties of the individual emitters, confirmation of their incorporation within the n1 and n2 nanoparticles, and characterization of the n1 and n2 nanoparticles are reported elsewhere.45 Briefly, AMMA was incorporated as the initial donor in the triple-emitter nanoparticles, where the AMMA scintillator exhibits blue emission upon X-ray excitation. When the AMMA donor is within close proximity to the NMMA acceptor, the excited-state AMMA donor nonradiatively transfers energy to the ground-state NMMA acceptor. Subsequently, the NMMA emitter acts as a donor when in close proximity to the RMMA acceptor, where the excited-state NMMA donor nonradiatively transfers energy to the ground-state RMMA acceptor. In this way, two sequential energy transfers occur within the n1 and n2 nanoparticles upon X-ray excitation (cf. Fig. S2, ESI†), where the AMMA/NMMA pair and NMMA/RMMA pair are known to exhibit FRET.42–44 The synthesized n1 and n2 nanoparticles are monodisperse and colloidal stable such that they spontaneously self-assembled into a CCA.453.1 Nanophotonic manipulation of radioluminescence and X-ray-induced FRET efficiency
Manipulation of the radioluminescent spectral properties of the n1 and n2 nanoparticles assembled in a liquid ordered structure (OS) by the photonic bandgap effect compared to that of a precisely comparable disordered structure (DS) is presented in Fig. 1 and 3, respectively. It should be noted that all RL spectra of n1 assembled in an OS was acquired using a small amount of n1 nanoparticles (1.36% (v/v)) in a polystyrene-based CCA containing no copolymerized emitters due to the large quantity of emitters in the nanoparticles. As described elsewhere,45 the size and stability of the n1 nanoparticles and polystyrene-based nanoparticles with no copolymerized emitters were similar such that it was assumed that there was no preferential ordering of the particles when mixed and, thus, the placement of n1 nanoparticles within the OS lattice was determined by the respective volume fraction (1.36%). At this volume fraction, the likelihood that two n1 nanoparticles were located next to each other in the OS was 1 in 5405. Additionally, it should be noted that all RL spectra was obtained at the (111) face of the liquid n1 and n2 OS due to the alignment of the (111) face of the FCC crystal to the wall of a cuvette.50,51 Thus, the observed λrw of the OS can be ascribed to the {111} partial photonic bandgap and the interplanar spacing (d111), lattice parameter (a), and nearest neighbor spacing (ann) could be estimated and is reported elsewhere.45A tungsten (W) X-ray source operating at a tube voltage and current of 25 kV and 158 μA, respectively, was utilized for all RL spectral collections. Both the n1 and n2 OS exhibited emission attributed to AMMA, NMMA, and RMMA when excited with an X-ray source (cf. Fig. 1 and 3), where the RL spectral properties of n1 and n2 were similar to their respective PL spectral properties described elsewhere.45 In both n1 and n2, very minor emission attributed to AMMA was observed at ca. 405 nm and 425 nm. The maximum emission of n1 was at 602 nm and attributed to RMMA accompanied by a smaller peak at 500 nm attributed to NMMA. The maximum emission of n2 was at 510 nm and attributed to NMMA accompanied by a smaller peak at 590 nm attributed to RMMA. Additionally, there was a shoulder at ca. 650 nm in the RL spectra of n1 and n2 attributed to the polystyrene host polymer (cf. Fig. S3, ESI†) that overlapped the emission attributed to RMMA. Fig. 1A and 3A present the radioluminescent spectral characteristics of n1 and n2 nanoparticles assembled in an OS as the partial photonic bandgap, or observed λrw (indicated by a red arrow), was shifted through the emission of each respective OS by increasing the interparticle spacing of the OS through dilution with deionized water. Similar to observations in the photoluminescence of the n1 and n2 OSs,45 suppressed emission was detected where the λrw overlapped the emission of the n1 and n2 OS. The RL of the n1 and n2 nanoparticles assembled in an OS was compared to a reference disordered