DOI:
10.1039/D5TC01024A
(Paper)
J. Mater. Chem. C, 2025, Advance Article
Structure–property relationship of p-alkoxyazobenzenes as molecular solar thermal phase change material energy storage systems (MOST-PCM)†
Received
7th March 2025
, Accepted 15th May 2025
First published on 16th May 2025
Abstract
In recent years, azobenzene (AB) has been investigated for thermal energy storage applications. One approach to enhance the function of AB is to combine photoisomerization with a solid ↔ liquid phase transition enabling harvesting solar energy and ambient heat simultaneously. Thus, the photoisomerization of a crystalline (E)-isomer can be used to obtain a liquid (Z)-isomer. Upon reversible photoisomerization, the (E)-liquid → (Z)-crystal back reaction releases the stored energy as isomerization enthalpy (ΔHisom.) as well as crystallization enthalpy (ΔHcryst.). In order to optimize such MOST-PCM, a systematic series of p-alkoxy-AB with increasing chain lengths were prepared to investigate the structure–property relationship of the melting points, kinetics, photoisomerizability and optical absorption in the solid state and in solution. It could be shown that with increasing chain length the half-lives in solution increase by almost 30% depending on the chain length. However, the neat compounds show a 50% shorter half-life, probably due to autocatalysis. Both parameters exhibit an odd–even effect. Powder-XRD revealed a two stage kinetic behavior. Interestingly, the highest energy densities could be observed with medium chain lengths of about six to eight carbon atoms.
1. Introduction
Energy management plays a crucial role in optimizing energy consumption and the sustainable use of resources. In this context, solar energy, as one of the most important renewable energy sources, is becoming increasingly important.1 A possibility for storing solar energy is the conversion of photon energy into chemical energy.2,3 One promising approach is the use of molecular solar thermal energy storage (MOST) systems, which store solar energy in chemical energy through photochemical reactions.4 For such systems molecular photoswitches are used, capable of absorbing photon energy to enable photoisomerization, resulting in energetically excited metastable photoisomers. The energy difference of the respective photoisomers corresponds to the energy which can be stored in this system. The stored energy can be released as thermal energy during the backreaction.5 Several molecular photoswitches were developed and investigated in recent years, including azobenzenes,6–8 fulvalenes,9,10 norbornadienes11–16 or dihydroazulenes.17–19 Azobenzenes (AB) are favourable owing to their easy accessibility and high stability.20 They can be switched by irradiation from their thermodynamically stable (E)-isomers to their energetically excited metastable (Z)-isomers. However, the AB system suffers from low energy density compared to other systems.21 In the past years, there have been several approaches to expand the energy difference between the respective photoisomsers to increase the storage capacity.22–28 One promising approach to raise the energy storage density is the combination of the photoisomerization with a solid ↔ liquid phase transition (Fig. 1). This enables not only the storage of photon energy through photoisomerization, but also harvesting thermal energy from the ambient environment through a simultaneous melting process. In this context, we note that the storage of thermal energy in the form of latent heat utilizing organic phase change materials (PCMs) such as paraffines or fatty acids is a well-established concept, but that the combination with energy storing photoswitches offers an attractive way to enhance and control the phase change phenomena.29 Compared to other photoswitches, AB undergoes a significant change in its geometry from 9.0 Å to 5.5 Å during isomerization.30 Due to these geometric changes the photoisomers have on the one hand considerably different physical properties, which makes AB very interesting for this application. On the other hand, these changes also render photoisomerization in the solid state significantly more challenging.31
 |
| | Fig. 1 Concept of MOST-PCM and crucial parameters. | |
Nevertheless, it is crucial to differentiate between two distinct types of AB based MOST-PCM systems. In the first type, photoisomerization of the AB moiety is possible within the crystal structure due to a sufficiently large free pore volume, which allows an irradiation-induced (E)-crystal → (Z)-liquid phase transition under isothermal conditions. Recently, a variety of such AB-based MOST-PCMs have been developed, including AB with various substitution patterns and chain lengths,32–42 substituted azoheterocycles43–48 and composites with AB and traditional organic PCMs.49–53 For such photoliquefiable MOST-PCMs, the melting point of the (Z)-isomer needs to be low enough to form a liquid phase during irradiation of the solid (E)-isomer at ambient temperatures. As a solid–liquid phase transition can only occur if the melting point of the (Z)-isomer is sufficiently low and that of the (E)-isomer is sufficiently high, the melting points of the (E)- and (Z)-isomers limit the temperature range in which the system can be used. Conceptually, a high melting point of the (E)-isomer and a low melting point of the (Z)-isomer would be desirable, to use the system in a broad temperature range. After charging the MOST-PCM, the energy can be released by an external stimulus such as thermal energy. In the second type of MOST-PCM, photoisomerization of the solid AB is not feasible due to a lack of free pore volume, which prevents molecular movements within the crystal structure. Therefore, the solid (E)-isomer needs to be thermally liquefied to enhance molecular mobility. After photoisomerization of the (E)-isomer to the (Z)-isomer in the liquid phase the system must be cooled below the melting point of the (E)-isomer to enable recrystallization during the energy release. This type of MOST-PCM is usually combined with traditional organic PCMs to increase the mobility in a composite and give higher control of the phase change.49–51 Literature-known AB based MOST-PCMs are usually substituted in the para-position with alkoxy or carboxy alkyl chains of different lengths and functionalities.32–37,42,48,54 For such systems it has been shown that the position and the length of the alkyl chain or other substituents strongly influence the properties of the (E)- and the (Z)-isomer.33,36 However, the influence of chain length on properties such as the melting point of the photoisomers, half-lives or energy storage density, for example, does not need to have a linear behavior and is difficult to rationalize. For a better understanding of the structure–property relationship in such MOST-PCM systems, herein, a systematic study was conducted to investigate the influence of different chain lengths on the physical storage properties. Therefore, in this study not only the influence on the melting points of the (E)- and (Z)-isomers or thermodynamic parameters such as the energy storage density is quantified, but also kinetic investigations are carried out and complemented with X-ray diffraction studies. On the one hand, the half-life of the excited (Z)-isomer is an essential parameter, as it corresponds to the energy storage time. On the other hand, the charging speed of the MOST-PCM is a decisive parameter, which is why the lowest ambient temperature at which effective photoliquefaction occurs is also a critical factor.
