
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePressure- and temperature-driven transitions and conformational conversions of n-hexyl substituted perylene diimide (PDI-C6) crystals†
Paulina Ratajczyk a,
Szymon Sobczaka,
Michał Andrzejewskib,
Francesco Marinc,
Marianna Marchinic,
Lucia Maini*c and
Andrzej Katrusiak*a
a,
Szymon Sobczaka,
Michał Andrzejewskib,
Francesco Marinc,
Marianna Marchinic,
Lucia Maini*c and
Andrzej Katrusiak*a
aFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland. E-mail: katran@amu.edu.pl
bPaul Scherrer Institute, Forschungsstrasse 111, Villigen, CH-5232, Switzerland
cDepartment of Chemistry “G. Ciamician”, University of Bologna, via Selmi 2, 40126 Bologna, Italy. E-mail: l.maini@unibo.it
First published on 4th June 2025
Abstract
Perylene diimides (PDIs) are organic semiconductors with properties suitable for applications in electronic devices. This study presents a comprehensive investigation of PDI-C6, a perylene diimide modified with an n-hexyl alkyl chain at the imide position, under non-ambient conditions, with a focus on the rich diagram of structurally varying phases and their anisotropic strain. Five distinct polymorphs of PDI-C6 have been found. The ambient phase I transforms to phase II at 465 K, which at 521 K transforms to phase III. The perylene cores arranged in parallel stacks in phase I rotate in the scissor-opening motion by 33° in phase II. Under high pressure, phase I transforms to phase IV at 1.22 GPa and to phase V at 1.50 GPa. The transitions between phases I, IV and V stepwise modify the conformation of the n-hexyl substituents. Surprisingly, all five polymorphs are of space-group type P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , which is unprecedented as the symmetry of phases usually involves the space-group changes. These findings significantly enhance our understanding of the structural transformations of next-generation electronic materials and highlight the role of crystal engineering in constructing novel organic semiconductors with high sensitivity to environmental stimuli.
, which is unprecedented as the symmetry of phases usually involves the space-group changes. These findings significantly enhance our understanding of the structural transformations of next-generation electronic materials and highlight the role of crystal engineering in constructing novel organic semiconductors with high sensitivity to environmental stimuli.
Introduction
Organic charge-transfer complexes and π-conjugated compounds are prospective materials for efficiently harvesting solar energy, proposed in response to the pressing demand for new environment-friendly sustainable energy sources. Presently, the mass production of silicon and hybrid-perovskite solar photovoltaic (SPV) panels poses significant drawbacks. Silicon panels are expensive and energy-demanding in production, their disposal is environment polluting and recycling difficult and expensive; the main problems connected to the presently innovative hybrid perovskites, such as [CH3NH3]PbI3 (MAPbI3), are their contents of metals, often poisonous and contaminating the environment both at the production and disposal stages, not to mention a relatively high susceptibility to humidity, temperature changes and stress. The mass production of Si-based panels threatens the environment and biosphere and more environment-friendly materials are intensely sought.1,2Organic materials are attractive due to their practically infinite ways of modifications and tuning properties, production and disposal. π-Conjugated molecular materials have recently attracted significant attention in the field of organic electronics due to their potential for practical applications, driven by the promising combination of superior charge carrier mobility and robust stability.3 Recent extensive research has focused on designing and synthesizing new materials, understanding their structures, properties and emerging opportunities for developing innovative devices.4 Compared to their inorganic counterparts, organic semiconductors offer not only high efficiency of the photoelectric conversion but also new qualities, including high flexibility and light weight, making them attractive for next-generation electronic devices,5,6 such as photovoltaic tapes and foils for highly flexible photovoltaic panels and tissues, apart from substituting the roof and other panels. The envisaged applications for the organic photovoltaics (OPVs) panels can be similar to those already witnessed for similar organic light-emitting diodes (OLEDs) and organic thin-film transistors (OTFTs).7–10
Thermal expansion, compressibility and polymorphism are key issues for organic semiconductors, because different crystal forms of the same compound can significantly vary in properties additionally modified by the temperature and pressure-induced strain.11,12 Molecular engineering through chemical synthesis is a direct method to tailor the optoelectronic properties of materials,13,14 but they may be enhanced or deteriorated by the monotonic changes and phase transitions. The synthesis process is usually time-consuming, labor-intensive and expensive, but the polymorphism of products can improve or diminish envisaged applications.15,16 Most organic semiconductors crystallize in two or more polymorphic forms with different molecular packings in solid state,17–21 leading to different properties.22,23 The electronic, optical, mechanical, and charge transport properties of organic materials are heavily dependent on molecular packing, which is governed by various types of strong and weak intermolecular interactions.24,25 Thus, investigating and controlling crystal structures and polymorphism is crucial for tailoring optical and electrical properties of organic semiconductors. Application of pressure and high- or low-temperature efficiently modifies intermolecular interactions and reveal the polymorphic behaviour of molecular compounds.26,27 Including this important physical variables in the study of organic materials opens new exciting opportunities for investigating and developing high-performance semiconductors for optoelectronic devices.28–30
Perylene-3,4,9,10-tetracarboxydiimides, commonly known as perylene diimides (PDIs) are a class of compounds with polyaromatic π-conjugated flat molecules. PDIs offer a number of attractive characteristics, including high photo- and thermal-stability along with chemical inertness,31 and have been extensively studied as semiconductors for OFETs,32–35 and OPVs.36–38 The properties of these n-type semiconducting materials can be efficiently modified through appropriate functionalization of the PDI core, achieved either by introducing different substituents at the imide N-atom or by modifying the core at the bay or ortho positions.39 Such substitutions effectively alter crystal packing and solubility, influencing the optical and electrochemical properties.36,40 Particularly effective, in the context of enhanced solubility, is the imide-site substitution by long alkyl chains. Several PDI systems with different alkyl chain length are known, but their polymorphic behaviour remains underinvestigated.41–44
Here, we present our study on N,N′-dihexyl-3,4,9,10-perylenedicarboximide (denoted as PDI-C6), a commercially available PDI derivative functionalized with a hexyl alkyl moiety at the imide position (Scheme 1). The UV-Vis spectrum shows the high absorbance (Fig. S1, ESI†) characteristic of highly efficient photovoltaic materials. Although two high-temperature transitions of PDI-C6 were reported,32–35 they have not been thoroughly investigated, and the structures of those phases remain unknown. Recently, Madhu and coworkers reported the crystal structure of PDI-C6 phase I at 173 K, revealing its one-dimensional (1D) columns, characteristic of PDI alkyl derivatives, stabilized by π–π stacking and C–H⋯O hydrogen bonding interactions.45 Some of PDIs, such as PDI-C5, have been studied at both room and high temperature, showing intriguing phase transitions, including polymorph obtained at 494 K that is characterized by a criss-cross arrangement of the aromatic cores.46,47 However, high-pressure studies of perylene diimides, and other organic photovoltaic (OPV) materials remain exceptionally rare.9,48–53 To fill this gap, we explored the polymorphism of PDI-C6 at various temperatures and high pressure by combining multiple techniques, including synchrotron and in-lab X-ray diffraction, as well as differential scanning calorimetry (DSC).
