
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalent carbon nanodot-azobenzene hybrid photoswitches: the impact of meta/para connectivity and sp3 spacer on photophysical properties†
Paul P. Debes‡
 ab,
Dominic Schatz‡ac,
Yagmur Aydogan-Sund,
Juan Pablo Martíneze,
Michal Langerf,
Janis Hesslingg,
Jaime Gallegoab,
Enzo Mennah,
Bernd M. Smarslyab,
Monika Schönhoffg,
Silvio Osellae,
Josef Wachtveitld,
Hermann A. Wegner*ac and
Teresa Gatti*abi
ab,
Dominic Schatz‡ac,
Yagmur Aydogan-Sund,
Juan Pablo Martíneze,
Michal Langerf,
Janis Hesslingg,
Jaime Gallegoab,
Enzo Mennah,
Bernd M. Smarslyab,
Monika Schönhoffg,
Silvio Osellae,
Josef Wachtveitld,
Hermann A. Wegner*ac and
Teresa Gatti*abi
aCenter for Materials Research, Justus Liebig University, Heinrich-Buff-Ring 16, 35392, Giessen, Germany
bInstitute of Physical Chemistry, Justus Liebig University, Heinrich-Buff-Ring 17, 35392, Giessen, Germany
cInstitute of Organic Chemistry, Justus Liebig University, Heinrich-Buff-Ring 17, 35392, Giessen, Germany. E-mail: Hermann.A.Wegner@org.chemie.uni-giessen.de
dInstitute of Physical and Theoretical Chemistry, Goethe University, Max von Laue-Strasse 7, 60438, Frankfurt, Germany
eChemical and Biological Systems Simulation Lab, Centre of New Technologies, University of Warsaw, 2c Banacha Street, 02-097, Warszawa, Poland
fIT4Innovations, VSB – Technical University of Ostrava, 17.listopadu 2172/15, 70800, Ostrava-Poruba, Czech Republic
gInstitute of Physical Chemistry, University of Muenster, Corrensstrasse 28/30, 48149, Muenster, Germany
hDepartment of Chemical Sciences, University of Padova and INSTM UdR Padova, via Marzolo 1, 35131, Padova, Italy
iDepartment of Applied Science and Technology, Politecnico di Torino, C.so Duca degli Abruzzi 24, 10129, Torino, Italy. E-mail: teresa.gatti@polito.it
First published on 7th May 2025
Abstract
The covalent surface functionalization of carbon nanodots (CNDs) can facilitate the design and development of nanocarbon hybrids with photoswitching properties, which can be applied in a wide range of applications, including sensing, optoelectronics, and even bio-applications. This study underscores the potential utilization of these hybrids as photoresponsive materials, for potential application in optostimulation. In this study, we examine the characteristics of covalent azobenzene-functionalized CNDs, with a particular emphasis on the impact of meta and para connectivity and the additional introduction of a glycine spacer. The CND synthesis process comprises a bottom-up microwave condensation of ethylenediamine and citric acid. Amide coupling to azobenzenes is confirmed through NMR diffusion-ordered spectroscopy and diffusion decay analysis. A comprehensive investigation is conducted into the size and optical properties of the resulting hybrids. Moreover, time-dependent density functional theory computations are employed to understand absorption spectra and charge transfer events. Furthermore, advanced optical characterisation is utilised to examine energy/charge transfer between the constituents. Finally, the switching properties, fatigue resistance, and half-life of the hybrids are studied to evaluate their performance for prospective applications like in optostimulation.
Introduction
The field of optostimulation is receiving increased attention due to its potential to enable remote monitoring of cellular signaling with high precision.1 Furthermore, the utilization of nanoparticles for biomedical applications is an expanding area of research, with the primary objectives of enhancing imaging techniques and improving drug delivery systems.2 Both approaches serve as valuable tools for advancing our understanding of bacterial cell physiology in both fundamental and applied research contexts. In 2023, the group of Paternò demonstrated that an amphiphilic azobenzene (azo) unit can non-covalently attach to the plasma membrane of Bacillus subtilis bacteria.3 Upon irradiation and azo isomerization, the membrane structure relaxes and expands. It has been demonstrated that carbon nanodots (CNDs) are capable of localizing within biomembranes. Consequently, covalent functionalization of CNDs with azos could enable the development of a hybrid material capable of entering biomembranes, being sensed, and undergoing structural changes in response to light irradiation.4,5 Ultimately, this research aims to provide new tools for effective optostimulation and imaging applications in biomedical research.Combining different materials not only improves functionality, but also results in a wide range of properties that can be tailored for specific applications in microbial systems and beyond. Over the past decade, the field of carbon nanomaterials has witnessed a growing body of research. Fullerenes, CNDs, carbon nanotubes, and graphene are prominent examples of these carbon nanomaterials.6–10 Notably, CNDs have attracted attention due to their facile synthesis, low cost, excellent biocompatibility, easy functionalization, water solubility, and bright luminescence.11 Bottom-up CNDs are synthesized using small molecules as starting materials.12 Commonly utilized molecules for synthesis include citric acid (CA), urea, amino acids, and ethylene diamine (EDA). The resulting nanoparticles exhibit a quasi-spherical shape and are typically smaller than 10 nm in size.13 These nanoparticles consist of carbon, hydrogen, oxygen, and often nitrogen.14 CNDs are composed of a carbon core surrounded by functional groups.15 The type and quantity of functional groups on the surface of CNDs are influenced by the selected starting materials and their ratios.16 Alcohols, amines, and carboxylic acids are common functional groups that allow covalent functionalization of a wide range of materials.17,18 Among the innumerable possibilities, functionalization with photochromic molecules enables the production of carbon nanomaterials with the ability to respond to light irradiation.19 Photoswitchable molecules have the capability to undergo a light-induced reversible transformation from one species to another. Depending on the molecule, there are at least two states which are stable or metastable.20
One of the widely used photochromic motifs is the trans/cis isomerization of azos,21,22 stilbenes,23 hydrazones,24–26 and Schiff bases.27,28 Azos undergo a significant change in length and geometry through isomerization, making them useful in energy or information storage,29–34 photobiology,35,36 host–guest systems,37 as molecular wind-up meters,38 and molecular machines.39,40 The trans to cis isomer switch is usually induced by irradiation in the π–π* band, typically between 300 nm and 360 nm. Reversion to the thermodynamically favored trans isomer can be induced thermally or by n–π* excitation around 440 nm,40,41 or by various catalytic processes.42,43 The linkage of two azo units, either via the para or the meta position, has demonstrated that the resulting combinations exhibit distinct properties. Compared to the π-conjugation in the para-connected azo, the meta-connection resulted in a more independent behavior of the two entities.44–46 Even if the systems are separated through space and are not in conjugation with each other, they influence one another.47,48 For a better understanding of the connectivity and through-space interaction between azos and nanocarbons such as CNDs, we report in this work three different systems with either meta- or para-connectivity, and considering a sp3 spacer between the two moieties in one of the systems. The impact of functionalization type and inter-unit distance on physicochemical characteristics—specifically absorbance, photoluminescence (PL), quantum yield (QY), half-life, and fatigue resistance—has been comprehensively investigated. Moreover, time-correlated single photon counting (TCSPC) is utilized to investigate the energy/charge transfer occurring between these components. Additionally, theoretical analysis of absorbance properties was conducted using time-dependent density functional theory (TDDFT) computations to provide deeper insights.
These different CND–azo hybrids could serve as valuable platform for functional species in several light-triggered applications.
