
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recyclable chloride trapping and sustained nicotinate release monitored by photoluminescence of Eu3+-doped layered yttrium hydroxide†
Yoonjae
Choi
,
Myeongjin
Kang
and
Song-Ho
Byeon
 *
*
Department of Applied Chemistry, College of Applied Science and Institute of Natural Sciences, Kyung Hee University, Gyeonggi 17104, South Korea. E-mail: shbyun@khu.ac.kr
First published on 29th January 2025
Abstract
Nicotinic acid (NA; also known as niacin or vitamin B3), an essential nutrient for humans, was inserted into the interlayer spaces of Eu3+-doped layered yttrium hydroxide (LYH:Eu). This hybrid system, NA-LYH:Eu, demonstrated remarkable efficiency in the storage of NA, offering long-term stability, reusability, and controlled, sustained release capabilities. Notably, the intercalated NA sensitized the luminescence of Eu3+ ions via the transfer of UV-energy absorbed for its π → π* transition from the S0 ground state to the S1 excited state, leading to the characteristic 5D0 → 7FJ (J = 1–4) transitions. The resulting bright red emission could be reversibly quenched and restored through repeated NA release (chloride uptake) and re-inclusion cycles within the LYH:Eu interlayers, demonstrating the potential application as a chloride-induced corrosion inhibitor. The release rate of NA in a phosphate buffer solution could be modulated by tuning the crystallinity and particle size of the LYH:Eu host. When highly crystalline NA-LYH:Eu was employed, the in vitro release of NA continued beyond 5 days without reaching equilibrium. Concurrently, the red emission from a colloidal solution containing NA-LYH:Eu particles gradually diminished as the release progressed, providing a visually observable indicator. This luminescence ‘on/off’ behavior offers a practical method for in situ visual monitoring of NA inclusion and release.
1. Introduction
Nicotinic acid (NA), also known as niacin or vitamin B3, is a derivative of benzoic acid composed of a carboxyl group and a pyridine ring, a nitrogen-containing six-membered heterocycle with strong electronegativity. As an essential human nutrient and a highly water-soluble vitamin, NA plays a crucial role in the synthesis of key nucleotides such as nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP).1 NA and its derivatives are widely used in topical therapeutics and cosmetic products due to their antioxidant, anti-inflammatory, and immunomodulatory properties, as well as their dermatological benefits, including moisture retention and skin brightening.2,3 However, high doses of NA can lead to undesirable side effects, such as vasodilation, nausea, abdominal pain, diarrhea, cutaneous flushing, and itching.4,5 Additionally, NA's low thermal stability results in its degradation during high-temperature food processing, causing the loss of this essential vitamin.6 Thus, developing a delivery system that can protect NA from decomposition and provide sustained release is highly desirable to minimize its clinical and dermatological side effects.NA also exhibits catalytic properties due to its potential Lewis base (pyridine ring) and Brønsted acid (carboxyl group) functionalities.7 These characteristics enable its use as an organocatalyst, where immobilization on silica extends its functionality from homogeneous to reusable heterogeneous catalytic conditions for organic synthesis.8 Moreover, NA serves as a bridging ligand in metal–organic frameworks (MOFs), enhancing therapeutic efficacy through synergistic effects.9 The strong coordination ability of NA-related acids has also been utilized to immobilize lanthanide complexes in organic–inorganic hybrid materials, where the carboxyl group moderates reactivity toward hydrolysis and condensation and the pyridine ring coordinates with lanthanide ions, sensitizing their luminescence.10–12 Additionally, NA has demonstrated anti-corrosion effects on steel by forming a protective layer on γ-FeOOH surfaces and isolating the substrate from corrosive agents.13
In recent years, encapsulating organic molecules within inorganic matrices has emerged as an effective strategy to overcome their limitations under conventional conditions. These organic–inorganic hybrids have been utilized as delivery systems for therapeutic drugs, genes, cosmetic ingredients, and composite materials in polymeric matrices.14–17 Layered double hydroxides (LDHs) are prominent examples of inorganic matrices capable of releasing organic anions in a controlled and sustained manner.18 The positively charged layers of LDHs and their ion exchange capacity allow them to accommodate anionic molecules, including organic carboxylates.19,20 NA, a member of the pyridinecarboxylic acid group, can also be readily intercalated into the interlayer of LDHs to form NA-LDH hybrids. This characteristic can be applied to separate NA, which is discharged into wastewater systems each year, from aqueous solutions.21 The interlayer space of LDHs provides NA with significantly enhanced chemical, thermal, and photostabilities, as well as control and sustainability in its release. For instance, NA-LDH hybrids have been explored for sustained release in topical applications,22 as corrosion inhibitors in coastal construction,23 and for their organocatalytic selectivity and recyclability. However, high NA concentrations can disrupt the LDH layered structure, limiting its loading capacity.24
Recently, layered rare-earth hydroxides (LRHs) have gained attention as alternative inorganic matrices for organic–inorganic hybrid systems. Similar to LDHs, LRHs have a lamellar structure consisting of hydroxocation layers alternated with charge-compensating anion layers. Their general formula is represented as [RE2(OH)5·nH2O]+(Xm−)1/m (RE = rare earth elements, X = interlayer anions).25 LRHs are known for their ease of preparation, anion exchange capability, and high stability under varying pH and pressure conditions, making them suitable for applications such as bioimaging and environmental adsorption.26,27 Furthermore, the optical properties of RE ions in LRH layers have shown great potential for luminescent delivery systems of bioactive molecules.28–30 In this study, Eu3+-doped layered yttrium hydroxide (LYH:Eu) was investigated as a matrix for the efficient loading and sustained release of NA. Notably, interlayer NA ions acted as ‘antenna molecules’, effectively sensitizing Eu3+ luminescence. This luminescent ‘on/off’ behavior enabled real-time probing of NA inclusion and release within the interlayer spaces of LYH:Eu.
2. Experimental
2.1 Synthesis of Eu3+-doped layered yttrium hydroxynitrate
Eu3+-doped layered yttrium hydroxynitrate (LYH:Eu) with nitrate (NO3−) as the interlayer anion, Y1.80Eu0.20(OH)5NO3·nH2O, was synthesized via a precipitation method. Stoichiometric amounts of Y(NO3)3·6H2O (36 mmol) and Eu(NO3)3·6H2O (4 mmol) were mixed at a molar ratio of Y![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Eu = 9:1, along with an excess amount of NaNO3 (520 mmol). These were completely dissolved in 400 mL of deionized (DI) water, followed by the addition of hexamethylenetetramine (HMT, 5.606 g) under continuous stirring. To obtain LYH:Eu samples with different crystallinities and particle sizes, the resulting solution was heated at 45 °C for 6 h and at 90 °C for 12 h under mild stirring. After cooling to room temperature (RT), the LYH:Eu precipitate was collected via centrifugation and washed three times with DI water. The final product was dried at 60 °C for 12 h.