structure (DS) at the same nanoparticle density and emitter content for each λrw position. A precisely comparable DS was fabricated for each λrw position of the n1 and n2 OS by disrupting the long-range order of the self-assembled nanoparticles. This was achieved by adding a small amount of ionic impurity to the OS, which disintegrated the crystal structure while maintaining the concentration of nanoparticles and emitter content for a controlled investigation of purely photonic effects. By comparing the difference in RL (ΔRL) of the n1 OS compared to the DS at each λrw condition, a −71%, −77%, and −71% decrease in n1 OS emission at the λrw integrated over the spectral regime corresponding to the full width at half maximum (FWHM) of the reflectance peak was revealed when the λrw was at 441 nm, 497 nm, and 603 nm, respectively (cf. Fig. 1B–G). Through a similar comparison, a −40%, −54%, and −65% decrease in n2 OS emission at the λrw integrated over the spectral regime corresponding to the FWHM of the reflectance peak was detected when the λrw of the n2 OS was at 424 nm, 505 nm, and 592 nm, respectively (cf. Fig. 3B–G). The suppressed emission at the λrw coincidence is indicative of the reduced LDOS at the λrw and a manifestation of the Purcell effect.52 The Purcell effect describes the enhancement or suppression of spontaneous emission of an emitter as a result of the interaction between the emitter and its surrounding environment. When the partial photonic bandgap reduces the probability of the emitter to decay by radiative processes, such as photon emission, it is hypothesized that the likelihood of the emitter to decay by nonradiative processes, such as FRET, is enhanced. Investigations of the photoluminescence of the n1 and n2 OS supporting this hypothesis have been previously reported;45 however, the RL of the n1 and n2 OS has not been explored. Fig. 1H and 3H present the change in integrated RL (ΔIRL) attributed to each copolymerized emitter in the n1 and n2 OS compared to that of the precisely comparable DS, respectively, as the λrw of each OS was shifted through the emission spectrum. Additionally, the FRET efficiency (EFRET) of each donor/acceptor pair could be estimated by eqn (1) (cf. Tables S1–S4, ESI†), where the donor and acceptor emission intensity is denoted as ID and IA, respectively. The change in EFRET (ΔEFRET) as the λrw was shifted to overlap each emitter in the n1 and n2 OS is presented in Fig. 1I and 3I, respectively.
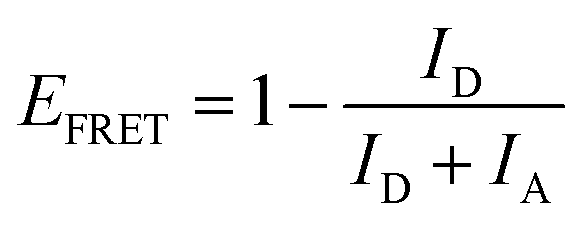 | (1) |
To confirm that the trends observed in the n1 nanoparticles were due to photonic effects, the integrated RL attributed to each emitter in the n1 OS was compared to that of corresponding DSs as the λrw of the OS was shifted from ca. 451 nm to 810 nm (cf. Fig. 2A–C). For this study, a n1 DS was fabricated at the same initial particle density of the n1 OS and each structure was diluted by deionized water in similar quantities such that the nanoparticle densities remained closely matched. The integrated RL presented in Fig. 2 was normalized by the number of nanoparticles in the colloid at each dilution. It is important to note that both the sampled area and volume of the structures remained invariant through the dilution. Thus, as the structures were diluted, a decreased number of nanoparticles were optically sampled. This resulted in a decreased amount of scattering and increased amount of light able to pass through the colloid at each dilution. While the integrated RL of each emitter in the DS exhibited a relatively steady positive increment with increasing dilution, the integrated RL attributed to each emitter in the OS did not exhibit this same trend. Instead the OS exhibited fluctuations of increased or decreased emission, depending on the position of the partial photonic bandgap. It is important to note that the integrated RL of AMMA did not exhibit a drop below the DS trendline at the initial λrw condition owing to the λrw located at 451 nm, which is slightly red-shifted with respect to the spectral region attributed to AMMA emission and yielded a slightly enhanced