Results and discussion
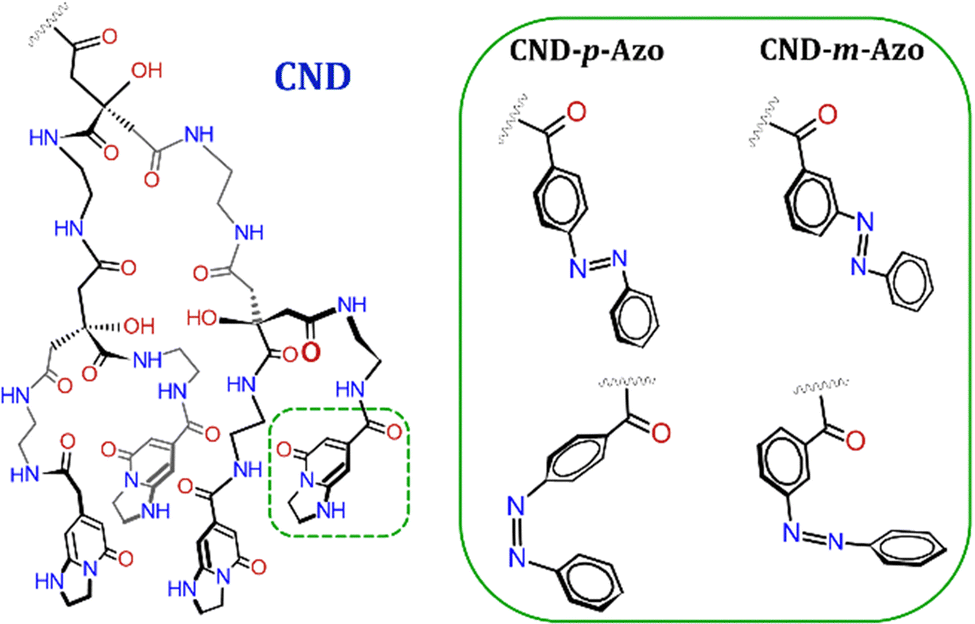
CNDs were synthesized via a microwave (MW)-assisted reaction between CA and EDA in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, followed by purification through filtration and dialysis. The synthesized CNDs were thoroughly characterized using atomic force microscopy (AFM), UV-vis absorbance, PL, and NMR spectroscopy to confirm their presence and gain insight into their properties. In a previous study, we reported the presence of CNDs featuring surface-exposed primary amine groups, which are available for further functionalization via an amide coupling reaction.16 Three distinct azo derivatives were synthesized utilizing the Baeyer–Mills reaction, each containing a carboxylic acid functional group. The first derivative has meta (m) connectivity, the second has para (p) connectivity, and the third has a glycine spacer in the meta position (m-Gly-Azo). Subsequently, the azo compounds were covalently bonded to the CNDs via an amide coupling, as illustrated in Fig. 1. To provide comprehensive evidence of the successful functionalization of the CND with the respective azo diffusion-ordered spectroscopy (DOSY) NMR was used. A similar approach was employed in 2018 by Prato and co-workers to demonstrate covalent functionalization of CNDs.49 Bare m-Azo, pristine CND, a physical mixture of CND and m-Azo, and the three hybrids were subjected to 2D DOSY NMR (see Fig. S1 to S6, ESI†) as well as an analysis of the diffusion decays (see Fig. 2). In the DOSY spectra m-Azo, CND, and the hybrids exhibit a single monomodal distribution, indicating that the constituents of these samples are present in a monodispersed state. In contrast, the physical mixture of CND and azo shows two separate diffusion distributions, which proves that the two moieties move separately in the mixture. For a more precise quantification, the diffusion coefficients were derived from the diffusion echo decays (see Fig. 2A) by applying the Stejskal–Tanner equation,50 and assuming a log-normal distribution of the diffusion coefficients (see ESI,† eqn (S2) and (S3)).
:1 ratio, followed by purification through filtration and dialysis. The synthesized CNDs were thoroughly characterized using atomic force microscopy (AFM), UV-vis absorbance, PL, and NMR spectroscopy to confirm their presence and gain insight into their properties. In a previous study, we reported the presence of CNDs featuring surface-exposed primary amine groups, which are available for further functionalization via an amide coupling reaction.16 Three distinct azo derivatives were synthesized utilizing the Baeyer–Mills reaction, each containing a carboxylic acid functional group. The first derivative has meta (m) connectivity, the second has para (p) connectivity, and the third has a glycine spacer in the meta position (m-Gly-Azo). Subsequently, the azo compounds were covalently bonded to the CNDs via an amide coupling, as illustrated in Fig. 1. To provide comprehensive evidence of the successful functionalization of the CND with the respective azo diffusion-ordered spectroscopy (DOSY) NMR was used. A similar approach was employed in 2018 by Prato and co-workers to demonstrate covalent functionalization of CNDs.49 Bare m-Azo, pristine CND, a physical mixture of CND and m-Azo, and the three hybrids were subjected to 2D DOSY NMR (see Fig. S1 to S6, ESI†) as well as an analysis of the diffusion decays (see Fig. 2). In the DOSY spectra m-Azo, CND, and the hybrids exhibit a single monomodal distribution, indicating that the constituents of these samples are present in a monodispersed state. In contrast, the physical mixture of CND and azo shows two separate diffusion distributions, which proves that the two moieties move separately in the mixture. For a more precise quantification, the diffusion coefficients were derived from the diffusion echo decays (see Fig. 2A) by applying the Stejskal–Tanner equation,50 and assuming a log-normal distribution of the diffusion coefficients (see ESI,† eqn (S2) and (S3)).