:Eu = 9:1, along with an excess amount of NaNO3 (520 mmol). These were completely dissolved in 400 mL of deionized (DI) water, followed by the addition of hexamethylenetetramine (HMT, 5.606 g) under continuous stirring. To obtain LYH:Eu samples with different crystallinities and particle sizes, the resulting solution was heated at 45 °C for 6 h and at 90 °C for 12 h under mild stirring. After cooling to room temperature (RT), the LYH:Eu precipitate was collected via centrifugation and washed three times with DI water. The final product was dried at 60 °C for 12 h.
2.2 NA intercalation and chloride trapping in the interlayer space of LYH:Eu
NA-intercalated LYH:Eu, Y1.8Eu0.2(C6H4NO2)n·mH2O (NA-LYH:Eu), was prepared via an exchange reaction between nitrate and NA anions in an aqueous solution. Equimolar amounts of C6H5NO2 (5.0 mmol) and NaOH (5.0 mmol) were dissolved in 50 mL of DI water under moderate stirring at RT. Subsequently, 0.5 g of LYH:Eu powder (molar ratio of nitrate:NA = 1:3) was dispersed in the prepared NA solution. The colloidal mixture was stirred continuously at 100 °C for 24 h. The NA-LYH:Eu powder was recovered by centrifugation, washed several times with DI water, and dried at 60 °C for 12 h. For reusability tests, NA-LYH:Eu was reacted with a 0.5 M aqueous NaCl solution for 12 h at RT. The recovered powder was then reused by reacting it with a 0.1 M aqueous NA solution for 12 h at RT.
2.3 Release of NA from NA-LYH:Eu in PBS
The release profile of NA from the interlayer of NA-LYH:Eu was investigated in phosphate buffer solution (PBS, pH 6.5). A known amount of NA-LYH:Eu powder was added to PBS to achieve a concentration of 1.0 g L−1. The resulting suspension was stirred constantly at RT, and the supernatant was collected at specific intervals (1 min, 10 min, 1 h, 3 h, 12 h, 1 day, 2 days, 3 days, 4 days, and 5 days) using a 0.45 μm syringe filter. The UV absorbance of the filtrates was measured at λmax = 262 nm to determine the concentration of released NA. The concentration was calculated using a calibration curve, and all measurements were performed in triplicate. The percentage of NA release was calculated based on the initial NA content in NA-LYH:Eu and the measured concentration of released NA.2.4 Characterization
Powder X-ray diffraction (XRD) patterns of NA-LYH:Eu were recorded before, during, and after NA intercalation and release using a Bruker D8 Advance diffractometer with Cu-Kα radiation. The size and morphology of NA-LYH:Eu were observed using field-emission scanning electron microscopy (FE-SEM) on a Carl Zeiss LEO SUPRA 55 electron microscope operated at 30 kV. Samples for FE-SEM analysis were coated with Pt–Rh for 180 s under vacuum. Thermogravimetric analysis (TGA) was conducted in air to estimate the amounts of interlayer nitrate, NA, and water, using a Rigaku STA8122 at a heating rate of 5 °C min−1. Fourier-transform infrared (FT-IR) spectra were recorded over the range of 4000–400 cm−1 using the KBr pellet technique on a Thermo Scientific Nicolet iS10 spectrophotometer. UV absorbance measurements were performed using Shimadzu Multispec-1501 and LAMBDA 35 UV-vis spectrophotometers. Photoluminescence spectra of NA-LYH:Eu before, during, and after NA release were obtained using an FP-6600 spectrophotometer (JASCO) equipped with a xenon flash lamp.3. Results and discussion
3.1 Synthesis of LYH:Eu
The LRH family is known for its biocompatibility, low cytotoxicity, and highly efficient, stable luminescence.26,30 Based on these attributes, LYH was chosen as a representative ion–exchangeable carrier to accommodate NA anions within its interlayer space in this study. In particular, LYH was doped with Eu3+ to enable the in situ monitoring of NA inclusion and release within the interlayer space of the LYH carrier, as NA effectively sensitizes luminescence when coordinated with trivalent RE ions.10,11 Although Tb3+ is also commonly adopted as an activator, Eu3+ was selected in this study to avoid the effect of chloride ions on its photoluminescence.
Fig. 1a shows the XRD pattern of the prepared LYH:Eu host. The series of strong and sharp (00l) reflections indicate a well-crystallized typical layered structure. Variations in reaction time, HMT concentration, and/or solution temperature during synthesis led to noticeable differences in the crystallinity and particle size of LYH:Eu. The observed average interlayer distance, approximately 9.2 Å, is consistent with previously reported values for LYH (9.18 Å).31 Minor discrepancies could be attributed to Eu3+ doping within the LYH layers and variability in the amount of interlayer water. The experimental molar ratio determined by ICP analysis, Y:Eu = 0.903(1):0.104(2), was close to the nominal composition of LYH:Eu. Fig. 2a presents the TGA curve of LYH:Eu, which reveals three distinct weight-loss events. These are centered at approximately 100 °C, 300 °C, and 520 °C and correspond to the loss of surface and interlayer water, the dehydroxylation of hydroxide layers, and the decomposition of nitrate anions to form Y2O3:Eu.32 The total observed weight loss of 36.2% is in close agreement with the calculated value of 36.17%, corresponding to the thermal decomposition of Y1.8Eu0.2(OH)5NO3·2.0H2O leaving Y1.8Eu0.2O3. The FT-IR spectra of LYH:Eu are displayed in Fig. S1a (ESI†). Broad peaks observed at approximately 3300–3500 cm−1 and 1638 cm−1 are assigned to the stretching and bending modes of hydroxyl groups and interlayer water molecules, respectively. The strong band at around 1380 cm−1 corresponds to the asymmetric stretching (ν3) mode of nitrate within the interlayer space.33
