
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Liquid-crystalline dual fluorescent dyes for white light emission†
Alfiya F.
Suleymanova
 a,
James R.
Fortwengler
b,
Xinrui
Chen
d,
Rafal
Czerwieniec
c,
Yafei
Wang
*d,
Marsel Z.
Shafikov
*c and
Duncan W.
Bruce
*a
a,
James R.
Fortwengler
b,
Xinrui
Chen
d,
Rafal
Czerwieniec
c,
Yafei
Wang
*d,
Marsel Z.
Shafikov
*c and
Duncan W.
Bruce
*a
aDepartment of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: duncan.bruce@york.ac.uk; Tel: +44 1904 324085
bDepartment of Chemistry, University of Southern California, Los Angeles, California 90089, USA
cInstitut für Physikalische und Theoretische Chemie, Universität Regensburg, Universitätsstrasse 31, Regensburg, D-93053, Germany. E-mail: shafikoff@gmail.com
dSchool of Materials Science & Engineering, Changzhou University, Changzhou 213164, P. R. China. E-mail: qiji830404@hotmail.com
First published on 7th May 2025
Abstract
Three isomeric phenanthrolines- 2,9-CzPhen, 3,8-CzPhen and 4,7-CzPhen - are formed when the heterocyclic core is functionalised 2,9-, 3,8- or 4,7- with 3,6-bis-(3,4-dioctyloxybenzene)-9H-carbazole. All are liquid crystalline and show a single columnar hexagonal phase, although that of 2,9-CzPhen has a very much higher clearing point (308 °C) compared to 3,8-CzPhen and 4,7-CzPhen (175 °C and 158.5 °C). This is rationalised through the sterically driven disposition of the carbazole groups on the phenanthroline ring alongside likely columnar packing motifs. Photophysical properties were investigated in toluene solution and in a polystyrene film. All compounds are luminescent (λmax 440–455 nm) with decent (40%, 3,8-CzPhen) to modest (4,7-CzPhen, 13% and 2,9-CzPhen, 18%) solution fluorescence quantum yields. However, in a polystyrene film, 3,8-CzPhen and 4,7-CzPhen also emit at 520 nm and 570 nm, representing a still relatively rare example of anti-Kasha emission. The 520 nm emission of 4,7-CzPhen shows a low-intensity decay component that involves a triplet state and is assigned to a thermally activated delayed fluorescence (TADF). At high emitter loadings in an OLED device, two emission frequencies result in the appearance of white light. Dual emission is understood through the relevant theory and considers rates of internal conversion following photoexcitation that populates the two emitting states – a carbazole-localised, singlet 1[πCzπ*Cz] state and a charge-transfer 1[πCzπ*phen] state. The rate and efficiency of internal conversion (IC) from the Cz-localised excited state to the charge-transfer state, involving electron transfer from the substituent(s) to 1,10-phenanthroline, appear relatively slow so that fluorescence from the higher-lying state competes. This is consistent with anti-Kasha behaviour as a result of a slow internal conversion between electronically decoupled states.
Introduction
Tailoring new molecules to combine multiple physical properties can afford materials that allow new applications or improve those already in existence. For instance, designed to feature liquid-crystallinity, photoemissive materials based on multi-resonant TADF can be deposited with preferential horizontal orientation to have a bulk aligned transition dipole moment which, applied in organic light emitting diodes (OLEDs), has the potential to afford improved light outcoupling.1,2 Materials combining white luminescence with mesogenic properties could therefore be beneficial for lighting application of OLEDs, for example as polarised white backlights for liquid crystal displays. Designing materials to show white emission, however, is not a trivial task. Covering the blue and red ranges of the spectrum typically requires dual emission, the occurrence of which in molecules is limited by Kasha's rule,3 which requires emission only from the lowest-energy excited state of a given multiplicity and relies on fast, efficient, non-radiative relaxation processes between the higher excited states. A rare exception is azulene4–8 where the two lowest excited singlet states are vibrationally decoupled due to the large energy gap. This reduces the efficiency of internal conversion so that dual fluorescence is observed. Known strategies for obtaining dual emissive materials are: (i) by designing molecules with two non-conjugated chromophores;9 (ii) combining the aggregate (excimer) emission with monomolecular emission10,11 and (iii) designing molecules with relatively slow intersystem crossing (ISC) so that both fluorescence and phosphorescence are seen.12–17 Recently, however, Li et al. showed18 that dual emissive behaviour is achievable in molecules with a donor–acceptor–donor (D–A–D) unit, where the excited states local to the donor (LE – local excitation) and the donor–acceptor charge-transfer (CT) excited states are decoupled electronically. This reduces the relaxation efficiency between the different states, affording dual emission that combines fluorescence from the donor-localised 1LE state and thermally activated delayed fluorescence (TADF) from thermal equilibration of the singlet and triplet CT states. This design of dual emissive materials holds much promise for the development of white-emitting OLED materials, especially as TADF can utilise both singlet and triplet excitons formed in the emissive layer of the OLED.In this regard, we have recently shown a working molecular design approach affording D–A–D type TADF molecules, which comprise terephthalonitrile or isophthalonitrile as the acceptor unit and alkoxyaryl-substituted carbazoles as the donor units and which are also liquid crystalline (Fig. 1).19,20 To develop this approach towards the design of dual emissive liquid-crystalline materials, 1,10-phenanthroline seemed an interesting and ubiquitous acceptor unit that offers the potential for functionalisation at different combinations of positions with donor units that may allow modulation of the internal conversion efficiency from the LE states of the donor units to the states of CT character. Due to its efficient electron-transporting properties,21 1,10-phenanthroline and its derivatives are also well known in the field of TADF materials, although in most cases they play a role of ligands in Cu(I) and Ag(I) complexes.22–26 While there are several examples of pyrazinophenanthrolines showing TADF behaviour, there is, however, only one such report of simply functionalised 1,10-phenanthrolines substituted in the 2,9-, 3,8- or 4,7 positions with an N-phenoxazine (Fig. 2).27 In toluene solution, all three compounds exhibited emission via a TADF mechanism with efficiencies ranging from 45% (2,9-isomer) to 82% (3,8-isomer) and emission wavelengths in the range 535 to 554 nm. An OLED device was constructed based on the 3,8-isomer with an external quantum efficiency of ≈19%.
 | ||
| Fig. 1 Examples of LC TADF materials prepared with phthalonitriles as the acceptor and a functionalised carbazole as the donor. | ||
 | ||
| Fig. 2 Purely organic phenanthroline-based TADF compounds. | ||
1,10-Phenanthroline may also be derivatised to generate liquid-crystalline materials and examples exist with 4,7-,22 2,9-23,28 and 3,8-disubstitution.29,30 Given the potentially attractive device properties of 1,10-phenanthrolines and the possibility to generate emissive LC derivatives by using the derivatised carbazoles (Scheme 1), we undertook the preparation and characterisation of three isomeric derivatives. The results of this study and now described.
 | ||
| Scheme 1 Synthesis of 4,7-CzPhen, 3,8-CzPhen and 2,9-CzPhen. | ||
Results and discussion
Synthesis
As shown in Scheme 1, the 4,7- and 2,9-disubstituted 1,10-phenanthroline derivatives (4,7-CzPhen and 2,9-CzPhen) were obtained via nucleophilic substitution by reaction of dichloro- and dibromo-1,10-phenanthrolines with 3,6-bis-(3,4-dioctyloxybenzene)-9H-carbazole. However, due to the lower electrophilicity of 3- and 8-positions, the same protocol did not work for preparation of 3,8-CzPhen which therefore was obtained using a Pd-catalysed Buchwald–Hartwig amination reaction of 3,8-dibromo-1,10-phenanthroline with the same carbazole.31 Unoptimised yields for the coupling reactions were between ca. 30% to 45%. The carbazole was prepared as described previously with details provided in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 The structure and purity of the compounds were verified via1H and 13C{1H} NMR spectroscopy, elemental analysis and mass spectrometry (ESI†).
20 The structure and purity of the compounds were verified via1H and 13C{1H} NMR spectroscopy, elemental analysis and mass spectrometry (ESI†).