2. Experimental
2.1 Chemicals
The chemicals used in this work are as follows: aniline (TCI, 98%), phenol (TCI, 99%), 1-bromobutane (TCI, 98%), 1-bromopentane (Sigma, 98%), 1-bromohexane (TCI, 98%), 1-bromoheptane (TCI, 98%), 1-bromooctane (TCI, 98%), 1-bromononane (BLD Pharm, 99%), 1-bromodecane (TCI, 98%), 1-bromoundecane (BLD Pharm, 98%), 1-bromododecane (TCI, 98%), natriumnitrit (Sigma, 97%), and Oxone® (Sigma). All chemicals were used as received without further purification.
2.2 Synthesis of p-alkoxy-azobenzenes
The investigated p-alkoxy-AB were prepared via literature known synthesis routes (see the ESI† for details). For the synthesis of p-alkoxy-AB with longer chain lengths, unsubstituted aniline was converted into the corresponding diazonium salt under acidic conditions with sodium nitrite in the first step of the synthesis (Fig. 2). Subsequently, the diazonium salt was reacted with phenol in an azo coupling under basic conditions to obtain the p-hydroxy-AB. Afterwards, p-hydroxy-AB was treated with the respective bromo alkanes under basic conditions to obtain the p-alkoxy-AB in a Williams etherification.
 |
| | Fig. 2 Synthesis of p-alkoxy-AB via azo-coupling reaction followed by Williamson etherification. | |
2.3 Investigation of the photochemical properties of neat p-alkoxy-AB for the (E)-crystal ↔ (Z)-liquid phase transition
Investigation of the solid ↔ liquid phase transition of the neat compounds does not allow the use of traditional analytical techniques. Therefore, new analytical methods were developed. To investigate the influence of different chain lengths on the efficiency of photoliquefication and energy storage time, the respective compounds were irradiated and measured via HPLC. The composition of the neat compounds was determined during the measurements. For this purpose, aliquots of the neat compound were taken, dissolved and analyzed via HPLC. Also HPLC measurements were performed to determine the composition of the (E)- and (Z)-isomers at PSS for irradiation of the neat compounds at 365 nm. Therefore, the neat compounds were irradiated at room temperature at 365 nm until no further change in composition was observed. To illustrate the process of photoliquefaction, time-resolved powder X-ray diffraction (PXRD) spectra were recorded during irradiation of the neat compounds. To study the changes in the absorption spectra during charging, discharging and thermal isomerization, a thin film of the neat compounds was prepared by thermally melting a small amount of the compound between two glass slides using capillary action and analyzed via UV-vis spectroscopy. To estimate the temperature dependency at which an effective photoliquefication or charging is possible the compounds were irradiated at different ambient temperatures.
2.4 Investigation of isomerization and crystallization enthalpy of (E)- and (Z)-isomers via DSC
Differential scanning calorimetry (DSC) measurements were performed to determine the effective energy that the respective p-alkoxy-AB can store or release during the (Z)-liquid ↔ (E)-crystal phase transition. For the determination of the melting point Tm and the crystallization temperature Tc of the (E)-isomers, as well as the crystallization enthalpy ΔHcry., DSC measurements were carried out with the respective p-alkoxy-AB in the ground state. To determine the isomerization enthalpy ΔHiso., the melting and crystallization temperatures or the glass transition temperatures of the (Z)-isomer, the (E)-isomers of the respective p-alkoxy-AB were converted into the respective (Z)-isomer by irradiation. For this purpose, a solution of the (E)-isomer in ACN was irradiated at 340 nm to obtain a solution with a high content of the (Z)-isomer. Photoisomerization was detected via UV-vis spectroscopy and continued until no further change in the absorption spectra was observed. The composition of (E)- and (Z)-isomers was determined via HLPC. The solvent was removed at room temperature under continued irradiation. The obtained liquid was weighed, and DSC measurements were carried out. To obtain the relevant parameters, a cooling curve was first recorded down to −100 °C to identify a potential crystallization temperature of the liquid AB and to prevent thermal back-isomerization. Subsequently, a heating curve was recorded to determine the glass transition temperature, as well as the isomerization temperature and enthalpy. During this process, the samples were heated to 200 °C to ensure that only the (E)-isomers were present. The samples were then cooled again to −100 °C to obtain the crystallization temperature and enthalpy. Finally, the samples were reheated to 200 °C to determine the melting point and melting enthalpy.