 | ||
| Fig. 1 Schematic representation of the microwave-assisted bottom-up synthesis of the CND from CA and EDA (1:1) in aqueous solution, followed by the covalent functionalization via amide coupling (EDC, NHS in DMF) with three different azos (m-Azo, p-Azo, and m-Gly-Azo) forming three distinct CND–azo hybrids, namely: CND–m-Azo, CND–p-Azo, and CND–m-Gly-Azo. | ||
 | ||
| Fig. 2 (A) 1H diffusion decay signal, integrated from 8.5 ppm to 7.3 ppm for the m-Azo, CND, a physical mixture of m-Azo and CND (1:10), CND–m-Azo, CND–p-Azo, and CND–m-Gly-Azo hybrids. (B) Distribution of diffusion coefficients according to the method of Lenoch et al.27 (see description in ESI†) for m-Azo, CND, a physical mixture of m-Azo and CND (1:10), CND–m-Azo, CND–p-Azo, and the CND–m-Gly-Azo. The diffusion coefficient of m-Azo is depicted as straight dotted line due to lack of a size distribution. | ||
Since no separate m-Azo resonances could be identified in the hybrid samples (see Fig. S1–S6, ESI†), the integral over the chemical shift region from 8.5 to 7.3 ppm was evaluated in all cases. m-Azo was used as a representative Azo moiety to compare the molecular diffusion coefficients to those of CND and the hybrids. The diffusion coefficient of m-Azo in solution is depicted as a dotted red line at 2.93 × 10−10 m2 s−1, as it does not possess a size distribution, in contrast to the CND or the hybrids (Fig. 2B). The diffusion coefficient distribution for pristine CND exhibits a maximum at D = 1.05 × 10−10 m2 s−1. Measurements of the hybrids revealed a 20% reduction in the diffusion coefficient, with values of 0.84 × 10−10 m2 s−1 observed for both the CND–m-Azo and the CND–m-Gly-Azo hybrids. The CND–p-Azo hybrid exhibited a diffusion coefficient of 0.93 × 10−10 m2 s−1, which corresponds to a decrease of 11% in comparison to pristine CND. A decrease of the diffusion coefficients accompanies an increase in particle size, which is consistent with successful functionalization of the CNDs. In contrast, the physical mixture of CNDs with m-Azo shows two independent peaks in the diffusion domain of the 2D spectrum (Fig. S3, ESI†), which can be viewed as separate peaks in the distribution of diffusion coefficients in Fig. 2B. Notably, the fast-diffusing component, attributed to m-Azo, exhibits a slightly slower diffusion in the presence of CNDs as compared to bare m-Azo solution, but generally agrees with the diffusion coefficient of bare m-Azo. Furthermore, the diffusion coefficient of the CNDs in the mixture, represented by the shoulder with the lower diffusion coefficient, agrees very well to that of bare CND. This indicates that the physical mixture consists of m-Azo and CND as distinct species. In contrast, the hybrid samples do not exhibit any component with a fast diffusion coefficient, suggesting the absence of individual m-Azo molecules. Additionally, the reduced diffusion of the hybrid CND particles reflects an increased radius, consistent with functionalization. In consideration of the Stokes–Einstein equation (see ESI,† eqn (S4)), the hydrodynamic diameter can be determined from the diffusion coefficient at the maximum. The size of the CND is at around 1.9 nm and the CND–m-Azo at around 2.4 nm, which is in good agreement with the measured AFM size (see Fig. S7, S8, and S11A and B, ESI†). Accordingly, the size of the remaining hybrids can be given by the hydrodynamic diameter as well. CND–p-Azo possess a size of 2.1 nm, while the CND–m-Gly-Azo measures 2.4 nm (Table 1). Whereas AFM measurements indicate a size of 4.9 nm for the CND–p-Azo and 4.8 nm for the CND–m-Gly-Azo, which can be attributed to potential agglomerates on the mica surface (see Fig. S9, S10, and S11C and D, ESI†).51
| Sample | D/m2 s−1 | Hydrodynamic diameter/nm |
|---|---|---|
| a The physical mixture corresponds to a weight ratio of 1:10 of m-Azo to CND. |
||
| m-Azo | 2.93 × 10−10 | 0.68 |
| CND | 1.05 × 10−10 | 1.90 |
| m-Azo + CND (physical mix)a | 2.32 × 10−10 | 0.86 |
| 1.03 × 10−10 | 1.90 | |
| CND–m-Azo | 0.84 × 10−10 | 2.40 |
| CND–p-Azo | 0.93 × 10−10 | 2.10 |
| CND–m-Gly-Azo | 0.84 × 10−10 | 2.40 |
Thermogravimetric analysis (TGA) suggests the occurrence of functionalization; however, the CND morphology precludes the possibility of making a precise statement regarding the degree of functionalization based on TGA results (Fig. S12, ESI†). As illustrated in Fig. 3, the absorption and emission spectra of the three hybrids are displayed in comparison to the pristine CND and the absorption of the corresponding azo compound (which are non-emissive). Table 2 presents the individual maxima of the absorption λAbs and emission λEm, the Stokes shift, as well as the PLQY (Fig. S13 and S14, ESI†).
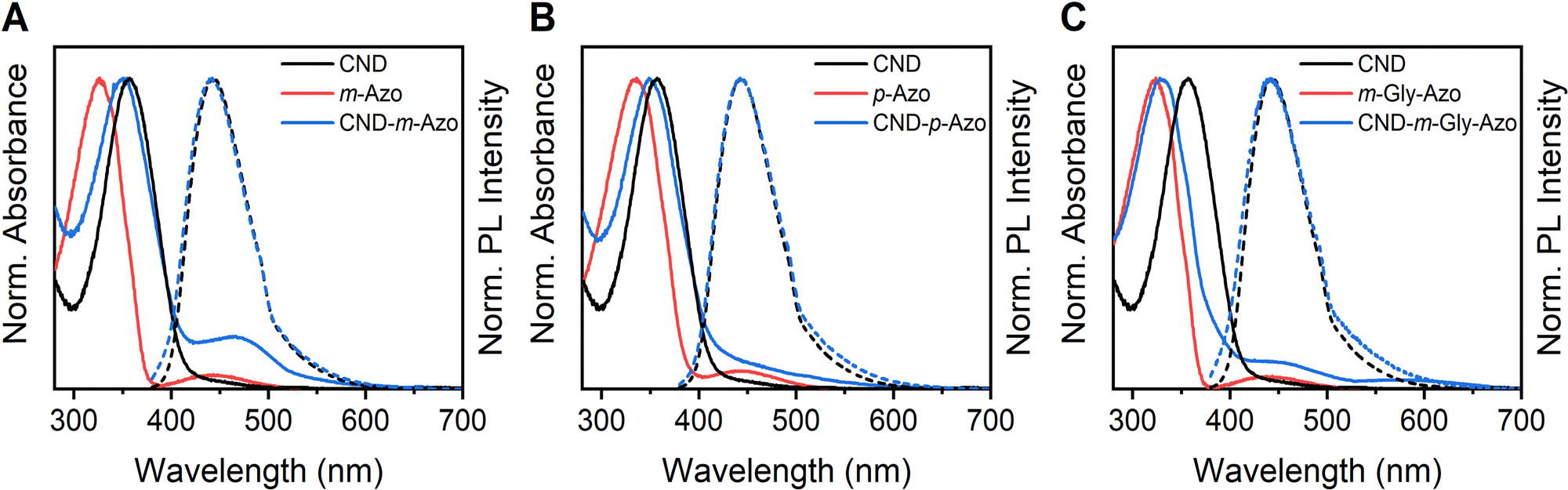 | ||
| Fig. 3 Normalized absorbance (red solid lines) of (A) m-Azo, (B) p-Azo, (C) m-Gly-Azo, normalized absorbance (blue solid lines) and PL (blue dotted lines) of (A) CND–m-Azo, (B) CND–p-Azo, (C) CND–m-Gly-Azo compared to pristine CNDs. | ||
| Sample | λAbs/nm | λEm/nm | Stokes shift/nm | PLQY/% |
|---|---|---|---|---|
| m-Azo | 326 | |||
| p-Azo | 335 | |||
| m-Gly-Azo | 323 | |||
| CND | 357 | 443 | 86 | 37 |
| CND–m-Azo | 350 | 440 | 90 | 18 |
| CND–p-Azo | 349 | 443 | 94 | 11 |
| CND–m-Gly-Azo | 329 | 441 | 112 | 1 |
| CND + m-Azo (mix) | 349 | 443 | 94 | 31 |
| CND + p-Azo (mix) | 343 | 443 | 100 | 18 |
| CND + m-Gly-Azo (mix) | 338 | 443 | 105 | 23 |
A comparative analysis of the absorption spectra of the three hybrids revealed that CND–m-Azo exhibited the smallest blue shift compared to the CND, with an absorption maximum at 350 nm versus 357 nm for CND. In contrast, CND–m-Gly-Azo displayed the largest blue shift (maximum at 329 nm), while CND–p-Azo (maximum at 349 nm) showed a shift similar to that of CND–m-Azo. Qualitative analysis of the 1H NMR spectra indicates that CND–m-Gly-Azo has a greater quantity of m-Gly-Azo on its surface, as evidenced by more pronounced azo group peaks compared to CND–m-Azo and CND–p-Azo (Fig. S57–S59, ESI†). This finding suggests that the extent of functionalization significantly influences the observed shifts of the absorption maxima. Although CND–p-Azo and CND–m-Azo exhibit comparable blue shifts relative to pure CND, p-Azo shows a larger red shift (335 nm) than m-Azo (323 nm), contributing to the minor blue shift in CND–p-Azo. To quantify surface functionalization, UV-vis spectra were deconvoluted using two Lorentzian functions, allowing for calculation of azo concentrations via Beer–Lambert law.52 The absorbance coefficients of the azo groups were considered (Fig. S15, ESI†), along with the maximum absorbance values derived from the deconvolution, which represent the respective azo species (Fig. S16, ESI†). This methodology has already been applied in a similar way to functionalized CNDs in one of our previous studies.53 The azo content was found to be 3.4% by mass for CND–m-Azo and 4.5% for CND–p-Azo, while CND–m-Gly-Azo exhibited a significantly higher value of 24%. Corresponding primary amine amounts were calculated as 150 μmol g−1 for CND–m-Azo, 200 μmol g−1 for CND–p-Azo, and 850 μmol g−1 for CND–m-Gly-Azo, consistent with previous findings on amine functionalization on these CNDs. In a previous study, the maximum amount of primary amines measured on the surface of the CND was measured to be 1670 μmol g−1.16 It is a well-known phenomenon that complete functionalization of primary amines on the CND's surface is rarely achieved. However, to increase the degree of functionalization, it is possible to change the starting materials of the CND to more nitrogen-rich precursors, such as arginine, or to increase the EDA content to obtain a larger amount of primary amines on the surface of the CND.54