Liquid-crystalline properties
The thermal properties of the new compounds (Fig. 3) were studied by polarised optical microscopy, differential scanning calorimetry (DSC) and small-angle X-ray scattering (SAXS). Transition temperatures were first obtained by optical microscopy before being confirmed using DSC, which then informed the temperature range over which X-ray were collected. None of the complexes was obtained as a crystalline solid on isolation from solution nor from cooling the sample on the microscope and so a supercooled mesophase was present at ambient temperature, even if visually the samples appeared solid-like. Initial identification of the phases by optical microscopy showed in each case textures characteristic of a columnar phase on cooling of the sample from the isotropic fluid (Fig. 3, top row) and, in the case of 2,9-CzPhen, the texture alone allowed identification of the phase as Colh. In fact, SAXS (Fig. 3, bottom row) showed that indeed all of the phases had the same symmetry, confirmed in each case by the observation of higher-order (11) and (20) reflections as well as (21) for both 4,7-CzPhen and 2,9-CzPhen. The d-spacings and hexagonal lattice parameters are found in Table 1.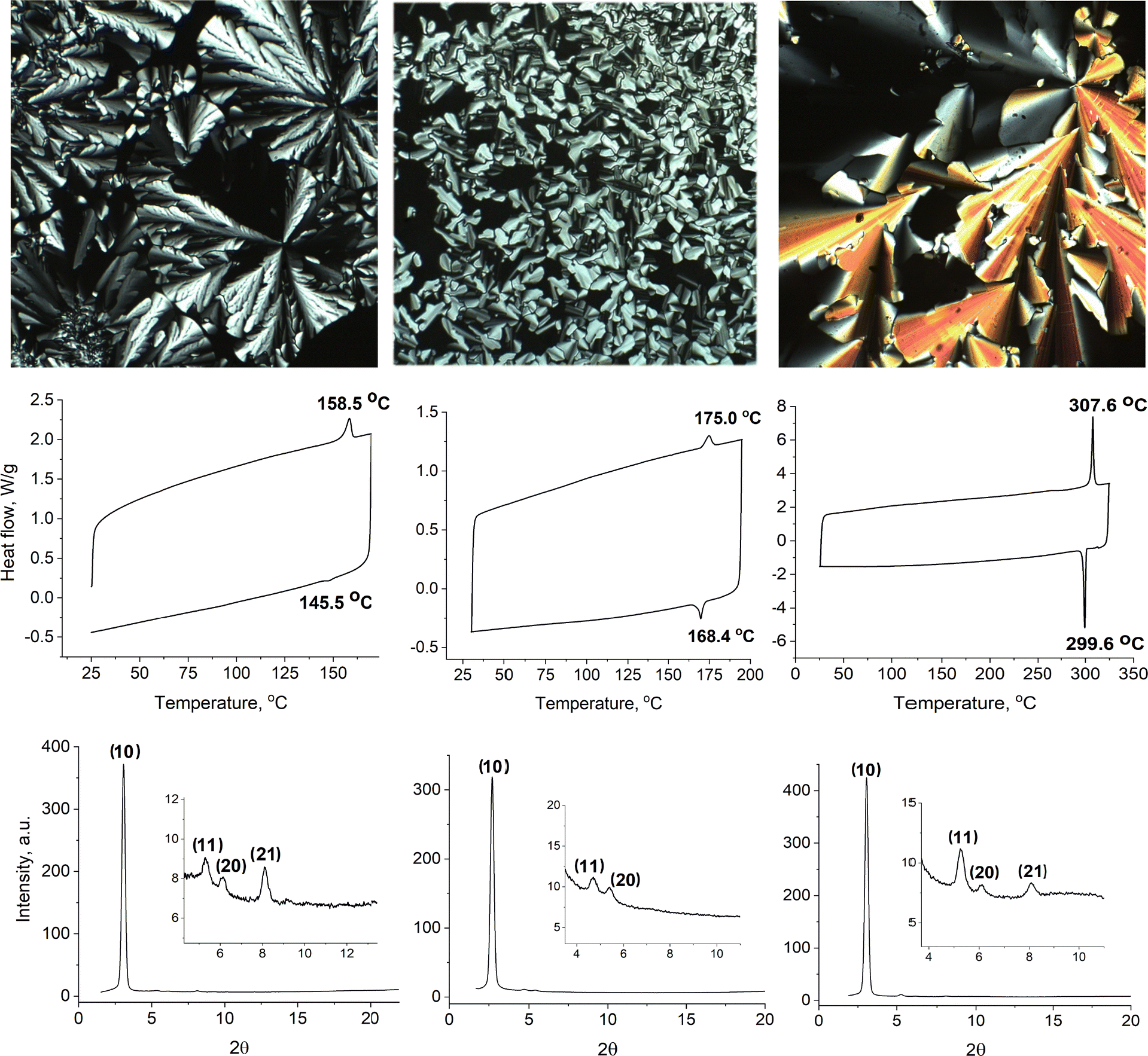 | ||
| Fig. 3 Liquid crystal characterisation of the new phenanthrolines. Top row (left to right): Polarised optical microscopy (magnification of 200×): 4,7-CzPhen at 83 °C on cooling; 3,8-CzPhen at 156 °C on cooling; 2,9-CzPhen at 304 °C on cooling. Middle row (left to right): DSC traces (at 10 K min−1) 4,7-CzPhen; 3,8-CzPhen; 2,9-CzPhen. Bottom row (left to right): SAXS data 4,7-CzPhen at 124 °C on heating; 3,8-CzPhen at 82 °C on heating; 2,9-CzPhen at 209.6 °C on heating. | ||
| Compound | Miller indices, (hk) | d obs/Å | a/Å |
|---|---|---|---|
| 4,7-CzPhen | 10 | 28.7 | 33.1 |
| 11 | 16.6 | ||
| 20 | 14.4 | ||
| 21 | 10.8 | ||
| 3,8-CzPhen | 10 | 32.5 | 37.5 |
| 11 | 18.8 | ||
| 20 | 16.3 | ||
| 2,9-CzPhen | 10 | 28.7 | 33.1 |
| 11 | 16.7 | ||
| 20 | 14.5 | ||
| 21 | 10.9 |
Aspects of the thermal behaviour require further comment. Thus, while both 2,9-CzPhen and 3,8-CzPhen appear solid at room temperature, under the polarising microscope it is evident that they are soft and deformable, consistent with the absence of a melting transition in the DSC trace (Fig. 3, middle row). 3,8-CzPhen melts at ca. 175 °C and then the mesophase re-forms around 168 °C, with the enthalpies measured on heating and cooling being effectively the same. This general behaviour is mirrored in 2,9-CzPhen with clearing around 308 °C with the mesophase re-forming around 300 °C, again with matching enthalpy change (the much higher temperatures found for 2,9-CzPhen are discussed below). Both compounds show a hysteresis in the clearing temperature of 7–8 °C and, while this is much larger than would be expected in simple calamitic systems, it is not unduly large for a columnar system.
However, things are a little different with 4,7-CzPhen and contemplation of the DSC trace shows two apparently anomalous yet related features. Thus, on heating there is an endothermic event with an onset temperature of 158.5 °C and then on cooling there is a much weaker exothermic event with an appreciably lower onset temperature of 145.5 °C. The difference in the two enthalpy changes is appreciable. When viewed under the polarising microscope at room temperature, again the material is not crystalline and can be deformed, its pliability increasing with temperature. There is evident birefringence but as expected on heating a sample of this nature, there is no discernible texture. Clearing occurs at ≈158 °C and then whatever the rate of cooling (from 10 °C min−1 to 0.1 °C min−1), the texture does not re-appear before ca. 145 °C and, on re-heating, persists again to ≈158 °C. Obtaining an optical texture on cooling is not, however, straightforward and the one shown in Fig. 3 resulted from a combination of very slow cooling and isothermal annealing.
The degree of hysteresis in the clearing temperature and the difference in the two enthalpy changes is reminiscent of monotropic behaviour, but this is not consistent with the persistence of the mesophase texture to 158 °C on re-heating. In addition, examination of the SAXS data (Fig. S7, ESI†), collected at 10 °C intervals on heating and cooling, shows behaviour that is ‘symmetric’ about the clearing temperature, inasmuch as the shape and position of the main (10) reflection at 2θ ≈ 2.7° is almost exactly the same at the same temperature on heating and cooling. As such, it seems that the observed hysteresis is due to a slow re-constitution of the columnar structure after clearing, which leads to a broad and supercooled transition.