 | ||
| Scheme 1 Structural formula of N,N′-dihexyl-3,4,9,10-perylenedicarboximide (PDI-C6). | ||
Results and discussion
Previous calorimetric studies of PDI-C6 revealed two phase transitions during heating, occurring around 464 and 519 K, and two during cooling, at 494 and 409 K,34 but they were not further investigated. To maintain consistent labelling with that previously introduced for PDI-C5,47 a PDI derivative with an n-pentyl chain, we will refer to the form stable under room conditions (296 K/0.1 MPa) as phase I, with subsequent phases labelled according to their order of appearance.PDI-C6 at room conditions
At room conditions, PDI-C6 phase I crystallizes in the triclinic space group P, with the PDI-C6 molecule located on an inversion center (Z′ = 0.5). This observation agrees with the structure reported by Madhu et al. at 173 K (CSD refcode: TIXTUS).45 In this structure, the molecules π-stacked into columns along the [100] crystal direction (Fig. S2, ESI†). The distance between π-stacked molecular cores (dπ–π) at room conditions is 3.3809(15) Å, somewhat shorter than the typical range 3.4–3.6 Å usually reported for π-stacked aggregates.54
The perylene core of the PDI molecules is planar, while the alkyl chains protrude off the plane in trans conformation. The inclination of the n-hexyl chain to the perylene core can be described by torsion angle C1–N1–C13–C14 (τ1), while the conformation of the alkyl chains is characterized by four torsion angles (Scheme 1): N1–C13–C14–C15 (τ2), C13–C14–C15–C16 (τ3), C14–C15–C16–C17 (τ4) and C15–C16–C17–C18 (τ5). In analogy to the ethane-like molecules, the hexyl chain conformers can be represented using four-letter descriptors.55 In phase I, the alkyl chains adopt anti-anti-anti-anti conformation denoted by descriptor AAAA, corresponding to angles τ2, τ3, τ4, τ5 all approaching 180°. The conformations involving gauche+ and gauche− torsions (60° and −60°, respectively), present in subsequent phases, are denoted e.g. G+AG−A. The eclipsed conformation (E) describes a molecular arrangement in which adjacent methylene or methyl groups are in close proximity, with the torsion angles between 0° and ± 30°, or close to 120°. The overall packing consists of alternating layers, with interdigitated alkyl chains and layers composed of rigid aromatic cores.
Temperature effect on PDI-C6
The thermal behaviour of PDI-C6 has been investigated by several complementary techniques, including differential scanning calorimetry (DSC); thermogravimetric analysis (TGA); hot-stage microscopy (HSM); variable-temperature synchrotron X-ray powder diffraction (VT-PXRD) and in-lab single-crystal X-ray diffraction (VT-SCXRD). These methods allowed us to explore a broad temperature range from 100 K up to the decomposition at 823 K with TGA (in a N2 atmosphere, Fig. S3 in ESI†) or at 673 K using VT-PXRD (in air).During the heating cycle, according to the DSC thermogram, phase I transforms to phase II at TI→II = 465 K, and at TII→III = 521 K phase II transforms to phase III (Fig. 1). These transitions are consistent with the VT-SCXRD and HSM. The thermograms collected for the PDI-C6 cooled at a rate of 20 K min−1 indicate that the transitions from phase III to phase II and from phase II to phase I are reversible, as previously reported.34
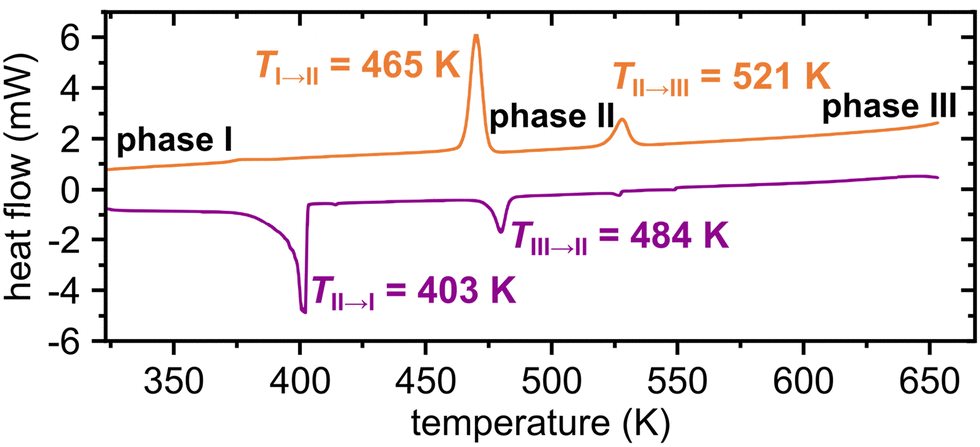 | ||
| Fig. 1 Differential scanning calorimetry (DSC) thermogram of PDI-C6 in the heating (top) and cooling (bottom) cycles. Temperatures of phase transitions are indicated. | ||
The HSM revealed intriguing behaviour during the heating and cooling of the crystals related to the transition between phases I and II. The transitions occurred at slightly different temperatures for individual crystals, which is characteristic of 1st-order reconstructive transitions. The phase I to phase II transition observed upon heating was characterized by clearly visible propagating fronts that induced wrinkle formation on the crystal surface (as visible for crystal 1 in Fig. 2a and b). A different behaviour was observed upon cooling, at approx. 392 K, all the PDI-C6 needles elongated (Fig. 2c and d), leading to the movement, jumping and bending of some crystals (Fig. S18e and f, ESI†). Similar polymorphism and thermomechanical behaviour were observed for other organic semiconducting materials.56,57 These macroscopic changes in single-crystal morphology can be attributed to the mutual rotation of the perylene cores, where the transformation of stress accumulated due to molecular rearrangement is converted into mechanical force.47
 | ||
| Fig. 2 Hot stage microscopy of PDI-C6 crystals during the heating cycle: (a) phase I at 470 K, (b) transition from phase I to phase II at 473 K; and during cooling cycle: (c) phase II at 423 K, (d) transition from phase II to phase I at 363 K, reshaping the crystals. Additional images are provided in Fig. S18 and S19 of the ESI.† | ||
Our VT-PXRD analysis shows that, on approaching the transition to phase II, simultaneously the reflections of both phases II and III appear, while above TII→III, at 538 K only phase III is present (Fig. 3). The coexistence of phases II and III is evident from two distinct reflections at 2θ below 1.25° in the PXRD pattern collected at 475 K (Fig. S4, ESI†). Because the transition from phase I to phase II is accompanied by a strong deformation, the unexpected appearance of phase III at 469 K can be associated with this strong mechanical effect ‘pulling’ small portions of the crystals into the adjacent higher-temperature phase III. The observation of phases outside their stability regions is occasionally experienced for other compounds, too, e.g. resorcinol or benzimidazole.58,59 This recourse of a portion of the fine-powder sample to phase III at 469 K contrasts with all our experiments on single crystals and DSC measurements, all of which transformed to phase II, as expected. Upon cooling, phase III demonstrates strong hysteresis, persisting up to 353 K, when a simultaneous transition into phases II and I occurs (Fig. S9, ESI†). This extended hysteresis of phase III in the cooling run may result from a substantial thermal contraction, as indicated by thermal-expansion tensor plot (Table S7, ESI†), potentially hindering the spatial reorganization of molecules during the transition to phases II/I. Notably, a significant difference is observed in the phase transition temperature across different thermal analyses (cf. Fig. S8, ESI†). This suggests that crystal morphology, particularly crystal size, plays a crucial role in influencing the kinetics of phase transition events, as it was observed for other materials.60
 | ||
| Fig. 3 (a) Temperature-resolved powder X-ray diffractograms of PDI-C6 upon heating (left). The dashed lines indicate the transition temperatures identified in the DSC measurement: TI→II = 465 K and TII→III = 521 K. The color scale represents intensity variations. Crystal structures of PDI-C6 phase I (b) and II (c) showing: the packing arrangement along the 1D columnar alignment (left) and the columnar stacking, including conformer descriptors (right). Molecules located at the inversion center with a trans conformation are depicted in purple, while those in general positions with a cis conformation are shown in orange. | ||
In order to better understand the structural changes and the temperature effect on PDI-C6 crystals, we performed a series of VT-SCXRD experiments up to 470 K. Heating phase I from 100 K to TI→II, gradually elongates the unit-cell parameters aI and cI (the index indicates the phase). Parameter bI initially elongates up to 338 K; on further heating to 430 K this trend is reversed and bI shortens; and between 430 K and 465 K, bI hardly changes with temperature (Fig. 4a). This unexpected behaviour can be attributed to the conformational changes of the n-hexyl chains. The π-conjugated stacking columns of PDI-C6 molecules aligned along direction aI in phase I can be described by the pitch and roll inclinations relative to the ideal cofacial π-stack, as reported by Curtis et al.61 In this analysis, the pitch (P) angle corresponds to the displacement caused by the shift of adjacent molecules along the long molecular axis, whereas the roll (R) angle refers to the relative shift between adjacent molecules perpendicular to the long axis of the molecule (or along its short molecular axis). As the temperature increases in phase I, the P angle decreases from 42.88(6)° at 100 K to 42.31(4)° at 440 K, while the R angle slightly increases from 20.49(7)° to 21.11(5)° over the same temperature range (Fig. 5a). The distance between π-stacked molecular planes (dπ–π) increases from 3.321(2) Å at 100 K up to 3.4397(13) Å at 440 K (Table S2, ESI†). The calculated volumetric thermal expansion coefficient (αV) in the 100–354 K range, is 162 × 10−6 K−1, which is close to the typical value of 170 × 10−6 K−1 observed for organic compounds.62 However, between 368 K and 461 K, the αV increases sharply, reaching 446 × 10−6 K−1, which is nearly three times the value observed in the previous temperature range (see the ESI† for more details).
 | ||
| Fig. 4 Temperature (a) and pressure (b) dependence of the unit-cell dimensions a, b, c (top) and molecular volume (Vm = V/Z, bottom) of PDI-C6. Estimated standard deviations (ESDs) are smaller than plotted symbols. The data at 173 K (squares) were taken from Madhu et al.45 The unit-cell parameters of phase III determined during the cooling cycle are marked with a lighter color. The data measured at 296 K is highlighted in blue. Lines joining the points are for guiding the eye only. The triclinic angles are plotted in Fig. S21 (ESI†). | ||
 | ||
| Fig. 5 Temperature (a) and pressure (b) effects on the π-stacking of PDI-C6 molecules under pressure: orange and purple symbols represent the pitch and roll angles and grey squares mark the distance between π-stacked molecules (dπ–π).61 Inset: Graphical representation of P, R and dπ–π. The alkyl substituents and H-atoms are skipped for clarity. The corresponding numerical values can be found in Tables S2 and S10 of the ESI.† | ||
The phase transition from phase I to phase II fractures the single crystals, which complicated the accurate determination of high-temperature structures. Despite these difficulties, we successfully solved the structure of phase II at 470 K, revealing its triclinic symmetry of space group P. The unit-cell parameters of PDI-C6 phase II closely resemble those of high-temperature phase III of PDI-C5 (a = 11.624 Å, b = 11.617 Å, c = 17.415 Å, α = 98.21°, β = 71.25°, γ = 116.06°, CSD refcode: DICMUX01) and its number of molecules in the unit cell (Z = 3) is the same.47 These similarities extend to the molecular packing and in PDI-C6 phase II one molecule lies at an inversion center, while the other occupies a general position. The alkyl chains of the Ci-symmetric molecule are in trans conformation (i.e. they protrude to the opposite sides of PDI core), whereas the alkyl chains in the molecule at general position are cis (they protrude to the same side). The trans-molecule adopts the AAEA conformation, while the cis molecule has AAAE and AEAE conformers (Fig. 3c). Within the columns, every two parallel cis-molecules are followed by a trans-molecule, whose perylene core is rotated by approximately 33° about the column axis and slightly tilted relative to the cis-molecules. This results in an alternating sequence of two molecules cis, criss-cross π-stacked to one molecule trans, followed by two molecules cis, etc. Above 521 K, the strong reconstructive transition hampered our attempts to determine the structure of PDI-C6 phase III. The unit-cell parameters of phase III are reported in Table 1.
| p (GPa)/T (K) | Phase | Unit cell | V (Å3) | Z/Z′ | dπ–π (Å) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | |||||
| 0.0001/298 | I | 4.7568(10) | 8.5487(16) | 17.576(2) | 81.678(14) | 84.850(15) | 83.169(16) | 700.3(2) | 1/0.5 | 3.3809(15) |
| 0.0001/470 | II | 11.443(7) | 11.988(8) | 19.506(9) | 96.05(5) | 73.16(5) | 114.90(6) | 2323(3) | 3/1.5 | 3.579(8) |
| 0.0001/537 | III | 7.676(2) | 10.148(1) | 21.559 (2) | 84.35(2) | 83.34(4) | 90.52(3) | 1665.5(5) | 2/1 | — |
| 1.41/296 | IV | 4.5308(1) | 16.9532(3) | 17.887(1) | 66.484(3) | 95.489(2) | 96.287(1) | 1250.21(8) | 2/1 | 3.2643(6) |
| 3.2044(5) | ||||||||||
| 1.95/296 | V | 4.4643(1) | 8.6151(1) | 15.8664(8) | 90.750(3) | 92.236(3) | 97.490(1) | ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 604.46(3) 604.46(3) |
1/0.5 | 3.2049(6) |
PDI-C6 under pressure
High-pressure single-crystal X-ray diffraction measurements were performed on PDI-C6 at room temperature, up to 4.61 GPa. The discontinuities in the unit-cell dimensions as well as the doubling of parameter bI at 1.22 GPa and its subsequent halving at 1.52 GPa evidence two pressure-induced phase transitions (Fig. 4b). Phase I is stable up to 1.05 GPa and then transforms to phase IV, which at 1.52 GPa transforms to phase V. The high-pressure phases IV and V are of the same triclinic space group type P as phases I, II and III. In phases I and V, the asymmetric part of the unit cell comprises half of the PDI-C6 molecule, while Z′ = 1 in phase IV. The crystallographic data are summarized in Table 1 and the crystal and experimental data are detailed in Table S16 in ESI.†
The compression of the phase I significantly reduces the aI and cI dimensions, which is associated with decreased π–π distances and deformations of the alkyl chains. The parameter bI is hardly affected by the pressure and upon the compression to 0.73 GPa, it decreases by only −0.14%. Interestingly, at 1.05 GPa, just before the transition to phase IV, the bI is approximately +0.01% longer compared to its length at 0.1 MPa. These changes correlate with variations in the shortest hydrogen bond CH4⋯O2, which shortens from 2.4752(15) Å at 0.1 MPa to 2.37(3) Å at 0.73 GPa, before lengthening slightly to 2.39(2) Å at 1.05 GPa. At 0.1 MPa, the P angle is 42.53° and the R angle is 20.75°. The compression of phase I from 0.1 MPa to 1.05 GPa of PDI-C6 has a small impact on the P angle, reducing it by only 0.33°. In contrast, the changes of the R angle and π–π stacking distance (dπ–π) are much more pronounced. The compression to 1.05 GPa decreases the R angle by 1.75° and shortens the dπ–π distance to 3.278(6) Å from 3.3882(4) Å measured at 0.1 MPa (Fig. 5b).
The unit cell of phase IV clearly corresponds to that of phase I, as evident from compression of parameters (Fig. 4b) and the structural comparison presented in Fig. 6. The nearly monotonic compression of a reflects the gradual contraction of π–π stacking distance between adjacent PDI-C6 molecules within the stacks. The unit-cell vectors of phases I (aI, bI, cI) and IV (aIV, bIV, cIV) are related by the following eqn (1):
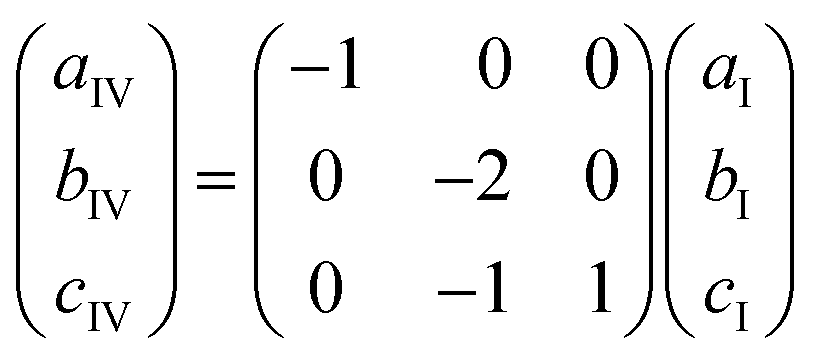 | (1) |
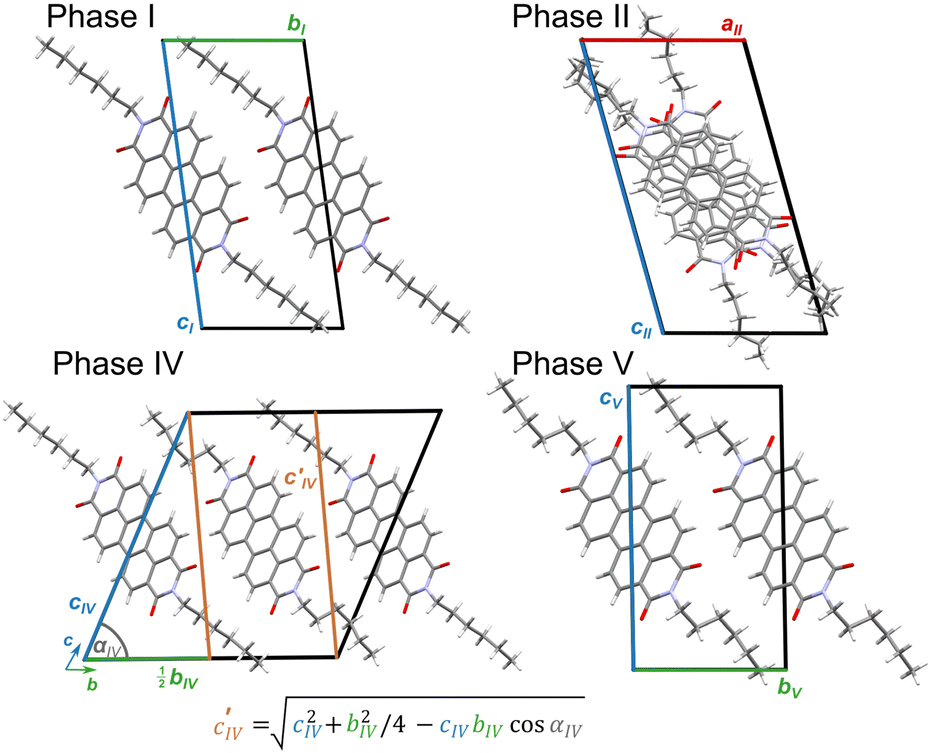 | ||
| Fig. 6 Lattice relations between phases I (0.1 MPa/298 K), II (0.1 MPa/470 K), IV (1.41 GPa/296 K), and V (1.95 GPa/296 K) in PDI-C6, illustrated for maintained orientations of the π-stacked molecules; the orange axes in phase IV indicate the corresponding directions in phases I and V. | ||
Apart from doubling of the b parameter (bIV = 2bI), the transition from phase I to IV leads to an abrupt reduction of the c′IV parameter by −2.20%, which can be recalculated in relation to the cI parameter of phase I with the eqn (2):
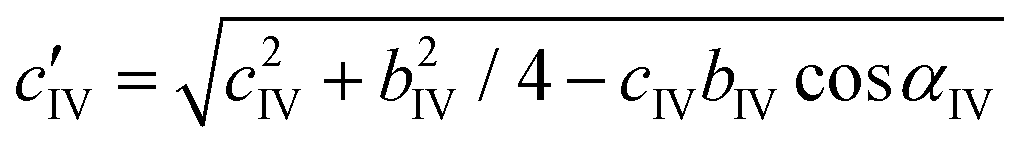 | (2) |
The transformation to phase IV releases the structural strain accumulated during compression by lowering the crystal symmetry and doubling the number of independent molecules. As a result, due to conformational alternation of the n-hexyl chains, the Z′ in phase IV increases from 0.5 to 1. One of the molecules retains Ci symmetry, maintaining the AAAA conformation of alkyl chains, as observed before in phase I, with τ2–τ5 torsion angles close to 180°. The other molecule adopts a gauche+/gauche− (G+/G−) conformation, where τ3 and τ5 remain near 180°, while τ2 and τ4 twist to approximately ±60°. Consequently, the conformers descriptors for two independent molecules present in phase IV are AAAA/AAAA and G+AG−A/G−AG+A (Fig. 7). The discrimination of molecules into two independent conformers affects their π-stacking in phase IV and significantly impacts the elastic response of the crystal. The pressure-induced deformation of the AAAA stacks follows the trends observed in phase I, with the P angle decreasing by 0.71°, and dπ–π by 0.42% compared to their values at 1.05 GPa. Under the same conditions, the R angle increases by 1.5°. The accommodation of new conformers G+AG−A/G−AG+A requires the P angle to increase by 1.73° and R angle to decrease by 0.93°, compared to the AAAA conformer at 1.41 GPa. As a result, the enhanced flexibility of G+AG−A/G−AG+A conformers allows for more efficient packing and the dπ–π distance shorter by 1.84% compared to that between conformers AAAA at 1.41 GPa.
 | ||
| Fig. 7 Conformational changes in PDI-C6 molecules in phases I (grey), IV (orange) and V (violet) illustrated by (a) superimposing fragment of the perylene core, and (b) torsion angles τ1–5 plotted as a function of pressure. In the description of the torsion angles, asterisks refer to the atoms in phase IV. Hydrogen atoms are omitted for clarity. | ||
At 1.5 GPa, a drop in the parameter cIV length and the halving of the parameter bIV mark another phase transition, changing the unit-cell vectors to resemble those of phase I:
 | (3) |
In phase V, the Z′ number is reduced back to 0.5, and all molecules adopt the G+AG−A/G−AG+A conformation (Fig. 7). As the pressure increases from 1.74 to 4.61 GPa, the aV parameter shortens by −4.12%, and cV by −3.62%, while parameter bV is hardly reduced (−0.48%). This unusual effect results from the preserved characteristic motif of π-stacking into columns, similarly as in phase I. The G+AG−A/G−AG+A conformation effectively accommodates compression, as evidenced by a negligible change in dπ–π distance up to 2.1 GPa. Above this pressure, dπ–π decreases significantly with compression, by 9.09% at 4.61 GPa compared to 0.1 MPa. The deformation of the π-stacked columns in phase V becomes more pronounced, with the P angle increasing by 0.22° and the R angle decreasing by 2.95° between 1.74 and 4.61 GPa (Table S10, ESI†).
Conclusions
We comprehensively investigated the solid-state phase transitions of the semiconductor PDI-C6, employing a combination of differential scanning calorimetry, hot-stage microscopy, and X-ray diffraction under high pressure and various temperatures. Our research highlights an unprecedented polymorphic diversity of PDI-C6, identifying reversible transformations between five distinct polymorphic forms. This includes two high-temperature polymorphs, previously observed only through DSC analysis, and two new high-pressure phases. Notably, all identified polymorphs crystallize in the same triclinic space-group type P.
The transition from phase I to phase II involves a scissor-like rotation of the perylene cores, accompanied by a significant conformational rearrangement of the alkyl chains. However, the structure retains its 1D columnar π-stacking and the interdigitation of the alkyl chains. PDI-C6 phase II is isomorphic to phase III of PDI-C5. The close structural relationship is further evidenced by similar mechano-responsive behaviour of crystals upon cooling from phase II back to phase I, when single crystals move, jump and reshape. Due to the reconstructive phase transition, the structure of phase III could not be determined; however, we were able to establish the unit-cell parameters and characterize its thermal contraction upon cooling. This significant thermal strain may hinder the spatial rearrangement of molecules, leading to a high hysteresis upon cooling as observed in the VT-PXRD.
The volume changes observed in PDI-C6 phase I on lowering the temperature from 298 to 100 K, on average, correspond to the compression from ambient pressure to ca. 0.5 GPa, fulfilling pressure-temperature correspondence rule.63 Conversely, in the temperature range from 100 to 440 K, the PDI-C6 phase I follows the rule of the inverse relation of the thermal expansion and compressibility, exemplified by thermal expansion along [z],64 which corresponds to the strongest compressibility in the same direction. While the thermal phase transition from phase I to II is dominated by the rotation of the aromatic core, the experimental and theoretical computations consistently indicate that the conformational flexibility of the n-hexyl substituents at the imide position is the driving factor of pressure-induced phase transitions. The stepwise emergence of conformers G+AG−A/G−AG+A during compression in phase IV, where half of the molecules preserve the conformation AAAA/AAAA, while in phase V all molecules adopt the G+AG−A/G−AG+A conformation, indicates that such conformational conversion facilitates more efficient molecular packing and thus effectively reduces the π-stacking distance by 9.09% between 0.1 MPa and 4.61 GPa.
Experimental methods
Crystallization
PDI-C6 was purchased from Aldrich and used without additional purification. Single crystals of PDI-C6 were obtained using a solvothermal reactor. The process involved preparing a supersaturated solution of PDI-C6 in toluene, p-xylene, CH2Cl2, or CHCl3, heating it to 453 K (or 373 K for chlorinated solvents) overnight, and then gradually cooling it to room temperature.Differential scanning calorimetry (DSC)
The DSC measurement was performed with PerkinElmer PyrisDiamond DSC-7 equipped with a PII intracooler, in N2 atmosphere and a scanning speed of 20 K min−1. The temperatures of phase transitions were determined using the method of intersecting slope and baseline lines.Hot-stage microscopy (HSM)
The HSM analysis was performed using an OLYMPUS BX41 microscope equipped with a VISICAM 5.0 and a NIKON DS FI3 camera. A Linkam TMS-94 stage was used for the temperature control. The images were taken under polarized light to highlight modifications in the crystals due to solid-state transition, with a 40× magnification.Variable-temperature single-crystal X-ray diffraction (VT-SCXRD)
The single-crystal measurements were performed on Xcalibur diffractometers with microfocus sources (CuKα = 1.54178 Å and MoKα = 0.71073 Å) and an Oxford Cryojet attachment. For data collection and processing, the CrysAlisPro software was employed. The crystal structures were initially solved using SHELXT and subsequently refined using SHELXL within the Olex2 suite.65,66 All non-H atoms were refined with anisotropic thermal parameters. H atoms were located in the difference Fourier map and from the molecular geometry. All crystal structures were analysed and compared using the software Mercury.67 We conducted measurements at 100, 298, 440 and 470 K. Additionally, we determined the unit-cell parameters between 450 and 465 K from quick-experiments. The final crystal data are summarized in Table 1 and have been deposited in the CCDC with numbers 2421994, 2422224, 2412030 and 2421130.†Variable temperature X-ray powder diffraction (VT-PXRD)
VT-PXRD experiments were conducted in the 312–537 K range at the ALBA Synchrotron and Powder Diffraction beamline BL04-MSPD equipped with the High-throughput Position Sensitive Detector (PSD) MYTHEN and an FMB Oxford hot air blower. Indexing of the high-temperature phases was achieved using a 30-second X-ray exposure, λ = 0.41235(1) Å, over the 2θ range of 0.8°–15°. Fig. 3a presents the VT-PXRD patterns obtained upon heating the material, while Fig. S9 (ESI†) contains a temperature-resolved diffractogram collected during cooling the PDI-C6 powder. Indexing of the PXRD data collected at 499 K was hampered. However, we successfully indexed the data collected at 537 K, which allowed the determination of the triclinic primitive unit-cell parameters of high-temperature polymorph III of PDI-C6. Furthermore, the data for PDI-C6 phase III at 537 K were refined using Pawley fitting in Topas-Academic V5 (TOPAS).68 The final crystallographic data are summarized in Table 1.High-pressure single-crystal X-ray diffraction
All high-pressure experiments were conducted using a modified Merrill-Bassett diamond anvil cell (DAC) with diamond anvils supported on steel discs.69 The diamond culets were 0.8 mm in diameter. The gasket was made of 0.3 mm thick tungsten foil with spark-eroded holes 0.4 mm in diameter. Strongly elongated thin plate crystals were prepared by cutting them to the dimensions required for the experimental chamber. The DAC chamber was loaded with the single-crystal sample and a ruby chip. Daphne Oil 7575 was used as the pressure-transmitting medium to maintain hydrostatic conditions.70,71 Subsequently, the DAC was closed and compressed. Pressure was generated by controlled tightening of three screws. The experimental details are shown in Fig. S20 of the ESI.† Pressure was calibrated before and after the measurement using the ruby fluorescence method with the Photon Control spectrometer, affording an accuracy of 0.02 GPa.72,73 Single-crystal X-ray diffraction measurements were conducted on a 4-circle diffractometer equipped with a CCD detector and MoKα X-ray source (λ = 0.71073 Å) and with synchrotron radiation (see below Synchrotron X-ray Diffraction Measurements for details). The DAC chamber was centered using the gasket-shadowing method.74 The CrysAlisPro software (ver. 43.98a) was used for the data collection, reduction and absorption correction. The structures were solved with direct methods using SHELXS and refined using least-squares methods with SHELXL, all within the Olex2 software suite.66,75 Anisotropic displacement parameters were applied to all non-hydrogen atoms. The hydrogen atoms were initially identified through different Fourier maps and recalculated based on the molecular geometry after each refinement cycle. In-lab X-ray diffraction measurements were conducted at pressures of 1.05, 1.74 and 2.41 GPa. Additionally, quick experiments were carried out at 1.22, 1.33, 1.47, and 1.52 GPa, where only the unit-cell parameters were determined. All reference images collected during the diffraction measurements were consistent, testifying that no transformations took place during these experiments.Synchrotron X-ray diffraction measurements
X-ray diffraction measurements were performed at the Materials Science beamline of the Swiss Light Source76 using synchrotron radiation of approx. 0.4920 Å, focused and collimated to 150 μm (exact wavelengths were placed in Table S16, ESI†). High-pressure diffraction data was collected for single crystals of the PDI-C6 systems placed in a DAC in a steel gasket (thickness ∼90 μm and a hole diameter 250 μm). A ruby fluorescent method was used to determine pressure inside the DAC. Daphne Oil 7575 was used as a pressure transmitting medium. We conducted measurements at 0.1 MPa, 0.5, 0.73, 1.41, 1.95, 2.05, 2.74, 3.43, and 4.61 GPa. The final crystal data from high-pressure measurements are summarized in Table 1 (cf. Table S16, ESI†) and have been deposited in CIF format in the Cambridge Crystallographic Database with numbers CCDC 2412018–2412029 (from in-lab and synchrotron measurements).†Author contributions
Sample preparation: F. M., L. M., investigation and analysis of high-pressure and high-temperature SCXRD: P. R., S. S., M. A., F. M., A. K.; investigation and analysis of DSC, HSM, VT-PXRD: F. M., M. M., L. M.; theoretical investigation: M. A.; funding acquisition: P. R., L. M., A. K.; visualization: P. R., supervision: S. S., L. M., A. K.; original draft preparation: P. R., S. S., L. M., A. K.; review and editing: all authors. All the authors have given their approval to the final version of the manuscript.Data availability
Data for this article, Have been included as part of the ESI,† and the crystallographic data for compound PDI-C6 has been deposited at the CCDC with numbers 2421994, 2422224, 2412030 and 2421130 (varied-temperature data) and 2412018–2412029 (high-pressure data) and can be obtained in the form of CIF files from https://www.ccdc.cam.ac.uk/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Dr Nicola Casati for providing access to the synchrotron at PSI, Villigen. PR is grateful to the Polish Ministry of Education and Science (Diamentowy Grant DI2019/0160/49) for their financial support.Notes and references
- S. Preet and S. T. Smith, J. Cleaner Prod., 2024, 448, 141661 CrossRef CAS.
- X. Wang, X. Tian, X. Chen, L. Ren and C. Geng, Sol. Energy Mater. Sol. Cells, 2022, 248, 111976 CrossRef CAS.
- W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489–1502 RSC.
- A. B. Koren, M. D. Curtis, A. H. Francis and J. W. Kampf, J. Am. Chem. Soc., 2003, 125, 5040–5050 CrossRef CAS PubMed.
- L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596–1608 CrossRef CAS PubMed.
- S. Park, T. Kim, S. Yoon, C. W. Koh, H. Y. Woo and H. J. Son, Adv. Mater., 2020, 32, 1–29 CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- H. Dong, X. Fu, J. Liu, Z. Wang and W. Hu, Adv. Mater., 2013, 25, 6158–6183 Search PubMed.
- P. Ratajczyk, S. Sobczak, P. Woźny, A. Wcisło, T. Poręba and A. Katrusiak, J. Mater. Chem. C, 2023, 11, 11055–11065 Search PubMed.
- G. Zhang, F. R. Lin, F. Qi, T. Heumüller, A. Distler, H. J. Egelhaaf, N. Li, P. C. Y. Chow, C. J. Brabec, A. K. Y. Jen and H. L. Yip, Chem. Rev., 2022, 122, 14180–14274 Search PubMed.
- Y. G. Zhen, H. L. Dong, L. Jiang and W. P. Hu, Chin. Chem. Lett., 2016, 27, 1330–1338 CrossRef CAS.
- M. Mas-Torrent and C. Rovira, Chem. Rev., 2011, 111, 4833–4856 Search PubMed.
- M. Zambianchi, L. Favaretto, M. Durso, C. Bettini, A. Zanelli, I. Manet, M. Gazzano, L. Maini, D. Gentili, S. Toffanin, F. Gallino, M. Muccini, M. Cavallini and M. Melucci, J. Mater. Chem. C, 2015, 3, 121–131 RSC.
- L. Maini, F. Gallino, M. Zambianchi, M. Durso, M. Gazzano, K. Rubini, D. Gentili, I. Manet, M. Muccini, S. Toffanin, M. Cavallini and M. Melucci, Chem. Commun., 2015, 51, 2033–2035 Search PubMed.
- D. Liu, C. Li, S. Niu, Y. Li, M. Hu, Q. Li, W. Zhu, X. Zhang, H. Dong and W. Hu, J. Mater. Chem. C, 2019, 7, 5925–5930 Search PubMed.
- J. Bernstein, Polymorphism in molecular crystals, OxfordUniversity Press, New York, 2002 Search PubMed.
- H. Chung and Y. Diao, J. Mater. Chem. C, 2016, 4, 3915–3933 Search PubMed.
- Y. Diao, K. M. Lenn, W. Y. Lee, M. A. Blood-Forsythe, J. Xu, Y. Mao, Y. Kim, J. A. Reinspach, S. Park, A. Aspuru-Guzik, G. Xue, P. Clancy, Z. Bao and S. C. B. Mannsfeld, J. Am. Chem. Soc., 2014, 136, 17046–17057 Search PubMed.
- C. Wang, Y. Yu, Z. Chai, F. He, C. Wu, Y. Gong, M. Han, Q. Li and Z. Li, Mater. Chem. Front., 2019, 3, 32–38 RSC.
- P. Pandey, N. Demitri, L. Gigli, A. M. James, F. Devaux, Y. H. Geerts, E. Modena and L. Maini, Cryst. Growth Des., 2022, 22, 1680–1690 CrossRef CAS.
- I. de Oliveira Martins, F. Marin, E. Modena and L. Maini, Faraday Discuss., 2022, 235, 490–507 RSC.
- C. Cappuccino, L. Catalano, F. Marin, G. Dushaq, G. Raj, M. Rasras, R. Rezgui, M. Zambianchi, M. Melucci, P. Naumov and L. Maini, Cryst. Growth Des., 2020, 20, 884–891 CrossRef CAS.
- C. Cappuccino, S. Canola, G. Montanari, S. G. Lopez, S. Toffanin, M. Melucci, F. Negri and L. Maini, Cryst. Growth Des., 2019, 19, 2594–2603 Search PubMed.
- G. R. Desiraju, Crystal engineering: the design of organic solids, Elsevier Science Publishers B.V., Amsterdam, 1989 Search PubMed.
- C. Gao, X. Li, Y. Qu, R. Dai, Z. Wang, H. Li and Z. Zhang, J. Phys. Chem. C, 2019, 123, 8731–8739 Search PubMed.
- E. V. Boldyreva, Acta Crystallogr., Sect. A:Found. Crystallogr., 2008, 64, 218–231 Search PubMed.
- A. Katrusiak, Cryst. Res. Technol., 1991, 26, 523–531 CrossRef CAS.
- Y. S. Guan, Z. Zhang, J. Pan, Q. Yan and S. Ren, J. Mater. Chem. C, 2017, 5, 12338–12342 RSC.
- M. Szafrański and A. Katrusiak, J. Phys. Chem. Lett., 2017, 8, 2496–2506 CrossRef PubMed.
- M. Szafrański and A. Katrusiak, J. Phys. Chem. Lett., 2016, 7, 3458–3466 CrossRef PubMed.
- A. Datar, K. Balakrishnan and L. Zang, Chem. Commun., 2013, 49, 6894–6896 RSC.
- T. B. Singh, S. Erten, S. Günes, C. Zafer, G. Turkmen, B. Kuban, Y. Teoman, N. S. Sariciftci and S. Icli, Org. Electron., 2006, 7, 480–489 CrossRef CAS.
- R. Centore, L. Ricciotti, A. Carella, A. Roviello, M. Causà, M. Barra, F. Ciccullo and A. Cassinese, Org. Electron., 2012, 13, 2083–2093 CrossRef CAS.
- L. I. Kuznetsova, A. A. Piryazev, D. V. Anokhin, A. V. Mumyatov, D. K. Susarova, D. A. Ivanov and P. A. Troshin, Org. Electron., 2018, 58, 257–262 Search PubMed.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724–6746 Search PubMed.
- E. Kozma and M. Catellani, Dyes Pigm., 2013, 98, 160–179 CrossRef CAS.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- W. S. Shin, H. H. Jeong, M. K. Kim, S. H. Jin, M. R. Kim, J. K. Lee, J. W. Lee and Y. S. Gal, J. Mater. Chem., 2006, 16, 384–390 RSC.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- F. Marin, A. Zappi, D. Melucci and L. Maini, Mol. Syst. Des. Eng., 2023, 8, 500–515 RSC.
- H. Langhals, S. Demmig and T. Potrawa, J. Prakt. Chem., 1991, 333, 733–748 CrossRef CAS.
- A. J. McKerrow, E. Buncel and P. M. Kazmaier, Can. J. Chem., 1993, 71, 390–398 CrossRef CAS.
- M. Carmen Ruiz Delgado, E. G. Kim, D. A. Da Silva Filho and J. L. Bredas, J. Am. Chem. Soc., 2010, 132, 3375–3387 Search PubMed.
- Y. Geng, H. Bin Li, S. X. Wu and Z. M. Su, J. Mater. Chem., 2012, 22, 20840–20851 RSC.
- J. Pitchaimani, A. Kundu, S. P. Anthony, D. Moon and V. Madhu, ChemistrySelect, 2020, 5, 2070–2074 CrossRef CAS.
- E. Hadicke and F. Graser, Acta Crystallogr., 1986, 42, 189–195 Search PubMed.
- F. Marin, S. Tombolesi, T. Salzillo, O. Yaffe and L. Maini, J. Mater. Chem. C, 2022, 10, 8089–8100 RSC.
- P. Ratajczyk, A. Katrusiak, W. Bogdanowicz, K. A. Przybył, P. Krysiak, A. Kwak and A. Iwan, Mater. Adv., 2022, 3, 2697–2705 RSC.
- K. A. Bogdanowicz, S. Lalik, P. Ratajczyk, A. Katrusiak, P. Krysiak, A. I. Pawłowska, M. Marzec and A. Iwan, Sci. Rep., 2023, 13, 13240 Search PubMed.
- W. Shi, T. Deng, G. Wu, K. Hippalgaonkar, J. S. Wang and S. W. Yang, Adv. Mater., 2019, 31, 1–8 Search PubMed.
- M. Knaapila and S. Guha, Rep. Prog. Phys., 2016, 79, 066601 Search PubMed.
- S. Bergantin, M. Moret, G. Buth and F. P. A. Fabbiani, J. Phys. Chem. C, 2014, 118, 13476–13483 CrossRef CAS.
- R. Li, M. Wang, H. Zhao, Z. Bian, X. Wang, Y. Cheng and W. Huang, J. Phys. Chem. Lett., 2020, 11, 5896–5901 Search PubMed.
- R. Thakuria, N. K. Nath and B. K. Saha, Cryst. Growth Des., 2019, 19, 523–528 CrossRef CAS.
- A. Półrolniczak, S. Sobczak and A. Katrusiak, J. Mater. Chem. C, 2023, 11, 16992–17002 RSC.
- M. Dharmarwardana, R. P. Welch, S. Kwon, V. K. Nguyen, G. T. McCandless, M. A. Omary and J. J. Gassensmith, Chem. Commun., 2017, 53, 9890–9893 Search PubMed.
- M. Dharmarwardana, S. Pakhira, R. P. Welch, C. Caicedo-Narvaez, M. A. Luzuriaga, B. S. Arimilli, G. T. McCandless, B. Fahimi, J. L. Mendoza-Cortes and J. J. Gassensmith, J. Am. Chem. Soc., 2021, 143, 5951–5957 Search PubMed.
- F. Safari and A. Katrusiak, J. Phys. Chem. C, 2021, 125, 23501–23509 Search PubMed.
- W. Zieliński and A. Katrusiak, Cryst. Growth Des., 2013, 13, 696–700 Search PubMed.
- A. Katrusiak, M. Rusek, M. Dušek, V. Petříček and M. Szafrański, J. Phys. Chem. Lett., 2023, 14, 3111–3119 CrossRef CAS PubMed.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318–4328 CrossRef CAS PubMed.
- A. van der Lee and D. G. Dumitrescu, Chem. Sci., 2021, 12, 8537–8547 RSC.
- M. Kaźmierczak, E. Patyk-Kaźmierczak and A. Katrusiak, Cryst. Growth Des., 2021, 21, 2196–2204 CrossRef.
- R. M. Hazen and L. W. Finger, Comparative Crystal Chemistry: Temperature, Pressure, Composition and the Variation of Crystal Structure, Wiley, London, 1982 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr. Sect. C Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 Search PubMed.
- C. F. MacRae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 Search PubMed.
- A. A. Coelho, J. Appl. Crystallogr., 2018, 51, 210–218 CrossRef CAS.
- L. Merrill and W. A. Bassett, Rev. Sci. Instrum., 1974, 45, 290–294 Search PubMed.
- D. Staško, J. Prchal, M. Klicpera, S. Aoki and K. Murata, High Press. Res., 2020, 40, 525–536 CrossRef.
- K. Murata and S. Aoki, Rev. High Press. Sci. Technol./Koatsuryoku No Kagaku Gijutsu, 2016, 26, 3–7 Search PubMed.
- G. J. Piermarini, S. Block, J. D. Barnett and R. A. Forman, J. Appl. Phys., 1975, 46, 2774–2780 Search PubMed.
- H. K. Mao, J. Xu and P. M. Bell, J. Geophys. Res., 1986, 91, 4673 Search PubMed.
- A. Budzianowski and A. Katrusiak, High-Pressure Crystallographic, 2004, vol. 140, pp. 101–112 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. R. Willmott, D. Meister, S. J. Leake, M. Lange, A. Bergamaschi, M. Böge, M. Calvi, C. Cancellieri, N. Casati, A. Cervellino, Q. Chen, C. David, U. Flechsig, F. Gozzo, B. Henrich, S. Jäggi-Spielmann, B. Jakob, I. Kalichava, P. Karvinen, J. Krempasky, A. Lüdeke, R. Lüscher, S. Maag, C. Quitmann, M. L. Reinle-Schmitt, T. Schmidt, B. Schmitt, A. Streun, I. Vartiainen, M. Vitins, X. Wang and R. Wullschleger, J. Synchrotron Radiat., 2013, 20, 667–682 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2421994, 2422224, 2412030, 2421130 and 2412018–2412029. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5tc00809c |
| This journal is © The Royal Society of Chemistry 2025 |