The degree of azo functionality on the surface of the CNDs may be influenced by the nature of the established connectivity. Preliminary observations suggest that m-Azo may exhibit greater steric demand compared to p-Azo; however, this assertion requires further investigation. In contrast, the introduction of a glycine spacer appears to reduce this steric demand, thereby facilitating a higher degree of functionalization within the meta-connected hybrids. This indicates that the connectivity and spacer design can modulate the optical properties. Meta-Functionalization is expected to lead to a more independent behavior of the CND and azo, while para connectivity may allow conjugation between the two systems.15,44 As a matter of fact, the hybrids exhibited frontier molecular orbitals localized on either the CND or azo fragments, but the conjugation is not extended on both (Fig. 4). Although para-functionalization should enable conjugation and thus strong intramolecular interaction, this is not evident in the absorption spectra. This implies that the azo moiety remains spatially separated from the fluorophores of the CND and only engages through space.
 | ||
| Fig. 4 Structures of the CND–azo hybrids used in the computation. | ||
Computational investigations were further employed to understand the UV-vis spectra presented in Fig. 3. First, molecular dynamics (MD) simulations were performed to develop a thermally stable model representing the molecular structure of the CND (Fig. S17 and S20, ESI†). Under high-temperature conditions, precursor molecules undergo polymerization and dehydration, forming complex polymer/carbon hybrid structure of CDs, where molecular fluorophores may be covalently attached to CDs.10 Unfortunately, the structural complexity of CDs makes it difficult to build exact model structures, thus forcing researchers to reach for ad hoc models.55–59 Our ad hoc model represents the polymeric CND model synthesized in the conditions before reaching full-carbonization, where small fluorophores may still dominate the optical properties of CNDs.57,60,61 The MD simulations revealed that the system self-assembled into the CND shaped particle within the simulation time (Fig. S17 Sections b–f, ESI†), with the average radius of gyration 1.0 nm (Fig. S18a, ESI†), thus the estimated hydrodynamic radius of this model shall be around 2.5 nm62 (see the note in the ESI†). This CND model was observed to be stabilized not only by hydrogen-bond formation (Fig. S18b, ESI†), but especially through the stacking of the 12 IPCA molecules (5-oxo-1,2,3,5-tetrahydroimidazo-[1,2-α]-pyridine-7-carboxylic acid), which are covalently embedded within the CND (Fig. S19, ESI†).
Subsequently, a quantum mechanics/molecular mechanics (QM/MM) approach based on tight-binding DFT63,64 and TD-ωB97XD/6-31G(d) methods were benchmarked to calculate UV-vis spectra in DMSO (Table S1 and Fig. S21 and S22, ESI†). The absorption spectra for CND shows that the most intense and lowest-energy transition S1 (357 nm, f = 0.27, Fig. 5) is in excellent agreement with the experimental λAbs = 357 nm value reported in Table 2. Motivated by these results, the CND model was chemically functionalized with one azo derivative at different sites at the surface of the CND (Table S2, ESI†). The most thermodynamically stable constitutional isomer of CND–azo was selected for TDDFT computations (Fig. S24, ESI†).
 | ||
| Fig. 5 The lowest-energy electron transitions in the CND (top) and CND–p-Azo (bottom, f = oscillator strength, % of molecular-orbital contributions). The azo moiety is in the trans configuration. Only the fragment simulated at the TDFFT level is illustrated for the sake of clarity. The calculated spectrum is also illustrated. | ||
Specifically, CND–p-Azo was selected as a representative example for the analysis of the TDDFT results, while calculated spectra and electron transition schemes for both para and meta conformers of cis/trans azo compounds, and the respective CND–azo hybrids, are presented in Fig. S25–S30 (ESI†). To confirm that the wB97xD/6-31G(d) functional and basis set combination used is a good choice to reproduce the experimental spectra, we performed benchmark experiments (see ESI† for details). The lowest-energy electronic transitions in CND–p-Azo incorporating the trans isomer of the azo fragment are illustrated in Fig. 5. The first strong peak obtained is attributed to the S1 n–π* transition at 513 nm, localized on the azo fragment, CND–p-Azo* (where the * indicates the moiety that is excited). The second peak/shoulder relates to the S2 π–π* transition at 397 nm, exhibiting a strong CT character, from the CND to the Azo moiety (CNDs+–p-Azo−). Furthermore, the calculated maximum of absorption intensity at 336 nm (f = 1.08) is in very good agreement with the experimental value of 349 nm. This peak is assigned to the π–π* transition S3 localized on the azo moiety, CND–p-Azo*, which is analogous to S2, although they differ in their orbital contributions. The π–π* transitions S4 and S5 are localized excitations on the nanodot fragment, CND*–p-Azo, which are blue shifted by at least 26 nm compared with the peak of 357 nm in pristine CND. Furthermore, the system is prone to CT events, since either S4 or S5 may undergo internal conversion to CT S2 state. The corresponding CT reactions CND*–p-Azo → CND+–p-Azo− are associated with favorable driving forces (0.6 eV for S4 → S2 and 0.8 eV for S5 → S2, see Fig. 5), assuming neglecting entropic effects, which is valid under the Franck–Condon approximation. In summary, considering CND–azo incorporating the azo fragment in para/meta and cis/trans configurations, electronic transitions in the UV region occur in both fragments (CND–azo* and CND*–Azo) for both para and meta conformers (see Fig. 5 and Fig. S30, respectively, ESI†) in the trans azo isomer. Conversely, for the cis isomer, excitations are primarily characterized by CND*–Azo in the UV region (Fig. S26 and S27, ESI†). We observed that the spectra of the hybrids incorporating the trans configuration of the azo moiety exhibit closer alignment with the experimental evidence (Fig. S28, ESI†). These findings suggest that the trans isomer of azo in the CND–azo hybrid is predominant in the electron transitions. In this regard, experiments determined that a wavelength of 340 nm induces the conversion to the cis isomer in the hybrids (Fig. 7). We additionally examined CT excitation energies (ω) in the hybrids, which shifted by 72 nm and 65 nm for trans CND–p-Azo and CND–m-Azo compared with the cis conformer, respectively (see Table 3). Nonetheless, CT excited states can be also accessed in the hybrids containing the cis isomer, albeit with smaller values. In this context, this cis/trans CT activity may be envisioned for use in photoswitching (on/off), data storage (0, 1), or molecular transistors (gate open/close).