3. Results and discussion
3.1 Photochemical propertied of p-alkoxy-AB in solution
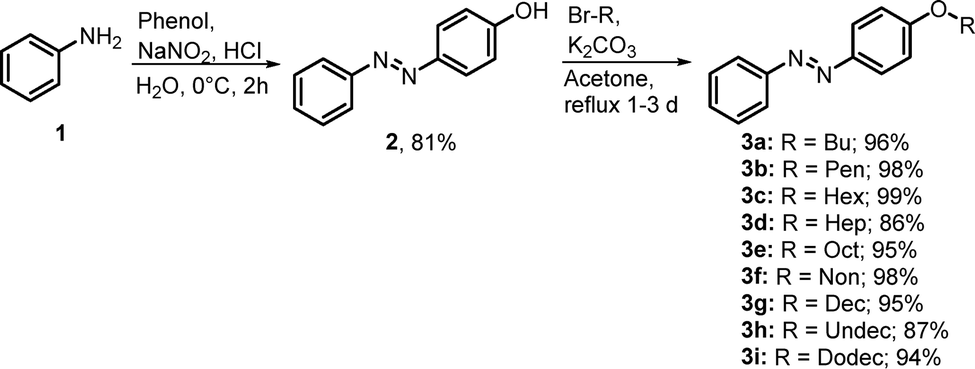
To investigate the photochemical properties and the influence of different chain lengths UV-vis absorption spectra in acetonitrile were measured for each compound to determine the absorption maximum. All p-alkoxy-AB showed a red shifted absorption maximum of the π–π* band at 347 nm and an absorption maximum of the n–π* band at around 440 nm in the ground state (Fig. 3a). Different wavelengths were tested to obtain the most effective photoisomerization. The highest amount of the (Z)-isomer in the photostationary state was obtained by irradiation with a wavelength of 340 nm. For the (Z) → (E) isomerization irradiation 448 nm was the most effective wavelength. The composition of the respective photoisomers at the photostationary states at 340 nm and 448 nm was determined via HPLC (Fig. 3b). It was observed that with increasing chain length the effectiveness of the photoisomerization for the (E) → (Z) isomerization slightly decreases from around 76% to around 73%. This decrease could be attributed to the increasing steric hinderance of the alkoxy chains, which lowers the mobility for inversion and rotation during the photoisomerization. For the (Z) → (E) photoisomerization at 448 nm the amount of the (E)-isomer slightly increases with longer chains at the respective photo stationary state. Furthermore, the variation in the composition of the photostationary states with increasing chain length could be due to solubility effects. The increasing non-polar tail of the p-alkoxy-AB may influence the stabilization of the respective isomers in a polar solvent. Kinetic studies of the p-alkoxy-AB in acetonitrile at 25 °C revealed that the half-life generally increases with increasing length of the alkoxy chain (Fig. 3c). A noticeable phenomenon is the alternating increase in half-lives, which could be explained by the odd–even effect due to the different interaction strengths of the alkoxy chains.55 The occurrence of the odd–even effect in azobenzene containing liquid crystals is a frequently observed phenomenon and illustrates the impact on physical properties.56–60 In a recent publication, a similar trend was observed for p-alkoxy-AB possessing only even chain lengths of C6, C8, C10, and C12, however, omitting the influence of the even–odd effect on the half-life.34 The increase of the half-life appears to saturate at a certain chain length. The increasing half-life could also be attributed to the increasing steric demand or by attractive interactions between the alkoxy chains, that could stabilize the (Z)-isomer and increasing the activation energy for the thermal back conversion. Alkoxy chains with an even number of carbons exhibit a longer half-life, whereas those with an odd number have a shorter half-life. The influence of the solvent polarity on the thermal half-life was not investigated. However, it can be assumed that the choice of solvent polarity could strongly affect the odd–even effect. In recent publications it has been shown that not only aliphatic substituents with increasing steric demand can enhance the thermal half-life due to stabilization of the (Z)-isomer, but also the present of polar or non-polar solvents can have a significant influence on intra- and intermolecular interactions, which are crucial for van der Waals based interactions.61 It was demonstrated that polar solvents provide greater stabilization compared to non-polar solvents.62
 |
| | Fig. 3 (a) UV-vis absorption spectra in ACN of p-OHex-AB 3c as example. (b) Content of (E)- and (Z)-isomer at the PSS 340 nm and 448 nm in ACN and neat PSS 365 nm. (c) Half-lives in ACN at 25 °C. (d) UV-vis thin film absorption spectra of solid of p-OHex-AB (blue) and photoliquefied AB (red). (e) Change of thin film absorption spectra during irradiation with 365 nm and (f) 448 nm of p-OHex-AB. | |
3.2 Reversible solid → liquid phase transition
To investigate the effect of the chain length on the irradiation induced phase transition from the solid (E)- to the liquid (Z)-isomer of different p-alkoxy-AB compounds, various wavelengths were tested to achieve the most effective photoisomerization in the solid state. However, irradiation of the neat compounds with 340 nm, which corresponds to the absorption maximum of the π–π* band at 347 nm in solution, does not lead to a successful photoisomerization. Instead, irradiation with 365 nm resulted a light-induced phase transition from solid to liquid, which was observed for all synthesized p-alkoxy-AB compounds. The red shift of the absorption maxima of the π–π* transition band in the solid state can be attributed to intramolecular interactions between the AB. The energy release and liquid to solid phase transition from the (Z)- to the (E)-isomer were induced by irradiation with a wavelength of 448 nm. For better understanding of the photophysical properties thin film UV-vis spectroscopy was performed of the corresponding AB (Fig. 3d). In the UV-vis absorption spectrum of the solid-state (E)-isomer an absorption maximum is observed at approximately 320 nm with a small shoulder at about 370 nm. Additionally, a low intensity absorption maximum at 444 nm was noted. Irradiation of the solid AB with 365 nm leads to a significant increase in the intensity of the absorption maximum at 444 nm, along with an increase in intensity of the shoulder at approximately 370 nm and at 260 nm (Fig. 2e). The absence of an isosbestic point is conspicuous. After an irradiation time of about 340 s, the intensity of the absorption maximum at 444 nm remains almost unchanged. Interestingly, no further increase in the intensity of the shoulder was observed with further irradiation; instead, there was a decrease in intensity. An isosbestic point appeared after an irradiation duration of 300 s. After 420 s of irradiation with 365 nm, the photostationary state was reached. Back switching of the photoliquefied (Z)-isomer was performed with 448 nm, which showed an opposite progression and more efficient photoisomerization (Fig. 2f). Comparing the absorption spectra between the initial state and the photostationary state at 365 nm shows that the intensity of the shoulder at 370 nm remains approximately the same and irradiation of the solid (E)-isomer causes an unusually strong increase in the n–π* band. This phenomenon could be attributed to anisotropic properties of the solid AB. The transition dipole moment of the n–π* band is in the plane of the AB. Assuming that the orientation of the AB is parallel to that of the substrate surface, the absorption is measured perpendicular to the transition dipole moment of the n–π* band resulting in low absorption at the beginning of the measurement. With increasing irradiation, the solid AB becomes randomly oriented, reducing the anisotropy. Thus, absorption increases with irradiation and decreasing anisotropy. This effect is observed as long as a certain amount of anisotropy is present in the solid or the mixed phase. This also explains the absence of an isosbestic point. Once the AB has completely converted to the liquid phase, the anisotropic properties disappear, which is confirmed by the presence the of an isosbestic point. There is only a slight change in the n–π* band. Additionally, a decrease in the intensity of the shoulder at 370 nm can be observed, which corresponds to a typical change in the n–π* absorption band of AB in solution.
For the investigation of photoliquefication at 365 nm, the p-alkoxy-AB was even distributed as a fine powder on a glass plate and irradiated (Fig. 4a). During irradiation with 365 nm at room temperature, it was observed that not all p-alkoxy-ABs were converted into the liquid (Z)-isomer with the same efficiency, even if the ambient temperature is above the melting point of the respective (Z)-isomer. 1H-NMR analysis was applied to investigate the composition of the photoliquefied AB after irradiation with 365 nm at room temperature for a certain period (Fig. 4b). The composition of the photoliquefied AB is nearly constant for moderate chain lengths and that the content of the (Z)-isomer decreases with increasing chain length. The AB with a chain length of C9 and C11 stand out, as they had not entered the liquid phase after the irradiation period. The analyzed samples contained approximately 33% and 38% of the (Z)-isomer. Comparing the melting points of the AB with chain lengths of C9 and C11, revealed that these have the highest melting points compared to the other chain lengths. Consequently, the ambient temperature required for an effective phase transition increases with melting points and chain lengths. To compensate the increased melting point of the respective (E)-isomer a higher mole fraction of the respective (Z)-isomer must be obtained to obtain a liquid phase due to the larger temperature difference between the ambient temperature and respective melting point. Since the liquid phase is only obtained at higher mole fractions, the movement of the molecule is hindered for an extended period due to the crystal structure. This observation introduces another critical parameter for the efficiency of a MOST-PCM, which is the lowest ambient temperature that allows an efficient photoliquefication. Assuming that photoliquefaction is independent of the mobility of the respective AB in the crystal structure, the lowest temperature required to reach a liquid phase depends on the melting points of the (E)- and (Z)-isomers and the respective mole fraction. Therefore, as the melting point of the (E)-isomer increases, the requirement for a higher molar fraction or a lower melting point of the (Z)-isomer also increases. To investigate this, the respective AB were irradiated with 365 nm at different ambient temperatures. It was found that AB with a higher melting point of the (E)-isomer required a higher ambient temperature to be completely photoliquefied within a certain period because at higher ambient temperatures, molecular movement is improved, and the necessary mole fraction of (Z)-isomers is reduced. However, this also favours the thermal back reaction, potentially preventing to obtain the required mole fraction needed to photoliquefy the AB. However, these processes depend not only on ambient temperature or melting points of the (E)- and (Z)-isomers but also on the degree of mobility of the AB within the crystal structure and irradiation intensity. In addition, the photostationary states of the neat compounds after irradiation with 365 nm were determined by HPLC, which showed that the photoisomerization of the neat compounds is almost quantitative, even if the required irradiation time is different for the respective compounds (Fig. 3b). As a result, a higher photoisomerizability is observed in the neat state than in solution. However, the photoisomerizability of the neat compounds with 365 nm is not directly comparable to the dissolved compounds as two different wavelengths were used.
 |
| | Fig. 4 (a) Photoliquefication due irradiation with 365 nm. (b) Photoisomerization after irradiation with 365 nm at a distance of 15 cm of neat p-alkoxy-AB for 18 h at rt. (c) Rate of photoisomerization of neat p-OHex-AB with 365 nm at rt with a distance to the LED of 10 cm (black) and 5 cm (red). (d) PXRD before (blue) and after (red) irradiation with 365 nm für 90 min at rt. (e) Change of reflex by PXRD during irradiation with 365 nm. (f) Change of the max. intensity of reflex vs. time. | |
3.3 Kinetic investigation of the irradiation induced solid → liquid phase transition
For the quantification of the effectiveness of such a system, both thermodynamic properties, and the kinetic aspects are important as previously demonstrated. To quantify the efficiency of photoliquefication of the respective AB, time-resolved HPLC measurements were performed during irradiation of the neat compounds with 365 nm. As an illustration of the influence of radiation intensity, one p-alkoxy-AB was irradiated with a distance of 5 cm as well as 10 cm to the radiation source (Fig. 4c). It was found that by reducing the distance to the half, the time required to achieve an equivale amount of (Z)-content was reduced by half as well. In addition, it was found that with increasing photoisomerization, the photoisomerization efficiency increases, which confirms the assumption, that irradiation of the solid state strongly inhibiting the photoisomerization. Furthermore, it seems that the photoisomerization significantly increases after reaching a certain (Z)-isomer concentration, which is likely to be the point at which the system has completely changed into the liquid phase. Therefore, it is practical to divide the photoliquefication or the charging process into two separate processes. The first process, involving the phase transition from solid to liquid, is likely to be the rate determining process. The second process is the unhindered photoisomerization in the liquid phase. Additionally, an attempt was made to assess the efficiency of photoliquefaction by irradiating thin films and determining the (Z)-isomer content over time using time-resolved HPLC at room temperature. It should be noted, however, that the rate of change in (Z)-isomer content over time shows a strong dependence on factors such as film thickness, particle size, and the crystallinity of the respective compounds, which makes a direct comparison and reproducibility not feasible. In addition, time-resolved measurements were conducted using powder XRD to visualize the speed of photoliquefaction (Fig. 4d). To minimize the influence of the different penetration depths of X-rays and UV radiation, it is beneficial to measure a reflex at a small angle.63 It was observed that the intensity of the reflexes decreased with longer irradiation time of the crystalline (E)-isomer, indicating an increasing degree of photoliquefaction (Fig. 4e). However, AB with longer chain length exhibit significantly reduced crystallinity, which makes these measurements not possible for all compounds. Nevertheless, it can be generally concluded that the efficiency of photoliquefaction is highly dependent on the melting points of the (E)- and (Z)-isomers.
3.4 Kinetic investigation of the thermal half-life of the liquid → solid phase transition
Since measuring the kinetics for the thermal half-life of the respective AB in solution is difficult to compare with the kinetics of the neat state, different analytic techniques were used to determine the half-life. In the investigation of the kinetics for MOST-PCM, both the kinetics for the back isomerization of the liquid AB, and the crystallization processes from its mixed phase must be considered, posing a significant challenge for a meaningful analysis. Secondary effects such as autocatalysis must also be considered. Analyzing the thermal half-life for MOST-PCMs, again two different processes must be considered. The first process relates to the kinetics of the fully photoliquefied and charged AB. One aspect for the first process is the influence of thermal energy released during back isomerization, which could accelerate the process. In conventional measurements of the thermal half-life, the respective AB are measured in high dilution, so that the thermal energy released due the back isomerization has only a marginal influence on the medium. From a certain mole fraction, crystallization processes start, which release additional thermal energy, accelerating the reverse reaction. For a better understanding of the effects of thermal isomerization on absorption, thin film UV-vis spectroscopy was performed. This also showed that the complete change in the absorption spectrum over time can be divided into two separate processes (Fig. 5a). The first process involves the thermal relaxation in the fully liquid phase. The presence of an isosbestic point indicates that changes in the absorption spectrum are caused only by progressive back-isomerization (Fig. 5b). An increase in the π–π* band at 365 nm occurs and a decrease in the n–π* band at 444 nm, which also corresponds to a characteristic change of the absorption spectrum in solution. In the second process of thermal relaxation, the unusual change in the absorption spectrum indicates a beginning recrystallization (Fig. 5c). No isosbestic point is recognizable in this spectrum, which indicates that not only thermal isomerization is taking place. The strong decrease of the n–π* band and the decrease of the π–π* band thus indicate the increase of anisotropic properties due to crystallization. To illustrate the changes in the π–π* band and n–π* band, the variation in absorbance over time at 365 nm and 444 nm was plotted against time. The alteration in absorption at a wavelength of 365 nm shows an increase in the π–π* band in the liquid phase, which is a typical for the π–π* band in solution (Fig. 5d). After about 25 h a drastic change occurs with a strong decrease of the π–π* band. The change in absorption at 444 nm shows a moderate decrease in intensity at the beginning. However, this decrease drastically increases after approx. 25 h. The significant change in the spectrum is probably caused by the beginning of crystallization and the occurrence of anisotropic effects. However, using thin film UV-vis spectroscopy, kinetic measurements were attempted for the first process of thermal isomerization in the liquid phase, assuming that the presence of an isosbestic point indicates isolated photoisomerization. However, no reproducible half-lives could be obtained using this method, as during thermal isomerization there are likely to be strong volume changes and orientation of the molecules in the liquid phase to each other to proceed to the process of crystallization. However, kinetic measurements were carried out using HPLC (Fig. 5f). A decrease in the half-life with increasing chain length was recognised. Furthermore, as in solution, an alternating behaviour can also be observed which suggests an odd–even effect. Furthermore, p-alkoxy-AB with an even chain length showed a higher half-life than those with an odd chain length. Interestingly, the half-lives for the neat substances are significantly shorter than for those in solution. This effect could be caused by autocatalysis, as the thermal energy released in the neat substance can only be poorly transferred to the environment, such as for kinetics in solution. Since the half-lives have only been measured for a short period of time, the half-lives could still increase after a certain period of time with increasing crystallinity. Thus, the half-lives for the thermal isomerization in the solid substance could be significantly longer than for those in the liquid phase.
 |
| | Fig. 5 (a) UV-vis absorption spectra of the thermal back conversion of liquefied p-OHex-AB. (b) Absorption spectra of thermal back conversion in the liquid phase. (c) Absorption spectra of thermal back conversion during phase transition. (d) Change of intensity at 365 nm. (e) Change of intensity at 444 nm. (f) Comparison of thermal half-lives of liquefied AB's. | |
3.5 Thermal and crystallization properties
Differential scanning calorimetry (DSC) was utilised to investigate the thermal characteristics of p-alkoxy-ABs providing insights into the temperature ranges and exothermic processes involved in the phase-changing phenomena by photoisomerization (Fig. 6). Analysis revealed that the (E)-AB compounds display rapid crystallisation and melting processes. An increase in carbon chains results in a general increase in transition temperature (Tc) and melting temperature (Tm). The corresponding (Z)-AB compounds exhibit a slow melting process, likely influenced by the variation in molecular structure once photoizomerised. This is evident by the sharp peaks present for the (E)-AB and the broad peaks for the (Z)-AB demonstrating the slower crystallisation process. By running the DSC measurements starting from −80 °C broad peaks can sometimes be observed, allowing the assignment of a glass transition temperature (Tg). The DSC results confirm that these materials behave as phase-changing photoswitching systems as they have melting temperatures above room temperature (rt) in the (E)-state, while after being irradiated by light inducing the isomerization form liquids with melting temperatures below rt. For the MOST-PCM purpose, the linear alkyl chain was varied to find a system, where the (E)-compound has a Tc below room temperature for rapid crystallisation and a large Tm difference between the photoisomers, while the (E)-compounds should have a melting temperature is below rt. In this series, the material that fits these parameters best is 3d (heptyl chain) as the there is a large difference between the Tm and Tg and the Tc is above RT. In some cases, the Tg of the photoisomer may surpass room temperature. Due to the amorphous nature of these materials, these broad peaks usually crystalize below rt. Additionally, if the sample is not maintained at −80 °C for an extended period, crystallization may not occur. For the sample where crystallisation has been observed, the energy of isomerization may also contribute to the melting of the frozen samples.
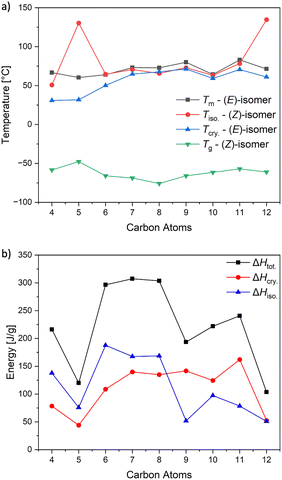 |
| | Fig. 6 (a) Melting point Tm (black) and crystallization temperature Tcry. (red) of (E)-isomers and glass transition temperature Tg (blue) and isomerization temperature Tiso. (green) of (Z)-isomers. (b) Isomerization enthalpy ΔHiso. of (Z)-isomers (blue), crystallization enthalpy ΔHcry. of (E)-isomers (red) and total storage enthalpy ΔHtot. (black) measured via DSC. | |
3.6 Energy densities
The two exothermic peaks, the isomerisation enthalpy ΔHiso. and the crystallisation enthalpy ΔHcry. can be evaluated to determine the amount of energy ΔHtot. that can be stored in a MOST-PCM material. The sharp exothermic peaks observed in the cooling curves of the trans-AB were attributed to the rapid crystallisation revealing ΔHcry. values ranging from 44.06 to 162.15 J g−1 (Table 1). The ΔHcry. of these organic phase-change materials falls within the range of thermal energy typically harvested from the ambient environment. This implies that they effectively capture a significant amount of ambient heat while maintaining their capacity to store photon energy. The ΔHiso. can be calculated by integrating the broad exothermic peaks resulting in ΔHiso of about 97.48 to 188.49 J g−1. To account for the photostationary state established upon irradiation at 340 nm, these values were adjusted, resulting in a corrected range of 51.00 to 187.75 J g−1 for 100% conversion. Upon irradiation or thermal back conversion, the total energy released (ΔHtot.), comprising both isomerization enthalpy (ΔHiso.) and crystallisation enthalpy (ΔHcry.), ranged from 103.45 to 307.42 J g−1. Previous studies included ΔHcry. twice in their calculations leading in same cases to deviating values.34 Notably, the p-OHep-AB 3d derivative exhibited the highest energy storage density, suggesting it as the most promising candidate for MOST-PCM applications and aligning with the targets proposed for solar thermal storage systems.
Table 1 Enthalpies measured by DSC normalized to the composition of the photoliquefied neat p-alkoxy-AB at PSS for 365 nm
| Compound |
ΔHcry [J g−1] |
ΔHiso [J g−1] for 100% |
ΔHtot [J g−1] for 100% |
| p-OBu-AB |
78.49 |
137.80 |
216.29 |
| p-OPen-AB |
44.06 |
75.82 |
119.88 |
| p-OHex-AB |
108.80 |
187.75 |
296.55 |
| p-OHep-AB |
139.90 |
167.52 |
307.42 |
| p-OOct-AB |
135.01 |
168.63 |
303.64 |
| p-ONon-AB |
141.70 |
51.89 |
193.59 |
| p-ODec-AB |
124.46 |
97.48 |
221.94 |
| p-OUndec-AB |
162.15 |
78.59 |
240.74 |
| p-ODodec-AB |
52.45 |
51.00 |
103.45 |
4. Conclusion
In summary, a series of p-alkoxy-AB with different chain lengths were prepared for investigation of the influence of several parameters for even and especially for odd chain lengths, which are crucial for MOST-PCM. All p-alkoxy-AB can be photoliquefied via irradiation at 365 nm. Photoliquefaction with 340 nm, which corresponds to the absorption maximum in ACN, was not possible and can be attributed to a change in absorption in the neat state. Besides thermodynamic properties also the kinetic aspects for charging and energy release were considered. In addition, properties were investigated in solution and of the neat compounds to illustrate the changes in their properties. Thereby even and odd chain lengths were investigated to emphasize the in influences on the odd–even-effect. The respective p-alkoxy-AB showed an alternating increase in half-life with increasing chain length in ACN. Thereby p-alkoxy-AB with an even chain length showed a general higher half-life compared to the odd chain lengths. This means that the half-life in solution of p-OPen-AB is around 10 h shorter than that of p-OBu-AB. In comparison, neat p-alkoxy-AB showed a decreasing half-life with increasing chain length. The half-life of p-OBu-AB is approx. 28 h, which is approx. 50% longer than that of p-ODec-AB. A slight influence of the odd–even effect can also be recognized, whereby odd chain lengths have a shorter half-life compared to the even chain length. However, the half-lives were generally lower compared to those in solution. This effect could also be attributed to autocatalysis due to the heat development during the thermal backisomerization. Furthermore, p-alkoxy-AB show strong differences in photoisomerizability. (Z)-isomer contents of over 92% were achieved for the neat compounds at the PSS365. The average (Z)-isomer content in solution was approx. 75% at the PSS340. For the determination of the efficiency of photoliquefaction or the charging process of p-alkoxy-AB, it was shown that these measurements are highly dependent on particle size, layer thickness, and crystallinity, making it difficult to perform systematic investigations with reproducible measurements. However, it was demonstrated that the melting points of the (Z)-isomers and especially of the (E)-isomers of the respective AB significantly influence the speed of the charging process. This correlation can be explained by the fact that with increasing content of (Z)-isomer during irradiation, resulting in a higher degree of photoliquefaction, the mobility and transparency increases, thereby enhancing the efficiency of photoliquefaction. Thus, at higher melting points of the (E)-isomer, the required higher proportion of the (Z)-isomer to compensate temperature difference between melting point of (E)-isomer and ambient temperature increases. For example, in the photoisomerization of neat p-ONon-AB 3f with a melting point of about 78 °C at room temperature under moderate irradiation, only a (Z)-content of 33% was achieved after 18 h. This shows that photoisomerization in the solid phase is not as effective as in the liquid phase, which was demonstrated by the photoisomerization process, in which photoisomerization increased significantly at content above 30–40% (Z)-isomer. The investigation of the energy storage density using DSC revealed that the p-alkoxy-ABs with an intermediate chain length have the highest storage capacities. For 100% photoisomerization of the respective neat p-OHep-AB 3d showed the highest energy storage density with 307.42 J g−1. Taking into account other parameters such as thermal half-lives and suppressed photoliquefication due to high melting points, the p-alkoxy ABs with a chain length of OHex to OOct are the most attractive candidates for a MOST-PCM systems. This concept is highly interesting due to the ability of using neat compounds as energy storage, which is an advantage compared to solvent depended MOST systems, to increases the total energy storage capacity, which offers an great potential application for coating surfaces or enabling usage of liquid flow systems.33,34,47 Furthermore, the total energy storage capacity is increased by harvesting additional crystallization enthalpy ΔHcry. due to the phase transition during photoisomerization. This systematic study will provide valuable data also for other molecule based functional materials.
Author contributions
C. A. and H. A. W. conceptualized the project and prepared the manuscript. C. A. synthesized all compounds and collected all experimental data bedside the DSC data. M. S. preformed DSC measurements and prepared the associated part of the manuscript. K. M. offered significant scientific input. All authors were involved in discussing the data. All authors have given approval to the final version of the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge financial support by the Deutsche Forschungsgemeinschaft (DFG) within the Research Unit FOR 5499 ‘‘Molecular Solar Energy Management – Chemistry of MOST Systems’’ (project 496207555), the Swedish Research Council – Vetenskapsrådet and the Göran Gustafsson Foundation “Göran Gustafssons Stiftelse för naturvetenskaplig och medicinsk forskning.”
Notes and references
- M. Perez and R. Perez, Sol. Energy Adv., 2022, 2, 100014 CrossRef.
- J. R. Bolton, Science, 1978, 202, 705–711 CrossRef CAS PubMed.
- G. Jones, T. E. Reinhardt and W. R. Bergmark, Sol. Energy, 1978, 20, 241–248 CrossRef CAS.
- Z. Yoshida, J. Photochem., 1985, 29, 27–40 CrossRef CAS.
- Z. Wang, P. Erhart, T. Li, Z.-Y. Zhang, D. Sampedro, Z. Hu, H. A. Wegner, O. Brummel, J. Libuda, M. B. Nielsen and K. Moth-Poulsen, Joule, 2021, 5, 3116–3136 CrossRef CAS.
- Z. Wang, R. Losantos, D. Sampedro, M. Morikawa, K. Börjesson, N. Kimizuka and K. Moth-Poulsen, J. Mater. Chem. A, 2019, 7, 15042–15047 RSC.
- A. R. Ibrahim, M. F. Khyasudeen, J. Husband, S. M. Alauddin, N. F. K. Aripin, T. S. Velayutham, A. Martinez-Felipe and O. K. Abou-Zied, J. Phys. Chem. C, 2021, 125, 22472–22482 CrossRef CAS.
- A. Kunz, A. H. Heindl, A. Dreos, Z. Wang, K. Moth-Poulsen, J. Becker and H. A. Wegner, ChemPlusChem, 2019, 84, 1145–1148 CrossRef CAS PubMed.
- A. Lennartson, A. Lundin, K. Börjesson, V. Gray and K. Moth-Poulsen, Dalton Trans., 2016, 45, 8740–8744 RSC.
- Y. Kanai, V. Srinivasan, S. K. Meier, K. P. C. Vollhardt and J. C. Grossman, Angew. Chem., Int. Ed., 2010, 49, 8926–8929 CrossRef CAS PubMed.
- J. Orrego-Hernández, A. Dreos and K. Moth-Poulsen, Acc. Chem. Res., 2020, 53, 1478–1487 CrossRef PubMed.
- A. Dreos, Z. Wang, J. Udmark, A. Ström, P. Erhart, K. Börjesson, M. B. Nielsen and K. Moth-Poulsen, Adv. Energy Mater., 2018, 8, 1601622 Search PubMed.
- A. U. Petersen, A. I. Hofmann, M. Fillols, M. Mansø, M. Jevric, Z. Wang, C. J. Sumby, C. Müller and K. Moth-Poulsen, Adv. Sci., 2019, 6, 1900367 CrossRef PubMed.
- M. Quant, A. Lennartson, A. Dreos, M. Kuisma, P. Erhart, K. Börjesson and K. Moth-Poulsen, Chem. – Eur. J., 2016, 22, 13265–13274 CrossRef CAS PubMed.
- F. Waidhas, M. Jevric, L. Fromm, M. Bertram, A. Görling, K. Moth-Poulsen, O. Brummel and J. Libuda, Nano Energy, 2019, 63, 103872 CrossRef CAS.
- M. J. Kuisma, A. M. Lundin, K. Moth-Poulsen, P. Hyldgaard and P. Erhart, J. Phys. Chem. C, 2016, 120, 3635–3645 CrossRef CAS PubMed.
- M. Cacciarini, A. B. Skov, M. Jevric, A. S. Hansen, J. Elm, H. G. Kjaergaard, K. V. Mikkelsen and M. Brøndsted Nielsen, Chem. – Eur. J., 2015, 21, 7454–7461 CrossRef CAS PubMed.
- Z. Wang, J. Udmark, K. Börjesson, R. Rodrigues, A. Roffey, M. Abrahamsson, M. B. Nielsen and K. Moth-Poulsen, ChemSusChem, 2017, 10, 3049–3055 CrossRef CAS PubMed.
- C. Schøttler, S. K. Vegge, M. Cacciarini and M. B. Nielsen, ChemPhotoChem, 2022, 6, 94993 CrossRef.
- D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- H. Taoda, K. Hayakawa, K. Kawase and H. Yamakita, J. Chem. Eng. Jpn., 1987, 20, 265–270 CrossRef CAS.
- A. H. Heindl, J. Becker and H. A. Wegner, Chem. Sci., 2019, 10, 7418–7425 RSC.
- W. Pang, J. Xue and H. Pang, Sci. Rep., 2019, 9, 5224 CrossRef PubMed.
- E. N. Cho, D. Zhitomirsky, G. G. D. Han, Y. Liu and J. C. Grossman, ACS Appl. Mater. Interfaces, 2017, 9, 8679–8687 CrossRef CAS PubMed.
- A. M. Kolpak and J. C. Grossman, Nano Lett., 2011, 11, 3156–3162 CrossRef CAS PubMed.
- A. M. Kolpak and J. C. Grossman, J. Chem. Phys., 2013, 138, 34303 CrossRef.
- J. Huang, Y. Jiang, J. Wang, C. Li and W. Luo, Thermochim. Acta, 2017, 657, 163–169 CrossRef CAS.
- L. Schweighauser, D. Häussinger, M. Neuburger and H. A. Wegner, Org. Biomol. Chem., 2014, 12, 3371–3379 RSC.
- K. Pielichowska and K. Pielichowski, Prog. Mater. Sci., 2014, 65, 67–123 CrossRef CAS.
- G. S. Kumar and D. C. Neckers, Chem. Rev., 1989, 89, 1915–1925 CrossRef CAS.
- O. S. Bushuyev, A. Tomberg, T. Friščić and C. J. Barrett, J. Am. Chem. Soc., 2013, 135, 12556–12559 CrossRef CAS.
- H. Liu, Y. Feng and W. Feng, Compos. Commun., 2020, 21, 100402 CrossRef.
- J. Gao, Y. Feng, W. Fang, H. Wang, J. Ge, X. Yang, H. Yu, M. Qin and W. Feng, Energy Environ. Mater., 2024, 7, e12607 CrossRef CAS.
- J. Ge, M. Qin, X. Zhang, X. Yang, P. Yang, H. Wang, G. Liu, X. Zhou, B. Zhang, Z. Qu, Y. Feng and W. Feng, SmartMat, 2024, 68, 693 Search PubMed.
- Z. Wu, C. Ji, X. Zhao, Y. Han, K. Müllen, K. Pan and M. Yin, J. Am. Chem. Soc., 2019, 141, 7385–7390 CrossRef CAS PubMed.
- Y. Yang, S. Huang, Y. Ma, J. Yi, Y. Jiang, X. Chang and Q. Li, ACS Appl. Mater. Interfaces, 2022, 14, 35623–35634 CrossRef CAS PubMed.
- Y. Shi, M. A. Gerkman, Q. Qiu, S. Zhang and G. G. D. Han, J. Mater. Chem. A, 2021, 9, 9798–9808 RSC.
- J. Hu, S. Huang, M. Yu and H. Yu, Adv. Energy Mater., 2019, 9, 1901363 CrossRef CAS.
- K. Griffiths, N. R. Halcovitch and J. M. Griffin, New J. Chem., 2022, 46, 4057–4061 RSC.
- J. Hu, M. Yu and H. Yu, J. Mater. Chem. C, 2025, 35, 4621 CrossRef.
- H. Akiyama and M. Yoshida, Adv. Mater., 2012, 24, 2353–2356 CrossRef CAS PubMed.
- A. Gonzalez, Q. Qiu, J. Usuba, J. Wan and G. G. D. Han, ACS Mater. Au, 2024, 4, 30–34 CrossRef CAS PubMed.
- L. Burg, L. Kortekaas, A. Gibalova, C. Daniliuc, J. Heßling, M. Schönhoff and B. J. Ravoo, Appl. Interfaces, 2025, 2, 373–380 RSC.
- L. Kortekaas, J. Simke, D. W. Kurka and B. J. Ravoo, ACS Appl. Mater. Interfaces, 2020, 12, 32054–32060 CrossRef CAS PubMed.
- M. A. Gerkman, R. S. L. Gibson, J. Calbo, Y. Shi, M. J. Fuchter and G. G. D. Han, J. Am. Chem. Soc., 2020, 142, 8688–8695 CrossRef PubMed.
- R. Liang, B. Yuan, F. Zhang and W. Feng, Angew. Chem., Int. Ed., 2025, 137, 2393 Search PubMed.
- Z. Shangguan, W. Sun, Z.-Y. Zhang, D. Fang, Z. Wang, S. Wu, C. Deng, X. Huang, Y. He, R. Wang, T. Li, K. Moth-Poulsen and T. Li, Chem. Sci., 2022, 13, 6950–6958 RSC.
- Z.-Y. Zhang, Y. He, Z. Wang, J. Xu, M. Xie, P. Tao, D. Ji, K. Moth-Poulsen and T. Li, J. Am. Chem. Soc., 2020, 142, 12256–12264 CrossRef CAS PubMed.
- H. Liu, J. Tang, L. Dong, H. Wang, T. Xu, W. Gao, F. Zhai, Y. Feng and W. Feng, Adv. Funct. Mater., 2021, 31, 8 Search PubMed.
- G. G. D. Han, H. Li and J. C. Grossman, Nat. Commun., 2017, 8, 1446 CrossRef PubMed.
- G. G. D. Han, J. H. Deru, E. N. Cho and J. C. Grossman, ChemComm, 2018, 54, 10722–10725 RSC.
- Y. Jiang, J. Liu, W. Luo, X. Quan, H. Li, J. Huang and W. Feng, Surf. Interfaces, 2021, 24, 101071 CrossRef CAS.
- X. Liu, H. Wang, X. Zhang, C. Ruan and H. Shi, J. Energy Storage, 2025, 108, 115084 CrossRef.
- J. Tang, Y. Feng and W. Feng, Compos. Commun., 2021, 23, 100575 CrossRef.
- J. C. S. Costa and L. M. N. B. F. Santos, J. Chem. Eng. Data, 2019, 64, 2229–2246 CrossRef CAS.
- Y. Ma, X. Cheng, H. Ma, Z. He, Z. Zhang and W. Zhang, Chem. Sci., 2022, 13, 13623–13630 RSC.
- D. Gnatek, S. Schuster, J. Ossowski, M. Khan, J. Rysz, S. Krakert, A. Terfort, M. Zharnikov and P. Cyganik, J. Phys. Chem. C, 2015, 119, 25929–25944 CrossRef CAS.
- T. Kobayashi and T. Seki, Langmuir, 2003, 19, 9297–9304 CrossRef CAS.
- A. Roviello and A. Sirigu, Makromol. Chem., 1982, 183, 895–904 CrossRef CAS.
- A. Blumstein and O. Thomas, Macromolecules, 1982, 15, 1264–1267 CrossRef CAS.
- L. Schweighauser, M. A. Strauss, S. Bellotto and H. A. Wegner, Angew. Chem., Int. Ed., 2015, 54, 13436–13439 CrossRef CAS PubMed.
- M. A. Strauss and H. A. Wegner, Angew. Chem., Int. Ed., 2021, 60, 779–786 CrossRef CAS PubMed.
- B. E. Jones, C. Blayo, J. L. Greenfield, M. J. Fuchter, N. Cowieson and R. C. Evans, J. Org. Chem., 2024, 20, 2005–2015 CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  cdef and
Hermann A. Wegner
cdef and
Hermann A. Wegner