AMMA emission. Nonetheless, regions of decreased integrated RL compared to that of the DS were observed for the NMMA and RMMA emitters as the λrw was shifted to overlap their respective regimes of emission. Additionally, regions of enhanced integrated RL compared to that of the DS were observed for all three emitters when the λrw overlapped their respective FRET pair. Specifically, when the λrw was near the emission attributed to the donor AMMA, enhanced emission attributed to the acceptor/donor NMMA and acceptor RMMA was evident. As the λrw was red-shifted to overlap the emission of acceptor/donor NMMA, increased emission attributed to the donor AMMA and acceptor RMMA was observed. By further shifting the λrw to overlap the emission attributed to the acceptor RMMA, increased emission attributed to the donor AMMA and acceptor/donor NMMA was detected. Finally, as the λrw of the OS was red-shifted past the RL spectrum of the n1 nanoparticles at ca. >700 nm, the integrated RL of AMMA, NMMA, and RMMA closely matched that of the DS. The enhancement and suppression of FRET between the copolymerized emitters in the undiluted n1 OS is presented in Fig. 2D and E, where photographs of a liquid droplet of the n1 OS under white light and X-ray excitation are presented, respectively. The left half of the n1 OS droplet had a λrw at 450 nm and exhibited an optically brighter output due to the enhanced total emission when excited with an X-ray source. The right half of the n1 OS droplet had a λrw at 600 nm, resulting in an optically dimer output due to the suppressed total emission when excited with an X-ray source.
Corroborating the results observed in investigations of the photoluminescence of the n1 and n2 OSs,45 the RL of the FRET acceptor in n1 and n2 was enhanced when that of the corresponding FRET donor was suppressed by the partial photonic bandgap. In the opposite case, the RL of the FRET donor n1 and n2 was enhanced when that of the corresponding FRET acceptor was suppressed by the partial photonic bandgap.
4 Conclusions
These results support and expand upon previously reported evidence that the nonradiative transfer of energy between multiple emitters can be manipulated and controlled by the photonic bandgap effect. Similar to observations of the photoluminescence of the nanoparticle series investigated in this work, modulations of the efficiency and direction of the X-ray-induced FRET between two FRET pairs (AMMA/NMMA and NMMA/RMMA) copolymerized within two radioluminescent nanoparticle series (n1 and n2) assembled into CCAs were demonstrated. The efficiency of the X-ray-induced FRET between a donor/acceptor pair could be enhanced by overlapping the donor emission with the partial photonic bandgap. However, by overlapping the acceptor emission with the partial photonic bandgap, the efficiency of X-ray-induced FRET between a donor/acceptor pair could be suppressed. These results support the connection between the LDOS and FRET between emitters and present evidence that this phenomenon can be leveraged in radioluminescent systems to methodically manipulate the quantum interactions between multiple emitters. To the best of our knowledge, this is the first example of FRET manipulation by the photonic bandgap effect in a radioluminescent CCA.Author contributions
H. J. contributed to the conceptualization, methodology, investigation, data curation, visualization, writing – original draft, and writing – review & editing. Y. B. contributed to the resources and methodology. S. F. contributed to the conceptualization, methodology, data curation, visualization, writing – original draft, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Gregg-Graniteville Foundation and the National Science Foundation (OIA-1632881) for financial support.References
- I. L. Medintz, A. R. Clapp, H. Mattoussi, E. R. Goldman, B. Fisher and J. M. Mauro, Nat. Mater., 2003, 2, 630–638 CrossRef CAS PubMed.
- R. D. Fritz, M. Letzelter, A. Reimann, K. Martin, L. Fusco, L. Ritsma, B. Ponsioen, E. Fluri, S. Schulte-Merker and J. van Rheenen, Sci. Signaling, 2013, 6, rs12 CrossRef PubMed.