| Hybrid | ω [nm (eV)] | qazo (e) | qCND (e) | d (Å) |
|---|---|---|---|---|
a d stands for the distance between the phenyl centroid in trans- or the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group in cis-azo and the closest centroid of IPCA in the CND. N group in cis-azo and the closest centroid of IPCA in the CND. |
||||
| trans CND–p-Azo | 397 (3.12) | –0.72 | +0.72 | 3.34 |
| trans CND–m-Azo | 375 (3.30) | –0.64 | +0.64 | 3.44 |
| trans CND–m-Gly-Azo | 374 (3.31) | –0.76 | +0.76 | 3.41 |
| trans CND–m-Gly-Azo | Inhibited | 7.95 | ||
| cis CND–p-Azo | 325 (3.81) | –0.74 | +0.74 | 3.24 |
| cis CND–m-Azo | 310 (4.00) | –0.85 | +0.85 | 3.90 |
| cis CND–m-Gly-Azo | 351 (3.53) | –0.85 | +0.85 | 2.86 |
| cis CND–m-Gly-Azo | Inhibited | 17.02 | ||
Similar trends are observed for the incorporation of a molecular spacer, CND–m-Gly-Azo (Fig. S31–S35, ESI†). Nonetheless, the flexibility provided by the molecular spacer either induces (when d < 4 Å) or inhibits (when d > 6 Å) the formation of CT states (presented in Table 3). However, the flexibility of the molecular spacer allows access to various structural configurations, indicating that CT states are not strictly inhibited.
Further, functionalization of the CND did not significantly change the emission maximum, as observed in the excitation–emission maps and excitation spectra of the CND and the hybrids (Fig. S37 and S38, ESI†). This aligns with the computational assumption put forth in this study and in our previous investigation, where an aggregated molecular fluorophore, IPCA, was identified as the primary PL source of the CNDs.10,16 It is notable that the molecular fluorophores present in the CND are not directly covalently linked with the azos; rather, they interact through space. Moreover, a through-bond mechanism appears to be unlikely for the lowest excited states, as the excitation energies are localized on the individual units, as illustrated in Fig. 5.
Accordingly, a through-space mechanism, such as Förster resonance energy transfer (FRET) or electron transfer, is a viable option, given the close proximity of the units (see Table 3).65–68 FRET relies on a dipole–dipole interaction, where the resonance between the donor's emission and the acceptor's absorbance and the relative orientation of their transition dipole moments plays a crucial role.69 In contrast, electron transfer requires a substantial overlap between the donor and acceptor orbitals, a prerequisite that is plausible based on our computed model.70,71 The potential of our synthesized hybrids to undergo both charge transfer and FRET suggests that quenching efficiency may be influenced by both mechanisms. As shown in Fig. 3, the CND emission strongly overlaps with the n–π* absorbance of the azo moieties, making a FRET from CND to the azo moieties possible. This was further confirmed by overlap integral calculations, which were used to determine the critical Förster radius (R0) (see ESI† for details). The R0 values vary slightly among the hybrids, averaging around 2.1 nm. Using these values and the distances listed in Table 3, FRET efficiencies were calculated, revealing a near-complete (99.9%) transfer efficiency for all three hybrids. This high efficiency results from the short CND-to-azo distance, which at 0.33 nm is well below the Förster radius.
However, the PLQY measurements indicate that this high efficiency is valid only for CND–m-Gly-Azo, as the other hybrids still exhibit a significant amount of CND fluorescence. To account for these differences, the quenching efficiency was determined using PLQY, since both FRET and electron transfer typically reduce the fluorescence quantum yield and lifetime of the donor. The resulting quenching efficiencies were 51% for CND–m-Azo, 70% for CND–p-Azo, and 97% for CND–m-Gly-Azo. Notably, quenching efficiency depends on the acceptor concentration within the system. Due to the undefined structural configuration of the hybrids and the unknown number of azo moieties per CND, accurately quantifying the relative contributions of different quenching processes is not possible.67
The elevated quenching efficiencies should also correspond to reduced fluorescence lifetimes of the CND moiety. To verify this, the fluorescence lifetimes of the pure CND and the hybrids were measured using TCSPC. All four systems exhibited three distinct fluorescence lifetimes, reflecting the complex surface structure of the CNDs. For the simplicity, these lifetimes were combined into an amplitude-weighted average lifetime. As expected, the pure CND holds with 5.70 ns the longest average fluorescence lifetime, followed by 3.60 ns and 3.45 ns for CND–p-Azo and CND–m-Azo, respectively. In contrast, with only 2.24 ns, the linker in CND–m-Gly-Azo clearly leads to the shortest fluorescence lifetime (Fig. 6 and Table S3, ESI†). While these lifetimes can also be used to estimate the quenching efficiencies of the hybrids, the obtained values are less reliable (see ESI† discussion for more details). The superior quenching efficiency of CND–m-Gly-Azo, in comparison to the hybrids that lack a linker, can be attributed to its higher density of functionalization. A greater density of azo moieties on the CND surface increases the probability of quenching. Additionally, our computational study demonstrated that the larger spacer in CND–m-Gly-Azo enhances flexibility and brings the azo units into closer proximity to the CND, further facilitating charge transfer processes.
 | ||
| Fig. 6 Decay curves of the fluorescence transients recorded with TCSPC. While the raw data are shown as circles, the respective fits consist of solid curves in a slightly darker colour. | ||
To confirm the retained photoswitching properties of the azos after covalent bonding to CNDs, the hybrids and bare azos were irradiated with a 325 nm LED for two minutes, followed by a 448 nm LED irradiation for 30 seconds, followed by measurement of the absorption spectra before and after each illumination (Fig. S39, ESI†). Among the hybrids, CND–m-Azo exhibited the smallest absorbance change upon trans-cis isomerization, whereas CND–m-Gly-Azo showed the largest, consistent with their respective degrees of functionalization. Among the pristine azos, the p-Azo displayed the smallest decrease in absorbance when irradiated with the 325 nm LED (Fig. S39, ESI†). However, the CND–p-Azo, demonstrated a greater change in absorbance than the CND–m-Azo, indicating that its 1.1% higher degree of functionalization outweighs the smaller change in absorbance observed in pure p-Azo. Additionally, pure azos exhibited greater changes in absorbance compared to hybrids, likely due to the significant contribution of the CND component to overall absorbance. The PL response to photoswitching indicated no significant reversible change in PLQY from the trans to the cis-isomers of the covalently linked azos. However, a slight decrease in PL intensity post-illumination prompted an investigation into fatigue resistance across multiple photoswitching cycles and associated PL properties (Fig. 7A–C). The selection of wavelengths was based on the absorption maxima of the hybrids, specifically 340 nm and 448 nm. The 340 nm wavelength was chosen to balance sufficient excitation energy while minimizing photodegradation. Each hybrid was irradiated for five minutes at these wavelengths to achieve the photostationary state (PSS) of the respective isomer (see Fig. S40, ESI†). CND–m-Azo underwent 13 trans–cis–trans cycles before reaching 50% of its original absorbance (Z50).