However, while the clearing point for 4,7-CzPhen (158.5 °C) is not dissimilar to that of 3,8-CzPhen (175 °C), both are very much lower than that observed for 2,9-CzPhen, an observation that has been checked very carefully. In considering why this might be, it is instructive to contemplate how the compounds might self-organise in the columnar phase and this is helped by considering the 3D structures of the complexes, which have been calculated for the methoxy (as opposed to octyloxy) derivatives as part of the quantum chemical study described below. The complete structures are found as Fig. S8 (ESI†), whereas the dimethoxyphenyl units have been removed from those in Fig. 4 for clarity.
 | ||
| Fig. 4 Calculated structures (both dimethoxyphenyl groups removed from each structure for clarity) for (a) 3,8-CzPhen, (b) 4,7-CzPhen and (c) 2,9-CzPhen. In all three cases, the representations (left to right) are: along the axis containing the 3- and 8-ring carbon atoms; perpendicular to the plane of the phenanthroline; proposed self-organisation of the molecules in the Colh phase. | ||
Considering first 3,8-CzPhen (Fig. 4a), then viewed along an axis containing the 3- and 8-carbons of the ring, it is evident that steric clashes with the 2-, 4-, 7- and 9-ring hydrogen atoms will twist the carbazole rings strongly out of plane (≈53°), while viewed from above it is easy to see how two molecules might stack one upon the other in pairs being disposed at right angles (the out-of-plane twist of the carbazole groups would prevent a simple stacking arrangement of multiple molecules). In 4,7-CzPhen (Fig. 4b), once more the carbazole units are also twisted well out of the plane of the phenanthroline ring (≈60° – steric clash with ring 3-, 5-, 6- and 8-hydrogen atoms) and in this case the possibility would exist for pairs of molecules to stack, although it is noted that the dipoles of the molecule (calculated as 2.06 D as part of the study below) may in part compromise the stability of this arrangement. It is then reasonable on the basis of these two arrangements that the clearing points are not dissimilar, although the columnar phase of 3,8-CzPhen would be expected (as observed) to be a little more stable given the slightly better packing available. However, in 2,9-CzPhen (Fig. 4c), there is only a steric clash with the 3- and 8-ring hydrogen atoms and so the twisting out of plane of the carbazoles is very much less at ca. 32°, meaning that a much more compact and anisotropic arrangement of ring…ring dimers is possible and the back-to-back nature of the arrangement means that the very large dipole moments (4.32 D) will be antiparallel, which will stabilise the arrangement. The combination of these factors results in a very much more stable columnar phase.
Finally, it is noted that each of the arrangements shown in Fig. 4 will lead to a repeating unit of very similar lateral dimensions, consistent with the observed hexagonal lattice parameters, which are all rather similar.
Photophysical properties
The three novel compounds were examined for their photophysical properties and key data are collected in Table 2.| 3,8-CzPhen | 4,7-CzPhen | 2,9-CzPhen | |
|---|---|---|---|
| Absorption | |||
| λ max/nm (ε/M−1 cm−1) | 485 (870), 390 (29000), 290 (110000) |
385 (7500), 355 (14700), 295 (110000), 285 (107000) |
390 (30300), 370 (32300), 345 (34400), 295 (102000) |
| Photoluminescence in degassed toluene at 300 K | |||
| λ max/nm | 440 | 455 | 450 |
| ϕ PL/% | 40 | 13 | 18 |
| τ/ns | 4.7 | 7.5 | 4.8 |
| Photoluminescence in polystyrene at 300 K | |||
| λ max/nm | 420, 570 | 420, 570 | 425 |
| ϕ PL/% | 25 | 25 | 13 |
| τ/ns | 2.5 (det. at 420 nm) | 5.9 (det. at 420 nm) | 3.4 |
| 5.6 (det. at 570 nm) | 21 (det. at 515 nm, fast component) | ||
 | ||
| Fig. 5 (a) Absorption (blue) spectrum measured under ambient conditions and emission (red solid for 300 K and red dashed at 77 K) spectra of 3,8-CzPhen in toluene (≈10−5 M). (b) Emission decay profile of 3,8-CzPhen sample in degassed in toluene (≈10−5 M) measured with detection at 440 nm (blue dots) and in doped PS film (≪1 wt%) measured with detection at 420 nm, both at ambient temperature. (c) Emission spectrum (red) and excitation spectra of 3,8-CzPhen doped in polystyrene (≪1 wt%) measured with detection at 420 nm (solid blue) and at 570 nm (dashed blue). (d) Emission decay profile of 3,8-CzPhen doped in polystyrene (≪1 wt%) measured with detection at 570 nm under nitrogen atmosphere at ambient temperature. | ||
The emission from a degassed sample (≈10−5 M) measured at T = 300 K and centred at 440 nm (Fig. 5a) is intense with a quantum yield ΦPL = 40% and is to higher energy than the charge-transfer absorption band (ca. 485 nm). This indicates that the observed emission is associated with a state of local character to one of the chromophores. 1,10-Phenanthroline has been reported to fluoresce at notably shorter wavelengths with the maximum in the range 360–380 nm depending on the solvent,33,37 whereas the emission spectrum taken for the precursor alkoxyaryl carbazole (Scheme 1) in toluene appears with a maximum at 397 nm (Fig. S9, ESI†). Therefore, in the light of the above evidence, we conclude that the emission of 3,8-CzPhen in toluene is associated with transitions on the alkoxyaryl carbazole units (the largest chromophores in the molecule) affected by the central 1,10-phenanthroline unit, rather than arising from the alkoxyaryl carbazole-to-1,10-phenanthroline charge-transfer transition (vide infra). The emission at T = 77 K in toluene appears in the same spectral range with resolved vibrational progression of 1460 cm−1 (Fig. 5a). Such vibrational resolution of emission is indeed characteristic of excited states with local excitation character or, in other words, with a relatively small electron density redistribution within the molecule when compared to the ground state. The decay profile of this emission at T = 300 K, obtained by time-correlated single photon counting (TCSPC), shows a fast decay component with a time constant of 4.7 ns (Fig. 5b), which is reproduced for both degassed and air-saturated samples. A nanosecond-scale decay time and the absence of sensitivity to molecular oxygen are characteristic of spin-allowed, radiative transitions with high oscillator strength and hence this emission is assigned to the fluorescence stemming from π → π* character singlet state on alkoxyaryl carbazole unit(s) (1[πCzπ*Cz]).
Polystyrene (PS) has been chosen as a solid matrix that can suppress charge-separation processes due to its non-polar nature (similar to toluene), thus facilitating efficient emission excited states with local character in competition with relaxation to the lower-lying charge-transfer state. Thus, measured in a doped PS film at loadings much less than 1 wt%, two distinct emission bands are observed with maxima at 420 nm and 570 nm (Fig. 5c), and with a combined emission quantum yield of ΦPL(PS) = 25%. The band centred at 420 nm is assigned as having the same origin as the emission observed in toluene solution, e.g. from a state local to an alkoxyaryl carbazole unit, but is blue-shifted owing to the different solvation by, and perhaps rigidity of, the polymer film, which limits the reorganisation-driven stabilisation of the emitting excited state. As a further supporting argument for this assignment, we note that the emission spectrum taken for the precursor alkoxyaryl carbazole (Scheme 1) doped in a PS film appears very similar with a slightly blue-shifted maximum at 408 nm (Fig. S9, ESI†). The emission decay profile of the 420 nm-centred emission obtained from TCSPC gives a decay time of 2.5 ns, which is shorter than that measured in degassed toluene solution (Fig. 5b) and may indicate a faster non-radiative relaxation of the emitting state (1[πCzπ*Cz]) to the charge-transfer state (1[πCzπ*Phen]) in the PS film medium.
The longer-wavelength emission band centred at 570 nm is relatively broad, indicating significant electron density redistribution and, consequently, stronger geometric reorganisation between the emitting and ground states. It is, therefore, assigned to a CT state arising from the transition from an alkoxyaryl carbazole to the 1,10-phenanthroline core, mentioned above as weakly absorbing at ca. 485 nm. It is noted that three control experiments, in which the dopant concentration in the film was consequently halved starting from c ≪ 1 wt%, did not change the spectral ratio and characteristics of this emission ruling out the involvement of excimers which also would not show up on the absorption spectrum. The TCSPC-measured decay profile of this emission gives a short lifetime of only 5.6 ns (Fig. 5d), indicative of the singlet multiplicity of the emitting state (1CT or 1[πCzπ*phen]) as well as, judged from the low intensity of the corresponding absorption band that suggests slow, radiative decay of the CT state as well as its fast and efficient non-radiative decay to the ground state.
The excitation spectra measured for the two emission bands in the polystyrene film at 420 nm and 570 nm, have very similar spectral features (Fig. 5c), evidenced by their features in overlapping range (300–400 nm), which further supports the view that they indeed belong to the same emitting species. The excitation spectrum obtained for the 570 nm emission in addition reproduces the charge-transfer band observed in the absorption spectrum, which, however, now appears with a higher intensity ratio with respect to the shorter wavelength bands. This is because the corresponding transitions evidently populate the state emitting at 570 nm more efficiently than the higher-energy transitions local to the chromophores. The observation of this CT band in the excitation spectrum further supports the assignment of the emission centred on 570 nm to a charge-transfer 1[πCzπ*phen] state. Importantly, the excitation spectrum measured with detection at 420 nm has relatively low intensity at ca. 335 nm compared both to the absorption spectrum and to the excitation spectrum taken with detection at 570 nm. This shows that the excited states absorbing at ca. 335 nm relax relatively efficiently to the charge-transfer state that emits at 570 nm and are more decoupled (weak electronic coupling and/or vibrational overlap with) from the state emitting at 420 nm. The energy of the state populated at 335 nm in the absorption spectrum agrees very closely with the energy of the lowest excited singlet state of 1,10-phenanthroline reported in handbooks36 and with the position of the 0–0 transition in its fluorescence spectrum.33,34 This (local) excited state of 1,10-phenanthroline (1[πphenπphen*]) would then indeed be decoupled from the 1[πCzπCz]* states of the alkoxyaryl carbazole units (which emit at 420 nm) by the wavefunctions of both unpaired particles, the hole and the excited electron, which then could indeed strongly limit the internal conversion (by Dexter energy transfer) efficiency between them (vide infra).