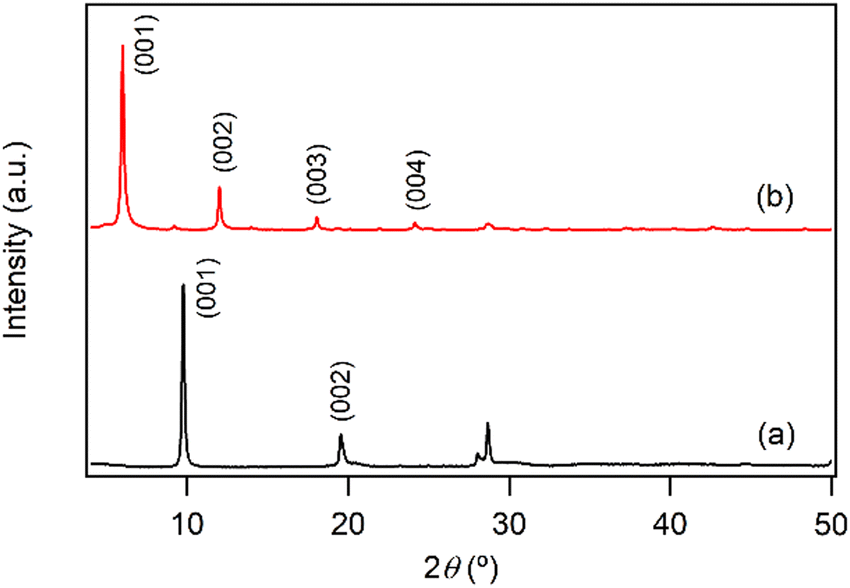 | ||
| Fig. 1 Powder XRD patterns of (a) LYH:Eu and (b) NA-LYH:Eu. | ||
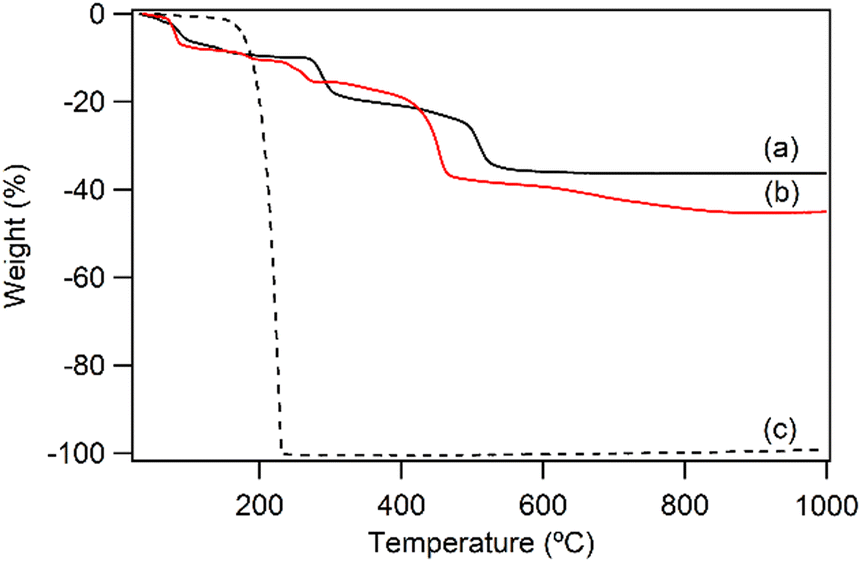 | ||
| Fig. 2 TGA curves of (a) LYH:Eu, (b) NA-LYH:Eu, and (c) nicotinic acid for comparison. | ||
3.2 Anion exchange reaction in the interlayer space of LYH:Eu
The anion exchange reaction between nitrate and NA anions in the interlayer space of LYH:Eu proceeded efficiently, forming NA-LYH:Eu in an aqueous solution. Given the significant quantities of NA produced and discharged into wastewater systems, LRHs, similar to LDHs, could serve as effective materials for NA removal from aqueous solutions.34As shown in Fig. 1b, the strong (00l) reflections in the XRD pattern of NA-LYH:Eu confirmed the preservation of the typical layered structure after the reaction between LYH:Eu and aqueous NA. A systematic shift of (00l) reflections to lower angles, corresponding to an increased interlayer distance from 9.2 Å to approximately 14.7 Å, was observed, indicating the successful replacement of nitrate by NA anions. The TGA curves of NA-LYH:Eu and pure nicotinic acid, compared with LYH:Eu, are shown in Fig. 2. NA-LYH:Eu exhibited three main weight-loss steps (Fig. 2b). The initial weight loss below 100 °C was attributed to the evaporation of water. For comparison, pure nicotinic acid decomposed completely in a single-step process below 230 °C (Fig. 2c). A slight weight loss in NA-LYH:Eu centered at similar temperature range likely resulted from the removal of NA molecules adsorbed on the particle surface. The dehydroxylation of NA-LYH:Eu layers occurred at around 270 °C, slightly lower than that of pristine LYH:Eu, while the decomposition of interlayer NA began at approximately 420 °C. Thus, the interlayer environment provided by LYH:Eu enhanced the thermal stability of NA through electrostatic attractions and effective intermolecular interactions, protecting the interlayer NA. This thermal stability suggests that the hybrid material could mitigate the loss of thermally unstable nicotinic acid (vitamin B3) during high-temperature food processing.6 The amounts of interlayer water and surface-adsorbed NA were determined based on weight losses at approximately 100 °C (7.3%) and 190 °C (3.2%), respectively. Considering that Y2O3:Eu oxide was obtained after thermal decomposition of NA-LYH:Eu at 1000 °C, the chemical formular could be determined by the total weight loss, including interlayer water evaporation, layer dehydroxylation, and interlayer NA decomposition. Thus, the observed total weight loss of 45.2% closely matched the calculated value of 45.8% for Y1.8Eu0.2(OH)5(interlayer NA)(surface NA)0.1·1.7H2O. This agreement suggests nearly complete replacement of nitrate by NA anions. The NA loading capacity in LYH:Eu, measured as approximately 305 mg g−1, exceeded previously reported capacities in Zn/Al LDHs with similar structures.22 This high loading capacity highlights the potential of LYH as an effective reservoir or carrier for NA. Fig. S1b (ESI†) compares the FT-IR spectra of NA-LYH:Eu and LYH:Eu host, showing significant differences. The appearance of strong bands at approximately 1560 cm−1 and 1392 cm−1, corresponding to the antisymmetric (νas) and symmetric (νs) stretching vibrations of –COO− groups, respectively, confirmed the presence of interlayer NA after the exchange reaction. Additionally, a strong band at approximately 1606 cm−1 was attributed to the C–C stretching vibration (νC–C) of the pyridine ring.35 In general, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band of the –COOH group in free acids appears around ∼1690 cm−1 in their spectra. Therefore, the shoulder observed in a similar region in the NA-LYH:Eu spectrum supports the presence of free nicotinic acid adsorbed on the surface.
O stretching band of the –COOH group in free acids appears around ∼1690 cm−1 in their spectra. Therefore, the shoulder observed in a similar region in the NA-LYH:Eu spectrum supports the presence of free nicotinic acid adsorbed on the surface.
Subtracting the layer thickness of 6.5 Å from the observed interlayer distance of 14.7 Å for NA-LYH:Eu suggests an available gallery height of approximately 8.2 Å for interlayer anions.36 This height, significantly larger than the molecular length (∼6.5 Å) of NA, which has a planar structure with a pyridine ring and a carboxyl group, implies that NA anions are arranged in a partially interdigitated bilayer mode within the interlayer space. Similar to the arrangement of interlayer benzoate anions,37 the carboxyl groups alternate in orientation toward adjacent hydroxide layers to form hydrogen bonds, while the pyridine rings are centrally positioned within the interlayer space, facilitating effective π–π stacking. A long-range hydrogen-bonding network may also form through the nitrogen atoms of the pyridine ring in this model.
3.3 Photoluminescence properties of NA-LYH:Eu
Many Eu3+-doped Y compounds, such as Y2O3:Eu and YF3:Eu, exhibit excellent red emission under UV light due to the efficient internal conversion of energy absorbed by the Y matrices into the excited state of Eu3+.38 However, as shown in Fig. 3a, the photoluminescence (PL) spectra of the LYH:Eu host showed no significant excitation or emission behavior. The abundance of hydroxyl groups within the layers and water molecules in the interlayer space likely increases the probability of nonradiative relaxation, significantly quenching the photoemission.39 When monitored at λem = 613nm, the excitation spectrum displayed several weak peaks in the 350–400 nm range. These were attributed to intra-4f6 transitions from the 7F0 ground state to the 5L6, 5GJ, and 5D4 states of Eu3+.40 Excitation at 7F0 → 5L6 (392 nm) produced weak emission peaks corresponding to the 5D0 → 7FJ (J = 1 – 4) transitions of Eu3+, resulting in a faint red emission that was barely visible under a commercial UV lamp (Fig. 3a, inset). The intrinsically low efficiency of these transitions is due to the low extinction coefficients of the forbidden 4f–4f intraconfigurational radiative transitions in RE3+ ions.41
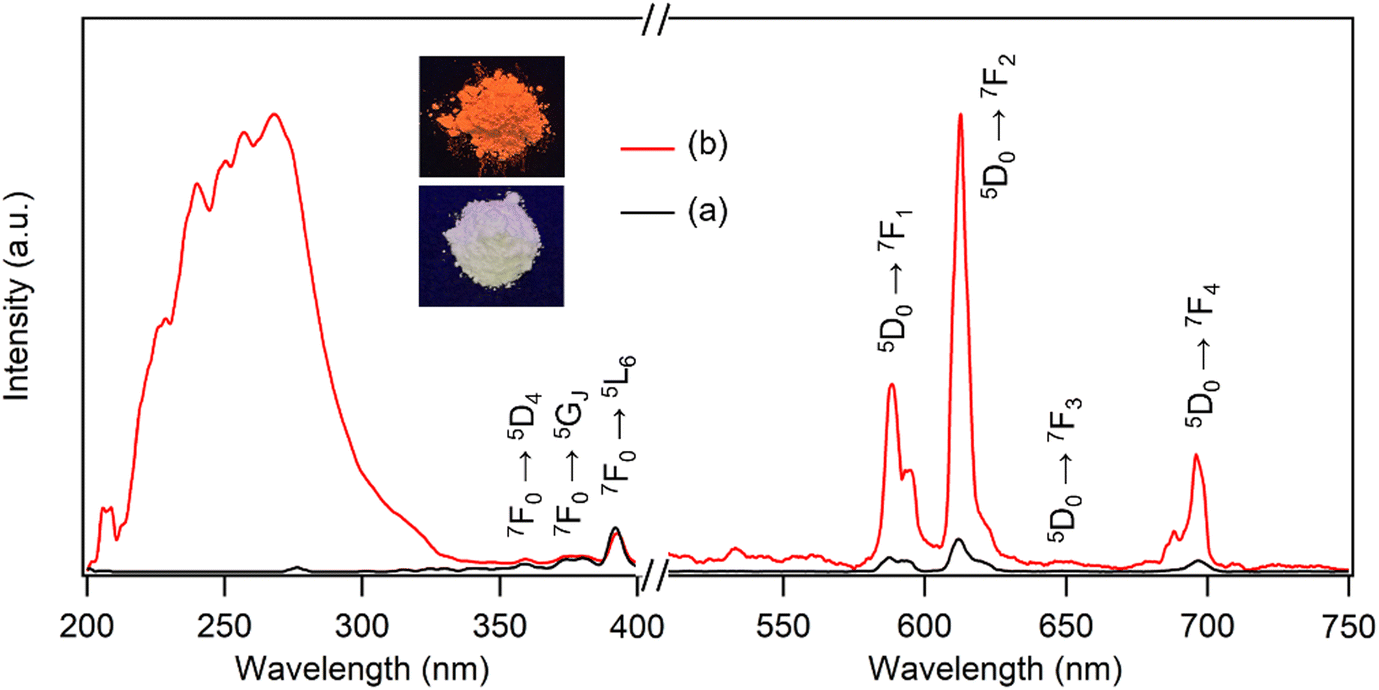 | ||
| Fig. 3 Excitation and emission spectra of (a) LYH:Eu (λem = 613 nm, λex = 392 nm) and (b) NA-LYH:Eu (λem = 613 nm, λex = 268 nm). Inset: Photographs of LYH:Eu and NA-LYH:Eu under a 254 nm UV lamp. | ||
It is well known that UV energy absorbed by organic ‘antenna’ molecules can be transferred to the lowest excited state of RE3+ emitting centers through intersystem crossing from their S1 excited states to the lowest triplet (T1) states.42 This bypasses the forbidden transition rule and enhances the 4f–4f luminescence efficiency. Pure NA in an aqueous solution exhibits a strong absorption band centered at approximately 262 nm, attributed to the π → π* transition from the S0 ground state to the S1 excited state.43 Accordingly, the inclusion of NA in the LYH:Eu matrix was expected to sensitize the luminescence of Eu3+. Fig. 3b compares the PL spectra of LYH:Eu and NA-LYH:Eu. The excitation spectrum of NA-LYH:Eu, monitored at the 5D0 → 7F2 transition (613 nm), showed a strong broad band in the 220–300 nm range that was absent in the LYH:Eu host. This band, with significantly increased intensity, was attributed to the S0 → S1 (π, π*) transition of interlayer NA. The maximum intensity wavelength (approximately 268 nm) was close to the 262 nm π → π* transition of pristine NA, supporting this assignment. As shown in Fig. S2 (ESI†), the UV absorption band of NA remaining unchanged after the inclusion into the interlayer of LYH:Eu, coupled with the excitation at a similar wavelength, indicated minimal interaction between interlayer NA and the hydroxide layer. Upon excitation at 268 nm, the emission intensities related to the 5D0 → 7FJ transitions of Eu3+ increased significantly after NA inclusion (Fig. 3b). The bright red emission, predominantly from the 5D0 → 7F1 and 5D0 → 7F2 transitions near 600 nm, was distinctly visible under a standard 254 nm UV lamp (Fig. 3b, inset). This demonstrates that UV absorption by interlayer NA is followed by energy transfer to the 5D0 excited state of Eu3+, effectively sensitizing and enhancing Eu3+ luminescence in the NA-LYH:Eu layer via the “antenna” effect.
Interestingly, weak intra-4f6 transitions from the 7F0 ground state of Eu3+ were still observed in the 350–400 nm range, despite the dominance of the S0 → S1 transition of interlayer NA in the excitation spectrum of NA-LYH:Eu. This feature suggests that Eu3+ was excited not only through sensitization by NA but also directly via intra-4f6 transitions, highlighting a relatively low efficiency of energy transfer from the NA sensitizer to the Eu3+ emitting center. Effective energy transfer from a sensitizer to Eu3+ requires energy-level matching. The T1 state of NA (26385 cm−1) is higher than the 5D0 excited state of Eu3+ (17250 cm−1),44,45 enabling energy transfer to occur. However, optimal energy transfer efficiency is achieved when the energy gap between the sensitizer's T1 state and the emitter's lowest excited state is within 2000–5000 cm−1, as larger gaps increase nonradiative transition pathways.46 The substantial energy difference (ΔE ∼ 9135 cm−1) between the T1 state of NA and the 5D0 state of Eu3+ likely contributes to the relatively inefficient energy transfer observed.
3.4 Photoluminescence ‘on/off’ behavior of LYH:Eu via NA inclusion/chloride trapping
The difference in photoluminescence intensity observed upon inclusion and release of NA in the interlayer space of LYH:Eu is significant enough to enable visual monitoring. For instance, LDHs have been proposed as effective materials to remove NA discharged into wastewater systems.21,34 Conversely, NA-LDHs have also been studied as corrosion inhibitors in marine concrete structures. By releasing interlayer NA through ion exchange with chloride ions, NA forms a protective layer that efficiently delays chloride-induced corrosion of rebars. The uneven binding force and relatively low binding energy of NA within the interlayer space facilitate its release from NA-LDHs via exchange with chloride ions.23 Given its high NA-loading capacity, LYH:Eu could be employed for similar applications. In these contexts, the ‘luminescence-on’ behavior of LYH:Eu (upon NA inclusion) could be used for visual monitoring of NA uptake, while the ‘luminescence-off’ behavior (upon NA release) could visually indicate chloride trapping.To investigate the exchangeability of interlayer NA with chloride ions, NA-LYH:Eu powder was reacted with a 0.5 M NaCl aqueous solution to simulate seawater conditions. As shown in Fig. 4, the XRD pattern of the sample after 24 h at RT revealed significant changes. The layered structure was preserved, as evidenced by the presence of (00l) reflections; however, the interlayer distance decreased from 14.7 Å to approximately 8.2 Å, consistent with the interlayer distance of LYH intercalated with chloride anions.25 TGA curves before and after the reaction (Fig. S3, ESI†) further supported this exchange. The weight loss attributed to the decomposition of interlayer NA at approximately 420 °C was nearly eliminated, while a new weight loss associated with the liberation of interlayer chloride appeared above 650 °C. The total observed weight loss (approximately 33.4%) was close to the calculated value (30.4%) for a Cl-LYH:Eu formulation containing only chloride anions. The slightly higher observed weight loss likely results from residual NA due to incomplete release. These results indicate that NA anions in LYH:Eu were replaced by chloride anions via a topotactic ion–exchange mechanism. The weak interaction between interlayer NA and the hydroxide layers of LYH:Eu, as suggested by the absence of significant shifts in the π → π* absorption wavelength of NA before and after inclusion (Fig. S2, ESI†) and the similarity in excitation wavelengths of free NA and NA-LYH:Eu (Fig. 3), likely facilitates this exchange. The high NA-loading capacity of LYH:Eu also implies a high chloride-uptake capacity, making it a potential candidate for chloride-trapping inhibitor. Additionally, the exchange process can be visually monitored in situ. As shown in the photographs in Fig. 4 insets, the red luminescence of colloidal solution containing NA-LYH:Eu powder under a 254 nm UV lamp was significantly diminished after reaction with chloride anions, allowing for clear visual differentiation. Fig. S4 (ESI†) compares the PL spectra of NA-LYH:Eu powder before and after reaction in the NaCl solution. The excitation spectrum monitored at λem = 613nm showed a substantial decrease in the intensity of bands attributed to energy transfer from interlayer NA, indicating the near-complete removal of interlayer NA. Upon excitation at 268 nm, the emission associated with the 5D0 → 7F2 main transition was also significantly reduced. This luminescence quenching, linked to NA release (or chloride trapping), was visually distinguishable under a commercial UV lamp.
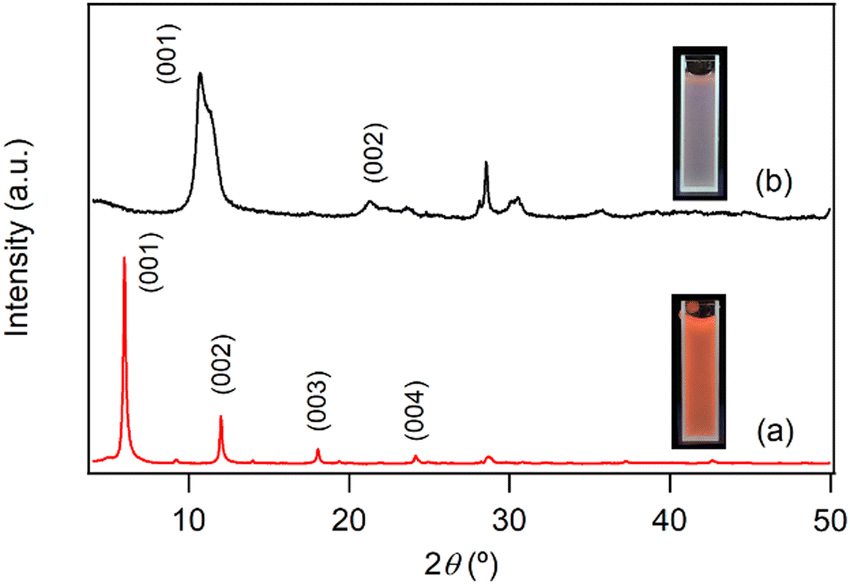 | ||
| Fig. 4 Powder XRD patterns of NA-LYH:Eu (a) before and (b) after reaction in a 0.5 M aqueous NaCl solution for 24 h. Insets: Photographs under a 254 nm UV lamp of colloidal solutions containing sample powders. | ||
The regeneration and recycling potential of LYH:Eu for repeated NA inclusion/release processes was also assessed. NA-LYH:Eu was reacted in the NaCl solution at RT to release interlayer NA, and the resulting powder was reused to uptake NA from an aqueous NA solution. This cycle was repeated 3 times, with the ‘luminescence-on/off’ behavior monitored under a 254 nm UV lamp. As shown in Fig. 5, the bright red emission of NA-LYH:Eu was restored after each regeneration, indicating successful reuptake of NA (‘luminescence-on’). Conversely, subsequent reaction in the NaCl solution quenched the red emission, reflecting NA release and chloride incorporation (‘luminescence-off’). Variations in the XRD patterns and TGA curves during repeated recycling processes are shown in Fig. S5 and S6 (ESI†). Despite the reduced crystallinity, the layered structure was preserved through three regeneration cycles. The relative weight loss gradually decreased with successive recycling, indicating that the regeneration of NA-LYH:Eu was not entirely effective. The NA loading capacity declined to approximately 85% after the third regeneration. Nevertheless, these results confirm that LYH:Eu retains its performance for NA inclusion and release, demonstrating its potential for multiple reuse cycles.
 | ||
| Fig. 5 Photographs under a 254 nm UV lamp of LYH:Eu powder as a function of the ‘on/off’ behavior caused by the NA inclusion and chloride trapping, respectively. | ||
3.5 Release behavior of NA from NA-LYH:Eu in PBS
The LRH family can be utilized as nanocarriers to release interlayer organic molecules in a sustained manner. Thus, encapsulating NA (vitamin B3) within the LYH:Eu delivery system was expected to not only prevent direct skin contact with high concentrations of NA but also facilitate the controlled, sustained release of NA, minimizing potential dermatological side effects. To examine the characteristics of NA release from the LYH:Eu matrix, NA-LYH:Eu powder was dispersed in PBS, and the absorbance of the filtered solution at relevant time intervals was measured at 262 nm to determine the NA concentration. Fig. 6 illustrates the in vitro release profile of NA over 5 days. The overall profile shape closely resembled the release curves commonly observed in various drug delivery systems. A burst release of approximately 40% of NA from NA-LYH:Eu occurred in the initial 1 h, followed by a much slower sustained release. This rapid initial release was primarily attributed to the organic molecules adsorbed on the particle surface and interlayer edges of the matrix.47 It is also noteworthy that, when the NA concentration was measured immediately after dispersing NA-LYH:Eu in DI water, approximately 8% of NA was detected in the filtrate, significantly higher than the surface-bound NA (approximately 3.2%) estimated using TGA curves. This result suggests that some NA-LYH:Eu particles were not filtered by the 0.45 μm syringe filter, additionally contributing to the NA concentration in the filtrate, especially in the initial stage due to the high NA content in the particles. Despite the accumulated release exceeding 80% after 5 days, the release profile did not plateau but continued to release NA. Compared to the 74–77% release of NA within the first hour and 77–83% cumulative release within 2 h observed in Zn/Al LDH,22 the more sustainable release from NA-LYH:Eu offers potential for use as a reservoir for NA in skin care and other topical applications. As shown in the insets of Fig. 6, red emission from the colloidal PBS containing NA-LYH:Eu particles was clearly visible under a 254 nm UV lamp during the release process, with its intensity gradually decreasing over time, providing a visual indication of NA release from the system.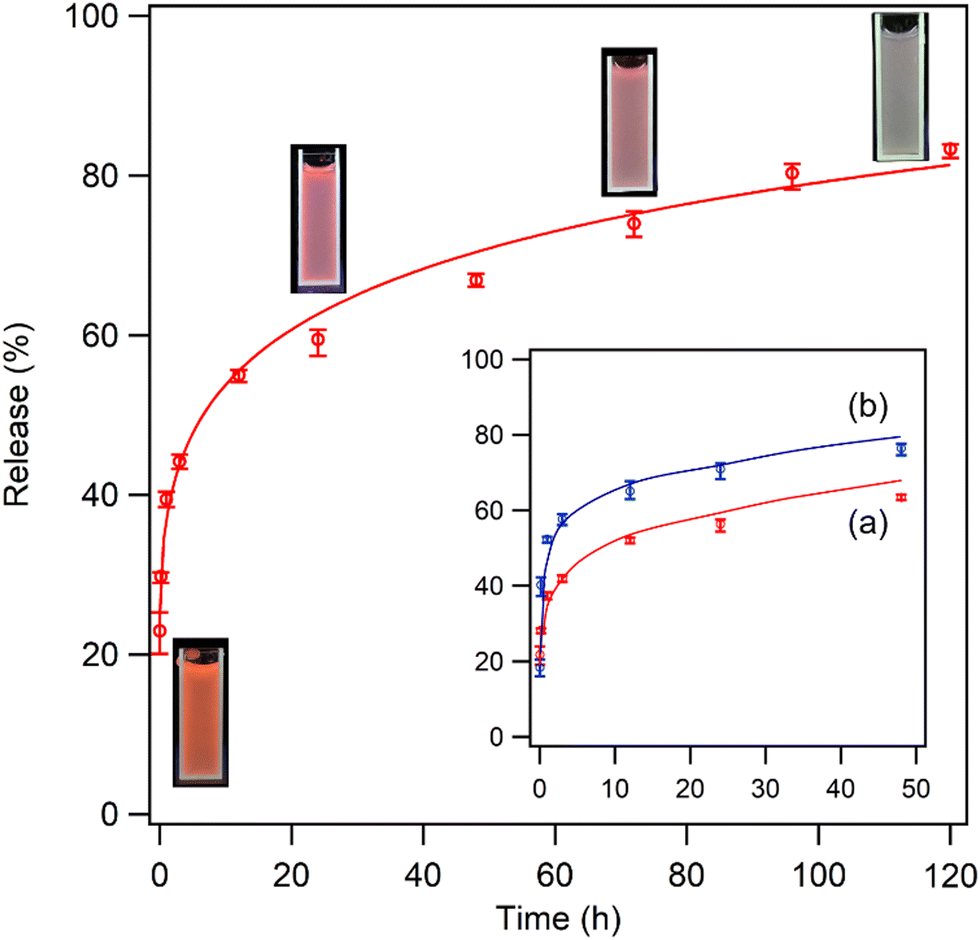 | ||
| Fig. 6 Release profile of NA from NA-LYH:Eu in PBS for 5 days. Inset: Photographs of corresponding solutions containing NA-LYH:Eu particles under a 254 nm UV lamp, and comparison of release profiles when LYH:Eu hosts obtained after reaction at (a) 90 °C for 12 h and (b) 45 °C for 6 h were used. | ||
The rate of NA release can be controlled by adjusting the crystallinity and size of LYH:Eu particles. When the host was synthesized under moderate conditions (45 °C for 6 h, as opposed to 90 °C for 12 h), the intensity of (00l) reflections in the XRD pattern was significantly reduced (see Fig. S7b, ESI†), indicating the formation of particles with lower crystallinity. Fig. S8 (ESI†) presents FE-SEM images of NA-LYH:Eu particles synthesized using hosts prepared under two different conditions. Under milder conditions, crystal growth was suppressed, resulting in much smaller particle sizes. As shown in the inset of Fig. 6, NA release was accelerated by approximately 20% when LYH:Eu host synthesized at 45 °C for 6 h was used. Therefore, NA-LYH:Eu with varying crystallinity and particle size can be designed to control the release rate of NA in PBS.
To further investigate the release behavior of NA, the XRD patterns of solid residues collected at 1, 2, 3, and 5 days after adding NA-LYH:Eu powder to PBS were analyzed. As depicted in Fig. 7, no shift was observed in the position of (00l) reflections until after 5 days, suggesting that the interlayer distance was preserved during the release process. This result contrasts with the significant shift in (00l) reflections observed after just 1 day in a NaCl solution (Fig. 4). In PBS, where H2PO4− and HPO42− phosphorous oxyanions are prevalent, a change in interlayer distance is inevitable for the replacement of interlayer NA by these oxyanions in the interlayer space. Therefore, it is evident that the release of interlayer NA from NA-LYH:Eu in PBS did not occur via an anion–exchange reaction that would normally alter the interlayer distance. Instead, the diffraction intensity of the (00l) reflections decreased progressively, eventually showing an amorphous-like pattern after 5 days (see inset of Fig. 7). As proposed previously,48 the formation of an amorphous phase would result from the reaction of layer Y3+/Eu3+ ions with phosphorous oxyanions, forming insoluble yttrium/europium hydrogen phosphate hydrate complexes from the outermost layer of NA-LYH:Eu particles. Consequently, interlayer NA is released via the gradual collapse of the hydroxide layer structure, rather than through the conventional anion–exchange process. FE-SEM images of NA-LYH:Eu particles before and after the release reaction, shown in Fig. 8, further support this observation. The initially clean surface morphology of the plate-shaped particles was significantly degraded, forming a porous structure after the release reaction in PBS for 24 h. It is also important to note that the high affinity of RE ions for phosphate facilitates the formation of orthophosphates with extremely low water solubility, with pKsp values ranging from 24 to 26.49 Thus, Y3+/Eu3+ ions in the LYH:Eu layer network likely react with phosphorous oxyanions, promoting inner-sphere complexation at the surface.
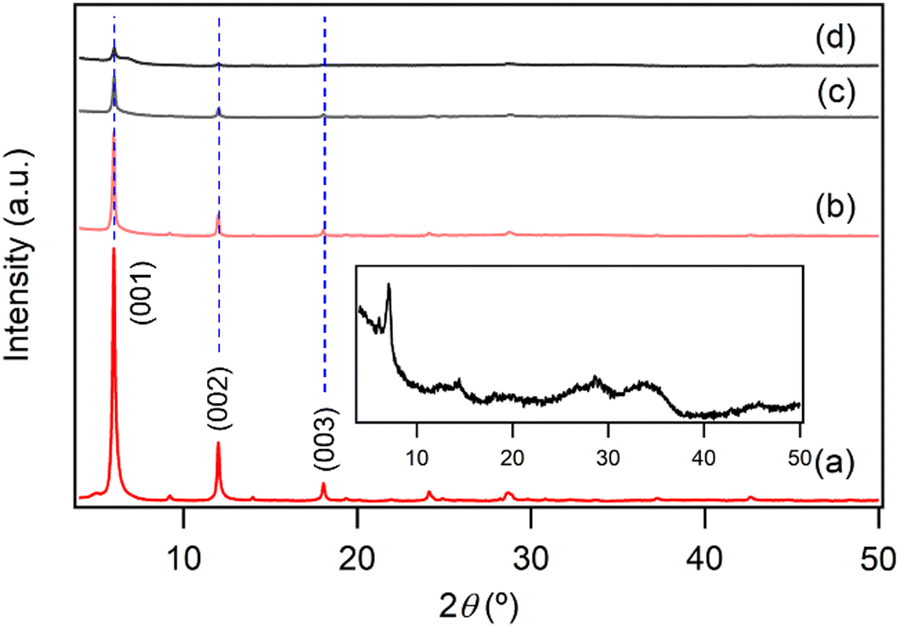 | ||
| Fig. 7 Powder XRD patterns of NA-LYH:Eu (a) before and after release reaction in PBS for (b) 24, (c) 48, and (d) 72 h. Inset: Enlarged XRD pattern of NA-LYH:Eu after release reaction in PBS for 120 h. | ||
 | ||
| Fig. 8 FE-SEM images of NA-LYH:Eu particles (a) before and (b) after release reaction in PBS for 24 h (bar = 100 nm). | ||
4. Conclusions
This study addressed several key aspects of the NA/LYH guest/host system, including loading capacity, long-term stability, reusability, and the sustained and controlled release of interlayer NA. This hybrid system demonstrated several advantages: First, a nearly complete exchange reaction achieved a loading capacity of over 300 mg g−1 of NA into the LYH matrix. As a result, the system's high chloride-trapping capacity, facilitated by the release of NA, could enhance durability against chloride-induced corrosion of rebars in marine construction concrete. Additionally, the system proved to be regenerable and reusable multiple times when employed to separate NA in aqueous solutions. The sustained release of interlayer NA in PBS over more than 5 days ensured improved stability and long-term effectiveness. Importantly, doping with Eu3+ enabled the transfer of UV energy absorbed by the interlayer NA, leading to the visually distinguishable 5D0 → 7F2 red emission of Eu3+ from the NA-LYH:Eu system. The photoluminescence ‘on/off’ behavior, resulting from the inclusion and release of NA, suggests potential applications of NA-LYH:Eu as a corrosion inhibitor and a luminescent carrier for sustained NA release.Author contributions
Yoonjae Choi: investigation, visualization, Myeongjin Kang: validation, formal analysis. Song-Ho Byeon: conceptualization, methodology, writing – reviewing and editing, supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2020R1A2C1006041).Notes and references
- L. A. Carlson, J. Intern. Med., 2005, 258, 94–114 CrossRef CAS PubMed.
- T. Hakozaki, L. Minwalla, J. Zhuang, M. Chhoa, A. Matsubara, K. Miyamoto, A. Greatens, G. G. Hillebrand, D. L. Bisset and R. E. Boissy, Br. J. Dermatol., 2002, 147, 20–31 CrossRef CAS.
- V. Gasperi, M. Sibilano, I. Savini and M. V. Catani, Int. J. Mol. Sci., 2019, 20, 974 CrossRef CAS PubMed.
- C. D. Meyers, V. S. Kamanna and M. L. Kashyap, Curr. Opin. Lipidol., 2004, 15, 659–665 CrossRef CAS.
- C. Minto, M. G. Vecchio, M. Lamprecht and D. Gregori, Nutr. Rev., 2017, 75, 471–490 CrossRef PubMed.
- A. Kondjoyan, S. Portanguen, C. Duchene, P. S. Mirade and G. Gandemer, J. Food Eng., 2018, 238, 44–53 CrossRef CAS.
- L. Albrecht, H. Jiang and K. A. Jorgensen, Chem. – Eur. J., 2014, 20, 358–368 CrossRef CAS.
- R. S. K. Singh, B. Kashyap and P. Phukan, Tetrahedron Lett., 2013, 54, 6687–6690 CrossRef CAS.
- M. Barjasteh, S. M. Dehnavi, S. A. Seyedkhani, M. Akrami and M. Rahimi, J. Drug Delivery Sci. Technol., 2024, 95, 105532 CrossRef CAS.
- B. Yan, X. Chen and J. Wu, Appl. Surf. Sci., 2007, 253, 8575–8580 CrossRef CAS.
- P. Liu, H. R. Li, Y. G. Wang, B. Y. Liu, W. J. Zhang, Y. J. Wang, W. D. Yan, H. J. Zhang and U. Schubert, J. Mater. Chem., 2008, 18, 735–737 RSC.
- X.-L. Wang and B. Yan, Colloid Polym. Sci., 2011, 289, 423–431 CrossRef CAS.
- Y. Zhang, Q. Xu, M. Sun, C. Xiong, P. Wang, Z. Chen, G. Sun, J. Guan, Z. Ding, M. Li and D. Hou, Constr. Build. Mater., 2021, 294, 123571 CrossRef.
- G. Sammartino, G. Marenzi, L. Tammaro, A. Bolognese, A. Calignano, U. Costantino, L. Califano, S. Tete and V. Vittoria, Int. J. Immunopathol. Pharmacol., 2005, 18, 55–62 CAS.
- V. S. Smitha, K. A. Manjumol, S. Ghosh, M. Brahmakumar, C. Pavithran, P. Perumal and K. G. Warrier, J. Am. Ceram. Soc., 2011, 94, 1731–1736 CrossRef CAS.
- P. Yang, S. Gai and J. Lin, Chem. Soc. Rev., 2012, 41, 3679–3698 RSC.
- S. H. Tolbert, P. D. McFadden and D. A. Loy, ACS Appl. Mater. Interfaces, 2016, 8, 3160–3174 CrossRef CAS.
- V. Rives, M. del Arco and C. Martín, Appl. Clay Sci., 2014, 88–89, 239–269 CrossRef CAS.
- X. Bi, H. Zhang and J. Dou, Pharmaceutics, 2014, 6, 298–332 CrossRef CAS PubMed.
- M. Lalikoglu, A. Gok, M. K. Gok and Y. S. AscI, J. Chem. Eng. Data, 2015, 60, 3159–3165 CrossRef CAS.
- A. Gok, J. Dispers. Sci. Technol., 2020, 41, 779–786 CrossRef CAS.
- P. E. Kleyi, P. Mudaly, S. K. Pillai and M. Beer, Appl. Clay Sci., 2021, 215, 106304 CrossRef CAS.
- E. Zhuang, J. Li, Z. Chen, B. Yu and Y. Nong, Composites, Part B, 2024, 277, 111414 CrossRef CAS.
- K. Karádi, Á. Kukovecz, Z. Kónya, P. Sipos, I. Pálinkó and G. Varga, J. Mol. Struct., 2022, 1261, 132868 CrossRef.
- F. Geng, R. Ma and T. Sasaki, Acc. Chem. Res., 2010, 43, 1177–1185 CrossRef CAS.
- Y. Yoon, B.-I. Lee, K. S. Lee, G. H. Im, S.-H. Byeon, J. H. Lee and I. S. Lee, Adv. Funct. Mater., 2009, 19, 3375–3380 CrossRef CAS.
- W. Li, W. Gu, F. Su, Y. Sun, G. Sun, S. Ma and X. Yang, Inorg. Chem., 2013, 52, 14010–14017 CrossRef CAS PubMed.
- Q. Gu, W. Chen, F. Duan and R. Ju, Dalton Trans., 2016, 45, 12137–12143 RSC.
- R. Ju and Q. Gu, Appl. Organomet. Chem., 2018, 32, e3926 CrossRef.
- H. Jung, H. Kim and S.-H. Byeon, ACS Appl. Mater. Interfaces, 2018, 10, 43112–43121 CrossRef CAS.
- L. J. McIntyre, L. K. Jackson and A. M. Fogg, J. Phys. Chem. Solids, 2008, 69, 1070–1074 CrossRef CAS.
- I. Schildermans, J. Mullens, J. Yperman, D. Franco and L. C. Vanpoucke, Thermochim. Acta, 1994, 231, 185–192 CrossRef CAS.
- J. T. Kloprogge and R. L. Frost, J. Solid State Chem., 1999, 146, 506–515 CrossRef CAS.
- D. Datta, S. Sah, N. Rawat and R. Kumar, J. Chem. Eng. Data, 2017, 62, 712–719 CrossRef CAS.
- Y. A. Chesalov, G. B. Chernobay and T. V. Andrushkevich, J. Mol. Catal. A: Chem., 2013, 373, 96–107 CrossRef CAS.
- N. Chu, Y. Sun, Y. Zhao, X. Li, G. Sun, S. Ma and X. J. Yang, Dalton Trans., 2012, 41, 7409–7414 RSC.
- Y. Chang, S.-Y. Cho and S.-H. Byeon, J. Lumin., 2023, 258, 119820 CrossRef CAS.
- J. G. Li, X. Li, X. Sun and T. Ishigaki, J. Phys. Chem. C, 2008, 112, 11707–11716 CrossRef CAS.
- A. Bahtat, M. C. Marco de DeLucas, B. Jacquier, B. Varrel, M. Bouazaoui and J. Mugnier, Opt. Mater., 1997, 7, 173–179 CrossRef CAS.
- S. Quici, G. Marzanni, M. Cavazzini, P. L. Anelli, M. Botta, E. Gianolio, G. Accorsi, N. Armaroli and F. Barigelletti, Inorg. Chem., 2002, 41, 2777–2784 CrossRef CAS PubMed.
- J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- W. T. Carnall, G. L. Goodman, K. Rajnak and R. S. Rana, J. Chem. Phys., 1989, 90, 3443–3457 CrossRef CAS.
- I. Chanda, R. Bordoloi, D. D. Chakraborty, P. Chakraborty and S. R. C. Das, J. Appl. Pharm. Sci., 2017, 7, 81–84 CAS.
- M. Hilder, M. Lezhnina, P. C. Junk and U. H. Kynast, Polyhedron, 2013, 52, 804–809 CrossRef CAS.
- V. I. Tsaryuk, K. P. Zhuravlev, V. F. Zolin, V. A. Kudryashova, J. Legendziewicz and R. Szostak, J. Appl. Spectrosc., 2007, 74, 51–59 CrossRef CAS.
- S. Maji and K. S. Viswanathan, J. Lumin., 2008, 128, 1255–1261 CrossRef CAS.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef CAS.
- S.-Y. Cho, Y. Chang and S.-H. Byeon, Appl. Clay Sci., 2023, 245, 107159 CrossRef CAS.
- F. Firsching and S. N. Brune, J. Chem. Eng. Data, 1991, 36, 93–95 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR and UV-vis spectra. Additional XRD patterns, TGA curves, PL spectra and FE-SEM images. See DOI: https://doi.org/10.1039/d4tc05448b |
| This journal is © The Royal Society of Chemistry 2025 |