 | ||
| Fig. 7 Top panel: Schematic illustration of the azo-functionalized CNDs (black spheres), showing the photoinduced trans-to-cis conversion under 340 nm irradiation and subsequent reversion to the trans-isomer via 448 nm exposure or thermal isomerization at 40 °C. Middle panel: Fatigue resistance of CND–azo hybrids over multiple photoisomerization cycles using alternating 340 nm and 448 nm irradiation. (A) CND–m-Azo retains 50% of its initial absorbance at 350 nm for 13 trans–cis–trans cycles. (B) CND–p-Azo shows stability for 18 cycles at 349 nm. (C) CND–m-Gly-Azo maintains more than 50% absorbance at 329 nm for more than 15 cycles. Bottom panel: Thermal cis-to-trans isomerization kinetics of the hybrids at 40 °C over 50 h. Pristine azo half-lives are included for comparison (see ESI† for details). (D–F) Evolution of the absorbance at 352 nm (CND–m-Azo), 350 nm (CND–p-Azo), and 330 nm (CND–m-Gly-Azo) during thermal reversion. Single-exponential fits were used to determine half-lives. Note: Graphs D and E are normalized to their respective isosbestic points at 363 nm and 397 nm respectively, as agglomeration accurse over the time scale of the kinetic experiment. Due to the higher loading of azo in the CND–m-Gly-Azo hybrid, no spectra processing was necessary (see ESI† for details). | ||
In contrast, CND–p-Azo maintained 50% of its original absorbance over 18 cycles, demonstrating greater stability than CND–m-Azo. CND–m-Gly-Azo exhibited an 18% decrease in absorbance after 15 cycles, indicating it is the most durable hybrid produced. Fig. S41 (ESI†) shows the PL spectra following fatigue resistance tests, showing a similar decline in PL intensity corresponding to absorbance degradation. This suggests that the switching properties of the azos on the CND surface remain unaltered, while contributions from absorption and emission of the CNDs diminish. This hypothesis is supported by fatigue resistance measurements conducted under identical conditions with the pure Azos (Fig. S42, ESI†). After 120 minutes of light exposure, no significant drop in absorbance can be observed. Azos exhibit high fatigue resistance due to minimal occurrence of side reactions during photoisomerization. The increased fatigue resistance of the CND–m-Gly-Azo hybrid may be due to a higher azo coverage of the CND surface by the introduction of a glycine spacer. In classical organic chemistry, undesired reactions involving a reactive intermediate often lead to the generation of side-products. By offering an inert environment for these intermediates, confinement can serve as a method to reduce side-product formation and enhance the fatigue resistance.72 Additionally, we assessed the thermal half-life of the cis-isomer of the hybrids at 40 °C in DMSO (Fig. 7D–F). A schematic representation of the azos on the CND surface, which is switched between trans- and cis-isomers via light or heat, is shown in the top panel of Fig. 7. Fig. S43–S48 (ESI†) display UV-vis spectra illustrating thermal conversion from cis- to trans-isomer alongside single exponential fits against time to determine the thermal half-lives (Table S6, ESI†). The hybrids tend to agglomerate during the 50-hour thermal cis-to-trans isomerization, which is potentially distorting the data. In order to compensate that, we normalized the absorption spectra of the CND–m-Azo and CND–p-Azo to the isosbestic points (see ESI† for details).
The CND–m-Azo variant exhibits minimal variation in half-life compared to pure m-Azo, while the CND–m-Gly-Azo hybrid displays a reduced half-life relative to m-Gly-Azo. For the meta-linked hybrids, it is assumed that they behave electronically independently from the CNDs. The observed differences can be attributed to steric effects and the degree of functionalization, resulting in comparable or shorter half-lives than pure azos. Further systematic studies are required to quantify these contributions. In contrast, the CND–p-Azo demonstrates an increased half-life compared to p-Azo, suggesting that the electronic properties of the hybrid – modulated by the exchange of the carboxylic acid for an amide – can also play a role in altering the half-life. Furthermore, the precise chemical background of the functionalization employed, namely the amine on the CND side, has the potential to exert different electronic influences, given the lack of knowledge regarding the CND structure.
A comparison of the obtained results with other carbon allotropes functionalized with azos in the literature reveals that the influence of CNDs on the switching properties of the azos is relatively weak. In the case of fullerene, a publication by Shirai et al.73 in 2008 demonstrated that direct conjugation of fullerene with an azo led to reduced photoisomerization yields, suggesting an efficient electronic energy transfer between the azo and fullerene. However, when fullerene and azo were electronically separated via a spacer, this effect was not observed. In our case, this further supports the hypothesis that the chemical nature of CNDs leads to electronic separation of the molecular fluorophores on CNDs and azo in all three of our investigated hybrids. Indeed, TDDFT results for CND–m-Gly-Azo revealed that the formation of CT states depends upon the distance between CND and azo, as previously concluded. In 2013, Feng et al.74 introduced a reduced graphene oxide (RGO)-azo hybrid for long-term solar thermal storage. By achieving high packing densities and incorporating additional ortho and para functionalization of the azos with moieties that facilitate the formation of H-bonds, they successfully attained extended half-lives of the cis-isomer. The quasi-spherical and amorphous character of CNDs prevents the establishment of a repeating structure, allowing the azo units to orient in various directions without forming ordered configurations as seen on a RGO surface. Furthermore, the limited amount of azo present on the surface likely suppresses interactions between different azo units on a single CND.
Conclusions
In this study, three distinct CND–azo hybrids were synthesized, featuring either meta- or para-linkages as well as an additional glycine spacer. These hybrids exhibit both photoswitching capability and emissive properties, allowing them to be sensed and optically stimulated in the same system. The incorporation of the glycine spacer resulted in an increase in fatigue resistance, likely due to the formation of an inert environment around the CND. Furthermore, a reduction in the half-life of the cis isomer was observed in the case of the hybrid with the glycine spacer. In contrast, the two hybrids without the spacer exhibited similar half-lives compared to their pristine azo counterparts. This shorter half-life renders the system useful in optical switches due to their fast response to external stimuli. Furthermore, investigations using TDDFT methods were performed to gain deeper insights into the experimental UV-vis absorption spectra and CT properties of the CND–azo hybrids. This approach enhanced our understanding of the influence of isomer configuration on electronic transitions, and it provided valuable guidance for the development of potential applications. The results demonstrate that trans-azo in CND–azo hybrids significantly stabilizes the LUMO energy, leading to enhanced CT activity compared to the cis counterpart. Specifically, trans isomers exhibit a lower CT excitation energy and more favorable driving forces for CT reactions, making them more effective for applications requiring efficient electron transfer. In contrast, cis isomers undergo CT processes but with reduced driving forces. This difference underscores the potential of functionalized CND–azo hybrids for use in photoswitches, data storage, and molecular transistors where CT is critical. The addition of molecular spacers, such as in CND–m-Gly-Azo, further illustrates that spacer flexibility influences the formation of CT states. Specifically, spacer distances greater than 6 Å inhibit CT states, while shorter distances can promote them, emphasizing the need for careful spacer design to optimize electronic properties. Lastly, the total emission quenching efficiencies for the hybrids were calculated from fluorescence lifetimes obtained with TCSPC measurements. The fluorescence lifetimes of the hybrids were observed to be shorter than those of the pristine CND, with the shortest lifetime noted for the CND–m-Gly-Azo hybrid. This is attributed to the higher concentration of azo groups present on the surface, as well as the higher flexibility of the spacer, which promotes CT processes. Overall, these findings highlight the potential of azo-functionalized CND hybrids in developing advanced light-responsive systems. This represents an intriguing point of departure for the design of novel scaffolds that can be integrated into devices, sensors/actuators, or living cells that are remotely controlled through light irradiation.Author contributions
P. P. D.: writing original draft, conceptualization, investigation; D. S.: writing original draft, conceptualization, investigation; Y. A.-S.: investigation, conceptualization; J. P. M.: writing original draft, conceptualization, investigation in the computational section; M. L.: writing original draft, investigation, computations; J. H.: investigation; J. G.: writing – review & editing, conceptualization; E. M.: investigation, supervision, resources; B. M. S.: supervision, resources; M. S.: supervision, resources; S. O.: writing – review & editing, conceptualization, resources; J. W.: supervision, resources; H. A. W.: writing – review & editing, conceptualization, project administration; T. G.: writing – review & editing, conceptualization, project administration.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the financial support of the DFG and of the National Science Center, Poland through the joint project “Low-Dimensional Nano-Architectures for Light Emission and Light-to-Electricity Conversion” (LOW-LIGHT, grant no. UMO/2020/39/I/ST4/01446). D. S. thanks the European and Hessian Government for funding the “Innovationslabor Prozessdiagnostik” (EFRE 21031934). J. P. M. acknowledges the funding of a post-doctoral contract by the LOW-LIGHT grant. T. G. acknowledges the European Research Council for the project JANUS BI (grant agreement no. [101041229]). M. L. acknowledges that this article has been produced with the financial support of the European Union under the REFRESH – Research Excellence For Region Sustainability and High-tech Industries project number CZ.10.03.01/00/22_003/0000048 via the Operational Programme Just Transition. This work was supported by the Ministry of Education, Youth and Sports of the Czech Republic through the e-INFRA CZ (ID: 90254). J. W. gratefully acknowledges the European Union for funding the TCSPC laser system through the European Regional Development Fund as part of the Union's response to the COVID-19 pandemic – REACT-EU, IWB-EFRE-Programm Hessen, \#20008794. The authors thank Heike Hausmann (JLU, Giessen) for performing the NMR 1H and 13C spectra and Pascal Schweitzer (JLU, Giessen) for the support with the AFM measurements.References
- G. M. Paternò, G. Bondelli and G. Lanzani, Bioelectricity, 2021, 3, 136–142 CrossRef.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885 RSC.