- G. C. H. Mo, C. Posner, E. A. Rodriguez, T. Sun and J. Zhang, Nat. Commun., 2020, 11, 1848 CrossRef CAS PubMed.
- E. A. Jares-Erijman and T. M. Jovin, Nat. Biotechnol., 2003, 21, 1387–1395 CrossRef CAS PubMed.
- L. Cerdán, E. Enciso, V. Martín, J. Bañuelos, I. López-Arbeloa, A. Costela and I. García-Moreno, Nat. Photonics, 2012, 6, 621–626 CrossRef.
- S. Buckhout-White, C. M. Spillmann, W. R. Algar, A. Khachatrian, J. S. Melinger, E. R. Goldman, M. G. Ancona and I. L. Medintz, Nat. Commun., 2014, 5, 5615 CrossRef CAS PubMed.
- T. Förster, Ann. Phys., 1948, 437, 55–75 CrossRef.
- D. L. Andrews and J. Rodriguez, J. Chem. Phys., 2007, 127, 084509 CrossRef PubMed.
- P. Andrew and W. L. Barnes, Science, 2000, 290, 785–788 CrossRef CAS PubMed.
- H. T. Dung, L. Knoll and D. G. Welsch, Phys. Rev. A: At., Mol., Opt. Phys., 2002, 65, 043813 CrossRef.
- P. Ghenuche, J. de Torres, S. B. Moparthi, V. Grigoriev and J. Wenger, Nano Lett., 2014, 14, 4707–4714 CrossRef CAS PubMed.
- D. Weeraddana, M. Premaratne, S. D. Gunapala and D. L. Andrews, J. Chem. Phys., 2017, 147, 074117 CrossRef PubMed.
- S. Patra, J.-B. Claude and J. Wenger, ACS Photonics, 2022, 9, 2109–2118 CrossRef CAS.
- T. Nakamura, M. Fujii, S. Miura, M. Inui and S. Hayashi, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 045302 CrossRef.
- B. Kolaric, K. Baert, M. Van der Auweraer, R. A. L. Vallee and K. Clays, Chem. Mater., 2007, 19, 5547–5552 CrossRef CAS.
- L. Gonzalez-Urbina, K. Baert, B. Kolaric, J. Perez-Moreno and K. Clays, Chem. Rev., 2012, 112, 2268–2285 CrossRef CAS PubMed.
- M. J. A. de Dood, J. Knoester, A. Tip and A. Polman, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 115102 CrossRef.
- F. T. Rabouw, S. A. den Hartog, T. Senden and A. Meijerink, Nat. Commun., 2014, 5, 3610 CrossRef PubMed.
- C. Blum, N. Zijlstra, A. Lagendijk, M. Wubs, A. P. Mosk, V. Subramaniam and W. L. Vos, Phys. Rev. Lett., 2012, 109, 203601 CrossRef PubMed.
- M. Wubs and W. L. Vos, New J. Phys., 2016, 18, 053037 CrossRef.
- G. Rosolen, B. Maes, P. Y. Chen and Y. Sivan, Phys. Rev. B, 2020, 101, 155401 CrossRef CAS.
- B. Kolaric, B. Maes, K. Clays, T. Durt and Y. Caudano, Adv. Quantum Technol., 2018, 1, 1800001 CrossRef.
- K. H. Drexhage, J. Lumin., 1970, 1(2), 693–701 CrossRef.
- W. L. Barnes, J. Mod. Opt., 1998, 45, 661–669 CrossRef CAS.
- T. Okubo, Prog. Polym. Sci., 1993, 18, 481–517 CrossRef CAS.
- E. Yablonovitch, Phys. Rev. Lett., 1987, 58, 2059–2062 CrossRef CAS PubMed.
- S. John, Phys. Rev. Lett., 1987, 58, 2486–2489 CrossRef CAS PubMed.
- J. D. Joannopoulos, S. G. Johnson, J. N. Winn and R. D. Meade, Photonic Crystals: Molding the Flow of Light, Princeton University Press, 2nd edn, 2008 Search PubMed.
- P. A. Hiltner, Y. S. Papir and I. M. Krieger, J. Phys. Chem., 1971, 75, 1881–1886 CrossRef CAS.
- P. A. Hiltner and I. M. Krieger, J. Phys. Chem., 1969, 73, 2386 CrossRef CAS.
- N. Clark, A. Hurd and B. Ackerson, Nature, 1979, 281, 57–60 CrossRef CAS.
- P. Rundquist, P. Photinos, S. Jagannathan and S. Asher, J. Chem. Phys., 1989, 91, 4932–4941 CrossRef CAS.
- C. López, Adv. Mater., 2003, 15, 1679–1704 CrossRef.
- J. R. Lawrence, Y. Ying, P. Jiang and S. H. Foulger, Adv. Mater., 2006, 18, 300–303 CrossRef CAS.
- J. R. Lawrence, G.-H. Shim, P. Jiang, M. Han, Y. Ying and S. H. Foulger, Adv. Mater., 2005, 17, 2344–2349 CrossRef CAS.
- J. Martorell and N. M. Lawandy, Phys. Rev. Lett., 1990, 65, 1877–1880 CrossRef CAS PubMed.
- B. Tong, P. John, Y. Zhu, Y. Liu, S. Wong and W. Ware, J. Opt. Soc. Am. B, 1993, 10, 356–359 CrossRef CAS.
- M. Megens, J. E. G. J. Wijnhoven, A. Lagendijk and W. L. Vos, Phys. Rev. A: At., Mol., Opt. Phys., 1999, 59, 4727–4731 CrossRef CAS.
- K. Shibata, H. Kimura, A. Tsuchida and T. Okubo, Colloid Polym. Sci., 2006, 285, 127–133 CrossRef CAS.
- M. Khokhar, Priya and R. V. Nair, Phys. Rev. A, 2020, 102, 013502 CrossRef CAS.
- L. González-Urbina, J. Perez-Moreno, K. Clays and B. Kolaric, Mol. Phys., 2016, 114, 2248–2252 CrossRef.
- M. K. Burdette, Y. P. Bandera, G. M. Gray and S. H. Foulger, Adv. Opt. Mater., 2019, 7, 1801142 CrossRef.
- M. K. Burdette, H. W. Jones, Y. Bandera and S. H. Foulger, Opt. Mater. Express, 2019, 9, 1416–1429 CrossRef CAS.
- S. Mell, H. W. Jones, Y. P. Bandera and S. H. Foulger, Langmuir, 2022, 38, 10089–10097 CrossRef CAS PubMed.
- H. W. Jones, Y. Bandera and S. H. Foulger, J. Mater. Chem. C, 2025, 13, 1694–1703 RSC.
- S. Kubota, T. Motobayashi, J. Ruan, T. Murakami, J. Kasagi and T. Shimizu, IEEE Trans. Nucl. Sci., 1987, 34, 438–441 Search PubMed.
- H. W. Jones, Y. Bandera, I. K. Foulger, I. Luzinov and S. H. Foulger, Adv. Photonics Res., 2024, 5, 2300296 CrossRef CAS.
- M. E. Woods, J. S. Dodge, I. M. Krieger and P. E. Pierce, J. Paint Technol., 1968, 40, 541 CAS.
- Y. S. Papir, M. E. Woods and I. M. Krieger, J. Paint Technol., 1970, 42, 571–578 CAS.
- Y. Monovoukas and A. P. Gast, J. Colloid Interface Sci., 1989, 128, 533–548 CrossRef CAS.
- Y. Monovoukas and A. P. Gast, Phase Transitions, 1990, 21, 183 CrossRef CAS.
- E. Purcell, Phys. Rev., 1946, 69, 681 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc05291a |
| This journal is © The Royal Society of Chemistry 2025 |