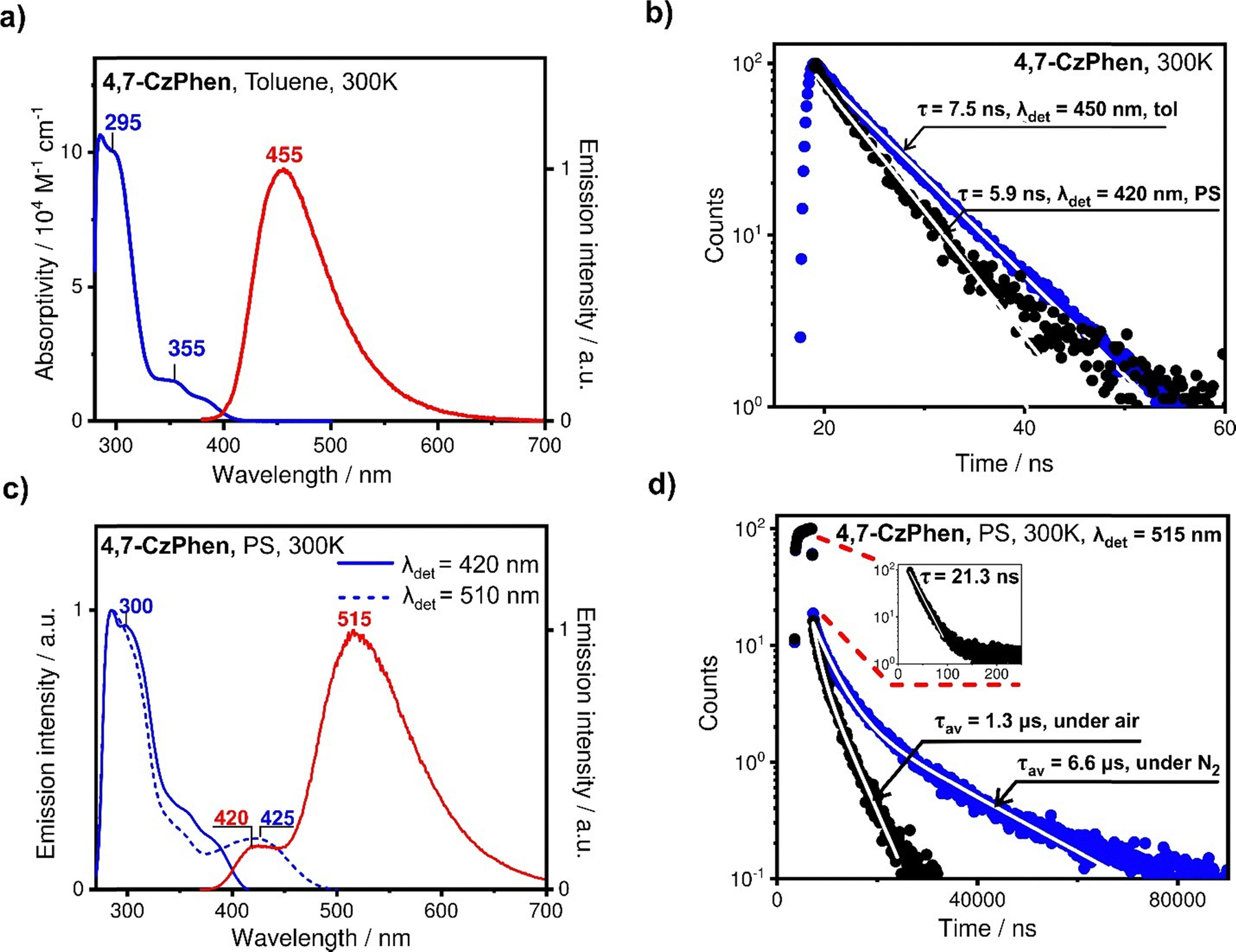 | ||
| Fig. 6 (a) Absorption (blue) and emission (red) spectra of 4,7-CzPhen in toluene (≈10−5 M) under ambient conditions. (b) Emission decay profiles of 4,7-CzPhen in degassed toluene (≈10−5 M) taken with detection at 455 nm under ambient temperature (blue dots) and in doped PS film (≪1 wt%) black dots measured with detection at 420 nm under ambient conditions. (c) Emission spectrum (red) and excitation spectra of 4,7-CzPhen doped in polystyrene (≪1 wt%) measured with detection at 420 nm (solid blue) and at 510 nm (dashed blue). (d) Emission decay profiles of 4,7-CzPhen doped in polystyrene (≪1 wt%) at ambient temperature under air (black dots) and under nitrogen atmosphere (blue dots) measured with detection at 515 nm. | ||
The emission spectrum of 4,7-CzPhen in toluene under ambient conditions is centred at 455 nm and is characterised by a TCSPC-measured decay time of 7.5 ns, assigned to fluorescence stemming from a state local to the alkoxyaryl carbazole moieties (1[πCzπ*Cz]). In a doped polystyrene film, emission is observed from two distinct bands centred at 420 nm and 515 nm (Fig. 6c), the former being assigned to the same emission observed in toluene although blue shifted in the PS film. The emission decay time of τ = 5.9 ns of the band at 420 nm is close to that in toluene solution. The broad emission band centred on 515 nm is assigned to charge-transfer state(s) arising from transitions from the alkoxyaryl carbazole moiety to the 1,10-phenanthroline ([πCzπ*phen]) and the decay curve consists of two components (Fig. 6d). The fast component, assigned to prompt fluorescence from the 1[πCzπ*phen] CT state, accounts for about 95% of the overall intensity and has a decay time of 21 ns (Fig. 6d inset), while the slow component shows non-monoexponential behaviour and is oxygen sensitive. The biexponential fit of this slow component gives the intensity-averaged decay time of 1.3 μs in air-saturated conditions, increasing to 6.6 μs under a nitrogen atmosphere (Fig. 6d). The sensitivity of the slow component to oxygen and the microsecond decay time implies involvement of a triplet state in the emission mechanism. This is most likely to be thermally activated delayed fluorescence (TADF), as was reported for analogous N-phenoxazine substituted phenanthrolines.27 Here, due to the weak exchange interaction in the charge-transfer state, which defines the energy separation of singlet and triplet states of the same orbital origin,38 the 1[πCzπ*phen] singlet and 1[πCzπ*phen] triplet states appear energetically close to degenerate and can be equilibrated via ISC and thermally activated reverse intersystem crossing (RISC) processes. It is noted that, typical for organic compounds, ISC and RISC processes between the singlet and triplet state are probably too slow for fast thermal equilibration, so that on the TCSPC curve, TADF appears as a separate decay component with a small, almost negligible, share of the intensity amounting to only 5% of the total measured intensity in deoxygenated conditions. We note that in principle, this slow component of the decay curve could also arise from phosphorescence from a lower-lying triplet state, although in this scenario a much longer decay time would be expected and it would be also observed for 3,8-CzPhen, so this is regarded as unlikely. It is noted that the relatively long decay time of the fast component and the appearance of a slow component involving a triplet state, is indicative of relatively slow and inefficient non-radiative decay of the S1 (1[πCzπ*phen]) state and perhaps T1 (3[πCzπ*phen]) state of 4,7-CzPhen to the ground state in a PS film as compared to 3,8-CzPhen compound. This may be related to the higher energy of the CT states (1[πCzπ*phen] and 3[πCzπ*phen]) of 4,7-CzPhen resulting in their relatively weaker vibrational overlap with the ground state. Indeed, emission from the 1[πCzπ*phen] state of 4,7-CzPhen is centred at 515 nm and is blue shifted compared to that of 3,8-CzPhen (570 nm), which may also be due to the more twisted geometry and consequently limited conjugation in 4,7-CzPhen, thus resulting in a relatively destabilised CT state. In common with the observations for 3,8-CzPhen, the excitation spectra of the two emission bands at 420 nm and 515 nm, are found similar in the range where each is measured (Fig. 6c), again showing that both originate from the same species. It is noted that the excitation spectrum of the emission centred at 420 nm does not reduce at ca. 335 nm to the extent found in 3,8-CzPhen, which might be due to the overlap of the excitation transition of the lowest local singlet state of 1,10-phentholine with bands of other transitions involving the alkoxyaryl carbazole moieties or faster Dexter energy transfer between local states of alkoxyaryl carbazole moieties and of 1,10-phenanthroline, so that internal conversion between them can be relatively efficient. Finally, the excitation spectrum obtained with detection at 515 nm reveals the charge-transfer transition band, centred at ca. 425 nm (Fig. 6c), which was not detectable in the absorption spectrum.
Interestingly, the emission quantum yield of 4,7-CzPhen increases from 13% in degassed toluene to 50% in a PS film under nitrogen, in contrast to the case of 3,8-CzPhen where the opposite trend was found. Apparently in 4,7-CzPhen, the PS film suppresses the non-radiative decay processes from the lower-energy charge-transfer state(s) to the ground state much more efficiently and the increase in overall emission quantum yield is associated with drastically increased efficiency of emission from the CT 1[πCzπ*phen] state. Apart from the effect of rigid media of the PS film, the higher energy of the CT state in 4,7-CzPhen compared to that in 3,8-CzPhen, may result in a slower and less efficient non-radiative decay to the ground state in accordance with the energy gap law.39 The lower efficiency of the non-radiative decay is also reflected in a significantly longer decay time of the lower-energy emission of 4,7-CzPhen in PS (515 nm, 21 ns), as compared to 3,8-CzPhen (570 nm, 5.6 ns) (compare Fig. 5b and 6b). Notably, the decay time of the emission stemming from the 1[πCzπ*Cz] state of 4,7-CzPhen, local to alkoxyaryl carbazole units (420 nm), does not decrease as significantly from a toluene solution to a PS film as in the case of 3,8-CzPhen, indicating that there is less reduction in emission efficiency of the 1[πCzπ*Cz] state from internal conversion.
 | ||
| Fig. 7 (a) Absorption (blue) and emission (red) spectra of 2,9-CzPhen in toluene (≈10−5 M) under ambient conditions. (b) Emission (red) and excitation (blue) spectra of 2,9-CzPhen doped in PS film (≪1 wt%). (c) Emission decay profiles of 2,9-CzPhen in toluene (≈10−5 M) at ambient temperature (blue dots) and as doped in PS film (black dots) (≪1 wt%). | ||
Quantum chemical calculations
Electronic structures of the three materials were examined with quantum chemical calculations using a DFT approach. To simplify the calculations, the alkoxy chains were truncated to methoxy groups and the corresponding structures are denoted as 2,9-CzPhen′, 3,8-CzPhen′, 4,7-CzPhen′. Calculations were carried out using Gaussian G09 code40 with the M11L41,42 DFT functional, the def2-SVP basis set41,43 and the C-PCM solvation model44 using parameters for toluene. The molecules were optimised in their ground-state electronic configuration and the given excited state energies represent vertical excitation energies with respect to the ground state. Iso-surface contour plots for the frontier orbitals are collected in Fig. 8, while Cartesian coordinates of the atomic positions for the optimised geometries are given in the ESI.† | ||
| Fig. 8 Iso-surface contour plots (iso-value = 0.05) of the frontier orbitals of 2,9-CzPhen′, 3,8-CzPhen′, 4,7-CzPhen′, each in its optimised ground state geometry. Hydrogen atoms are omitted for clarity. | ||
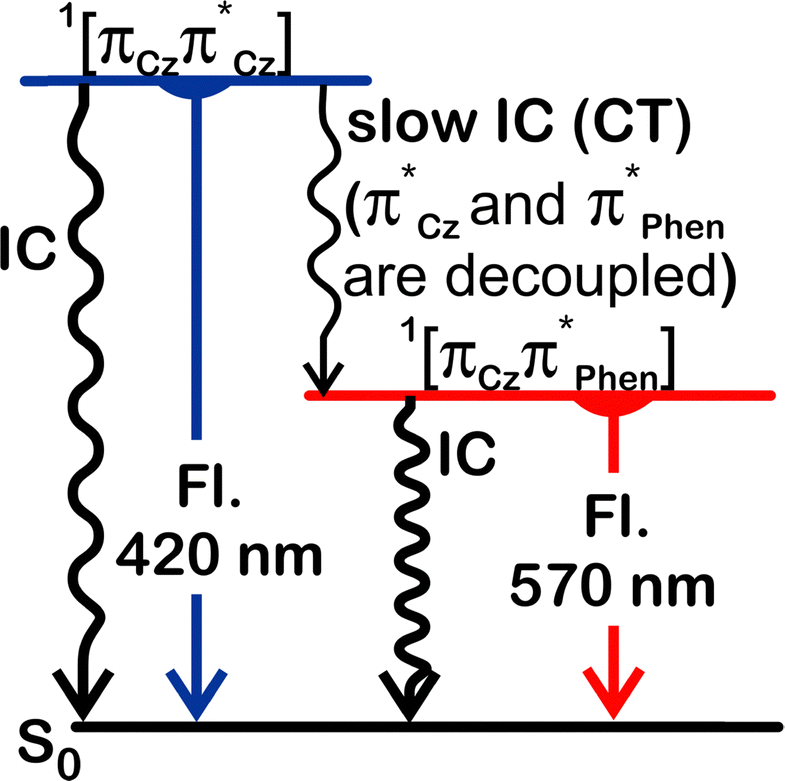 | ||
| Fig. 9 Simplified Jablonski diagram summarising luminescent properties of 3,8-CzPhen in a PS film (Fl. = fluorescence). A slightly more detailed diagram is found in the ESI† (Fig. S10). | ||
It is noted that this computational result is specific to the level of theory employed and that the actual character and properties of the 1[πCzπ*Cz] emitting state of 2,9-CzPhen will be modified by the relaxation and the specific conditions of the experiment.
Discussion
The photophysics of the three isomeric compounds are rich and also somewhat involved. Therefore, in order to understand the experimental observations and contextualise them in terms of the results of the quantum chemical calculations, it is helpful to appreciate the theory underlying the different processes involved. The discussion that follows is predicated on the properties of 3,8-CzPhen, from which the behaviour of the other isomers may be understood and relates to the Jablonski diagram in Fig. 9, which shows the different emission and deactivation pathways.The appearance of two spectrally well-separated fluorescence emissions in a rigid polymer medium, where excited-state molecular geometry reorganisation is relatively limited, is indicative of a limited rate of internal conversion (IC) of the higher-energy emitting state. Indeed, the fact that the absorption band assigned to charge transfer from the alkoxyaryl carbazoles to the phenanthroline has almost vanishing intensity in 3,8-CzPhen (485 nm) and is so weak that it is not detected in 4,7-CzPhen and 2,9-CzPhen, points to the fact that the two units are electronically well decoupled in all three isomers. Note that the CT transition S0 → 1[πCzπ*phen] in 4,7-CzPhen has been detected only in the excitation spectrum taken for 515 nm emission in PS film. Consequently, the rate of internal conversion processes requiring electron transfer(s) between the chromophores, such as 1[πCzπ*Cz] → 1[πCzπ*phen], can indeed be strongly diminished in agreement with the observation of dual fluorescence, 1[πCzπ*Cz] → S0 and 1[πCzπ*phen] → S0, in the PS film (see also ref. 18).
The internal conversion between local state of 1,10-phenanthroline and a local state of an alkoxyaryl carbazole unit – 1[πphenπ*phen] → 1[πCzπ*Cz] – requires that not just one but two electrons are exchanged between the two decoupled chromophores (Dexter energy transfer) and therefore that the rate of such process can also be very low. This is especially true in a relatively rigid and π-stacked medium within a PS film, where both vibration and the reorganisation of the molecular geometry are restricted. Thus, the reduction in intensity of the peak at ca. 335 nm to near zero in the excitation spectrum obtained for 3,8-CzPhen in a PS film with detection at 420 nm, further supports the earlier assignment of the 420 nm-centred emission as the 1[πCzπ*Cz] states of the alkoxyaryl carbazole units. For comparison, in order that the 1πCzπ*phen CT state of 3,8-CzPhen, which emits at 570 nm, is populated from the 1[πphenπ*phen] state of 1,10-phenathroline requires a transfer of a single electron between the two decoupled chromophores and that internal conversion of 1[πphenπ*phen] → 1[πCzπ*phen] is more efficient than 1[πphenπ*phen] → 1[πCzπ*Cz]. This is indeed seen from comparison of excitation spectra taken using detection at 420 nm and 570 nm (Fig. 6c).
The electronic decoupling of the 1[πCzπ*Cz] and 1[πCzπ*phen] states is probably the result not only of two chromophores being twisted with respect to one other, but also a contribution from orbital topology. Thus, consider the lowest unoccupied orbitals of the alkoxyaryl carbazoles (π*Cz) and those of phenanthroline (π*phen) – the orbitals whose mutual overlap would mediate the electron transfer in the internal conversion from the 1[πCzπ*Cz] state to the CT 1[πCzπ*phen] state(s). The lowest unoccupied orbitals of the alkoxyaryl carbazole units (LUMO+2 and LUMO+3 orbitals in Fig. 8) have a node at the carbazole nitrogen atom – the linking point with the phenanthroline core – and so do those on the phenanthroline (LUMO and LUMO+1). Thus the lowest unoccupied orbitals of the two chromophores are isolated and do not overlap. Since this is found in the calculations for all three compounds, it is evidently not affected by differences in the twist angle between the alkoxyaryl carbazole and phenanthroline chromophores. This indicates that the vibronic coupling of the electronic wavefunctions of the two states (1[πCzπ*Cz] and 1[πCzπ*phen]), which arise from vibrations along the dihedral angles between the chromophores and could in principle greatly enhance the internal conversion rate by charge transfer compared to that predicted by a Franck–Condon approximation, cannot be significant.
While the appearance of the 570 nm-centred emission of 3,8-CzPhen in a PS film indicates lower efficiency of the non-radiative decay of the CT 1πCzπ*phen state due in part to the rigidity of the medium, the observed quantum yield of 25% is appreciably lower than the 40% found in toluene solution. This would be consistent with increased efficiency of internal conversion to the lower-energy, emitting 1[πCzπ*phen] state with concomitant reduction in the emission efficiency of the higher-energy 1[πCzπ*Cz] state. To understand why this might be, the processes that define the quantum yield of a system of two emitting states is analysed. Thus, the emission quantum yield for fluorescence (ΦPL) from an individual excited state is defined as the ratio of its rate of fluorescence (kr) to the sum of all decay rates radiative and non-radiative with 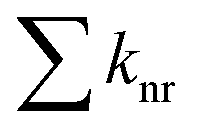 – representing the sum of the rates of all non-radiative decay paths so that
– representing the sum of the rates of all non-radiative decay paths so that  . Hence, with the assumption that the non-radiative decay of the 1[πCzπ*Cz] state consists both of a direct, non-radiative decay pathway to the ground state and of internal conversion leading ultimately to population in the lower energy emitting 1[πCzπ*phen] state, the overall quantum yield ΦPL(tot) of the system of these two states, initially excited to the 1[πCzπ*Cz] state, can be approximated as shown in eqn (1):
. Hence, with the assumption that the non-radiative decay of the 1[πCzπ*Cz] state consists both of a direct, non-radiative decay pathway to the ground state and of internal conversion leading ultimately to population in the lower energy emitting 1[πCzπ*phen] state, the overall quantum yield ΦPL(tot) of the system of these two states, initially excited to the 1[πCzπ*Cz] state, can be approximated as shown in eqn (1):
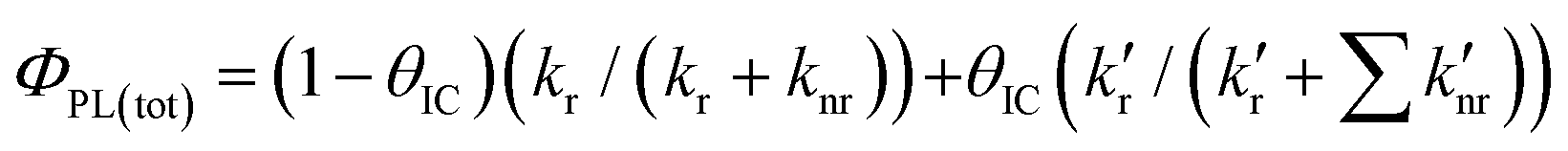 | (1) |
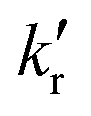 and
and  are the rates of fluorescence and sum of all non-radiative decay rates, respectively, of the lower-energy 1[πCzπ*phen] emitting state. θIC is the efficiency of the internal 1[πCzπ*Cz] → 1[πCzπ*phen] conversion, which is in turn defined as θIC = kIC/(kIC + kr + knr), with kIC being the rate of this internal conversion.
are the rates of fluorescence and sum of all non-radiative decay rates, respectively, of the lower-energy 1[πCzπ*phen] emitting state. θIC is the efficiency of the internal 1[πCzπ*Cz] → 1[πCzπ*phen] conversion, which is in turn defined as θIC = kIC/(kIC + kr + knr), with kIC being the rate of this internal conversion.
Indeed, the rate of fluorescence from this lower-energy, 1[πCzπ*phen] emitting state of CT character is expected to be much smaller in comparison to that from the higher-energy 1[πCzπ*Cz] emitting state, as can be judged form the relevant transition intensities in the absorption spectrum. This is due both to poorer overlap of the excited-state and ground-state wavefunctions due to spatial separation of the hole and electron, and a smaller value for Einstein's spontaneous emission coefficient,46 which is proportional to the cube of the emission frequency. In addition, the rate of non-radiative relaxation directly to the ground state is expected to be higher for the lower-energy 1[πCzπ*phen] state, due to the stronger vibrational overlap of the lower energy state with the ground state, as expressed through the energy-gap law.39 The CT character of the 1[πCzπ*phen] state will further enhance its vibrational overlap with the ground state due to the more extensive geometry reorganisation, when compared to that in the local character states. This is consequent on the significant redistribution of electron density in the charge-transfer state, such as 1[πCzπ*phen], that is manifest by the broad emission band. Hence this further increases the non-radiative decay rate (knr) of the 1[πCzπ*phen] state to the ground state. Further, in addition to the direct decay to the ground state, the 1[πCzπ*phen] state can be energetically close to the triplet 3[πCzπ*phen] state due to the small exchange energy between the states of CT character, which may add an additional non-radiative decay pathway via intersystem crossing. Therefore, for 3,8-CzPhen with 1[πCzπ*Cz] and 1[πCzπ*phen] states, it is expected that in eqn (1), 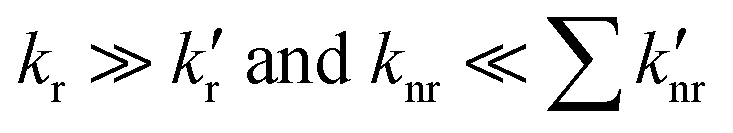 . Then the overall quantum yield of the system is expected to be greatest when the internal conversion from 1[πCzπ*Cz] → 1[πCzπ*phen] shows negligible efficiency, i.e. θIC ≈ 0 in eqn (1). Thus, as θIC increases, the overall emission quantum yield of the system is expected to decrease. Indeed, the observed decrease in the decay time of the higher-energy 1[πCzπ*Cz] emitting state from 4.7 ns in a toluene solution to 2.5 ns in a PS film (Fig. 6b and d) might be associated with an increase in the rate of internal conversion (greater kIC) and greater efficiency (greater θIC) for 1[πCzπ*Cz] → 1[πCzπ*phen] in the PS film. This could be due to the rigid medium of the PS film affecting the relaxed geometries and energies of the two excited states resulting in better vibrational overlap between them. As a result, the loss of quantum yield from the higher-energy 1[πCzπ*Cz] emitting state due to the increase of the internal conversion efficiency in the PS film, is not fully compensated by the quantum yield of the lower-energy emitting state. Consequently, the total fluorescence quantum yield of the system is lower compared to that in toluene solution.
. Then the overall quantum yield of the system is expected to be greatest when the internal conversion from 1[πCzπ*Cz] → 1[πCzπ*phen] shows negligible efficiency, i.e. θIC ≈ 0 in eqn (1). Thus, as θIC increases, the overall emission quantum yield of the system is expected to decrease. Indeed, the observed decrease in the decay time of the higher-energy 1[πCzπ*Cz] emitting state from 4.7 ns in a toluene solution to 2.5 ns in a PS film (Fig. 6b and d) might be associated with an increase in the rate of internal conversion (greater kIC) and greater efficiency (greater θIC) for 1[πCzπ*Cz] → 1[πCzπ*phen] in the PS film. This could be due to the rigid medium of the PS film affecting the relaxed geometries and energies of the two excited states resulting in better vibrational overlap between them. As a result, the loss of quantum yield from the higher-energy 1[πCzπ*Cz] emitting state due to the increase of the internal conversion efficiency in the PS film, is not fully compensated by the quantum yield of the lower-energy emitting state. Consequently, the total fluorescence quantum yield of the system is lower compared to that in toluene solution.
An important implication of the appearance of the two spectrally well-separated fluorescence emissions, where the higher-energy fluorescence from the 1[πCzπ*Cz] state has to compete with the internal conversion to the lower-energy, electronically decoupled 1[πCzπ*phen] state, is that the 1[πCzπ*Cz] emitting state in its own relaxed geometry is probably not the lowest excited state of singlet multiplicity. This would correspond to the textbook definition of the rare example where Kasha's rule3 is not followed (so-called anti-Kasha emission). Indeed, the three compounds exhibit fluorescence stemming from the 1[πCzπ*Cz] state in the relatively rigid medium of a PS film. This behaviour includes 4,7-CzPhen and 2,9-CzPhen where, due to the relatively greater steric hinderance in the former and due to the π–π overlap of the alkoxyaryl carbazole substituent in the latter, geometric reorganisations in the excited state are restricted compared to the case of 3,8-CzPhen. Moreover, 3,8-CzPhen also shows 1[πCzπ*Cz] fluorescence in the medium of frozen toluene, where excited-state geometry reorganisations are also diminished. These evidence that the 1[πCzπ*Cz] state emits while remaining in the Marcus inverted region with respect to the lower energy 1[πCzπ*phen] charge-transfer state and that its emission is indeed anti-Kasha in nature. The slow internal conversion of 1[πCzπ*Cz] to the charge-transfer state, affording dual fluorescence, is then interpreted as the result of both electronic decoupling of the states, as discussed above, as well as diminished vibrational overlap of states due to large energy gap in the Marcus inverted region (energy gap law).
Device performance
While dominated by singlet emission and not capable of efficient utilisation of triplet excitons (statistically 75% of all excitons), the observation of dual fluorescence (white emission) in two of the compounds prompted the fabrication of solution-processed OLED devices to explore their electroluminescent (EL) properties. The configuration used was: ITO/poly(3,4-ethylene-dioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) (40 nm)/emitter layer (50 nm)/1,3,5-tris(3-pyridyl-3-phenyl)benzene (TmPyPB) (45 nm)/LiF (0.5 nm)/Al (120 nm) (Fig. 10a). In this device structure, PEDOT:PSS and LiF are used as the hole-injection layer and the electron-injection layer, respectively while TmPyPB is used as the electron-transport layer. Lithium fluoride (LiF) and aluminum (Al) were utilised as electron-injection layer and cathode, respectively. The host matrix of the emitting layer is a blend of poly(N-vinylcarbazole) (PVK) and 1,3-bis-(5-(4-tert-butylphenyl)-1,3,4-oxadiazol-2-yl)benzene (OXD-7), used in a 7:3 ratio. The dopant concentrations used were 1 and 3 wt%, with the optimised emitter concentration being established as 1 wt%. The relevant EL data are summarised in Table S4 (ESI†).
 | ||
| Fig. 10 Device performance at 1 wt% loading of emitter. (a) Schematic of the energy levels and device structure; (b) EL spectra; (c) the EQE–current density curves; (d) the current density–voltage–luminance curves. | ||
As shown in Fig. 10b, all devices emit in the blue region of the spectrum with emission peaks in the range 440–470 nm, assignable to 1[πCzπ*Cz] state emission. The CIE coordinates are correspondingly found (0.25 ± 0.02, 0.29 ± 0.03) (Table S4, ESI†). The EL spectra demonstrate that there is a complete energy transfer from the host matrix to the dopant, with both electron and hole ending up on the alkoxyaryl carbazole chromophore, and reasonable turn-on voltages of between 6.0 and 8.0 V are observed for the devices.
When the emitter concentration was increased to 3 wt%, both 2,9-CzPhen and 3,8-CzPhen showed a decreased device performance, probably due to concentration quenching and so all data were collected using 1 wt% of emitter. Thus, consistent with the highest PLQY, the 3,8-CzPhen-based device is most efficient with an EQEmax of 3.01%, a maximum brightness (Lmax) of 145.6 cd m−2 and a current efficiency (CEmax) of 4.34 cd A−1 (Table S4, ESI† and Fig. 10). Devices prepared using 4,7-CzPhen and 2,9-CzPhen showed EQEmax = 0.32 and 0.60%, respectively.
The EL spectra of the devices prepared using 3,8-CzPhen do not show dual emission which is likely due to a poor population and/or efficient non-radiative decay of the charge-transfer 1[πCzπ*phen] state in the host employed, although a shoulder is seen at ca. 570 nm, whose intensity increases with increasing drive voltage (Fig. S11, ESI†).
The devices employing 3,8-CzPhen were then prepared using much higher emitter loadings of 20 wt% and 40 wt%, both emitted white light with the EL spectra featuring an additional, red-shifted band centred at ca. 570 nm (Fig. S11, ESI†). This new band, assigned as the 1[πCzπ*phen] charge-transfer state, intensifies relative to the blue band with increasing drive voltage. We believe this effect arises as in the matrix of the emitting layer at low loadings of 3,8-CzPhen, the charge-transfer 1[πCzπ*phen] state is not populated efficiently by internal conversion from the higher-lying state localized on alkoxy-aryl carbazoles (1[πCzπ*Cz]) whose sole emission is observed in the devices with lower doping levels. However, with much higher loadings, the phenanthroline core may start acting as the electron transport material, leading to direct population of the 1[πCzπ*phen] charge-transfer state without the need for internal conversion from the higher-lying 1[πCzπ*Cz] state. This is supported by the fact that photoluminescence of 3,8-CzPhen when doped at 20% into the host does not show signs of emission from the 1[πCzπ*phen] state (Fig. S13, ESI†). The sensitivity of the longer-wavelength band to the drive voltage of the device may then indicate the more efficient charge separation (electron transfer) needed to overcome the exciton binding energy, which is in turn necessary to populate the charge-transfer states from the local state of the alkoxyaryl carbazole substituents.
Conclusion
Despite the non-planar nature of the molecules, which arises from sterically induced twisting of the carbazole unit out of the plane of the central phenanthroline unit, all of the new compounds are liquid crystalline, each showing a single Colh mesophase. However, while the mesophase stabilities of 4,7-CzPhen and 3,8-CzPhen are rather similar, the Colh phase of 2,9-CzPhen is very much more stable, clearing some 130 °C higher (≈308 °C) than that of 3,8-CzPhen. From the calculated structures, it is possible to predict how the compounds are likely to self organise in the columnar structure and such an analysis is able to rationalise both the similarities between the clearing points of 4,7-CzPhen and 3,8-CzPhen and also the difference with 2,9-CzPhen.All three compounds – 2,9-CzPhen, 3,8-CzPhen and 4,7-CzPhen – show fluorescence in toluene solution centred at ca. 450 nm arising from a singlet state localised on the alkoxyaryl carbazole moieties (1[πCzπ*Cz]). In the doped PS film this emission shift to ca. 420 nm in case of 3,8-CzPhen and 4,7-CzPhen and is also accompanied by additional fluorescence bands centred at 570 nm and 515 nm, respectively, assigned to the charge-transfer singlet state (1[πCzπ*phen]). Such unusual dual fluorescent behaviour is rationalised by the electronic decoupling of the 1[πCzπ*Cz] and 1[πCzπ*phen] states, brought primarily by spatially isolated π*Cz and π*phen orbitals, where the wavefunction of the excited electron is well separated between the two states. This drastically limits the rate and efficiency of internal conversion between the two states and thus allows rare anti-Kasha emission from a higher-energy excited singlet state. Only for 4,7-CzPhen in a PS matrix could a very small share of the emission intensity (ca. 5% of emission from the 1[πCzπ*phen] state) be identified as arising with the participation of a triplet state, being assigned to TADF. This is associated with (i) the presence of two CT triplet states, T1 and T2, in very close energetic proximity to the S1 state (1[πCzπ*phen]) so that ISC and RISC processes can be relatively fast, and (ii) relatively high energy of the lowest singlet and triplet CT states limiting the efficiency of their non-radiative decay rate to the ground state, manifested by much longer decay time of 1[πCzπ*phen] prompt fluorescence (21 ns) compared to that of other two materials. Both photophysical parameters are understood to arise from the relatively twisted geometry of the alkoxyaryl carbazole substituents with respect to the phenanthroline, diminishing ΔE(S1–T1) and giving higher energies of S1 and T1 as well as a hindered geometry, so restricting reorganisation in the CT excited state. It is concluded, therefore, that molecular design strategies involving substitutions that would position the alkoxyaryl carbazoles at a larger angle with respect to the phenanthroline core and hinder geometry changes in the CT character excited state would be beneficial for the improvement of the TADF properties of these liquid-crystalline, white-luminescent materials. Additionally, complexation of the phenanthroline with transition metals could help enhance the spin–orbit coupling effect and thus afford more efficient ISC and RISC processes involved in TADF.
The use of 3,8-CzPhen as the emitter in an OLED device gave an EQE of ca. 3% which, while low by many published standards, actually represents a very efficient device in this instance. Thus, exciton multiplicity is defined purely statistically by the spins of injected electrons and holes, over which we have no control, resulting in 25% singlets and 75% triplets. The materials are then able to use only these (25%) singlet excitons and so the EQE is then understood in the context of a PLQY of 40% and a maximum outcoupling efficiency on the order of 30%. At high emitter loadings of 20 or 40 wt%, dual-emission, white-emitting devices are realised arising from the electron-transporting properties of the phenanthroline unit of the emitter materials, although the EQE suffers from quenching effects and the inability of the emitter to utilise triplet excitons. However, and as noted previously,47,48 the non-LC nature of the host materials and the loadings of LC emitter used means that LC properties are not expressed in the device, which is a facet that requires further attention.
However, while it is demonstrated here that dual emission affording white luminescence is readily achievable in a PS film, the choice of a suitable OLED host to transfer it to the device turned out not to be a trivial task. Comparison of photo- and electro-luminescence spectra of 3,8-CzPhen in PVK/OXD-7 shows that the main problem is the slow and inefficient population of the CT state by internal conversion from the higher-lying states local to the carbazoles. As such, CT emission intensity is low/vanishing at low loadings and appears significant only at high concentrations where the phenanthroline unit of the emitter start acting as an electron-transport material.
Nonetheless, the observation of both matrix- and device-based dual emission giving rise to white light is a rare18,49,50 and very significant finding and it is concluded that host blends that allow direct population of local states and CT states need to be tried and developed, an approach requiring emitter materials with better-developed TADF properties.
Author contributions
AFS, MZS and DWB conceived the project. AFS undertook all of the synthetic work and characterised the liquid crystal behaviour with DWB. Photophysical characterisation and the quantum chemical calculations were undertaken by MZS and JRF. XC made the OLED devices. Liquid crystal behaviour was analysed and interpreted by DWB, photophysical data were analysed and interpreted by MZS and YW analysed and interpreted the device data. AFS, MZS and YW wrote the first drafts of the manuscript, which they then helped refine with DWB. DWB and RC secured funding for the work.Data availability
The data supporting this article: (i) have been included as part of the ESI† and (ii) are available at https://doi.org/10.15124/e2cc8792-1bc6-43f2-914f-47b40a609b08.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the University of York (AFS), the Universität Regensburg (MZS), the NSF (GRFP Grant DGE-1842487 to JRF) and the German Research Foundation (DFG) (Project No. 389797483) for financial support, Dr Stephen Cowling (York) for assistance with the polarised optical microscopy and Xiaoyi Lai for assistance with some photoluminescence measurements.References
- D. Chen, F. Tenopala-Carmona, J. A. Knöller, A. Mischok, D. Hall, S. Madayanad Suresh, T. Matulaitis, Y. Olivier, P. Nacke, F. Gießelmann, S. Laschat, M. C. Gather and E. Zysman-Colman, Angew. Chem., Int. Ed., 2023, 62, e202218911 CrossRef CAS.
- C. Mayr, M. Taneda, C. Adachi and W. Brütting, Org. Electron., 2014, 15, 3031–3037 CrossRef CAS.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- D. Klemp and B. Nickel, Chem. Phys. Lett., 1986, 130, 493–497 CrossRef CAS.
- G. D. Gillispie and E. C. Lim, J. Chem. Phys., 1978, 68, 4578–4586 CrossRef CAS.
- G. Eber, F. Grüneis, S. Schneider and F. Dörr, Chem. Phys. Lett., 1974, 29, 397–404 CrossRef CAS.
- D. Huppert, J. Jortner and P. M. Rentzepis, Chem. Phys. Lett., 1972, 13, 225–228 CrossRef CAS.
- M. Beer and H. C. Longuet-Higgins, J. Chem. Phys., 1955, 23, 1390–1391 CrossRef CAS.
- T. Yoshihara, S. Murayama and S. Tobita, Sensors, 2015, 15, 13503–13521 CrossRef CAS.
- C. Si, T. Wang, Y. Xu, D. Lin, D. Sun and E. Zysman-Colman, Nat. Commun., 2024, 15, 7439 CrossRef CAS PubMed.
- M. Z. Shafikov, A. F. Suleymanova, D. N. Kozhevnikov and B. König, Inorg. Chem., 2017, 56, 4885–4897 CrossRef CAS PubMed.
- M. Shafikov, A. Suleymanova, R. J. Kutta, A. Gorski, A. Kowalczyk, M. Gapińska, K. Kowalski and R. Czerwieniec, J. Mater. Chem. C, 2022, 10, 5636–5647 RSC.
- M. Shafikov, A. Suleymanova, R. J. Kutta, F. Brandl, A. Gorski and R. Czerwieniec, J. Mater. Chem. C, 2021, 9, 5808–5818 RSC.
- M. Z. Shafikov, D. N. Kozhevnikov, M. Bodensteiner, F. Brandl and R. Czerwieniec, Inorg. Chem., 2016, 55, 7457–7466 CrossRef CAS.
- D. N. Kozhevnikov, V. N. Kozhevnikov, M. Z. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. Williams, Inorg. Chem., 2011, 50, 3804–3815 CrossRef CAS.
- T. Hofbeck, Y. C. Lam, M. Kalbáč, S. Záliš, A. Vlček and H. Yersin, Inorg. Chem., 2016, 55, 2441–2449 CrossRef CAS.
- Y. Liu, H. Guo and J. Zhao, Chem. Commun., 2011, 47, 11471–11473 RSC.
- X. Li, G. Baryshnikov, C. Deng, X. Bao, B. Wu, Y. Zhou, H. Ågren and L. Zhu, Nat. Commun., 2019, 10, 731 CrossRef CAS.
- A. Suleymanova, M. Shafikov, X. Chen, Y. Wang, R. Czerwieniec and D. W. Bruce, Phys. Chem. Chem. Phys., 2022, 24, 22115–22121 RSC.
- A. Suleymanova, M. Shafikov, A. C. Whitwood, R. Czerwieniec and D. W. Bruce, J. Mater. Chem. C, 2021, 9, 6528–6535 RSC.
- S. Naka, H. Okada, H. Onnagawa and T. Tsutsui, Appl. Phys. Lett., 2000, 76, 197–199 CrossRef CAS.
- H. Yersin, R. Czerwieniec, M. Z. Shafikov and A. F. Suleymanova, ChemPhysChem, 2017, 18, 3508–3535 CrossRef CAS.
- M. Z. Shafikov, A. F. Suleymanova, R. Czerwieniec and H. Yersin, Inorg. Chem., 2017, 56, 13274–13285 CrossRef CAS.
- M. Z. Shafikov, A. F. Suleymanova, R. Czerwieniec and H. Yersin, Chem. Mater., 2017, 29, 1708–1715 CrossRef CAS.
- M. Z. Shafikov, A. F. Suleymanova, A. Schinabeck and H. Yersin, J. Phys. Chem. Lett., 2018, 9, 702–709 CrossRef CAS PubMed.
- C. E. Housecroft and E. C. Constable, J. Mater. Chem. C, 2022, 10, 4456–4482 RSC.
- K. Wu, T. Zhang, L. Zhan, C. Zhong, S. Gong, Z.-H. Lu and C. Yang, Adv. Opt. Mater., 2016, 4, 1558–1566 CrossRef CAS.
- H. Norouzi-Arasi, W. Pisula, A. Mavrinskiy, X. Feng and K. Müllen, Chem. – Asian J., 2011, 6, 367–371 CrossRef CAS.
- S. J. P. Bousquet and D. W. Bruce, J. Mater. Chem., 2001, 11, 1769–1771 RSC.
- T. Cardinaels, J. Ramaekers, P. Nockemann, K. Driesen, K. Van Hecke, L. Van Meervelt, G. Wang, S. De Feyter, E. F. Iglesias, D. Guillon, B. Donnio, K. Binnemans and D. W. Bruce, Soft Matter, 2008, 4, 2172–2185 RSC.
- H. Suzuki, T. Kanbara and T. Yamamoto, Inorg. Chim. Acta, 2004, 357, 4335–4340 CrossRef CAS.
- K. M. Knötig, D. Gust, T. Lenzer and K. Oum, J. Photochem., 2024, 4, 163–178 CrossRef.
- B. N. Bandyopadhyay and A. Harriman, J. Chem. Soc., Faraday Trans., 1977, 73, 663–674 RSC.
- G. Accorsi, A. Listorti, K. Yoosaf and N. Armaroli, Chem. Soc. Rev., 2009, 38, 1690–1700 RSC.
- N. Armaroli, L. De Cola, V. Balzani, J.-P. Sauvage, C. O. Dietrich-Buchecker and J.-M. Kern, J. Chem. Soc., Faraday Trans., 1992, 88, 553–556 RSC.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, 3rd edn, 2006 Search PubMed.
- I. Berlman, Handbook of Fluorescence Spectra of Aromatic Molecules, Academic, 2nd edn, 1971 Search PubMed.
- A. Szabo and N. S. Ostlund, Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory, Dover Publications, 1989 Search PubMed.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- R. Peverati and D. G. Truhlar, J. Phys. Chem. Lett., 2012, 3, 117–124 CrossRef CAS.
- R. Peverati and D. G. Truhlar, Phys. Chem. Chem. Phys., 2012, 14, 11363–11370 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- R. C. Hilborn, Am. J. Phys., 1982, 50, 982–986 CrossRef CAS.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- R. R. Parker, D. Liu, X. Yu, A. C. Whitwood, W. Zhu, J. A. G. Williams, Y. Wang, J. M. Lynam and D. W. Bruce, J. Mater. Chem. C, 2021, 9, 1287–1302 RSC.
- S. Tang, Y. Tsuchiya, J. Wang, C. Adachi and L. Edman, Nat. Commun., 2025, 16, 653 CrossRef CAS PubMed.
- A. Maggiore, Y. Qu, R. Guillot, P. Pander, M. Vasylieva, P. Data, F. B. Dias, P. Audebert, G. Clavier and F. Miomandre, J. Phys. Chem. B, 2022, 126, 2740–2753 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5tc01393c |
| This journal is © The Royal Society of Chemistry 2025 |