- T. C. de Souza-Guerreiro, G. Bondelli, I. Grobas, S. Donini, V. Sesti, C. Bertarelli, G. Lanzani, M. Asally and G. M. Paternò, Adv. Sci., 2023, 10, 2205007 CrossRef CAS.
- B. Mavroidi, A. Kaminari, E. Sakellis, Z. Sideratou and D. Tsiourvas, Pharmaceuticals, 2023, 16, 833 CrossRef CAS PubMed.
- S. Zhang, C. Xiao, H. He, Z. Xu, B. Wang, X. Chen, C. Li, B. Jiang and Y. Liu, Environ. Sci.: Nano, 2020, 7, 880–890 RSC.
- M. Döbbelin, A. Ciesielski, S. Haar, S. Osella, M. Bruna, A. Minoia, L. Grisanti, T. Mosciatti, F. Richard, E. A. Prasetyanto, L. De Cola, V. Palermo, R. Mazzaro, V. Morandi, R. Lazzaroni, A. C. Ferrari, D. Beljonne and P. Samorì, Nat. Commun., 2016, 7, 11090 CrossRef.
- D. Niedzialek, I. Duchemin, T. B. De Queiroz, S. Osella, A. Rao, R. Friend, X. Blase, S. Kümmel and D. Beljonne, Adv. Funct. Mater., 2015, 25, 1972–1984 CrossRef CAS.
- S. Osella, M. Wang, E. Menna and T. Gatti, Opt. Mater. X, 2021, 12, 100100 CAS.
- A. M. Ross, S. Osella, V. R. Policht, M. Zheng, M. Maggini, F. Marangi, G. Cerullo, T. Gatti and F. Scotognella, J. Phys. Chem. C, 2022, 126, 3569–3581 CrossRef CAS.
- M. Langer, L. Zdražil, M. Medveď and M. Otyepka, Nanoscale, 2023, 15, 4022–4032 RSC.
- A. Banger, S. Gautam, S. Jadoun, N. K. Jangid, A. Srivastava, I. N. Pulidindi, J. Dwivedi and M. Srivastava, Catalysts, 2023, 13, 858 CrossRef CAS.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS.
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS.
- F. Arcudi, L. Ďorďević and M. Prato, Acc. Chem. Res., 2019, 52, 2070–2079 CrossRef CAS PubMed.
- F. Rigodanza, M. Burian, F. Arcudi, L. Đorđević, H. Amenitsch and M. Prato, Nat. Commun., 2021, 12, 2640 CrossRef CAS PubMed.
- P. P. Debes, M. Langer, M. Pagel, E. Menna, B. Smarsly, S. Osella, J. Gallego and T. Gatti, ChemNanoMat, 2024, 10, e202300471 CrossRef CAS.
- Y. Park, J. Yoo, B. Lim, W. Kwon and S.-W. Rhee, J. Mater. Chem. A, 2016, 4, 11582–11603 RSC.
- F. Arcudi, V. Strauss, L. Đorđević, A. Cadranel, D. M. Guldi and M. Prato, Angew. Chem., 2017, 129, 12265–12269 CrossRef.
- B. Liao, P. Long, B. He, S. Yi, B. Ou, S. Shen and J. Chen, J. Mater. Chem. C, 2013, 1, 3716 RSC.
- X. Zhang, L. Hou and P. Samorì, Nat. Commun., 2016, 7, 11118 CrossRef CAS.
- K. Nakatani, J. Piard, P. Yu and R. Métivier, in Photochromic Materials: Preparation, Properties and Applications, ed. H. Tian and J. Zhang, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016, pp. 1–45 Search PubMed.
- T. Nägele, R. Hoche, W. Zinth and J. Wachtveitl, Chem. Phys. Lett., 1997, 272, 489–495 CrossRef.
- J. L. Oudar, J. Chem. Phys., 1977, 67, 446–457 CrossRef CAS.
- M. J. Moran, M. Magrini, D. M. Walba and I. Aprahamian, J. Am. Chem. Soc., 2018, 140, 13623–13627 CrossRef CAS PubMed.
- L. Hegedüsová, N. Blaise, L. F. Pašteka, Š. Budzák, M. Medveď, J. Filo, B. Mravec, C. Slavov, J. Wachtveitl, A. M. Grabarz and M. Cigáň, Chem. – Eur. J., 2024, 30, e202303509 CrossRef.
- B. Mravec, Š. Budzák, M. Medved’, L. F. Pašteka, C. Slavov, T. Saßmannshausen, J. Wachtveitl, J. Kožíšek, L. Hegedüsová, J. Filo and M. Cigáň, J. Org. Chem., 2021, 86, 11633–11646 CrossRef CAS PubMed.
- J. C. Tobin, A. F. Hegarty and F. L. Scott, J. Chem. Soc. B Phys. Org., 1971, 2198–2202 RSC.
- A. I. A. Soliman, M. Sayed, M. M. Elshanawany, O. Younis, M. Ahmed, A. M. Kamal El-Dean, A.-M. A. Abdel-Wahab, J. Wachtveitl, M. Braun, P. Fatehi and M. S. Tolba, ACS Omega, 2022, 7, 10178–10186 CrossRef CAS PubMed.
- Z. Wang, R. Losantos, D. Sampedro, M. Morikawa, K. Börjesson, N. Kimizuka and K. Moth-Poulsen, J. Mater. Chem. A, 2019, 7, 15042–15047 RSC.
- L. Dong, Y. Feng, L. Wang and W. Feng, Chem. Soc. Rev., 2018, 47, 7339–7368 RSC.
- K. Kreger, P. Wolfer, H. Audorff, L. Kador, N. Stingelin-Stutzmann, P. Smith and H.-W. Schmidt, J. Am. Chem. Soc., 2010, 132, 509–516 CrossRef CAS PubMed.
- M. A. Gerkman and G. G. D. Han, Joule, 2020, 4, 1621–1625 CrossRef.
- Z. Wang, P. Erhart, T. Li, Z.-Y. Zhang, D. Sampedro, Z. Hu, H. A. Wegner, O. Brummel, J. Libuda, M. B. Nielsen and K. Moth-Poulsen, Joule, 2021, 5, 3116–3136 CrossRef CAS.
- D. Schatz, M. E. Baumert, M. C. Kersten, F. M. Schneider, M. B. Nielsen, M. M. Hansmann and H. A. Wegner, Angew. Chem., Int. Ed., 2024, 63, e202405618 CrossRef CAS PubMed.
- K. Hüll, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef.
- M. O. Lenz, A. C. Woerner, C. Glaubitz and J. Wachtveitl, Photochem. Photobiol., 2007, 83, 226–231 CAS.
- M. Liu, X. Yan, M. Hu, X. Chen, M. Zhang, B. Zheng, X. Hu, S. Shao and F. Huang, Org. Lett., 2010, 12, 2558–2561 CrossRef CAS PubMed.
- C. Averdunk, K. Hanke, D. Schatz and H. A. Wegner, Acc. Chem. Res., 2024, 57, 257–266 CrossRef CAS.
- Y. Norikane and N. Tamaoki, Org. Lett., 2004, 6, 2595–2598 CrossRef CAS.
- G. S. Hartley, Nature, 1937, 140, 281 Search PubMed.
- E. Merino and M. Ribagorda, Beilstein J. Org. Chem., 2012, 8, 1071–1090 Search PubMed.
- D. Schulte-Frohlinde, Ann. Chem., 1958, 612, 138–152 Search PubMed.
- A. Goulet-Hanssens, C. Rietze, E. Titov, L. Abdullahu, L. Grubert, P. Saalfrank and S. Hecht, Chem, 2018, 4, 1740–1755 CAS.
- C. Slavov, C. Yang, L. Schweighauser, C. Boumrifak, A. Dreuw, H. A. Wegner and J. Wachtveitl, Phys. Chem. Chem. Phys., 2016, 18, 14795–14804 RSC.
- S. Bellotto, R. Reuter, C. Heinis and H. A. Wegner, J. Org. Chem., 2011, 76, 9826–9834 CrossRef CAS.
- R. Reuter, N. Hostettler, M. Neuburger and H. A. Wegner, Eur. J. Org. Chem., 2009, 5647–5652 CrossRef CAS.
- R. Ziessel, P. Stachelek, A. Harriman, G. J. Hedley, T. Roland, A. Ruseckas and I. D. W. Samuel, J. Phys. Chem. A, 2018, 122, 4437–4447 CrossRef CAS.
- F. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327–336 RSC.
- I. J. Gomez, B. Arnaiz, M. Cacioppo, F. Arcudi and M. Prato, J. Mater. Chem. B, 2018, 6, 5540–5548 RSC.
- W. S. Price, NMR Studies of Translational Motion: Principles and Applications, Cambridge University Press, Cambridge, 2009 Search PubMed.
- S. Sarkar, S. Dinda, P. Choudhury and P. K. Das, Soft Matter, 2019, 15, 2863–2875 RSC.
- R. Luther and A. Nikolopulos, Z. Physiol. Chem., 1913, 82U, 361–384 CrossRef.
- P. P. Debes, M. Pagel, S. Muntean, J. Hessling, B. M. Smarsly, M. Schönhoff and T. Gatti, Photochem, 2025, 5, 1 CrossRef CAS.
- G. Filippini, F. Amato, C. Rosso, G. Ragazzon, A. Vega-Peñaloza, X. Companyó, L. Dell’Amico, M. Bonchio and M. Prato, Chem, 2020, 6, 3022–3037 CAS.
- M. Langer, T. Hrivnák, M. Medved and M. Otyepka, J. Phys. Chem. C, 2021, 125, 12140–12148 CrossRef CAS.
- M. Langer, M. Paloncýová, M. Medved and M. Otyepka, J. Phys. Chem. Lett., 2020, 11, 8252–8258 CrossRef CAS PubMed.
- M. J. Krysmann, A. Kelarakis, P. Dallas and E. P. Giannelis, J. Am. Chem. Soc., 2012, 134, 747–750 CrossRef CAS PubMed.
- L. Vallan, E. P. Urriolabeitia, F. Ruipérez, J. M. Matxain, R. Canton-Vitoria, N. Tagmatarchis, A. M. Benito and W. K. Maser, J. Am. Chem. Soc., 2018, 140, 12862–12869 CrossRef CAS PubMed.
- X. Zhao, J. Wei, T. Song, Z. Wang, D. Yang, X. Zhang, F. Huo, Y. Zhang and H.-M. Xiong, Chem. Eng. J., 2024, 481, 148779 CrossRef CAS.
- C. Xia, S. Zhu, T. Feng, M. Yang and B. Yang, Adv. Sci., 2019, 6, 1901316 CrossRef CAS PubMed.
- F. Ehrat, S. Bhattacharyya, J. Schneider, A. Löf, R. Wyrwich, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2017, 17, 7710–7716 CrossRef CAS PubMed.
- C. M. Kok and A. Rudin, Makromol. Chem., Rapid Commun., 1981, 2, 655–659 CrossRef CAS.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, WIREs Comput. Mol. Sci., 2021, 11, e1493 CrossRef CAS.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- I. Yildiz, M. Tomasulo and F. M. Raymo, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11457–11460 CrossRef CAS.
- M. Sykora, M. A. Petruska, J. Alstrum-Acevedo, I. Bezel, T. J. Meyer and V. I. Klimov, J. Am. Chem. Soc., 2006, 128, 9984–9985 CrossRef CAS.
- P. Moroz, Z. Jin, Y. Sugiyama, D. Lara, N. Razgoniaeva, M. Yang, N. Kholmicheva, D. Khon, H. Mattoussi and M. Zamkov, ACS Nano, 2018, 12, 5657–5665 CrossRef CAS PubMed.
- M. H. Stewart, A. L. Huston, A. M. Scott, A. L. Efros, J. S. Melinger, K. B. Gemmill, S. A. Trammell, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, ACS Nano, 2012, 6, 5330–5347 CrossRef CAS.
- D. W. Piston and G.-J. Kremers, Trends Biochem. Sci., 2007, 32, 407–414 CrossRef CAS.
- E. F. Valeev, V. Coropceanu, D. A. da Silva Filho, S. Salman and J.-L. Brédas, J. Am. Chem. Soc., 2006, 128, 9882–9886 CrossRef CAS.
- J. L. Brédas, J. P. Calbert, D. A. da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804–5809 Search PubMed.
- M. Canton, A. B. Grommet, L. Pesce, J. Gemen, S. Li, Y. Diskin-Posner, A. Credi, G. M. Pavan, J. Andréasson and R. Klajn, J. Am. Chem. Soc., 2020, 142, 14557–14565 CrossRef CAS PubMed.
- Y. Shirai, T. Sasaki, J. M. Guerrero, B.-C. Yu, P. Hodge and J. M. Tour, ACS Nano, 2008, 2, 97–106 CrossRef CAS.
- Y. Feng, H. Liu, W. Luo, E. Liu, N. Zhao, K. Yoshino and W. Feng, Sci. Rep., 2013, 3, 3260 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc00116a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |