
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tropospheric alkene ozonolysis chemistry: an extended computational chemistry assessment of structural effects†
Nathan A. I.
Watson
 *ab,
Mike J.
Newland
d,
Beth S.
Nelson
d,
Andrew R.
Rickard
de and
Joseph M.
Beames
ac
*ab,
Mike J.
Newland
d,
Beth S.
Nelson
d,
Andrew R.
Rickard
de and
Joseph M.
Beames
ac
aSchool of Chemistry, Cardiff University, Main Building, CF10 3AT, UK. E-mail: nathan.watson@manchester.ac.uk; nathan.atmos.phd@gmail.com
bDepartment of Earth and Environmental Sciences, University of Manchester, Simon Building, Manchester, M13 9PS, UK
cSchool of Chemistry, Main Building, Edgbaston, Birmingham, UK
dWolfson Atmospheric Chemistry Laboratories, Department of Chemistry, University of York, UK
eNational Centre for Atmospheric Science, Department of Chemistry, University of York, UK
First published on 9th January 2025
Abstract
Nineteen structurally different alkene ozonolysis reactions studied herein proceed via a 1,3-cycloaddition step to produce a short-lived primary ozonide, which then breaks down to form a Criegee intermediate (CI) and an aldehyde/ketone co-product. Both steps of each ozonolysis reaction are examined here using a high-level computational chemistry approach (DF-HF/DF-LCCSD(T)-F12a//B3LYP/aug-cc-pVTZ), and a rate constant and product branching ratio are produced for each reaction. The reactions are then categorized into broadly defined taxonomic groups on the basis of how the ozonolysis chemistry is affected by functional groups, steric bulk and the spatial arrangement of the substituent groups. The five alkene taxonomic groups used for classification are monosubstituted alkenes, trisubstituted alkenes, E-2-alkenes, Z-2-alkenes and haloalkenes. The general cycloaddition reactivity trend identified for these alkene groups is kTHEO (haloalkenes) < kTHEO (monosubstituted alkenes) < kTHEO (E-2-alkenes) ∼ kTHEO (Z-2-alkenes) < kTHEO (trisubstituted alkenes). Within these categories, one secondary trend was that if one or more substituents was small and rich in hyperconjugative α-H atoms, such as a methyl group, a higher alkene rate and a higher CI yield would be induced, compared to a bulky and α-H-poor substituent, such as a tert-butyl (tBu) group. Furthermore, bulky or electronegative substituents were also shown to prompt a reduction in syn-CI yields. Also highlighted in the study is the theoretical mechanism of how the ozonolysis of haloalkenes generates significant yields of tropospheric CF3CHO, a species which can undergo photolysis to produce the strong greenhouse gas fluoroform (CHF3).
Environmental significanceThis systematic computational chemistry analysis produces the rate constants and product branching ratios for the ozonolysis of nineteen different alkenes, showing that these reactions are important sinks for alkenes and ozone, as well as strong sources of Criegee intermediates and, consequentially, OH radicals, implicated in the breakdown of many toxic pollutants. This study further demonstrates that the ozonolysis of haloalkene refrigerants produces significant yields of gaseous trifluoroacetaldehyde, which can break down to produce the greenhouse gas fluoroform. Additionally, by comparing the structure–activity-based trends in this large array of alkenes, this study outlines a broad electronic and steric taxonomic framework that could become the basis of an extended, systematic way of determining the ozonolysis chemistry of multi-alkene environments. |
1 Introduction
1.1 Alkene ozonolysis background
Tropospheric emissions of volatile organic compounds (VOCs) are of considerable importance with respect to air quality and human health. They are implicated in several serious lung diseases (e.g., asthma and respiratory-related illnesses), with a sizable portion of VOC emissions being both biogenic and anthropogenic alkenes.1–4 Biogenic alkenes are estimated to account for ∼80% of the 760 Tg (C) year of these tropospheric VOC emissions. Folic emissions, from rainforest, shrubs and boric forests, are the dominant sources of the most prevalent alkene, isoprene.1,4 While larger alkenes like isoprene and α- and β-pinene dominate alkene emissions in locations like the rainforest, smaller alkenes are often more abundant in urban environments because they are produced from local anthropogenic sources, like ethene and propene evaporation from automobile fuel.3,5The key ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C
C![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) functional group is unsaturated and so has a high susceptibility to reaction, meaning that gaseous alkenes can be depleted via a large variety of sink mechanisms (e.g. via addition reactions with NO3 and Cl radicals).6–11 While reaction with OH radicals dominates alkene breakdown, reaction with ozone, referred to as alkene ozonolysis, is an important alkene removal pathway that is, for example, responsible for ∼10% of the depletion of tropospheric isoprene.12 Exploring ozonolysis depletion mechanisms is a vital area of research because it is a crucial non-photolytic source of radicals in the troposphere, particularly OH radicals.13,14 Furthermore, it is implicated in the formation of photochemical smog in urban areas, which can have significant impacts on human and ecosystem health.15–17
functional group is unsaturated and so has a high susceptibility to reaction, meaning that gaseous alkenes can be depleted via a large variety of sink mechanisms (e.g. via addition reactions with NO3 and Cl radicals).6–11 While reaction with OH radicals dominates alkene breakdown, reaction with ozone, referred to as alkene ozonolysis, is an important alkene removal pathway that is, for example, responsible for ∼10% of the depletion of tropospheric isoprene.12 Exploring ozonolysis depletion mechanisms is a vital area of research because it is a crucial non-photolytic source of radicals in the troposphere, particularly OH radicals.13,14 Furthermore, it is implicated in the formation of photochemical smog in urban areas, which can have significant impacts on human and ecosystem health.15–17
Alkene ozonolysis follows a two-step reaction sequence in which the initial 1,3-cycloaddition step forms a short-lived 5-membered ring, referred to as a primary ozonide (POZ), or a 1,2,3-trioxolane adduct. This is followed by almost instantaneous fragmentation of the POZ, due to excess energy produced in the initial ozonolysis step and the torsional strain within the POZ structure. This fragmentation involves fissures in both the original CC bond and one of the bonds in the O–O–O structure to form an aldehyde or ketone (R3R4CO) and a Criegee intermediate (R1R2COO). Carbonyl species, such as formaldehyde, can be chemically harmful to human health by causing skin irritation and even be carcinogenic and toxic if inhaled.18–20
However, the considerable importance of alkene ozonolysis in the troposphere is largely because it generates the group of products known as Criegee intermediates (CIs), a family of short-lived tropospheric species that possess the carbonyl oxide (COO) functional group.9 One of the reasons why these CIs are notable is that a large proportion of them (37–50%) emerge from the exothermic ozonolysis process with such a significant degree of internal excitation that these hot or excited CIs fragment extremely rapidly.21
This CI decay can proceed through multiple fragmentation pathways to generate highly reactive radical species, such as OH, HO2 & RO2, with one of the most prominent of these pathways being fragmentation via a vinyl hydroperoxide, known for a very high OH yield (see Section S8.4† for greater detail from the literature on these CI decay processes).5,22–25 OH radicals are often referred to as the ‘atmospheric detergent’ due to their capacity to deplete many different tropospheric pollutants, such as CO and NO2.15,26–31 A key daytime source of OH radicals is the photolysis of O3 in the presence of H2O, but the fragmentation of hot CIs is a valuable source of OH radicals in the morning, evening and into the night, due to the non-photolytic nature of the alkene ozonolysis.9,22,26,32,33
The remaining portion of CIs produced from alkene ozonolysis undergo collisional quenching to produce stabilised Criegee intermediates (sCIs), which can then react further with a wide range of tropospheric species, including H2O, NO2, HNO3 MeOH and organic acids.21,34–49 The full chemistry of the sCI is complex and depends on the composition and spatial position of substituents relative to the orientation of the terminal oxygen on the carbonyl oxide. This is best shown with monosubstituted CIs, such as CH3CHOO, where the two distinct conformers, syn-CH3CHOO and anti-CH3CHOO (see Fig. 1), which are separated by a high isomerisation barrier (∼160 kJ mol−1), have different unimolecular decay rates (136 and 53 s−1, respectively).5,50,51 The importance of the spatial positioning of the sCI substituents is further highlighted by the fact that the anti-CH3CHOO + H2O or MeOH rate constant (∼10−14 to 10−12 cm3 s−1) is several orders of magnitude larger than for the equivalent syn-CH3CHOO reactions (∼10−17 to 10−16 cm3 s−1).24,51–55 Given these factors, CIs can be grouped into broad categories of disubstituted CIs, anti-CIs and syn-CIs. Formaldehyde oxide, CH2OO, is rather unique for several reasons including that, it has no readily transferable α-H atoms that would assist a H-transfer decay mechanism, like with many syn-CIs. Furthermore, the barrier to the transfer of the substituent H atoms is extremely high as CHOOH is not stable (more details on CH2OO decay in ESI Section 8.4†).5 Furthermore, CH2OO does not possess any inductive or bulky substituent groups, which are known to affect the bimolecular chemistry of many anti-CIs, and therefore it is often grouped separately from these other categories.21,35
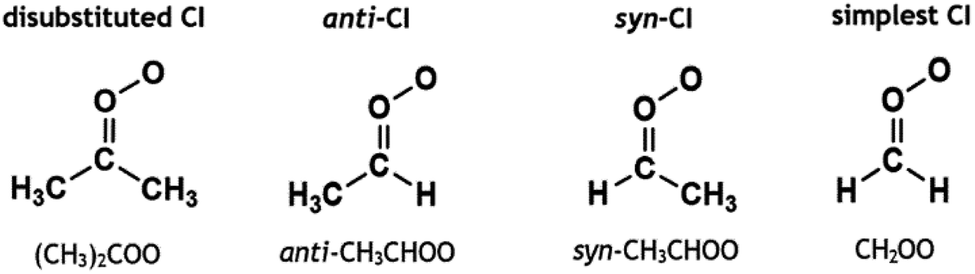 | ||
| Fig. 1 Schematic of three groups of sCIs (disubstituted, anti-sCI and syn-sCI) with examples of each; and the simplest sCI (CH2OO), which is often grouped separately. | ||
While the ozonolysis of alkenes has been examined in depth in the literature (e.g. Newland et al., 2022),56 one key complication is that determining the CI branching fractions is difficult because of their short-lived nature, and so these CI branching fractions are usually inferred from the yields of other primary or secondary products.8,9,32,57–68 This mainly consists of measuring the proportions of the different aldehyde/ketone co-products; however, this does not directly allow for differentiation between anti- & syn-CI yields.9,18–20 As anti- & syn-CI conformers often differ in fragmentation chemistry and OH radicals are often the product of such fragmentations, distinguishing anti- & syn-CI branching fractions can be inferred from the OH yield of alkene ozonolysis reactions (see Section 3.1.1 for more details).9 However, if the OH yield was used as an approach to try to quantify the branching proportions of syn-CI & anti-CI conformers, it would largely depend on assuming the OH yields from the decays of both the syn-CI and the anti-CI. Determining the validity of such an approach is beyond the scope of this study and so the OH yield is simply used here to infer the branching preference of syn-CI over that of the anti conformer.9,56 These difficulties in experimental analysis make an extended computational study of a range of these alkene ozonolysis reactions a worthwhile proposition and, while individual reactions have been computationally analysed, a general extended study has yet to be carried out.6,69–74
1.2 Alkenes involved in this study
A wide selection of alkenes (see Fig. 2) were studied, not only to consider the role of the number and position of the alkene substituents, but also to incorporate variation in the composition of the substituents, which include conjugative unsaturated features, hyperconjugative α-hydrogen (α-H) atoms, halogenated groups and sterically bulky components. By studying alkenes with a large variety of substituents, it can be determined how this range of structural and electronic attributes may have an activating/deactivating influence on the keyCC functional group. The –R1 substituent position for each of these alkenes is assigned to the largest substituent by mass. Given the relationship alkene reactivity has with both the structure and the chemical composition of substituent groups, the alkenes examined (labelled alkenes 1–19 in Fig. 2) are grouped into five categories: monosubstituted alkenes, trisubstituted alkenes, hydrofluoroolefins (also referred to as haloalkenes), E- & Z-2-alkenes and 2-methylpropene.
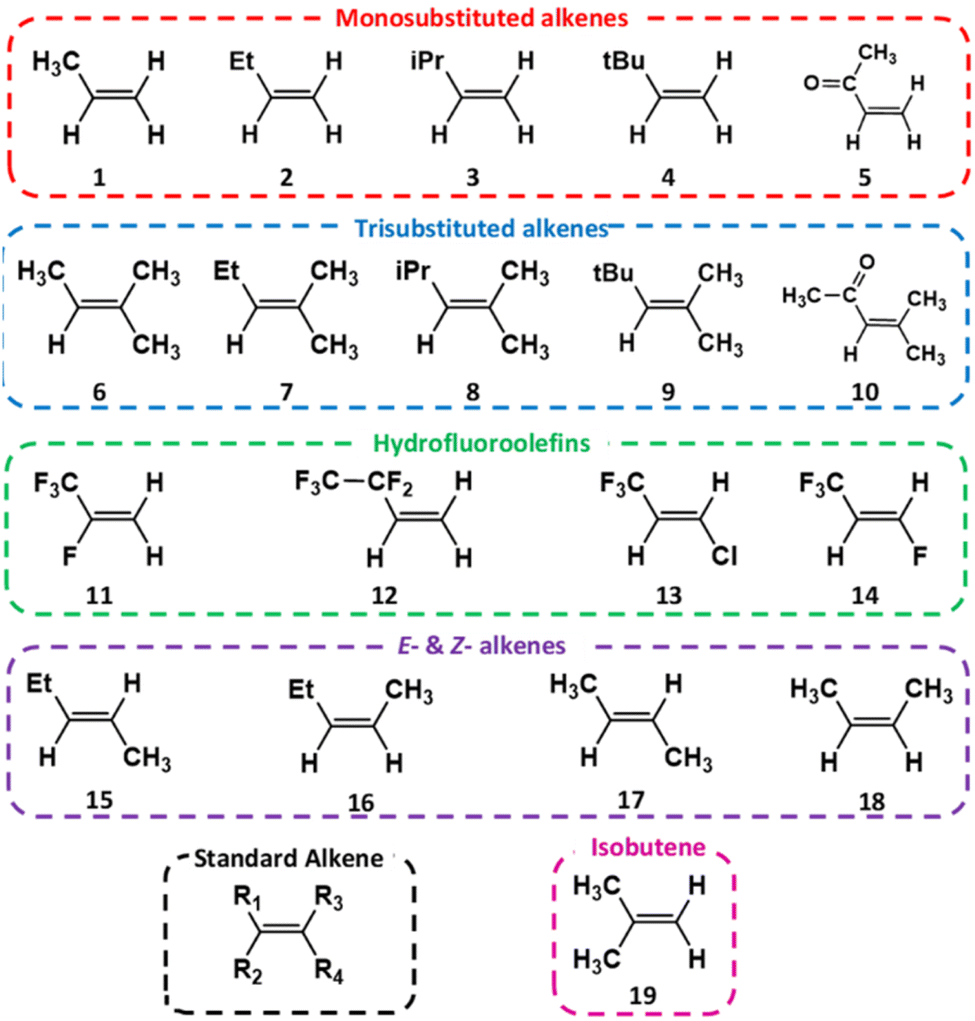 | ||
| Fig. 2 Alkenes, labelled 1–19 and grouped by structural factor. | ||
A connection between the characteristics and number of substituents could in the future pave the way to generating a more sophisticated taxonomic system of alkene groups, whereby the structure of the alkene could be used to forecast the overall ozonolysis chemistry. If such a predictive model could then be integrated into important atmospheric chemical mechanisms, the computational cost of modelling the atmospheric implications of these reactions would fall significantly. To explore this connection between alkene substituents and ozonolysis chemistry further, the master equation rate constant (kTHEO) & product branching fractions (ΓTHEO) for each system have been determined here through a thorough computational chemistry investigation into the ozonolysis of alkenes 1–19 (Fig. 2).
The first of these alkene groups, monosubstituted alkenes, comprises propene, 1-butene, 3-methyl-1-butene, 3,3-dimethyl-1-butene and methyl vinyl ketone (referred to as alkenes 1–5), all of which have the common structure of R1-CHCH2. These alkenes are ordered this way to see if the increasing size of the bulky and complex –R1 substituent, and the concurrent stepwise decline in the number of α-H atoms, correlates with changes in the overall ozonolysis chemistry. Although high boiling points restrict the tropospheric role of alkenes 3 & 4, the ozonolysis of the smaller monosubstituted alkenes is important in cities such as Porto Alegre, Brazil, where alkenes 1 & 2 are significantly abundant (28.3 and 7.8 ppb, respectively).5,75 Furthermore as a product of the breakdown of biogenic isoprene, alkene 5 has considerable populations in rural environments, like rainforests (∼1 ppb), giving the O3 + alkene 5 reaction a substantial tropospheric role.5,39,68,76
The second alkene grouping, trisubstituted alkenes, consists of 2-methyl-2-butene, 2-methyl-2-pentene, 2,4-dimethyl-2-pentene, 2,4,4-trimethyl-2-pentene and mesityl oxide (labelled alkenes 6–10 respectively), all of which have the common structure R1-CHC(CH3)2. Alkenes 6–10 are ordered using the same stepwise increase in the bulk and complexity in the –R1 substituent as for alkenes 1–5, therefore facilitating a direct comparison of reactivity trends between these two groups. This comparison can report if changes in the ozonolysis chemistry caused by this stepwise alteration of the –R1 group are replicated in both alkene sets. Also, if any inductive impact is brought about by the two additional –CH3 substituents, this can be quantified across five different reaction pairs (e.g., alkenes 1 & 6, alkenes 2 & 7…). While alkenes 8 & 9 are present in some urban areas (∼0.01 ppbv), the smaller alkenes 6 & 7 have larger typical urban concentrations of ∼0.1–1 ppb, with even greater abundances in cities like Porto Alegre (17 & 4 ppbv, respectively).5,75 Although it is unlikely to be present in significant quantity in the troposphere due to having a very high boiling point (130 °C) and no large emission sources, studying the ozonolysis of alkene 10 contributes to elucidating the role that conjugated carbonyl substituent groups have on overall alkene ozonolysis chemistry.
The halogenated alkenes 11–14, (2,3,3,3-tetrafluoropropene, 3,3,4,4,4-pentfluoro-1-butene, 1-chloro-3,3,3-trifluoropropene and 1,3,3,3-tetrafluoropropene respectively) are referred to as hydrochlorofluoroolefins (HCFOs) and hydrofluoroolefins (HFOs) and are referred to generically as HFOs throughout this study. These HFOs have emerged recently to replace the older haloalkane refrigerants, that have larger ozone depletion potentials (ODPs) and/or high global warming potentials (GWPs).77,78 As HFO production and use in a variety of countries, including the US and China, has grown, HFO emissions from factories and landfills have also risen.79,80 Studies of the O3 reactions with HFO are sparing because of the currently small tropospheric HFO concentrations in certain cities (∼1 ppqv to 1 pptv). But as HFO concentrations are projected to grow to ∼0.3 ppbv in some cities, the importance of analysing the role of HFO ozonolysis in the troposphere will increase.63,79–81 Analysing HFO ozonolysis also means that the haloalkyl substituents (–CXF2x+1) can be compared to the equivalent alkyl groups (–CXH2x+1), providing an opportunity to further assess the impact of hyperconjugative α-H atoms in alkene substituents. The ozonolysis of alkenes 13 & 14 is a subject of particular importance in the literature as it is known to produce CF3CHO and the photolysis of CF3CHO has been implicated in the production of fluoroform (CHF3), a potent greenhouse gas.82–84 Therefore, it is of increasing importance to determine the full mechanism of this process and at what rate and in what proportions CF3CHO is produced.
The study of the E- & Z-2-alkene group, E- & Z-2-pentene and E- & Z-2-butene (referred to as alkenes 15–18), provides important insights into how ozonolysis chemistry is impacted by both E- & Z-isomerisation and the decreasing number of α-H atoms in substituents. The atmospheric significance of the E- & Z-2-alkenes is greatest in urban environments due to their larger abundance in metropolitan zones like Mexico City (0.37–2.48 ppbv).5,75
The analysis of the ozonolysis of 2-methylpropene, referred to as alkene 19, is important because it exists in substantial abundance in a variety of cities including Taipei, Mexico City and Porto Alegre (0.51, 5.28 & 16.5 ppbv respectively).75,85 Additionally, due to the inductive impact of the additional α-H rich alkyl groups, it is anticipated that the ozonolysis of alkenes 6–10 can give large yields of (CH3)2COO compared to the equivalent yields of CH2OO produced from alkenes 1–5. Therefore, analysis of alkene 19 may help explicitly confirm and delineate the inductive impact of these alkyl groups on CI yield because it is the only alkene ozonolysis reaction here that produces primary yields of both CH2OO and (CH3)2COO directly. The tropospheric abundance levels of alkenes 1–19 in various locations can be found in greater detail in the ESI Section S5.†
2 Methods
2.1 Computational methods
Throughout this study, a density functional theory (DFT) approach is employed to optimise the geometries and determine the vibronic frequencies of all minima and transition state structures, utilizing a B3LYP hybrid functional and the dunning correlation-consistent aug-cc-pVTZ basis set.86–89 The energy pathways between the transition states and local minima are mapped using intrinsic reaction co-ordinate (IRC) calculations through a steepest descent algorithm.90,91 Where the IRC computations are carried out on larger transition states, such bulky structures induce a much higher computational cost and so, to reduce this cost, an approach using a lower-level basis set (B3LYP/aug-cc-pVDZ) is used.90,91 The computational chemistry suite Gaussian09 is used for all DFT calculations described in this study.92The molecular energies of the optimised geometries are then calculated through an explicitly correlated, density-fitted, local fitting, coupled-cluster approach (DF-HF/DF-LCCSD(T)-F12a/aug-cc-pVTZ).93,94 The molecular energy calculations use the MOLPRO software package.95 Energies are further zero-point corrected using a thermal correction factor from the equivalent DFT harmonic frequency calculations. This overall approach is scalable enough to apply to all structures across this study and combines low computational cost and energetic accuracy.
The open-source software Master Equation Solver for Multi Energy Well Reactions (MESMER) uses the results from these calculations to determine the computational rate constant (kTHEO) and product branching ratio (ΓTHEO) for each reaction.96 Unless otherwise stated, standard pressure and temperature are used when calculating all rate constants and product branching ratios discussed in the main body of this manuscript and similar results at other temperatures can be found in the ESI Section S2.† There is significant literature evidence to show that alkene ozonolysis does indeed take place in tropospheric conditions at standard temperature and pressure (see ESI Section S8.3.3†). In this study, where an intermediate product fragments through the same transition state to produce two final products at once, the post-reaction complex is used to determine the MESMER product branching fractions and a 100% dissociation of this complex is assumed (referred to as the “infinite sink approximation”). MESMER also incorporates the asymmetric Eckart function (κECKART), a non-ab initio method that takes into account the forward and reverse barrier heights and the imaginary frequency of the TS barrier, into the calculations to account for the contribution of quantum tunnelling.97
All MESMER-determined kTHEO & ΓTHEO values for bimolecular systems found herein involve using an inverse Laplace transform (ILT) capture rate coefficient of 1 × 10−10 cm3 s−1 and an excess reactant concentration of 1 × 1016 mol cm−3. A standard grain size of 10 cm−1 is used for the EGME calculations for smaller systems, although, as noted in ESI Section S2,† this was adjusted up for larger and more complex systems to ease the cost and intensity of such computations. The results from the MESMER calculations here are relatively insensitive to the small grain size changes within the parameters of this study. As with previous studies, the standard bath gas used was N2 with an “<ΔEdown>” collisional energy transfer factor of 300 cm−1.96,98,99 Additional details and literature background relating to the MESMER input variables are found in ESI Sections S8.1 and S11,† an example MESMER input file can be found.
Throughout this study, the kTHEO constant and ΓTHEO values for each reaction are compared both with experimental data and other theoretical data found in the literature to test the veracity of the theoretical method used in this study. The validity of this approach is also confirmed by a comparison of the rates and Gibbs free energies from this study and the literature in the sensitivity study in the ESI (Sections S8.2 & S8.3†). The sensitivity study finds that the Gibbs free energies of the alkene ozonolysis reaction barriers are usually within ∼4–5 kJ mol−1 of the equivalent experiment barriers, a level of consistency considered favourable by many other studies including studies of alkene ozonolysis.82,99–102 To underline the validity of this approach, a log–log plot of the literature experimental rate coefficients as a function of the results of the calculated rate constants is provided and discussed in ESI Section 8.3.2,† that shows close proximity of the theoretical rate constants produced in this study compared those found in the recent experimental literature.
Studies in the literature also show, when using multi-reference treatments as a benchmark, that both the coupled-cluster CCSD(T)-F12-based approaches and DFT-based approaches, like B3LYP, are high-performance computational approaches for determining the chemistry of the ozone + ethene reaction (this literature is discussed in further detail in ESI Section 8.2†).101–103 Considering both the authentication of this approach from the sensitivity study and the literature, there is appropriate evidence showing that this approach provides computational chemistry results which are consistent with literature studies of similar reactions at a manageable computational cost.24,94,104 While alkene ozonolysis has been occasionally examined in the computational chemistry literature, when addressing the chemistry of a previously unstudied alkene or potential new reaction pathways, this study is exceptional because of the comprehensive range of alkenes studied herein. This thorough investigation into the mechanism and yield of each reaction path, and the distinctive insights this delivers, provides a unique opportunity to produce a system of taxonomic alkene groups generated from these results.
3 Results and discussion section
3.1 General overview of alkene ozonolysis chemistry
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH2).
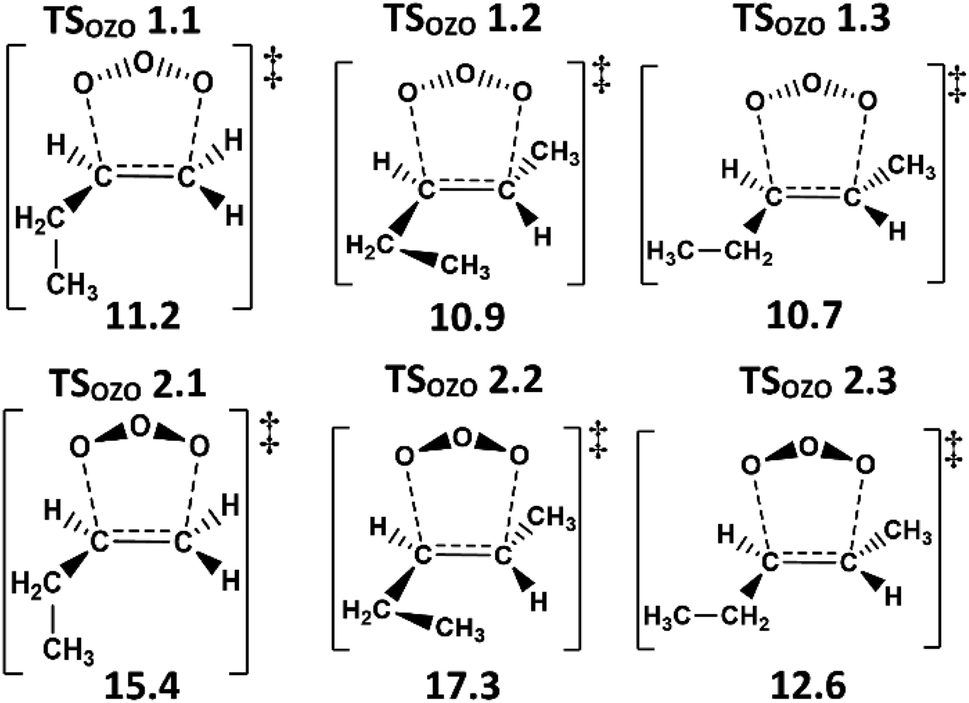
To determine kTHEO and ΓTHEO values for an alkene ozonolysis reaction, it is important to understand the main stationary points on the reaction potential energy surface, including key transition states and minima. The propene reaction (alkene 1) serves as a detailed example due to its simple structure and thorough study in the literature. O3 + propene react to produce two primary ozonide conformers (POZ 1 & 2) via two 1,3-cycloaddition transition states (TSOZO 1 & 2). TSOZO 1 & 2 differ due to the ozone orientation at the point of reaction.
CH2).
To determine kTHEO and ΓTHEO values for an alkene ozonolysis reaction, it is important to understand the main stationary points on the reaction potential energy surface, including key transition states and minima. The propene reaction (alkene 1) serves as a detailed example due to its simple structure and thorough study in the literature. O3 + propene react to produce two primary ozonide conformers (POZ 1 & 2) via two 1,3-cycloaddition transition states (TSOZO 1 & 2). TSOZO 1 & 2 differ due to the ozone orientation at the point of reaction.
The MESMER calculations for O3 + alkene 1 show that all the POZs subsequently fragment, almost instantaneously, without any statistically notable proportion of the POZ population being collisionally stabilised for any significant length of time. The results from the MESMER calculations of all other alkene ozonolysis reactions in this study also show POZs are not collisionally stabilized under atmospheric conditions. This is confirmed by work by Olzemann et al. and in many other studies in the literature, where they found that the collisional stabilization of POZs in ozonolysis of alkenes, like ethene and 2,3-dimethyl-2-butene, is negligible, and the POZ fragments swiftly after its initial formation.105–108
TSOZO 2 has a higher energy barrier than TSOZO 1 (Fig. 3), thanks to the increased steric interaction between the O3 and the –CH3 substituent, thereby reducing the computed POZ yield from TSOZO 2 (0.158) compared to TSOZO 1 (0.842). Experimental verification of this is problematic due to the rapid interconversion between POZs and the subsequent POZ fragmentation, however the similarity in experimental rate constants (kEXP ∼ 0.5–1.3 × 10−17 cm3 s−1) and the kTHEO value calculated here (3.24 × 10−17 cm3 s−1) helps to validate the veracity of this computational approach.67,109–118
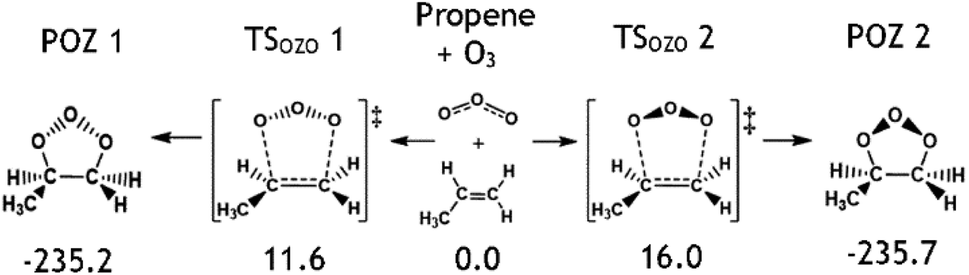 | ||
| Fig. 3 The molecular structures of the cycloaddition steps for the ozonolysis of alkene 1 with relative energies (kJ mol−1). | ||
POZ interconversion occurs over a low-energy pseudorotation isomerisation barrier (TSPOZ) such that all POZ conformers on the O3 + propene potential energy surface access all fragmentation pathways (Fig. 4). This allows the chemistry to be completely modelled by examining a single POZ structure, reducing the computational cost of examining these reactions. ESI Section S6.2† shows results obtained from this assumption in comparison to a full treatment for reactions of O3 with alkenes 1, 6, 11 & 13.
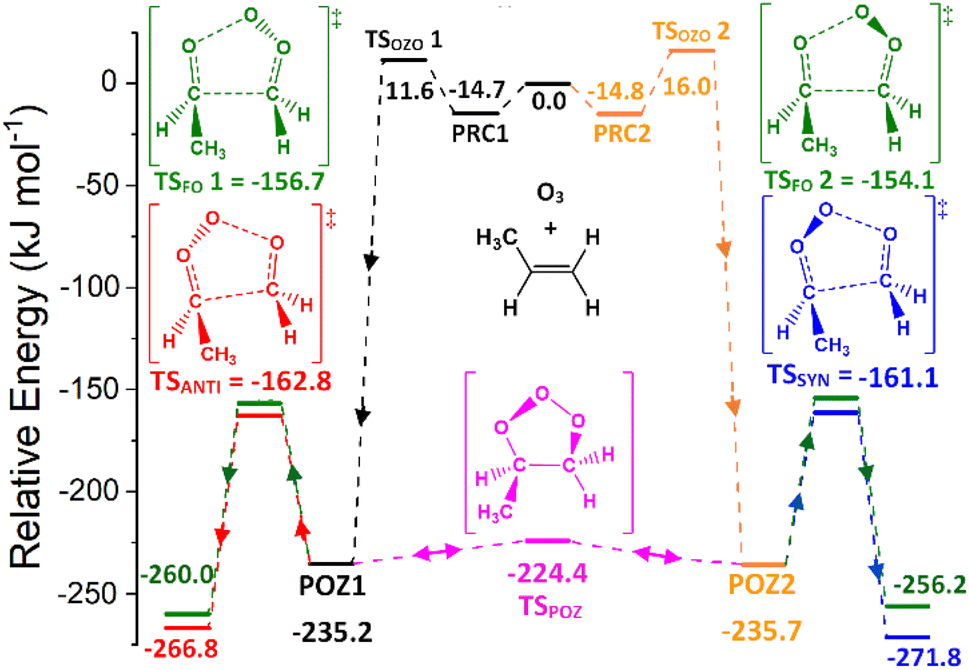 | ||
| Fig. 4 Potential energy surface of the O3 + alkene 1 reaction (propene). Energies are relative to initial reactants. The POZ 1 & 2 rings are not chemically distinct due to interconversion over the low TSPOZ barrier. | ||
The POZ fragments along several reaction pathways, including those that produce a Criegee intermediate (CI) and an aldehyde. As an example, this reaction produces formaldehyde oxide, referred to by the abbreviation “FO” or the chemical formula “CH2OO”, and acetaldehyde CH3CHO through TSFO 1 & 2 (Fig. 4). All transition states are labelled according to the CI produced: TSANTI produces anti-CH3CHOO + HCHO and TSSYN produces syn-CH3CHOO + HCHO. Throughout this study, the syn- & anti-categorisations are assigned to different isomers with reference to the position of the largest group by mass (demonstrated in Fig. 6, Section 3.1.2, using syn- & anti-CF3CFOO, the products of O3 + alkene 11). The computational examination of these fragmentation pathways does not give relative proportions of collisionally stabilised CIs to internally excited CIs and therefore they are referred to collectively as “CIs” when discussing the ΓTHEO values. Pre-reaction complexes (PRCs) and post-reaction complexes are included when running calculations using MESMER, but these complexes have negligible impact and are only discussed sparingly (all structures of these complexes can be found in the ESI).†
As CIs have short atmospheric lifetimes, experimentally determining branching ratios (ΓEXP) of CIs directly is challenging and so ΓEXP data for CIs are generally inferred from branching ratios of aldehyde/ketone co-products. However experimental yields of aldehyde & ketone co-products are not explicitly used here to differentiate between anti & syn conformers of the same CI. Nevertheless, CI species fragment via a variety of mechanisms and anti and syn conformers of the same CI often have very different favoured fragmentation routes. For example, mentioned prior were the unimolecular decompositions of (CH3)2COO, syn-CH3CHOO and syn-EtCHOO, which proceed via 1,4-alkyl-H-migration, producing a vinyl hydroperoxide (VHP) species that subsequently fragments, producing a significant yield of OH radicals.5,22–25 In contrast, anti-CH3CHOO and anti-EtCHOO largely decay via a 1,3-cyclisation mechanism that largely does not produce OH radicals. Anti-CH3CHOO and anti-EtCHOO have unimolecular decay pathways that could produce OH radicals, such as the 1,3-H-migration mechanism, but the rate constants for these fragmentation routes are orders of magnitude smaller, and thus the fraction proceeding through them is negligible, compared to 1,3-cyclisation.5
Given that hot anti- & syn-CIs are estimated to largely be generated with a similar range of internal energy it is likely that the CI conformer with the lower unimolecular rate constant, kUNI, (see Table 1), is less likely to decay and more likely to collisionally stabilise, when produced from the same reaction.119,120 Furthermore, a comparatively greater proportion of the hot anti-CIs are more likely to be stabilised by collision whereas a larger share of the hot syn-CH3CHOO & syn-EtCHOO are more likely to proceed via rapid unimolecular decay.120 This means the vast bulk of the OH yield emerges from syn-CH3CHOO or syn-EtCHOO as their anti-CI equivalents do not deplete via the VHP fragmentations (further discussion of the literature on OH yields from CI decay is found in ESI Section 8.4†). While these methods do not provide full ΓEXP values for many of the CIs produced from POZ fragmentation, they can be used to assess the veracity of the ΓTHEO values calculated in this study.
| k uni (s−1) | CH3CHOO | EtCHOO | CH2OO | Me2COO |
|---|---|---|---|---|
| Syn- | 136 | 205 | 0.3 | 478 |
| Anti- | 53 | 74 |
During the ozonolysis of propene, POZ fragmentation is more favourable via the low energy TSANTI & TSSYN pathways, as reflected by their high branching fractions, ΓANTI (0.452) & ΓSYN (0.276). The correspondingly low yields for TSFO 1 & 2 (0.157 & 0.115) lead to a collective αCH2OO yield of ∼0.272 that is quite modest (the αCH2OO term is used here for the collective CH2OO + CH3CHO yield, the sum of the yields from TSFO 1 & 2). The literature ΓEXP range for CH2OO + CH3CHO of ∼0.38–0.45 is somewhat higher than the computational αCH2OO value but both values agree that CH2OO does not make up a majority of the CI yield.9,39,121–124 As syn-CH3CHOO is the only CI produced from O3 + propene to have a high OH production rate for the reasons outlined above, the relatively low experimental OH yield range (0.32–0.33) seen during the ozonolysis of propene is broadly indicative of the same low to medium yield for syn-CH3CHOO seen here.9,39,121–124
Some studies explore the potential of additional “DeMore” and “O'Neal–Blumstein” channels and a set of epoxidation mechanisms too. The evidence in the literature shows that these mechanisms are likely to play some modest role in the reaction kinetics.73,74,106,125–131 Some restricted evaluations of these additional mechanism are trialled (see ESI Sections S6.4–S6.5†), but any definitive expanded study of these channels was deemed beyond the scope of this study.
The examination of this well-studied reaction shows the validity of the computational approaches and provides a framework for the discussion of more complex reaction systems. To further underline the veracity of this method in describing alkene ozonolysis, a sensitivity analysis of the computational results of a select group of the alkene ozonolysis reactions has been studied and this investigation can be found in ESI Section S8.3.†
 | ||
| Fig. 5 Multiple TSOZO 1 and TSOZO 2 subchannels for O3 + alkene 2 reaction with energies (in kJ mol−1) relative to the initial reactants. | ||
Some of the alkenes in this study have an unsaturated substituent (e.g., alkenes 5 & 10) or a bulky –R1 group (e.g., alkene 8) and this additional conjugation or the steric bulk may potentially inhibit substituent rotations (see Fig. 9 for an example) and increase the height of the isomerisation barrier (TSISO ∼ 20–21 kJ mol−1). It is also possible that the TSISO barrier could inhibit some of the cycloaddition pathways that react with the less energetically favourable alkene conformer, and this was studied in ESI Section S6.1.† However, this investigation into the impact of alkene interconversion on this ozonolysis chemistry, carried out on alkenes 5, 8 & 10, found that the TSISO barrier had no impact on this cycloaddition process.
As with O3 + alkene 1, the products of the alkene 2 ozonolysis reaction include a single set of syn- and anti-mono-substituted CIs, syn- & anti-EtCHOO (Fig. 6). One of the CI products of alkene ozonolysis in this study is “(CH3)2COO” and this can be referred to as dimethyl carbonyl oxide or dimethyl formaldehyde oxide (or DMFO), but here the term acetone oxide has been used and has been abbreviated to “AO” in much of the labelling through this study and the ESI.†
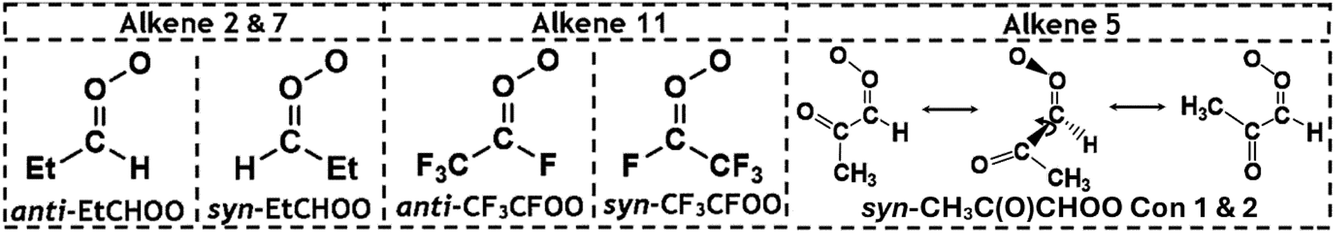 | ||
| Fig. 6 Schematic of syn- & anti-EtCHOO and syn- & anti-CF3CFOO grouped by the alkene ozonolysis reactions that produce them and a display of sterically hindered alkyl substituent rotations inside the Criegee intermediate syn-CH3C(O)CHOO. | ||
These sterically hindered alkyl substituent rotations also raise the isomerisation barriers both for the POZ conformers (∼9–14 kJ mol−1) and the Criegee intermediate and aldehyde final products (∼23–36 kJ mol−1). These kinds of low barriers to POZ interconversion have already been shown to be of negligible importance to overall alkene ozonolysis chemistry (see ESI Section S6.2†). Furthermore, with the exception of the barrier between anti- & syn-CIs, the isomerisation barrier between CI conformers (such as in Fig. 9 for example) is still relatively small and so they are likely to freely interconvert after ozonolysis (see ESI Section S6.3† for more details).
3.2 Results of the ozonolysis of alkenes 1–19
To establish the role that the substituent groups have on an alkene's ozonolysis chemistry, the theoretical (kTHEO & ΓTHEO values) results obtained in this study are assembled into several tables of reference. The similar fluctuations in relative reactivity between alkenes, represented by the energy barriers (ΔETS) and kTHEO values, may be observed to pair with common changes in substituent groups (R1, R2, R3 & R4), both found in Table 2. As mentioned previously in Section 2.1, the number of substituent α-H atoms may be linked with changes in ozonolysis chemistry and so the collective number of substituent α-H atoms are also listed in Table 2. A range of experimental (kEXP) rate constants from the literature are also collated in Table 2, to determine if the kTHEO values are reasonable and to confirm whether the observed alkene ozonolysis trends are correct.| #No. | α-H | Substituent groups | ΔETS (kJ mol−1) | k (10−18 cm3 s−1) | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | TSOZO 1 | TSOZO 2 | k THEO | k EXP | |||
| a The number label (#no.); the number of α-hydrogens (α-H); the identity of alkene substituents (R1, R2, R3 & R4); the lowest energy TSOZO 1 & 2 cycloaddition barriers (ΔETS); the master equation rate constants (kTHEO); and the comparative literature experimental rate constants (kEXP). b Conditions used for determination of kTHEO (such as grain size) are in ESI Section S2.1. | ||||||||||
| 1 | 3 | CH3 | H | H | H | 11.6 | 16.0 | 32.4 | 5.0–13.2 | 67, 109–118 and 132 |
| 2 | 2 | Et | H | H | H | 10.7 | 12.6 | 103 | 9.65–10.9 | 57 and 132 |
| 3 | 1 | iPr | H | H | H | 9.8 | 11.5 | 60.6 | 7.3–9.5 | 57 |
| 4 | 0 | t Bu | H | H | H | 10.3 | 17.3 | 22.5 | 3.8–3.9 | 66 |
| 5 | 0 | C(O)CH3 | H | H | H | 15.2 | 7.8 | 69.0 | 4.5–5.8 | 9, 68, 115, 116 and 133–136 |
| 6 | 9 | CH3 | H | CH3 | CH3 | −1.64 | 2.37 | 4336 | 386–797 | 65 |
| 7 | 8 | Et | H | CH3 | CH3 | −0.8 | −2.2 | 5114 | 406–454 | 8 |
| 8 | 7 | iPr | H | CH3 | CH3 | 3.2 | 4.8 | 245.9 | (223) – SAR | 8 |
| 9 | 6 | t Bu | H | CH3 | CH3 | 3.7 | 6.3 | 245.6 | 125–139 | 65 |
| 10 | 6 | C(O)CH3 | H | CH3 | CH3 | 12.8 | 10.4 | 1.57 | 8.1 ± 2.8 | 64 |
| 11 | 0 | CF3 | F | H | H | 31.3 | 27.5 | 0.0114 | (2.77 ± 0.21) × 10−3 | 63 |
| 12 | 0 | CF2CF3 | H | H | H | 26.5 | 27.3 | 0.0299 | 0.20–0.234 | 61 and 62 |
| 13 | 0 | CF3 | H | H | Cl | 26.2 | 28.6 | 0.0185 | (1.46 ± 0.12) × 10−3 | 60 |
| 14 | 0 | CF3 | H | H | F | 29.9 | 29.6 | 0.0081 | (2.81 ± 0.21) × 10−3 | 59 |
| 15 | 5 | Et | H | H | CH3 | 2.1 | 7.6 | 1127 | 159.2–315 | 58 |
| 16 | 5 | Et | H | CH3 | H | 0.6 | 4.1 | 2482 | 127–128.27 | 58 |
| 17 | 6 | CH3 | H | H | CH3 | 6.8 | 361 | 127.8–200 | 58 and 132 | |
| 18 | 6 | CH3 | H | CH3 | H | 3.1 | 10.9 | 1175 | 121.5–125 | 58 and 132 |
| 19 | 6 | CH3 | CH3 | H | H | 12.3 | 30.2 | 10.8–18.0 | 32, 57, 58 and 132 | |
The final products from all alkene ozonolysis reactions analysed in this study (except for alkenes 13–18) contain at least one set of disubstituted CIs, i.e., CIs with identical substituent groups. In Table 3, these disubstituted CIs are all denoted using the “X2COO” term (X = H or CH3) and the yields of these disubstituted CIs are represented both using a collective branching fraction (αX2COO), as well as the individual ΓTHEO values (ΓX2COO(1) or ΓX2COO(2)). To confirm the observed product branching fraction trends seen for these alkene ozonolysis reactions, the results of the different POZ fragmentation mechanisms analysed here (ΓANTI, ΓSYN & ΓX2COO(1) or ΓX2COO(2)) are displayed alongside the literature measurements related to such processes (αX2COO & OH yield). The OH yields are used here as an indirect method of determining the branching preference between syn- & anti-CI, as techniques to directly measure experimental syn- & anti-CI yields have only recently become available and hence such measurements are limited to very few studies and a small number of alkenes (see Section 3.1.1 for more details). In contrast, the collective theoretical X2COO yield (αX2COO) is compared to experimentally determined branching ratios (ΓEXP) of X2COO measured using co-reactant yields.
| #No. | α-H | Theoretical | Literature | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|
| Γ ANTI | Γ SYN | Γ X2COO(1) | Γ X2COO(2) | α X2COO | α X2COO | OH yield | |||
| a Note: O3 + alkene 19 yields no anti- & syn-CIs and ΓCH2OO & Γ(CH3)2COO values are presented differently herein. | |||||||||
| 1 | 3 | 0.438 | 0.239 | 0.175 | 0.148 | 0.323 | 0.35–0.45 | 0.18–0.39 | 9, 39, 121–124 |
| 2 | 2 | 0.427 | 0.201 | 0.199 | 0.173 | 0.373 | 0.35–0.36 | 0.29–0.41 | 118, 124, 137 and 138 |
| 3 | 1 | 0.431 | 0.115 | 0.267 | 0.187 | 0.454 | 0.49 | — | 124 |
| 4 | 0 | 0.470 | 0.059 | 0.303 | 0.168 | 0.471 | 0.68 | — | 124 |
| 5 | 0 | 0.046 | 0.020 | 0.588 | 0.345 | 0.934 | 0.65–0.95 | 0.13–0.16 | 68, 122, 136 and 139 |
| 6 | 9 | 0.216 | 0.072 | 0.509 | 0.203 | 0.712 | 0.65–0.70 | 0.81–0.98 | 9, 122 and 140–143 |
| 7 | 8 | 0.152 | 0.037 | 0.653 | 0.158 | 0.811 | — | — | — |
| 8 | 7 | 0.117 | 0.014 | 0.718 | 0.151 | 0.868 | 0.81 | — | 124 |
| 9 | 6 | 0.114 | 0.010 | 0.778 | 0.098 | 0.876 | 0.82 | — | 9 |
| 10 | 6 | 0.024 | 0.729 | 0.243 | 0.004 | 0.972 | — | — | — |
| 11 | 0 | 0.001 | <0.001 | 0.532 | 0.468 | 0.999 | — | — | — |
| 12 | 0 | 0.045 | 0.004 | 0.524 | 0.427 | 0.955 | 0.261 | — | 61 |
| 19 | 6 | Γ CH2OO: 0.158 | Γ (CH3)2COO: 0.842 | 0.842 | 0.68–0.75 | 0.60–0.84 | 118, 137, 138 and 144 | ||
The branching fractions for O3 + alkenes 13–18 are displayed in a similar way in Table 4, except with new ΓTHEO labelling, for anti- & syn-R1R2COO (ΓANTI-R1 & ΓSYN-R1) and anti- & syn-R3R4COO (ΓANTI-R3 & ΓSYN-R3). While the experimental yields of the individual anti- & syn-CI conformers are not included in Table 4, the ΓTHEO values can be verified by comparing the experimental collective branching fractions of anti- & syn-R1R2COO (αR1CR2OO) obtained from indirect measurements of R3R4CO co-reactant yields to theoretical αR1CR2OO values.
| #No. | α-H | Theoretical | Literature | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Γ ANTI-R1 | Γ SYN-R1 | Γ ANTI-R3 | Γ SYN-R3 | α R1CR2OO | α SYN | α R1CR2OO | OH yield | |||
| a The trends and comparisons with the experimental literature seen in these tables are discussed in Sections 3.3–3.6. | ||||||||||
| 13 | 0 | 0.604 | 0.197 | 0.108 | 0.091 | 0.728 | 0.397 | 0.63 | — | 145 |
| 14 | 0 | 0.473 | 0.450 | 0.050 | 0.027 | 0.923 | 0.477 | — | — | — |
| 15 | 5 | 0.215 | 0.206 | 0.256 | 0.322 | 0.422 | 0.578 | 0.46 | 0.46 | 140 and 143 |
| 16 | 5 | 0.395 | 0.034 | 0.510 | 0.060 | 0.429 | 0.169 | 0.48 | 0.27–0.29 | 140 and 143 |
| 17 | 6 | 0.520 | 0.480 | — | — | 1 | 0.480 | 1 | 0.19–0.64 | 9, 146 and 147 |
| 18 | 6 | 0.817 | 0.183 | — | — | 1 | 0.183 | 1 | 0.17–0.41 | 9, 39 and 138 |
3.3 Analysis of the ozonolysis of monosubstituted terminal alkenes
This section involves studying the ozonolysis of the monosubstituted terminal alkenes (alkenes 1–5 in Fig. 7) from the perspective of the step-by-step increase in the structural complexity of the –R1 group (from –CH3 & –Et to –iPr & –tBu). | ||
| Fig. 7 Chemical structures of alkenes 1–5. | ||
CH2).
The high kTHEO value for O3 + alkene 2 (1.03 × 10−16 cm3 s−1) is a result of the low energies of the TSOZO 1 & 2 barriers (TSOZO 1.3 ∼ 10.7 kJ mol−1 & TSOZO 2.3 ∼ 12.6 kJ mol−1), at least relative to the O3 + alkene 1 equivalents (TSOZO 1 ∼ 11.6 kJ mol−1 & TSOZO 2 ∼ 16.0 kJ mol−1). Although our calculations show O3 + alkene 2 to be more reactive than the experimental data suggest, this kTHEO value is still within an order of magnitude of the literature range of kEXP values (0.97–1.09 × 10−17 cm3 s−1). One important trend in the literature to highlight is that the TSOZO 1 barriers, calculated here for both alkenes 1 & 2, are lower than that seen in the theoretical literature for O3 + ethene (14.6 kJ mol−1), indicating that the inclusion of even one hydrocarbon –R1 group has an activating effect on the CC bond.
Vereecken et al. note that the sensitivity and the asymmetry of the π-bond on the alkene is crucial when considering the reaction kinetics between alkene and ozone.148 Here, the addition of the –CH3 group induces greater electron density around the alkene's π-bond, distorting the symmetry within the π-orbitals and therefore increasing the overall susceptibility of the alkene CC bond to ozonolysis.148 This is substantiated in the experimental literature by the higher kEXP values for O3 + alkenes 1 & 2 (∼10−17 cm3 s−1) compared to that of O3 + ethene (∼1.45–1.59 × 10−18 cm3 s−1).32,57,67
The POZ fragmentation in the O3 + alkene 2 reaction shows lower TS barriers for pathways that generate anti- & syn-CH3CHOO (TSANTI 3 = −164.4 kJ mol−1 & TSSYN 3 = −163.3 kJ mol−1) than those that produce CH2OO (TSFO 1.3 = −158.5 kJ mol−1 & TSFO 2.3 = −156.7 kJ mol−1). The lengthening of the –R1 group and the corresponding decline in the number of α-H atoms appears to lead to a reduction of the collective yield in R1CHOO CIs (0.627). Therefore, the corresponding theoretical yield of CH2OO for O3 + alkene 2 is significantly larger (αCH2OO ∼ 0.373) than alkene 1 (∼0.323). Although there is some variation in the αCH2OO values for O3 + alkene 1 found in the literature (0.35–0.45), an experimental study by Rickard et al. found that the αCH2OO values for O3 + alkenes 1 & 2 were both ∼0.35, which is very close to the theoretical branching fractions found in this study.118,121,124,137,138
The larger αCH2OO value for O3 + alkene 2 leads to a smaller ΓTHEO value for syn-EtCHOO (0.201) and this is attributed primarily to the TSSYN 2 mechanism having an energy barrier ∼12–13 kJ mol−1 higher than TSSYN 1 & 3. As illustrated in Fig. 8, this high barrier is a consequence of the specific orientation of the –Et substituent, unseen in the TSSYN 1 & 3 structures, placing the –Et group and an oxygen atom in close enough proximity to yield notable steric repulsion. This increased steric interaction within the TSSYN 2 structure has no equivalent in the other POZ fragmentation structures of O3 + alkene 2.
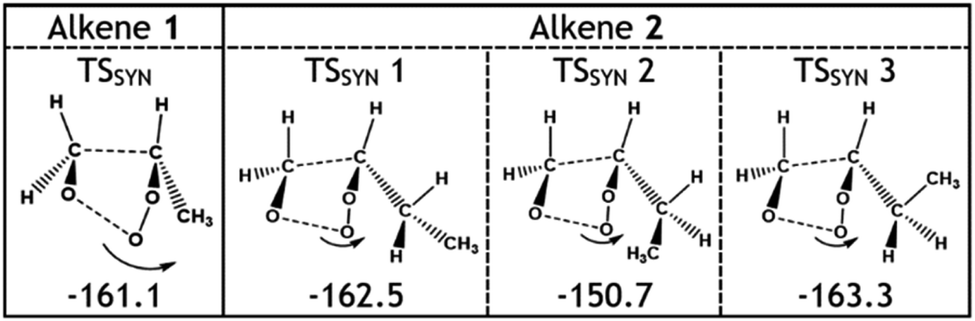 | ||
| Fig. 8 Geometries and energies of the different TSSYN structures of the O3 reactions with alkenes 1 & 2. The arrows in the figure show the movement that takes place as the molecule proceeds through the transition states and indicates what steric interactions might result. Energies (in kJ mol−1) are relative to raw reactants. | ||
This reduction in the ΓTHEO value for syn-EtCHOO + HCHO (0.201) compared to that of syn-CH3CHOO + HCHO for alkene 1 (0.276) would be expected to lead to O3 + alkene 2 having the lower OH yield. However, it is noted in the literature that the kUNI value for fragmentation via VHP formation for syn-EtCHOO is greater (205 s−1) than that of syn-CH3CHOO (74 s−1), implying that OH formation from syn-EtCHOO is the more favourable. Therefore, the higher experimental OH yield seen for alkene 2 (0.29–0.41) compared to alkene 1 (0.18–0.39) is well within the scope of expected results, even considering the marginally smaller ΓSYN value. In addition, this difference in the experimentally determined OH yield between alkenes 1 and 2 is difficult to authenticate due to the large ranges seen for the respective OH yields and both reactions see a similar overall ΓTHEO trend: ΓANTI > ΓSYN > ΓCH2OO(1) > ΓCH2OO(2).
CH2 & tBuCHCH2).
There are many TS structures accessible in the ozonolysis of alkene 2, which is also observed for the conformationally flexible alkene 3 but the increased steric bulk of the –iPr group has a deactivating impact on the CC bond. The lowest energy cycloaddition barriers of the O3 + alkene 3, TSOZO 1.1 & TSOZO 2.3, are lower in energy (9.8 & 11.5 kJ mol−1) than their alkene 2 equivalents, and, if only the lowest TSOZO barriers were considered, then alkene 3 would have the larger kTHEO value. However, the sum of all rate constants over all cycloaddition subchannels (TSOZO 1.1–1.3 & TSOZO 2.1–2.3) generates an overall kTHEO value for alkene 3 (6.06 × 10−17 cm3 s−1) which is smaller than the overall kTHEO of alkene 2 (1.03 × 10−16 cm3 s−1). This downward trajectory in kTHEO values seen for O3 + alkenes 2 & 3 continues for alkene 4 (2.24 × 10−17 cm3 s−1). This stepwise reduction in reactivity is consistent with the experimental data as a decline in kEXP values for O3 + alkene 3 (7.3–9.5 × 10−18 cm3 s−1) followed by O3 + alkene 4 (3.8–3.9 × 10−18 cm3 s−1).57,66,67 This decrease in ozonolysis rates from alkenes 2–4 corresponds to a stepwise decline in the number of α-H atoms in the –R1 substituent, demonstrating the activating effect α-H atoms have on the CC bond.
The increase in the αCH2OO value observed between the ozonolysis of alkenes 1 & 2, (see Fig. 9), continues in an upward trajectory for the αCH2OO values of O3 + alkenes 3 & 4 (0.454 & 0.471). This upward trajectory is replicated in the literature with experimental αCH2OO values for O3 + alkenes 3 & 4 of 0.49 to 0.68, respectively.124 Much of the previously observed ΓTHEO trends persist across all O3 reactions with alkenes 1–4, as shown by the significant fall in the ΓSYN values seen in Fig. 9 (in green).
 | ||
| Fig. 9 Branching ratios (ΓTHEO) of O3 + alkenes 1, 2, 3 & 4 reactions (R1 group refers to CH3, Et, iPr & tBu respectively). | ||
As for alkene 2, the main changes in the POZ fragmentation chemistry for O3 + alkenes 1–4 occur within the TSSYN channel. The energy barriers in the TSSYN channel see significant variation (−164 to −149 kJ mol−1) and the increased number of high-energy TSSYN barriers increases with the size of the –R1 substituent, as the steric repulsion between the O atom and the –R1 substituent increases. This contributes to the overall stepwise reduction in ΓSYN values across the O3 reactions with alkenes 1–4 (see Fig. 9).
Considering only steric interactions, the low TSSYN barrier height (∼−161 kJ mol−1) seen for O3 + alkene 1 would likely be more similar to the TSFO barrier (∼−155 kJ mol−1). We postulate that the low barrier heights seen for some TSSYN structures (such as TSSYN 2 in Fig. 10) are due to the α-H atoms in the –CHR2 substituent group providing stabilisation of the newly forming COO functional group. The role of hyperconjugative stabilisation has literature precedent as it is known that α-H atoms in a syn position reduce the ground state energy of syn-CH3CHOO to ∼14 kJ mol−1 lower than anti-CH3CHOO, which has no hyperconjugative α-H atoms in a syn position.24 The different energies of TSSYN 2 & 3 in Fig. 10 suggest that α-H atoms may only be hyperconjugative if they are in the gauche rather than the anti orientation, as one might anticipate. The impact of both this substituent effect and steric bulk for the barrier for TSSYN barriers is discussed in further detail in ESI Section 8.5.†
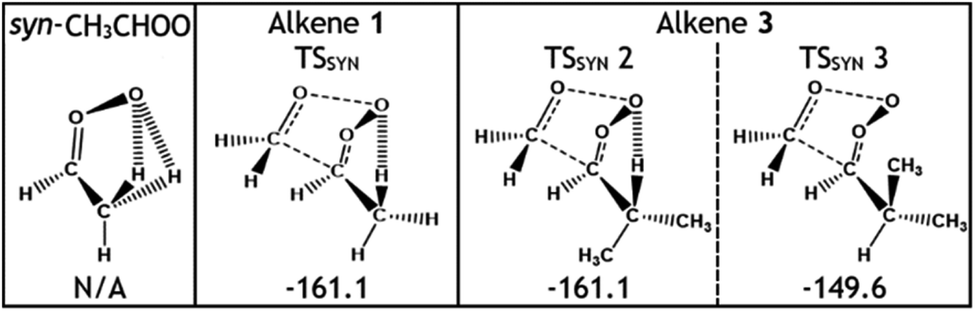 | ||
| Fig. 10 Schematic featuring the stabilising role of α-H atoms in a syn-CHR2 group in relation to the terminal oxygens in the carbonyl oxide group, using the ozonolysis of alkenes 1 & 3. Energies (in kJ mol−1) are relative to raw reactants. | ||
CH2).
Alkene 5, methyl vinyl ketone (MVK), which is abundant in biogenically influenced locations such as rainforests (ca. 1 ppbv), exhibits a significant difference in its ozonolysis chemistry to that of the anthropogenic alkenes 1–4 thanks to the strong electron-withdrawing nature of the –C(O)CH3 substituent.5,75 There are two planar isomers of alkene 5 both of which exhibit conjugation between –C(O)CH3 and the CC groups and these two minimum energy geometries, as well as all cycloaddition TSs, are divided by the same E- or Z-orientation in accordance with Cahn–Ingold–Prelog rules.
One notable observation in the cycloaddition of O3 + alkene 5 is that TSOZO 2.1 would be expected to have a high energy barrier due to a lot of steric repulsion between the central oxygen in the O3 and the R1 substituent (see Fig. 11). Additionally, the electronegative ketone oxygen is near the central oxygen of the O3 and these segments of the overall TS structure would usually coulombically repel each other, further increasing the barrier height. But TSOZO 2.1 is the lowest energy (7.8 kJ mol−1) of the cycloaddition mechanisms by a substantial margin. Furthermore, even though TSOZO 1.1 also assumes the same Z-orientation during cycloaddition, the difference in energy with the Z-orientated cycloadditions is only marginal (∼2–3 kJ mol−1). This leads to a cycloaddition process with a higher-than-expected kTHEO value (6.90 × 10−17 cm3 s−1), with ∼92% of this cycloaddition proceeding via the low energy TSOZO 2.1 structure. While this kTHEO rate constant is somewhat proximate to the kEXP value (4.5–5.0 × 10−18 cm3 s−1), the gap between the experimental and theoretical rate constants is larger here than those seen for alkenes 1–4.9,68 This inductive electronic effect promotes alkene 5 to have a greater reactivity than alkenes 3 & 4, which is likely due to the presence of their bulkier –iPr & –tBu substituents that inhibit reaction. However, despite this, alkene 5 has a much lower rate constant than alkene 2, without any significant increase in steric bulk, and therefore, it is likely that this decrease in reactivity is associated with the decrease in the number of hyperconjugative α-H atoms.
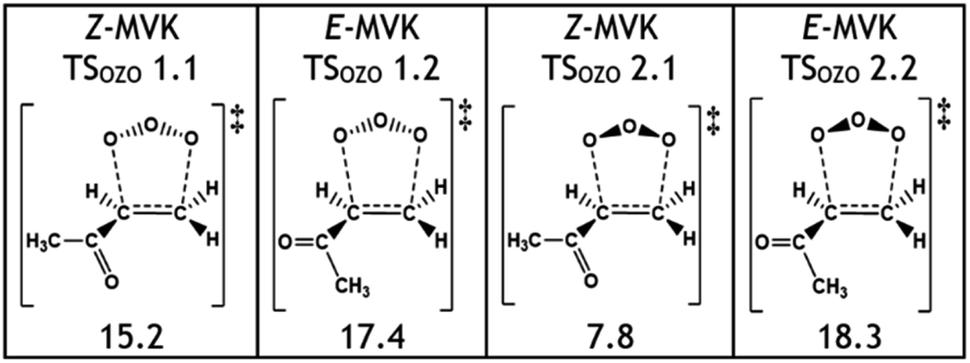 | ||
| Fig. 11 A schematic and the relative energies (kJ mol−1) of the different TSOZO structures of O3 + alkene 5 and the E- or Z-isomer of those alkene 5 structures. Energies (in kJ mol−1) are relative to raw reactants. | ||
During the POZ fragmentation process the –C(O)CH3 group can take orientations where the oxygen in the ketone functional group is either in a “cis” orientation or a “trans” orientation to the 1,2,3-trioxolane section of the POZ or TS structure (see Fig. 12 for examples). The cis conformer is usually ∼15–20 kJ mol−1 higher in energy because of the proximity between the oxygen in the ketone group and the 1,2,3-trioxolane. While all transition states are included in all calculations of POZ yields, the low ΓTHEO values of the cis-TSs (TSANTI 1, TSSYN 1, TSFO 1.1 & TSFO 2.1) means that only the trans-TSs (TSANTI 2, TSSYN 2, TSFO 1.2 & TSFO 2.2) are discussed in greater depth here.
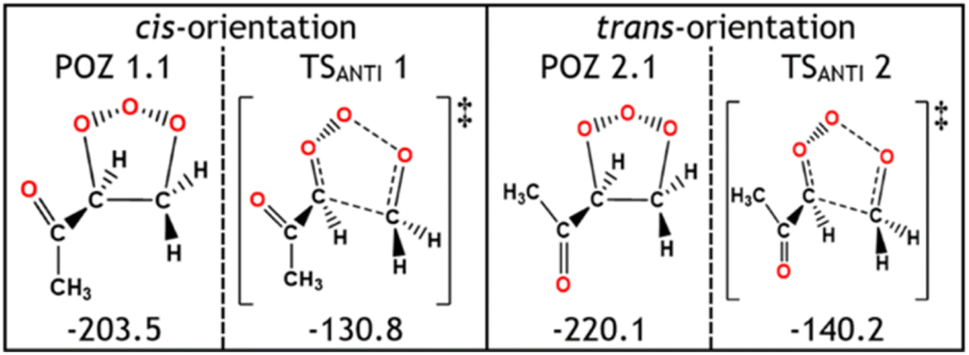 | ||
| Fig. 12 A schematic and the relative energies (kJ mol−1) of the different POZ and TSANTI structures of O3 + alkene 5 differentiated by the cis or trans orientation. Energies are relative to raw reactants. | ||
The low TSFO barrier seen for O3 + alkene 5 in Fig. 13 leads to a high ΓTHEO yield for CH2OO (∼0.934) and a corresponding drop in the yields for the other CI products (syn- & anti-methylglyoxal oxide) from the other TSSYN & TSANTI channels. This is in line with the high collective αCH2OO branching ratios seen in the experimental literature (0.65–0.95).68 H2 + CO2 and H2O + CO are the dominant products from the unimolecular decomposition of CH2OO (Stone et al., 2018; Petolta et al., 2020), with only a negligible OH yield, if any.122,139 The low literature OH yields seen for O3 + alkene 5 (0.13–0.16) are in the range that are expected, considering that O3 + alkene 5 produces high ΓTHEO yields for only CH2OO. The yield for the TSANTI channels for O3 + alkene 5 (ΓANTI ∼ 0.046) is lower than those observed for O3 + alkenes 1–4 (∼0.45) as the –C(O)CH3 group has a deactivating effect on the COO functional group. It is also thought that the low ΓSYN yield for O3 + alkene 5 (∼0.020), compared to alkenes 1–3 (∼0.11–0.28), is in part thanks to the lack of hyperconjugative α-H atoms in a syn position stabilising the COO group.
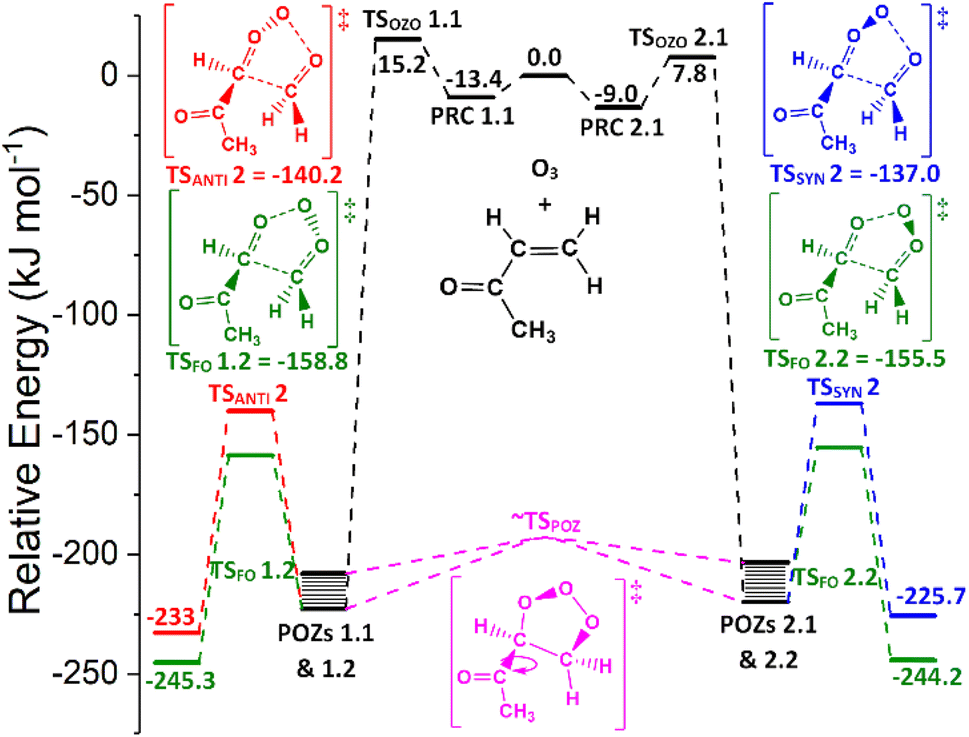 | ||
| Fig. 13 PES of ozonolysis of alkene 5, methyl vinyl ketone, showing structures and energies for the lowest energy for all major channels. POZs 1.1, 2.2, 2.1 & 2.2 all interconvert via various TSPOZ structures (see ESI† Sections S6.2 & S9.0). Energies are relative to raw reactants. | ||
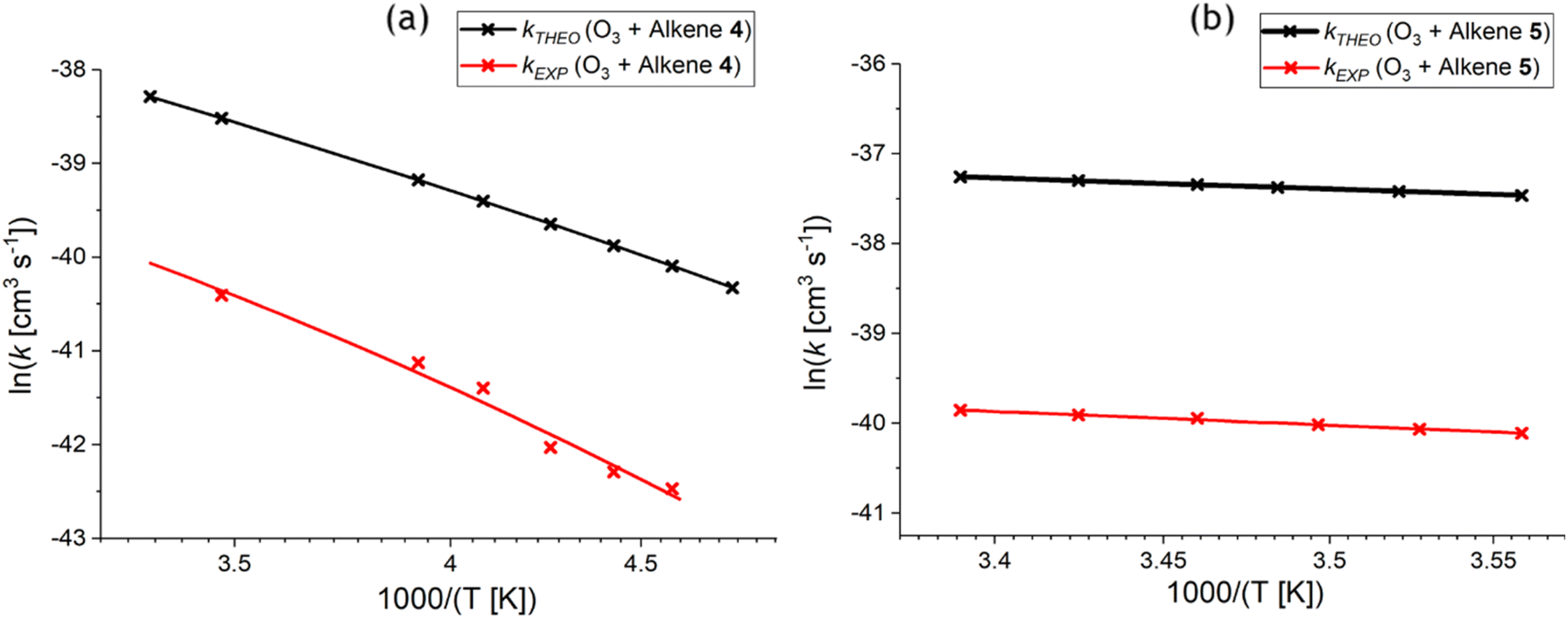 | ||
| Fig. 14 Arrhenius plot of the theoretical & experimental relationships between temperature and the rate constant (kTHEO & kEXP) for the ozonolysis of alkene 4 (a) and alkene 5 (b) (for full numerical details check ESI† Section S2.1).65,68 | ||
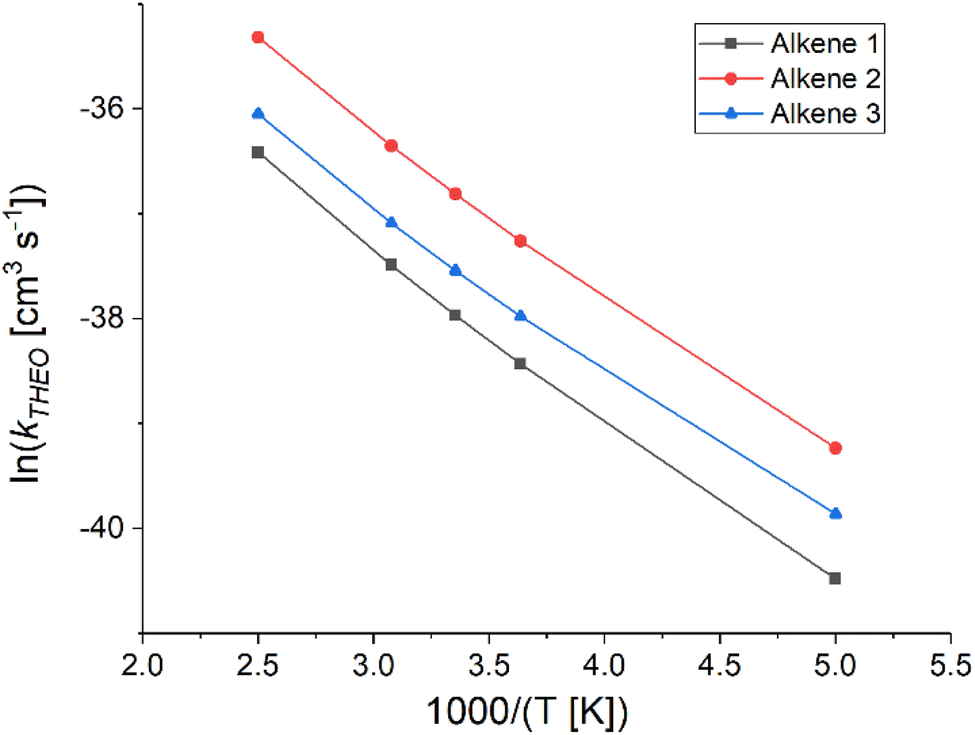 | ||
| Fig. 15 The Arrhenius plots of theoretical rate constants (kTHEO) for the O3 reactions with alkenes 1–3 at a range of temperatures. | ||
3.4 Analysis of the ozonolysis of trisubstituted alkenes
Alkenes 6–10 all have a common structural arrangement around theCC bond, featuring neighbouring –CH3 groups in the –R3 and –R4 positions, and with the –R1 substituent groups replicating the same stepwise increase in complexity seen for terminal alkenes 1–5 (see Fig. 16).
 | ||
| Fig. 16 Chemical structures of alkenes 6–10. | ||
C(CH3)2).
The ozonolysis of alkene 6 proceeds via two low-energy cycloaddition TSOZO 1 and 2 barriers (−1.64 and 2.37 kJ mol−1), producing a kTHEO value for alkene 6 of 4.34 × 10−15 cm3 s−1, which is larger than alkene 1 by just over two orders of magnitude, supported by a range of literature kEXP values for O3 + alkene 6 that are larger (∼3.86–7.97 × 10−16 cm3 s−1) than for alkene 1 (∼9.9–10.1 × 10−18 cm3 s−1). This indicates that the nine α-H atoms on the three –CH3 substituents have a significant inductive effect on the CC bond of alkene 6.9,65,67 These methyl groups also alter the POZ fragmentation chemistry substantially, as the (CH3)2COO yield for O3 + alkene 6 dominates the total CI yield (0.701). This agrees with the experimental (CH3)2COO yield (∼0.69) reported by Rickard et al., which was determined from the corresponding yield of the most substituted primary carbonyl, here acetone (∼0.31), which is also a carbonyl co-product in the syn- & anti-CH3CHOO generating pathways.121 Furthermore, this approach is used in the experimental literature to find a range for the product branching fraction of (CH3)2COO (0.65–0.70), again, which generally agree with the large (CH3)2COO yields found here.9,122,141,142 This higher (CH3)2COO yield leads to much-reduced yields of syn- & anti-CH3CHOO (ΓANTI ∼ 0.241 & ΓSYN ∼ 0.005) compared to those calculated for alkene 1.
Given that, much like with syn-CH3CHOO, the swift unimolecular fragmentation of (CH3)2COO via a short-lived vinyl hydroperoxide (VHP) is known to generate OH radicals, a high OH yield is expected from O3 + alkenes 6–10 when there is a large ΓTHEO of (CH3)2COO.5 While syn-CH3CHOO could contribute significantly to the OH yield here, the ΓTHEO value for this CI is very low and therefore the large (CH3)2COO yield is probably mostly responsible for the high OH yields from the ozonolysis of alkene 6 seen in the literature (0.81–0.98).9,121,138,140,141,150
C(CH3)2).
The stepwise change in structure between alkene 6 and alkene 7 is the same as the essential difference between alkenes 1 & 2, leading to analogous changes in reaction chemistry of the cycloaddition step, which principally is an increase in the reactivity of the CC group. Furthermore, as illustrated in Fig. 17, the TSOZO 1.1 & 1.3 barriers for O3 + alkene 7 are ∼12 kJ mol−1 below their alkene 2 equivalents. This shows that the inclusion of the two –CH3 substituents in the –R3 & –R4 positions of alkene 7 have the same activating impact on the CC bond seen in the analysis of alkene 6. One key difference in cycloaddition chemistry between alkenes 2 & 7 is that in the TSOZO 1.2 structure the –Et group in the alkene 2 adopts a low-energy eclipsed orientation relative to the CC bond TSOZO 1.2, that the addition of the two –CH3 substituents makes unfeasible for alkene 7. Instead, alkene 7 progresses via a pseudo-eclipsed TSOZO 1.2 geometry, where the –Et group is proximate to the O3 during reaction (see Fig. 17). These additional steric interactions raises the barrier height of TSOZO 1.2 (& TSOZO 2.2) to well above the other cycloaddition barriers (e.g. TSOZO 1.1, 1.3, 2.1, 2.3). Despite this, as a result of containing multiple inductive substituents, alkene 7 has the largest kTHEO constant (5.11 × 10−15 cm3 s−1) determined in this study, and the largest kEXP value in the literature of the O3 + alkene reactions studied here (4.06–4.54 × 10−16 cm3 s−1).8,9
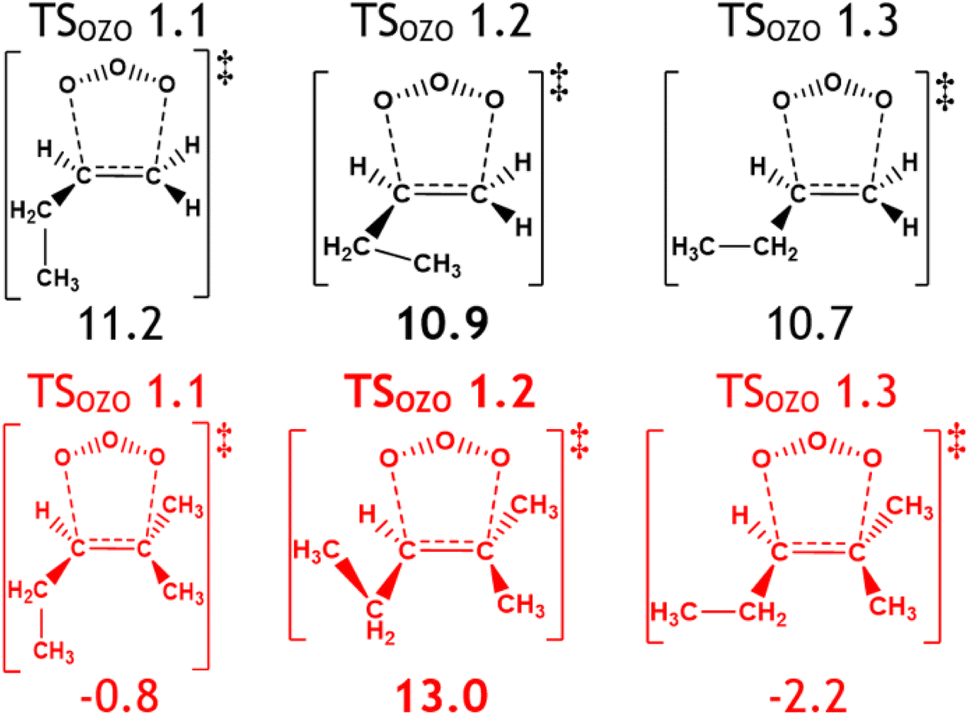 | ||
| Fig. 17 The effect of adjacent extra –CH3 substituents on TSOZO 1 subpathways by comparing alkenes 2 (black) and 7 (red). Energies (in kJ mol−1) are relative to raw reactants. | ||
The O3 + alkene 7 reaction produces a α(CH3)2COO value (0.811) that exceeds the experimental α(CH3)2COO value for O3 + alkene 6 (0.68), and is very similar to the α(CH3)2COO of alkene 8 (0.81). These results seem to be both consistent with a similar positive upward α(CH3)2COO trend in alkenes 1–4. The corollary of this is a decline in the ΓTHEO value for both anti- & syn-EtCHOO (0.152 & 0.037 respectively) for O3 + alkene 7 compared to alkene 6 (ΓANTI ∼ 0.223 & ΓSYN ∼ 0.076). This provides evidence that branching fractions favour CIs with greater numbers of α-H atoms on their alkyl substituents.
C(CH3)2 & tBuCHC(CH3)2).
As seen with alkenes 3 & 4, the increased steric bulk of the –iPr & –tBu groups in alkenes 8 & 9 has, a deactivating impact on the CC bond and this leads to a corresponding decrease in ozonolysis reactivity from alkenes 7–9. This decline in reactivity is illustrated in the increased heights of the TSOZO 1 & 2 barriers of O3 + alkene 8 (3.2 & 4.8 kJ mol−1), with further rises in the alkene 9 TSOZO 1 & 2 barrier heights (3.7 & 6.3 kJ mol−1). This leads to a corresponding decline in computational rates between alkenes 7–9 (see Fig. 18) but this stepwise decline in reactivity is difficult to experimentally corroborate as there are no kEXP values measured for O3 + alkene 8 in the literature. Therefore, the kTHEO value is compared to a rate constant (kSAR) from a structure–activity relationship, derived by McGillen et al., instead (see ESI Section S1.3† for more details).8,9 The computational kTHEO value for O3 + alkene 8 (2.459 × 10−16 cm3 s−1) and the kSAR value (2.2 × 10−16 cm3 s−1) are similar in absolute terms and both rate constants are significantly smaller compared to the respective computational and experimental O3 + alkene 7 rate constants, further illustrating the decline in reactivity across alkenes 7–9 noted earlier.
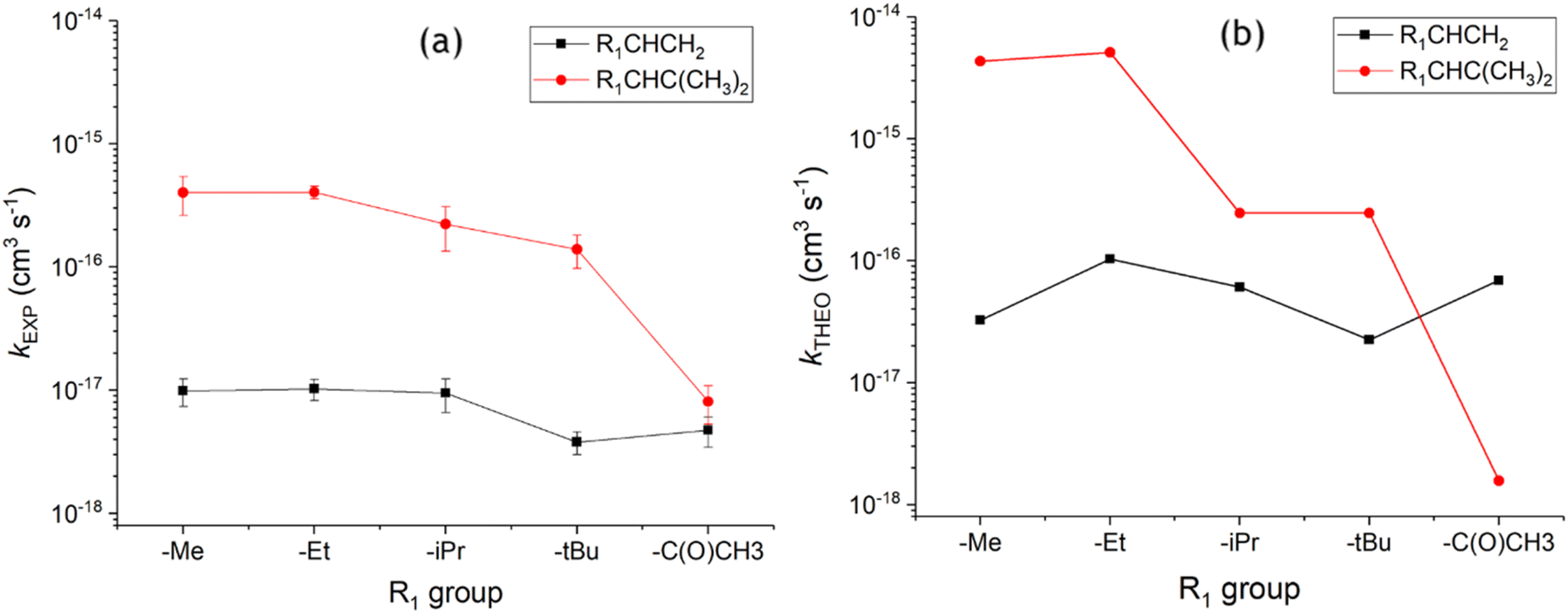 | ||
| Fig. 18 Comparison of ozonolysis for alkenes 1–10 using experimental rate constants (a) and theoretical rate constants (b).9 Note: iPrCHC(CH3)2kEXP rate constant is derived from the kSAR found in the text. | ||
Both the kTHEO value (2.46 × 10−16 cm3 s−1) and the literature kEXP range (1.25–1.39 × 10−16 cm3 s−1) of the O3 + alkene 9 reaction continue the downward trend seen across the ozonolysis rate constants of alkenes 6–10 (see Fig. 18), albeit slight. Fig. 18 shows that the kTHEO values show similar trends to the literature kEXP constants, and that kTHEO values seen for ozonolysis of alkenes 6–10 trend in the same way to O3 + alkenes 1–5. Additionally, it is also clear that the inductive impact of the two additional –CH3 substituents increases the rate constants of all ozonolysis reactions with alkenes 6–9 compared to those of alkenes 1–4.
As the size of –R1 substituent increases, the POZ fragmentation displays an increased preference for producing (CH3)2COO, as shown by the small increases in α(CH3)2COO values for O3 + alkene 8 (0.868) and O3 + alkene 9 (0.876) over the O3 + alkenes 6 & 7 reactions. The preference for (CH3)2COO production is supported by experimental literature α(CH3)2COO values for O3 + alkene 8 (0.81) and O3 + alkene 9 (0.82) in comparison to O3 + alkene 6.9,124 These trends correspond with an overall decline in both ΓANTI (in orange) & ΓSYN (in green) values (Fig. 19), consistent with a reduction in the number of α-H atoms in the –R1 group of the carbonyl oxide.
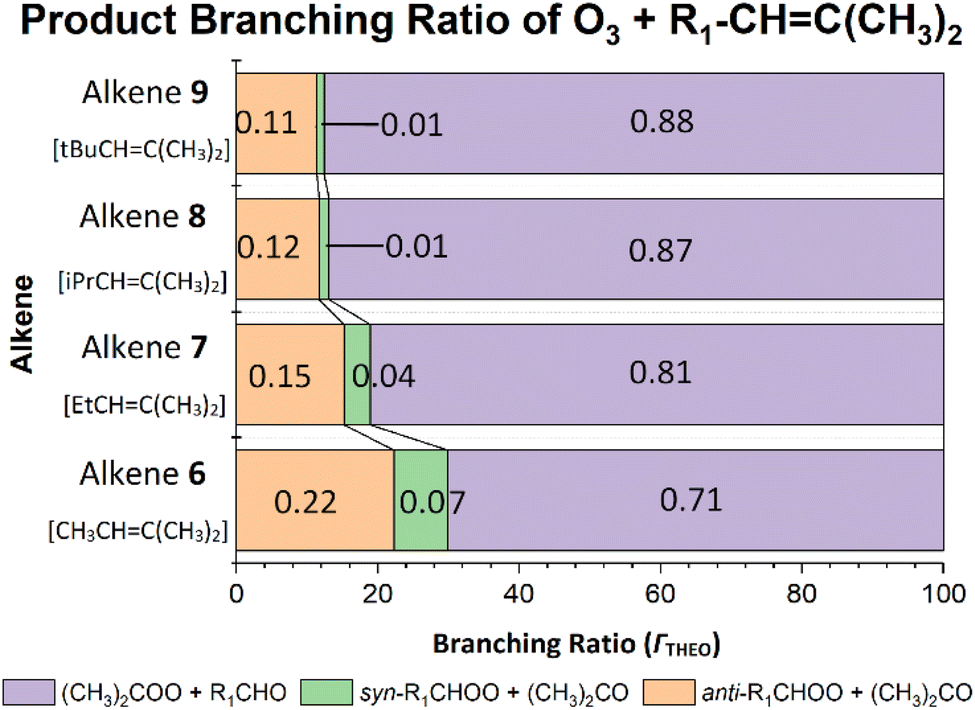 | ||
| Fig. 19 Branching ratios (ΓTHEO) for ozonolysis reactions of alkenes 6–9. | ||
C(CH3)2).
The –C(O)CH3 substituent is electron-withdrawing and contains no hyperconjugative α-H atoms, deactivating the CC bond in alkene 10, as reflected by its comparatively small ozonolysis rate constant (kTHEO ∼ 1.57 × 10−18 cm3 s−1). This is in stark contrast to the hydrocarbon analogue, alkene 7, where the electron-donating nature of the α-H-rich –Et substituent clearly has an activating effect leading to a much larger ozonolysis rate constant (5.11 × 10−15 cm3 s−1). This difference in ozonolysis rate constants is confirmed in the experimental literature where the kEXP of O3 + alkene 7 (4.06–4.54 × 10−16 cm3 s−1) is considerably larger than that of O3 + alkene 10 (8.1 × 10−18 cm3 s−1).8,9,64 This deactivating effect was also exhibited for alkene 5, which, due to the presence of the electron-withdrawing –C(O)CH3 group, also has a lower ozonolysis kTHEO value than its closest purely hydrocarbon analogue, alkene 2 (see Section 3.3.3).
Alkene 10 has the lowest ozonolysis kTHEO value of all the trisubstituted alkenes examined here, corroborated experimentally (see Fig. 18 in Section 3.4.3). Fig. 18 shows that the kTHEO value for O3 + alkene 5 (6.9 × 10−17 cm3 s−1) is higher than that for O3 + alkene 10. As for alkenes 6–9, POZ fragmentation from O3 + alkene 10, leads to a very high α(CH3)2COO value (0.972), largely due to the inductive impact of the α-H atoms present in the two –CH3 substituents. However, it is also worth noting that the low POZ fragmentation yields of syn- & anti-CH3C(O)CHOO calculated here for the ozonolysis of alkene 10 (0.004 & 0.024) are similar to those of O3 + alkene 5 (0.020 & 0.046). This implies that the steric bulk and the electron-withdrawing nature of the –C(O)CH3 group hinders the formation of these syn- & anti-CH3C(O)CHOO. There are no experimental measurements for either the product branching fractions or OH yields from O3 + alkene 10 but the higher branching fraction for (CH3)2COO calculated here is likely to produce a high OH yield, as it does for O3 + alkene 6 (OH yield ∼0.81–0.98).9
CH2) compared with the ozonolysis of monosubstituted and trisubstituted alkenes.
The ozonolysis of 2-methylpropene, (CH3)2CCH2 (alkene 19) is compared to alkenes 1 & 6 (Fig. 20) to provide direct evidence for (CH3)2COO preferential formation compared to CH2OO.
 | ||
| Fig. 20 Chemical structures of standard alkene and alkenes 1, 6 & 19. | ||
The ozonolysis of alkene 19 produces a kTHEO value (3.02 × 10−17 cm3 s−1) which is within an order of magnitude of the kEXP range found in the literature (1.08–1.14 × 10−17 cm3 s−1).32,57,58 O3 + alkene 19 also shows a lower rate constant than O3 + alkene 6 both in this study (kTHEO ∼ 4.3 × 10−15 cm3 s−1) and in the experimental literature.32,57,58,65 O3 + alkene 19 is a very similar reactivity to that of O3 + alkene 1 (kTHEO ∼ 3.24 × 10−17 cm3 s−1), an observation also noted in the literature.32,57,58,67
The POZ fragmentation chemistry of O3 + alkene 19 shows a much higher barrier to CH2OO formation (TSFO ∼ −158.4 kJ mol−1) than to (CH3)2COO formation (TSDMFO ∼ −172.8 kJ mol−1). The dominance of (CH3)2COO yield, seen both here (α(CH3)2COO ∼ 0.842) and in the experimental literature (α(CH3)2COO ∼ 0.75), and the comparatively low barrier for VHP unimolecular decomposition for (CH3)2COO is responsible for the high experimental OH yield (0.60–0.84).118,137,138,144 This definitively confirms that the inductive α-H atoms in the two –CH3 groups promote CI formation compared to –H substituents, strongly favouring (CH3)2COO vs. CH2OO.
3.5 Ozonolysis of E- and Z-2-alkenes
Both the disubstituted alkenes, 2-butene and 2-pentene, share the common structural feature of alkyl substituents located at either end of theCC bond, and therefore, have two geometric isomers. These two chemical structures have different spatial arrangements (see Fig. 21): an E isomer sees the two alkyl groups adopt the –R1 and –R4 positions, such as with alkenes 15 & 17; and the Z isomer has the two alkyl groups in the –R1 and –R3 positions, for instance with alkenes 16 & 18. These trans & cis isomers are chemically distinct, as the presence of the double bond eliminates any prospect of unimolecular isomerisation rotation along the CC bond axis at atmospheric temperatures.
 | ||
| Fig. 21 Chemical structures of alkenes 15–18. | ||
CHCH3).
Alkenes 17 & 18 are the simplest of the E- and Z-hydrocarbon alkenes and the ozonolysis is consistent with prior systems, with the small exception that for symmetric alkene 17 the ozonolysis reaction only produces one cycloaddition TSOZO structure. This means that the single TSOZO has a degeneracy of 2, doubling the raw kTHEO value of O3 + alkene 17 to 3.61 × 10−16 cm3 s−1. However, as seen in Fig. 22, the relative TSOZO energy barrier for O3 + E-2-butene (6.8 kJ mol−1) is higher than that of TSOZO 1 for O3 + Z-2-butene (3.1 kJ mol−1). This leads to a higher master equation rate constant for the ozonolysis of Z-2-butene (kTHEO ∼ 1.18 × 10−15 cm3 s−1) than that of O3 + E-2-butene. Based on rate constants from the experimental literature, O3 + Z-2-butene appears to have a lower rate constant (kEXP ∼ 1.25 ± 0.31 × 10−16 cm3 s−1) than that of O3 + E-2-butene (kEXP ∼ 1.90 ± 0.67 × 10−16 cm3 s−1).9,58 Although this appears to contradict the theoretical results observed here, such differences between kEXP values are once again minimal and the ranges of uncertainty for each of these kEXP values overlap.
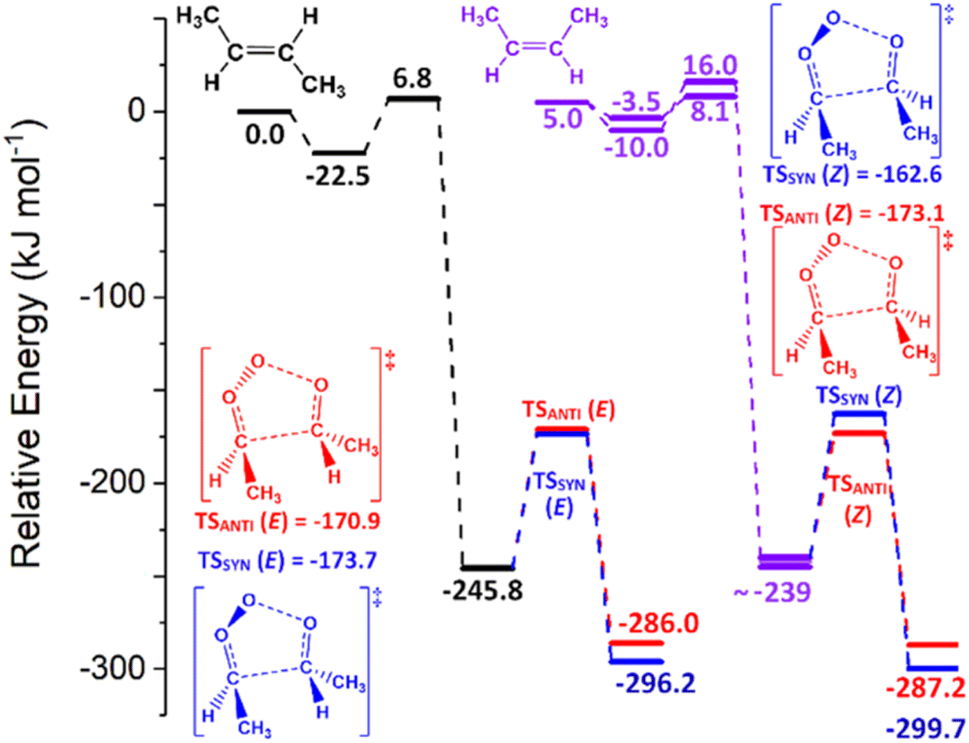 | ||
| Fig. 22 The PESs of the ozonolyses of alkene 17 (in black) and alkene 18 (in violet), with the POZ fragmentation transition states labelled according to the E or Z structure of the alkene involved ((TSANTI (E) & TSSYN (E) are part of the ozonolysis of alkene 17 and TSANTI (Z) & TSSYN (Z) are part of the ozonolysis of alkene 18)). The ozonolysis of alkene 18 produces two POZ conformers, but as they are not chemically distinct they are represented as one POZ at ∼−239 kJ mol−1. Energies are relative to the raw energy of O3 + alkene 17. | ||
During the ozonolysis of both E- & Z-2-butene, the POZs only fragment through a TSANTI & a TSSYN mechanism producing either anti- or syn-CH3CHOO, as well as a CH3CHO co-product. Although the TSANTI barrier is marginally higher than the TSSYN barrier for O3 + E-2-butene (Fig. 22), the subsequent product branching fraction for anti-CH3CHOO is slightly smaller (ΓANTI ∼ 0.49) than that of the syn conformer (ΓSYN ∼ 0.51). In contrast, the TSANTI pathway in the O3 + Z-2-butene reaction has a 10 kJ mol−1 lower barrier than TSSYN leading to a substantially greater yield for anti-CH3CHOO over the syn-CH3CHOO (0.872![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.128). The spatial arrangement of the substituents within the TSSYN structure of the Z-2-butene reaction brings the two –CH3 groups and the central oxygen of the ozonide into close proximity (see Fig. 22) and this increased steric interaction likely contributes to increasing the height of the TSSYN energy barrier. These results appear consistent with a computational study by Rathman et al., which also calculated that ozonolysis of alkene 17 had a lower TSSYN barrier (−116.6 kJ mol−1) than the TSANTI barrier (−109.3 kJ mol−1).151 Furthermore, the Rathman et al. study determined that the TSANTI barrier for O3 + alkene 18 (−119.6 kJ mol−1) is lower than the TSSYN barrier (−113.0 kJ mol−1), which is in agreement with the results found here.151
:0.128). The spatial arrangement of the substituents within the TSSYN structure of the Z-2-butene reaction brings the two –CH3 groups and the central oxygen of the ozonide into close proximity (see Fig. 22) and this increased steric interaction likely contributes to increasing the height of the TSSYN energy barrier. These results appear consistent with a computational study by Rathman et al., which also calculated that ozonolysis of alkene 17 had a lower TSSYN barrier (−116.6 kJ mol−1) than the TSANTI barrier (−109.3 kJ mol−1).151 Furthermore, the Rathman et al. study determined that the TSANTI barrier for O3 + alkene 18 (−119.6 kJ mol−1) is lower than the TSSYN barrier (−113.0 kJ mol−1), which is in agreement with the results found here.151
As discussed earlier, of the two CI products from these ozonolysis reactions, syn-CH3CHOO produces high yields of OH (more details in Section 3.1.1) and therefore a high OH yield from the ozonolysis reaction here is indicative of a high syn-CH3CHOO branching fraction.21 Rathman et al. study showed that O3 + alkene 17 had a clear preference for syn-CH3CHOO formation, whereas O3 + alkene 18 preferentially formed anti-CH3CHOO.151 This is reflected in significantly greater experimental OH yields seen for alkene 17 (0.24–0.64) compared to alkene 18 (0.14–0.41).9,39,138,140,141,147,151 O3 + alkene 17 consistently has a higher OH yield than alkene 18 as seen both in the IUPAC recommendations (0.64 & 0.33) and across a broader body of the literature including Alam et al. (0.63 & 0.26), Rickard et al. (0.59 & 0.37) and Orzechowska and Paulson (0.64 & 0.33).121,140,152,153
CHCH3).
The lengthy and flexible nature of the –Et group in alkene 16 leads to increased steric blocking of the active site than seen in the case of alkene 15. However, alkene 16 still has a greater ozonolysis rate constant (2.48 × 10−15 cm3 s−1) than alkene 15 (1.13 × 10−15 cm3 s−1), so one can postulate that Z-substituted alkenes have a higher overall reactivity with ozone than their E-counterparts. Furthermore, the additional inductive influence that the –Et substituent has is also evident, as alkenes 15 & 16 both have higher rates than alkenes 17 & 18 (3.61 & 11.8 × 10−16 cm3 s−1).
The kEXP values reported by Calvert et al., support this assessment, showing that O3 + alkene 15 (3.15 × 10−16 cm3 s−1) & alkene 16 (∼1.28 × 10−16 cm3 s−1) both have higher rates than alkene 17 (1.28–1.90 × 10−16 cm3 s−1) and alkene 18 (∼1.25 × 10−16 cm3 s−1).9 However, kEXP is generally larger for the E-isomer alkene 15 rather than the Z-isomer, alkene 16: once again the values are so close as to make definitive comparisons challenging though. The main underlying factor that distinguishes the ozonolysis of alkenes 15 & 16, is that the POZ fragmentation produces syn- & anti-EtCHOO, as well as the syn- & anti-CH3CHOO species.
During the POZ fragmentation for the O3 + alkene 15 reaction, the lowest-energy transition states to each product set all have similar energy barriers (−175 to −172 kJ mol−1) giving rise to a near-even distribution of products (Fig. 23). A slight exception is seen for the smaller yield of anti-EtCHOO + CH3CHO (∼0.22), which is likely due to the greater steric interaction between the bulkier –Et group and the ozonide moiety of the transition state structure, raising the energy to reaction (ESI Section S3.1†). In contrast, POZ fragmentation during the ozonolysis of alkene 16 is dominated by the anti-orientated CIs, anti-EtCHOO & anti-CH3CHOO (see Fig. 23), with around ∼83% of the product yield. This is because the formation of both syn-EtCHOO & syn-CH3CHOO requires the ozonide component of the POZ ring to be in close proximity with both the bulky –Et group and a cis-orientated –CH3 group. This greater steric interaction is reflected in the higher barriers to syn-CI formation, ∼−168 kJ mol−1, compared to the low barriers to syn-CI formation, ∼−180 kJ mol−1 (see ESI Section S3.1† for more details).
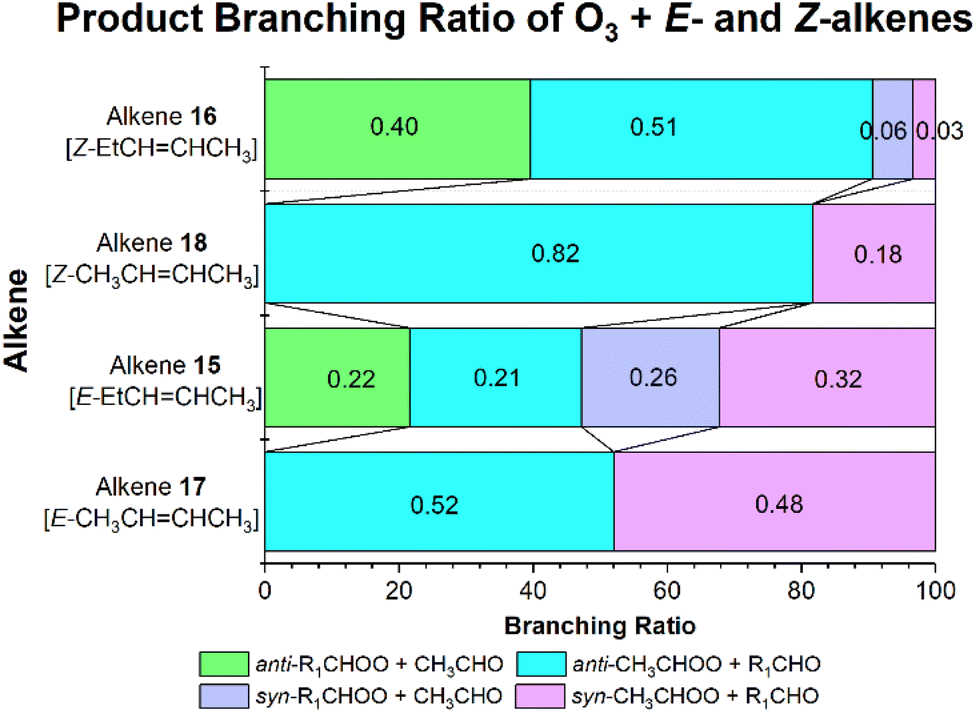 | ||
| Fig. 23 Branching ratios (ΓTHEO) of O3 + alkenes 15–18 reactions. | ||
The higher experimental OH yield of alkene 15 (0.46) vs. alkene 16 (0.27–0.29) is consistent with the higher syn-CI formation seen for alkene 15 in these calculations.140 Moreover, syn-CIs efficiently unimolecularly decompose via a VHP intermediate to form not only OH radicals but also secondary carbonyls and a variety of other atmospheric species.9,124,142,154 For example CH3CHOO can breakdown to produce OH + HCHO (as well as HO2 & CO). This secondary carbonyl generating channel also applies to syn-EtCHOO, where an α-H rich alkyl group in the syn position often facilitates a unimolecular 1,4-H-migration decay mechanism into a VHP intermediate. This VHP then reacts further via an excited hydroxycarbonyl species to produce in a secondary CH3CHO yield.143,154 Therefore, if alkene 15 has the higher syn-EtCHOO formation then it should also produce a significant excess CH3CHO yield. A study by Nelson et al. shows that O3 + alkene 15, does indeed produce a far greater excess CH3CHO yield than O3 + alkene 16 does, inferring that alkene 15 does generate a higher syn-EtCHOO formation.143
3.6 Ozonolysis of halogenated alkenes
The halogenated alkenes studied here (alkenes 11–14, seen in Fig. 24) are all part of a new generation of refrigerants, which are replacing previous atmospherically detrimental chlorofluorocarbon (CFC), hydrochlorofluorocarbon (HCFC) and hydrofluorocarbon (HFC) refrigerants.15,155 These haloalkenes, often referred to as hydrofluoroolefins (HFOs), are being phased in as commercial coolants because of their relative non-toxicity, short atmospheric lifetimes, low global warming potentials (GWP) and low ozone depletion potentials (ODP).15,77,80 | ||
| Fig. 24 Chemical structures of alkene 11–14 (also referred to as HFOs). | ||
As the EU, the USA and China are all in various stages of phasing in these HFO products on a large-scale in the industrial and commercial sector, their widespread use has led to large tropospheric emissions of HFOs both on a local and a global level.7,77,78,80,156 One outcome of this has been that the tropospheric decay of alkene 11 in cities like Beijing has led to an increase in tropospheric abundance of trifluoracetic acid (TFA), a tropospheric gas known to cause irritation to the human respiratory tract.157 It has also been shown that some HFO ozonolysis reactions produce CF3CHO, which can breakdown to produce the strong greenhouse gas, fluoroform (CHF3), although the yield is disputed.82–84 As HFOs usage and emissions are still on the increase, it is vital to understand the chemistry of their tropospheric decay and to determine both the primary and secondary products of HFO breakdown and their yields. These products have also included sCIs produced from O3 + HFO reactions and some computational studies have shown that bimolecular reactions with “HFO-sCIs” may deplete atmospheric toxins, such as SO2 and CH4.23,24,99,104,158–161
Alkene 12 (CF3CF2CHCH2) has limited commercial use, but it is examined here primarily to investigate whether if the inclusion of α-F atoms in the –CF2CF3 substituent has a de-activating impact on the CC group, in contrast to the activating effect that hyperconjugative α-H atoms have in alkene 2. Previous studies have shown that similar haloalkyl –CF3 groups have a de-activating impact on the COO functional group in anti-sCIs and so this deactivating impact may also apply to unsaturated CC bonds too.23,158,160 Some limited computational analysis of O3 + alkenes 11–13 is present in the literature, but the analysis carried out here is extended far wider to include many previously unidentified TSs and final products for each reaction.7,70,156
CH2).
Ozonolysis of alkene 11 (CF3CFCH2) proceeds via the two cycloaddition structures, TSOZO 1 and TSOZO 2, with noticeably higher energy barriers (31.3 & 27.5 kJ mol−1) than those observed with other alkenes. This gives rise to a kTHEO value of 1.14 × 10−20 cm3 s−1 [kEXP value (2.77 × 10−21 cm3 s−1)].63 The deactivating role of the halogen –F and haloalkyl –CF3 substituents in alkene 11 is clearly evident in the reduced kTHEO value for O3 + alkene 11 compared to those of alkenes 1–10 (10−18 to 10−15 cm3 s−1).
During the POZ fragmentation of O3 + alkene 11, the energy barriers to generating CH2OO + CF3CHO (TSFO 1 & 2) are very much lower than TSANTI & TSSYN that produce anti-CF3CFOO and syn-CF3CFOO (syn & anti designations are here given based on whether the –CF3 group is syn-periplanar or anti-periplanar to the terminal oxygen as seen in Fig. 25). A study by Paul et al., showed a similar difference between TSANTI (−120.5 kJ mol−1) and TSFO 1 (−187.8 kJ mol−1) to those observed in Fig. 25.7 This extremely large disparity between energetic barriers produces a very large collective CH2OO branching fraction (αCH2OO > 0.99). It appears that the electronegative nature of –F and –CF3 substituents and the lack of α-H atoms substantially reduces the ΓTHEO values for both anti-CF3CFOO (2.49 × 10−6) and of syn-CF3CFOO (3.31 × 10−7). The impact of –F and –CF3 substituents is analogous to the deactivating role of –C(O)CH3 in alkenes 5 & 10.
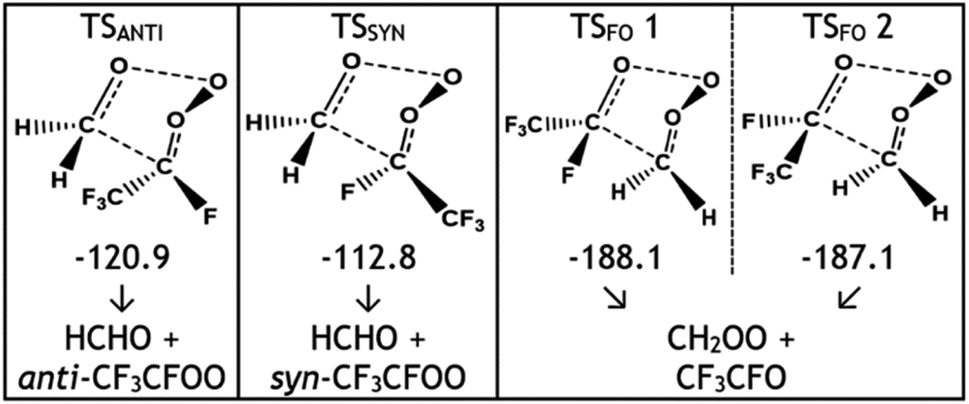 | ||
| Fig. 25 A schematic and the relative energies (kJ mol−1) of the TSANTI & TSSYN structures of O3 + alkene 11 in conjunction with the products these mechanisms generate. Energies are relative to raw reactants. | ||
CH2).
Alkene 12 (CF3CF2CHCH2) is more reactive with O3 than the other HFOs studied here but the energy of the lowest TSOZO barrier for alkene 12 is 15.8 kJ mol−1 higher than its purely hydrocarbon analogue, alkene 2 (EtCHCH2), as seen in Fig. 26. This results in a kTHEO value for alkene 12 (2.99 × 10−20 cm3 s−1) [kEXP range (2.0–2.34 × 10−19 cm3 s−1)] that shows that the –CF2CF3 substituted alkene has a much lower ozonolysis rate than the –Et substituted alkene 2 (1.03 × 10−16 cm3 s−1), demonstrating that the replacement of hyperconjugative α-H atoms with α-F atoms reduces the reactivity of the alkene.61,62 The substitution of the –Et group with a –CF2CF3 group also leads to a decline in β-H atoms too, but β-H atoms have less impact as they are less hyperconjugative than α-H atoms.23
 | ||
| Fig. 26 A comparative of the O3 + alkenes 2 and 12 potential energy surfaces using the lowest energy barriers for each channel only. The direction of the potential energy surface for O3 + alkene 2 reaction progresses from the centre of plot to the left of the plot and the direction of the potential energy surface for O3 + alkene 12 reaction progresses from the centre of plot to the right of the plot. Energies are relative to raw reactants. | ||
The removal of α-H atoms alters the POZ fragmentation chemistry by raising all the energetic barriers (Fig. 26). The increase in TSFO 1 & TSFO 2 barriers is small compared to TSANTI & TSSYN, biasing the product branching fraction more in favour of αCH2OO (0.955) than O3 + alkene 2 does (0.373). However, the only literature study found of this reaction produces a much lower collective αCH2OO value (0.261) for O3 + alkene 12.61 The corresponding theoretical yields for anti- & syn-CF3CF2CHOO are small (0.045 & 0.004) but not as marginal as seen for alkene 11 (both ≪0.01), probably because of the absence of the additional –F substituent seen in the –R2 position of alkene 11, which also deters CI formation.
CHCl) and alkene 14 (E-CF3CHCHF).
The structures of alkenes 13 & 14 both include a –CF3 group in the –R1 position and a halogen group (either –Cl or –F) in the –R4 position and are evaluated here simultaneously. The ozonolysis of alkene 13 has lower energy TSOZO 1 & 2 structures (26.2 & 28.6 kJ mol−1) than alkene 14 (29.9 & 29.6 kJ mol−1) and this difference leads to a larger ozonolysis kTHEO value for alkene 13 (1.85 × 10−20 cm3 s−1) than alkene 14 (8.1 × 10−21 cm3 s−1) [kEXP ∼ 1.46 & 2.81 × 10−21 cm3 s−1].59,60 This is because the –F group is more electron-withdrawing than the –Cl group and therefore has a greater deactivating impact on the CC bond.
Whereas the spatial arrangements of alkenes 11 & 12 placed their deactivating groups in –R1 & –R2 substituents at only one end of the alkene CC bond, alkenes 13 & 14 are E-orientated alkenes with electron-withdrawing groups on the opposing –R1 & –R4 positions. As this produces a slightly more complex potential energy surface, it is important to highlight that POZ fragmentation either proceeds via: TSANTI 1 and TSSYN 1 producing anti- & syn-CF3CHOO, or TSANTI 2 & TSSYN 2 producing anti- & syn-ClCHOO or anti- & syn-FCHOO, respectively. As the deactivating groups are now at either end of the CC bond, it can be calculated whether anti- & syn-CF3CHOO formation is preferred over anti- & syn-ClCHOO and anti- & syn-FCHOO in absolute terms, rather than just through comparative analysis.
While all POZ fragmentation channels are competitive, the formation of anti- & syn-CF3CHOO is favoured over the other CIs, as shown by the high overall αCF3CHOO value seen for O3 + alkene 13 (0.728). This observation, corroborated in the experimental literature (αCF3CHOO ∼ 0.63), demonstrates that the deactivating impact of a purely halogen substituent deters CI formation more than a haloalkyl substituent does.145 The preferences for anti-CIs continues for O3 + alkene 14, for both anti- & syn- CF3CHOO (0.604 & 0.197) and anti- & syn-ClCHOO (0.108 & 0.091). The greater electron-withdrawing nature of the –F substituent appears to have an intensified deactivating effect on CI formation by increasing this ΓTHEO preference for anti- & syn-CF3CHOO (0.473 & 0.450) over that of anti- & syn-FCHOO (0.050 & 0.027). As mentioned earlier, the CF3CHO co-product has been associated with the tropospheric production of the strong greenhouse gas fluoroform (CHF3) and this study shows that particularly the ozonolysis of alkenes 13 & 14 does indeed produce a modest CF3CHO yield (0.199 & 0.077).82–84 This not only provides evidence that additional investigation into the tropospheric breakdown of HFOs is desirable, but it also provides the important CF3CHO branching ratio data that will be needed in computer models that simulate the atmospheric impact that wider HFO use might have.
3.7 Summary
On the whole, this wide-ranging computational study shows that the alkene substituents provide several different key structural and electronic characteristics to theCC bond, that can therefore have a significant influence on both the cycloaddition and POZ fragmentation aspects of the alkene ozonolysis process. With respect to the rate determining step of this reaction, the cycloaddition step, the total number of alkyl substituents, particularly those rich in α-H atoms, has a significant inductive impact on the CC bond, raising the rate constant. This can be seen in the contrast between the high kTHEO values seen for the heavily substituted & α-H-rich alkenes 6–9 (∼10−15 cm3 s−1) and the low reactivity of the α-H-poor alkenes 1–4 (∼10−17 cm3 s−1). The inductive impact of additional alkyl substituents can also be seen in that the ozonolysis of the E- & Z-orientated alkenes, like alkenes 15 & 16 (E- & Z-EtCHCHCH3), had lower ozonolysis kME values than their trisubstituted counterpart, alkene 7 (EtCHC(CH3)2), but higher ozonolysis kME values than their monosubstituted equivalent, alkene 2 (EtCHCH2).
The important role of substituent α-H atoms is particularly highlighted, by the fact that the decline in the number of α-H atoms across monosubstituted alkenes 2–5, leads to a decline in ozonolysis kTHEO values, a pattern that also occurs across the ozonolysis of the tri-substituted alkenes 7–10. Additionally, by contrasting the ozonolysis of alkene 12, (CF3CF2CHCH2) with its purely hydrocarbon analogue alkene 2 (EtCHCH2), it can be seen that the substitution of hyperconjugative α-H atoms in the –Et substituent with α-F atoms was seen to significantly lower the alkene ozonolysis rate constant. The deactivating impact that both the halogen substituents (such as –F or –Cl) and haloalkyl groups (such as –CF3 or –CF2CF3) have on alkenes are evidently responsible for the smaller ozonolysis rates calculated for alkenes 11–14 (kTHEO ∼ 10−21 to 10−19 cm3 s−1).
During POZ fragmentation too, it is clear throughout this study that CI formation is favoured if the CI contains multiple alkyl substituents that are rich in α-H atoms. This is shown by the large yields of α-H rich (CH3)2COO from O3 + alkenes 6–9 & 19 (0.6–0.9) compared to the low CH2OO yields produced from the ozonolysis of alkenes 1–4 (0.2–0.5).
Furthermore, by comparing the ozonolysis reactions of alkenes 1–10, it is shown that not only is anti-R1CHOO formation usually preferred over syn-R1CHOO, but that by gradually increasing the size of –R1 substituents, from an –CH3 group to a –tBu group, the overall yield of the syn-CI, and the anti-CI to a lesser degree, declines in a stepwise manner. Also, the O3 + Z-alkene reactions produce much smaller yields of syn-CIs compared to the E-alkene reactions, a result that can largely be inferred from the experimental literature too. During the examination of O3 + alkenes 11–14, it appears that the deactivating effect of halogen and haloalkyl substituents significantly deters the formation of halogenated CIs and it was determined that halogenated CIs formation followed the subsequent ΓTHEO trend: CF3CHOO > ClCHOO > FCHOO.
4 Atmospheric implications
As with many studies, the tropospheric bimolecular reactions studied here are partially of interest because of their capacity at depleting one of the two reactants, with the focus here drawn to the depletion of alkenes. While the tropospheric mixing ratio of ozone is known to vary widely, it has a significant abundance in a wide range of environments, everywhere from rural forests (∼30 ppb) to highly polluted urban areas (80–200 ppb), frequently overlapping with regions where alkene concentrations are substantial (see Vereecken et al. study for more details).5While using the complex and fine-tuned atmospheric models significantly is outside the purview of this investigation, there are many other different approaches to assessing the comparative importance of reactions in the troposphere.162–164 One common approach is done by determining individual atmospheric lifetimes, which is obtained using the rate constants and the concentrations of the co-reactant, here ozone, and comparing these lifetimes against other main sinks for alkenes (e.g. reaction with OH & NO3).6,7,165–167 However, within the literature various “atmospheric lifetime” approaches can include other complexities and nuances to offset or incorporate the impact of factors such as the impact of secondary reactions, as well as variable ranges in temperature and co-reactant abundance.166,168–170
The effective rate constant (kEFF) is used here as a straightforward approach to assess the relative importance of reaction with O3 as a sink for alkenes compared to reaction with other important atmospheric oxidants (i.e. OH & NO3 radicals), traditionally associated with tropospheric alkene removal. The kEFF is calculated as the product of the oxidant's concentration and its reaction rate coefficient with the alkene.24,104,123,158,171 As the purpose of this section is simply to provide a preliminary assessment of the relative atmospheric significance of the alkene ozonolysis reactions included in this study, the uncomplicated nature of the kEFF methodology and the largely unambiguous definition of the effective rate constant in the literature, make the kEFF methodology well suited for an assessment of the tropospheric significance of the alkene ozonolysis reactions here.
Table 5 displays the kEFF values for several different alkenes, selected for their differing reactivity, functional groups and atmospheric relevance, and comparing their reactivity with O3, OH and NO3 and with their kEFF(Total), which is the sum of their kEFF(O3), kEFF(OH), and kEFF(NO3). Reaction rates with OH and NO3 and the typical atmospheric concentrations are taken from the existing literature.7,9,81,166,172–175 For the calculations of kEFF(O3) values in Table 5, both experimental and theoretical rate constants have been used. ESI Section S7† for a wider range of the kEFF values for alkene reactions with O3, OH & NO3 determined for this study.
| Alkene | No. | k EFF = k × [co-reactant] (10−5 s−1) | Ref. | |||
|---|---|---|---|---|---|---|
| OH | NO3 | O3 | k EFF(O3)/kEFF(Total) | |||
| a Note: [OH] ∼ 1 × 107 mol cm−3, [NO3] ∼ 5.7 × 108 mol cm−3, [O3]urban ∼ 1.9 × 1012 mol cm−3, [O3]rural ∼ 1.4 × 1012 mol cm−3.9,178kEFF (Total) = kEFF(O3) + kEFF(OH) + kEFF(NO3), where values are present. | ||||||
| CH3CHCH2 |
1 | 26.3 | 0.536 | 1.88 | 0.066 | 9 and 58 |
| 6.16 | 0.19 | This work | ||||
| CH3C(O)CHCH2 |
5 | 20.1 | — | 0.73 | 0.035 | 9, 173 and 176 |
| 9.66 | 0.32 | This work | ||||
| EtCHC(CH3)2 |
7 | 89 | 485 | 88.4 | 0.13 | 9 and 177 |
| 973 | 0.63 | This work | ||||
|
E-(CH3)CHCHCl |
13 | 0.44 | — | 0.00028 | 0.00063 | 9, 60 and 156 |
| 0.0035 | 0.0079 | This work | ||||
|
E-(CH3)CHCHF |
14 | 0.93 | — | 0.00053 | 0.00058 | 9 and 59 |
| 0.0015 | 0.0017 | This work | ||||
Alkenes 1 & 7 represent either end of the reactivity range of alkenes without heteroatoms, while still exhibiting abundances between 1–20 ppb in urban environments.5,75,85 Both of these species display a modest but still notable kEFF ozonolysis values, demonstrating that in certain urban environments ozone is likely to somewhat reduce the tropospheric alkene budget. alkene 5, methyl vinyl ketone (MVK), has a significant role in rural environments (e.g. Vereecken et al. estimates MVK ozonolysis to be responsible for up to 10% of CI generation in temperate forests) as it is produced from the breakdown of biogenic isoprene emissions, the kEFF values are calculated using the tropospheric abundance of species found in rural environments.5Table 5 shows that within the constraints of this analysis, ozonolysis will likely have some moderate impact on alkene 5's tropospheric budget.
In contrast, the kEFF values produced from both the theoretical and experimental ozonolysis rate constants found for the urban halogenated alkenes 13 & 14 are much less competitive (∼0.3–1.7% of the overall kEFF) than their non-halogenated equivalents. Previous studies estimate that ozonolysis is a sink for only 0.13–2.96% of tropospheric halogenated alkenes anyway, but these haloalkene ozonolysis reactions are more the subject of interest as they are sources of both halogenated CIs and the important CF3CHO species.82
The calculated POZ fragmentation yields that emerge from these reactions are also crucial, not only for reactions like O3 + alkene 7 where no experimental data on CI branching fractions currently exists in the literature, but also for reactions where certain CI conformer yields have not been calculated before. For instance, the POZ fragmentation yields from O3 + alkene 5 shows, not only the same branching preference for CH2OO (0.934) seen in the literature (0.65–0.95), but also produces a significant yield for both anti- & syn-CH3C(O)CHOO (0.04 & 0.020).
Calculating the branching fractions of both syn- and anti-CIs is important across most ozonolysis reactions here largely because the atmospheric fate of the CI will depend on the secondary reactions that the syn-/anti-CI favours and this is particularly highlighted for the analysis of the ozonolysis of E- & Z-2-alkenes (alkenes 15–18). Ozonolysis of E-2-butene and E-2-pentene produce significant yields of syn-CH3CHOO & syn-EtCHOO which can undergo rapid unimolecular decay via the 1,4-H-migration mechanism. This decay pathway produces highly reactive radical species, such as the OH, HO2 & RO2.5,9,22,24,30,49,124,142,147,154,179,180 In contrast, the ozonolysis of Z-2-butene and Z-2-pentene produce higher branching fractions of anti-CH3CHOO and anti-EtCHOO, which are much more likely to be collisionally stabilised.120 These CIs then react with atmospheric species, including water, SO2, NO2 and organic acids, but products of these sCI reactions with SO2 and organic acids often contribute to the nucleation of secondary organic aerosols.5,22,24,30,49,147,179,180
As stated prior, non-ozonolysis routes dominate the removal of tropospheric HFOs, such as alkenes 11–14, however yields of “HFO-sCIs” (anti and syn conformers of ClCHOO, FCHOO and CF3CHOO) are still quite likely, as these HFOs are typically emitted in more urban environments where there is also high ozone concentrations.157 While there are some literature computational studies on HFO ozonolysis, this extended study includes a much wider range of reaction paths and provides new insights into product yields.7,9,61,70,156 Within the context of halogenated species, CF3CHO has recently been heavily scrutinised due to a potential capacity to undergo photolytic dissociation and produce fluoroform, a very strong greenhouse gas.82–84,104 If the yield of this greenhouse gas turns out to be significant it may trigger a ban on certain blends involving this HFO and may also trigger a phase out by the EU in coming years.181,182 This paper shows that the ozonolysis of alkenes 13 & 14 produce noteworthy branching fractions of CF3CHO as a co-product (0.199 and 0.077 respectively). These rates and yields are vital in exploring tropospheric fluoroform production and may inform policy makers working in this space.
Lastly, many structure-related trends have been identified for the ozonolysis chemistry of alkenes within this manuscript and these can be used to generate a computationally-effective series of taxonomic alkene groups which we would tentatively propose to be monosubstituted alkenes, E-2-alkenes, Z-2-alkenes, trisubstituted alkenes and halogenated alkenes. Although these are reasonably general groupings, there are definitely still anomalies within each group depending on numbers of α-H atoms and/or steric hindrance within different substituents. This will form the basis of a more comprehensive future study on CI classes within atmospheric models.23
5 Conclusions
To aid understanding of the alkene ozonolysis process, a broad classification system is cautiously proposed to categorise alkenes 1–19 based on the number of substituents and the composition of the –R groups; it consists of monosubstituted alkenes, trisubstituted alkenes, E-2-alkenes, Z-2-alkenes and haloalkenes. When considering the effect substituents have on the cycloaddition step, the principal insight of this investigation is that the ozonolysis reactions of the substituent-heavy and α-H-rich trisubstituted alkenes 6–9, have large rate constants (∼10−15 cm3 s−1) compared to the α-H-poor monosubstituted alkenes 1–5 (∼10−17 cm3 s−1) and especially compared to the haloalkenes (∼10−21 to 10−20 cm3 s−1).This produces the following cycloaddition reactivity trend for these alkene groups: kTHEO (haloalkenes) < kTHEO (monosubstituted alkenes) < kTHEO (E-2-alkenes) ∼ kTHEO (Z-2-alkenes) < kTHEO (trisubstituted alkenes). Also noteworthy is that within both alkenes 2–4 and alkenes 7–9 groups, one secondary trend was that alkenes with smaller alkyl substituents, with many hyperconjugative α-H atoms, like –Et groups, were more likely to be susceptible to the cycloaddition reaction than alkenes with bulky and α-H-poor substituent tert-butyl (tBu) groups.
During the POZ fragmentation step of the trisubstituted set of alkenes, the high number of hyperconjugative α-H atoms in (CH3)2COO tends to distort the branching fraction in its favour (ΓTHEO ∼ 0.7–0.9), and this can be juxtaposed with the more balanced branching distribution between the CI products from the monosubstituted alkenes. Also, it can be seen across both alkenes 1–4 and alkenes 6–9, that if the α-H atoms in the –R1 substituent are substituted by bulkier groups there is a general reduction in the branching preference for CIs with that –R1 substituent (CH3CHOO > EtCHOO > iPrCHOO > tBuCHOO). Within this organisational structure, the two taxonomic groups E- & Z-alkenes differ primarily in CI yields, where the O3 + Z-alkene reactions have very high ΓTHEO values for anti-CIs (∼0.8) compared to the E-alkenes, where anti- & syn-CI yields are fairly even, an observation also inferred from the experimental literature.143
When considering the halogenated alkenes, it is clear that electronegative substituents were shown to prompt a reduction in the formation preferences for CIs with electron-withdrawing halogen or haloalkyl substituents, such as for the haloalkenes 11 & 12. However, as the halogenated substituents in alkenes 13 & 14 adopt the opposing –R1 and –R4 substituents, a more even distribution between R1CHOO & R3CHOO is determined. The balanced product distribution from O3 + alkenes 13 & 14 includes a sizable yield of the CF3CHO co-product (ΓTHEO ∼ 0.07–0.20), an important species that can break down through photolysis to produce the powerful greenhouse gas fluoroform (CHF3).82–84,104,158
The taxonomic model outlined has already been used to build a hybrid model that can be used to determine both rate constants and product branching ratios for the ozonolysis of some larger alkenes, at lower cost.23 This includes predicting yields of each CI conformer more easily, which is important because syn-CIs are more likely to be a highly efficient non-photolytic source of a variety of important radicals (e.g. OH, HO2 & RO2), whereas anti-CIs are known to react with pollutants, like SO2 and organic acids or to act as nucleation sources for secondary organic aerosols.5,9,22,24,30,49,124,142,147,154,179,180 Therefore, if this general structure–activity taxonomic framework is refined into an extended and systematic approach and then is integrated into an atmospheric model, it could significantly simplify modelling the impact of alkene ozonolysis in an intricate multi-alkene environment.
Abbreviations
| CI | Criegee intermediate |
| sCI | Stabilized Criegee intermediate |
| POZ | Primary ozonide |
| VHP | Vinyl hydroperoxide |
| HFO | Hydrofluoroolefin |
| Alkene 1 | Propene |
| Alkene 2 | 1-Butene |
| Alkene 3 | 3-Methyl-1-butene |
| Alkene 4 | 3,3-Dimethyl-1-butene |
| Alkene 5 | Methyl vinyl ketone |
| Alkene 6 | 2-Methyl-2-butene |
| Alkene 7 | 2-Methyl-2-pentene |
| Alkene 8 | 2,4-Dimethyl-2-pentene |
| Alkene 9 | 2,4,4-Trimethyl-2-pentene |
| Alkene 10 | Mesityl oxide |
| Alkene 11 | 2,3,3,3-Tetrafluoropropene |
| Alkene 12 | 3,3,4,4,4-Pentafluoro-1-butene |
| Alkene 13 | E-1-Chloro-3,3,3-trifluoropropene |
| Alkene 14 | E-1,3,3,3-Tetrafluoropropene |
| Alkene 15 | E-2-Pentene |
| Alkene 16 | Z-2-Pentene |
| Alkene 17 | E-2-Butene |
| Alkene 18 | Z-2-Butene |
| Alkene 19 | Isobutene |
Data availability
The data supporting this article have been included as part of the ESI.† This includes Cartesian coordinates and vibrational frequencies of all stationary points used, all Master equation rate constants, Canonical rate constants, product branching fractions (with the input conditions also listed). All rate constant and product branching fraction calculations were carried out using Master Equation Solver for Multi-Energy well Reactions (MESMER) Software v5.2 for this study. An example MEMSER input file also found in the ESI.† This MESMER software can be found at the following URL: https://sourceforge.net/projects/mesmer/.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was partially supported by Advanced Research Computing at Cardiff and High Performance Computing (HPC) Wales, a company formed between the Universities and the private sector in Wales which provides the UK's largest distributed supercomputing network. Dr J. M. Beames was supported in initiating this research through Marie Skłodowska Curie Individual Fellowship NPTC (701593). This work was partially supported by the Natural Environment Research Council (NERC) through the Integrated Research Observation System for Clean Air (OSCA) grant (NE/T001984/1). Mike Newland and Andrew Rickard also acknowledge support from the Mechanisms for Atmospheric chemistry: GeneratioN, Interpretation and FidelitY – MAGNIFY project, funded by the UK Natural Environment Research Council (NERC, via grant NE/M013448/1). Beth Nelson acknowledges the NERC SPHERES Doctoral Training Partnership (DTP) for her studentship. Information on the data underpinning the results presented here is shown in the ESI.†References
- K. Sindelarova, C. Granier, I. Bouarar, A. Guenther, S. Tilmes, T. Stavrakou, J.-F. Müller, U. Kuhn, P. Stefani and W. Knorr, Global Data Set of Biogenic VOC Emissions Calculated by the MEGAN Model over the Last 30 Years, Atmos. Chem. Phys., 2014, 14(17), 9317–9341, DOI:10.5194/acp-14-9317-2014.
- P. D. Goldan, W. C. Kuster, F. C. Fehsenfeld and S. A. Montzka, The Observation of a C5 Alcohol Emission in a North American Pine Forest, Geophys. Res. Lett., 1993, 20(11), 1039–1042, DOI:10.1029/93GL00247.
- R. C. Rhew, M. J. Deventer, A. A. Turnipseed, C. Warneke, J. Ortega, S. Shen, L. Martinez, A. Koss, B. M. Lerner, J. B. Gilman, J. N. Smith, A. B. Guenther and J. A. d. Gouw, Ethene, Propene, Butene and Isoprene Emissions from a Ponderosa Pine Forest Measured by Relaxed Eddy Accumulation, Atmos. Chem. Phys., 2017, 17(21), 13417–13438, DOI:10.5194/acp-17-13417-2017.
- S. Reimann and A. C. Lewis, Anthropogenic VOCs, in Volatile Organic Compounds in the Atmosphere, John Wiley & Sons, Ltd, 2007, pp. 33–81, DOI:10.1002/9780470988657.ch2.
- L. Vereecken, A. Novelli and D. Taraborrelli, Unimolecular Decay Strongly Limits the Atmospheric Impact of Criegee Intermediates, Phys. Chem. Chem. Phys., 2017, 19(47), 31599–31612, 10.1039/c7cp05541b.
- M. Antiñolo, I. Bravo, E. Jiménez, B. Ballesteros and J. Albaladejo, Atmospheric Chemistry of E- and Z-CF3CH–CHF (HFO-1234ze): OH Reaction Kinetics as a Function of Temperature and UV and IR Absorption Cross Sections, J. Phys. Chem. A, 2017, 121(43), 8322–8331, DOI:10.1021/acs.jpca.7b06174.
- S. Paul, R. C. Deka and N. K. Gour, Kinetics, Mechanism, and Global Warming Potentials of HFO-1234yf Initiated by O3 Molecules and NO3 Radicals: Insights from Quantum Study, Environ. Sci. Pollut. Res., 2018, 25(26), 26144–26156, DOI:10.1007/s11356-018-2633-7.
- M. R. McGillen, T. J. Carey, A. T. Archibald, J. C. Wenger, D. E. Shallcross and C. J. Percival, Structure–Activity Relationship (SAR) for the Gas-Phase Ozonolysis of Aliphatic Alkenes and Dialkenes, Phys. Chem. Chem. Phys., 2008, 10(13), 1757–1768, 10.1039/B715394E.
- J. G. Calvert, J. J. Orlando, W. R. Stockwell and T. J. Wallington, The Mechanisms of Reactions Influencing Atmospheric Ozone, Oxford University Press, 2015 Search PubMed.
- R. Atkinson and W. P. L. Carter, Kinetics and Mechanisms of the Gas-Phase Reactions of Ozone with Organic Compounds under Atmospheric Conditions, Chem. Rev., 1984, 84(5), 437–470, DOI:10.1021/cr00063a002.
- S. D. Razumovskii, Comparison of Reaction Rates for Ozone-Alkene and Ozone-Alkane Systems in the Gas Phase and in Solution, Russ. Chem. Bull., 1995, 44(12), 2287–2288, DOI:10.1007/BF00713595.
- T. B. Nguyen, G. S. Tyndall, J. D. Crounse, A. P. Teng, K. H. Bates, R. H. Schwantes, M. M. Coggon, L. Zhang, P. Feiner, D. O. Milller, K. M. Skog, J. C. Rivera-Rios, M. Dorris, K. F. Olson, A. Koss, R. J. Wild, S. S. Brown, A. H. Goldstein, J. A. de Gouw, W. H. Brune, F. N. Keutsch, J. H. Seinfeld and P. O. Wennberg, Atmospheric Fates of Criegee Intermediates in the Ozonolysis of Isoprene, Phys. Chem. Chem. Phys., 2016, 18(15), 10241–10254, 10.1039/C6CP00053C.
- Y. F. Elshorbany, R. Kurtenbach, P. Wiesen, E. Lissi, M. Rubio, G. Villena, E. Gramsch, A. R. Rickard, M. J. Pilling and J. Kleffmann, Oxidation Capacity of the City Air of Santiago, Chile, Atmos. Chem. Phys., 2009, 9(6), 2257–2273, DOI:10.5194/acp-9-2257-2009.
- B. S. Nelson, D. J. Bryant, M. S. Alam, R. Sommariva, W. J. Bloss, M. J. Newland, W. S. Drysdale, A. R. Vaughan, W. J. F. Acton, C. N. Hewitt, L. R. Crilley, S. J. Swift, P. M. Edwards, A. C. Lewis, B. Langford, E. Nemitz, Shivani, R. Gadi, B. R. Gurjar, D. E. Heard, L. K. Whalley, Ü. A. Şahin, D. C. S. Beddows, J. R. Hopkins, J. D. Lee, A. R. Rickard and J. F. Hamilton, Extreme Concentrations of Nitric Oxide Control Daytime Oxidation and Quench Nocturnal Oxidation Chemistry in Delhi during Highly Polluted Episodes, Environ. Sci. Technol. Lett., 2023, 10(6), 520–527, DOI:10.1021/acs.estlett.3c00171.
- Fifth Assessment Report – Climate Change 2013, https://www.ipcc.ch/report/ar5/wg1/, accessed 2018-08-26 Search PubMed.
- M. Hallquist, J. Munthe, M. Hu, T. Wang, C. K. Chan, J. Gao, J. Boman, S. Guo, Å. M. Hallquist, J. Mellqvist, J. Moldanova, R. K. Pathak, J. B. Pettersson, H. Pleijel, D. Simpson and M. Thynell, Photochemical Smog in China: Scientific Challenges and Implications for Air-Quality Policies, Natl. Sci. Rev., 2016, 3(4), 401–403, DOI:10.1093/nsr/nww080.
- U.S. Environmental Protection Agency Office of Air and Radiation, The Benefits and Costs of the Air Act from 1990 to 2020: Final Report – Rev. A, 2011, https://www.epa.gov/clean-air-act-overview/benefits-and-costs-clean-air-act-1990-2020-report-documents-and-graphics, accessed 2020-07-20 Search PubMed.
- A. Tiwary and I. Williams, Air Pollution: Measurement, Modelling and Mitigation, CRC Press, 4th edn, 2018 Search PubMed.
- L. Zhu, D. J. Jacob, F. N. Keutsch, L. J. Mickley, R. Scheffe, M. Strum, G. González Abad, K. Chance, K. Yang, B. Rappenglück, D. B. Millet, M. Baasandorj, L. Jaeglé and V. Shah, Formaldehyde (HCHO) As a Hazardous Air Pollutant: Mapping Surface Air Concentrations from Satellite and Inferring Cancer Risks in the United States, Environ. Sci. Technol., 2017, 51(10), 5650–5657, DOI:10.1021/acs.est.7b01356.
- J. C. Salamanca, J. Meehan-Atrash, S. Vreeke, J. O. Escobedo, D. H. Peyton and R. M. Strongin, E-Cigarettes Can Emit Formaldehyde at High Levels under Conditions That Have Been Reported to Be Non-Averse to Users, Sci. Rep., 2018, 8 DOI:10.1038/s41598-018-25907-6.
- D. Johnson and G. Marston, The Gas-Phase Ozonolysis of Unsaturated Volatile Organic Compounds in the Troposphere, Chem. Soc. Rev., 2008, 37(4), 699–716, 10.1039/B704260B.
- M. A. H. Khan, C. J. Percival, R. L. Caravan, C. A. Taatjes and D. E. Shallcross, Criegee Intermediates and Their Impacts on the Troposphere, Environ. Sci.: Processes Impacts, 2018,(20), 437–453, 10.1039/C7EM00585G.
- N. Watson, An Analysis of the Sources and Sinks for Criegee Intermediates: an Extended Computational Study, Cardiff University, Cardiff, UK, p. 2021 Search PubMed.
- N. A. I. Watson, J. A. Black, T. M. Stonelake, P. J. Knowles and J. M. Beames, An Extended Computational Study of Criegee Intermediate–Alcohol Reactions, J. Phys. Chem. A, 2019, 123(1), 218–229, DOI:10.1021/acs.jpca.8b09349.
- P. L. Luo, Y. Endo and Y. P. Lee, Identification and Self-Reaction Kinetics of Criegee Intermediates Syn-CH3CHOO and CH2OO via High-Resolution Infrared Spectra with a Quantum-Cascade Laser, J. Phys. Chem. Lett., 2018, 9(15), 4391–4395, DOI:10.1021/acs.jpclett.8b01824.
- A. M. Holloway and R. P. Wayne, Atmospheric Chemistry, Royal Society of Chemistry, 2010 Search PubMed.
- R. Atkinson, D. L. Baulch, R. A. Cox, R. F. Hampson, J. A. Kerr, M. J. Rossi and J. Troe, Evaluated Kinetic, Photochemical and Heterogeneous Data for Atmospheric Chemistry: Supplement V. IUPAC Subcommittee on Gas Kinetic Data Evaluation for Atmospheric Chemistry, J. Phys. Chem. Ref. Data, 1997, 26(3), 521–1011, DOI:10.1063/1.556011.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Volume I - Gas Phase Reactions of Ox, HOx, NOx and SOx Species, Atmos. Chem. Phys., 2004, 4(6), 1461–1738, DOI:10.5194/acp-4-1461-2004.
- Y. Fang, F. Liu, V. P. Barber, S. J. Klippenstein, A. B. McCoy and M. I. Lester, Communication: Real Time Observation of Unimolecular Decay of Criegee Intermediates to OH Radical Products, J. Chem. Phys., 2016, 144(6), 061102, DOI:10.1063/1.4941768.
- A. Novelli, L. Vereecken, J. Lelieveld and H. Harder, Direct Observation of OH Formation from Stabilised Criegee Intermediates, Phys. Chem. Chem. Phys., 2014, 16(37), 19941–19951, 10.1039/C4CP02719A.
- R. M. Harrison, J. Yin, R. M. Tilling, X. Cai, P. W. Seakins, J. R. Hopkins, D. L. Lansley, A. C. Lewis, M. C. Hunter, D. E. Heard, L. J. Carpenter, D. J. Creasey, J. D. Lee, M. J. Pilling, N. Carslaw, K. M. Emmerson, A. Redington, R. G. Derwent, D. Ryall, G. Mills and S. A. Penkett, Measurement and Modelling of Air Pollution and Atmospheric Chemistry in the U.K. West Midlands Conurbation: Overview of the PUMA Consortium Project, Sci. Total Environ., 2006, 360(1), 5–25, DOI:10.1016/j.scitotenv.2005.08.053.
- J. G. Calvert, The Mechanisms of Atmospheric Oxidation of the Alkenes, Oxford University Press, 2000 Search PubMed.
- A. Geyer, K. Bächmann, A. Hofzumahaus, F. Holland, S. Konrad, T. Klüpfel, H. W. Pätz, D. Perner, D. Mihelcic, H. J. Schäfer, A. Volz-Thomas and U. Platt, Nighttime Formation of Peroxy and Hydroxyl Radicals during the BERLIOZ Campaign: Observations and Modeling Studies, J. Geophys. Res., 2003, 108(D4), 8249, DOI:10.1029/2001JD000656.
- P. Neeb, O. Horie and G. K. Moortgat, The Ethene–Ozone Reaction in the Gas Phase, J. Phys. Chem. A, 1998, 102(34), 6778–6785, DOI:10.1021/jp981264z.
- R. I. Martinez and J. T. Herron, Stopped-Flow Studies of the Mechanisms of Ozone-Alkene Reactions in the Gas Phase: Trans-2-Butene, J. Phys. Chem., 1988, 92(16), 4644–4648, DOI:10.1021/j100327a017.
- H. Niki, P. D. Maker, C. M. Savage, L. P. Breitenbach and M. D. Hurley, FTIR Spectroscopic Study of the Mechanism for the Gas-Phase Reaction between Ozone and Tetramethylethylene, J. Phys. Chem., 1987, 91(4), 941–946, DOI:10.1021/j100288a035.
- D. Cremer, J. Gauss, E. Kraka, J. F. Stanton and R. J. Bartlett, A CCSD (T) Investigation of Carbonyl Oxide and Dioxirane. Equilibrium Geometries, Dipole Moments, Infrared Spectra, Heats of Formation and Isomerization Energies, Chem. Phys. Lett., 1993, 209(5), 547–556, DOI:10.1016/0009-2614(93)80131-8.
- R. Gutbrod, R. N. Schindler, E. Kraka and D. Cremer, Formation of OH Radicals in the Gas Phase Ozonolysis of Alkenes: The Unexpected Role of Carbonyl Oxides, Chem. Phys. Lett., 1996, 252(3), 221–229, DOI:10.1016/0009-2614(96)00126-1.
- R. Gutbrod, E. Kraka, R. N. Schindler and D. Cremer, Kinetic and Theoretical Investigation of the Gas-Phase Ozonolysis of Isoprene: Carbonyl Oxides as an Important Source for OH Radicals in the Atmosphere, J. Am. Chem. Soc., 1997, 119(31), 7330–7342, DOI:10.1021/ja970050c.
- O. Horie and G. K. Moortgat, Gas-Phase Ozonolysis of Alkenes. Recent Advances in Mechanistic Investigations, Acc. Chem. Res., 1998, 31(7), 387–396, DOI:10.1021/ar9702740.
- O. Horie and G. K. Moortgat, Decomposition Pathways of the Excited Criegee Intermediates in the Ozonolysis of Simple Alkenes, Atmos. Environ., 1991, 25(9), 1881–1896, DOI:10.1016/0960-1686(91)90271-8.
- F. Su, J. G. Calvert and J. H. A. Shaw, FTIR Spectroscopic Study of the Ozone-Ethene Reaction Mechanism in Oxygen-Rich Mixtures, J. Phys. Chem., 1980, 84(3), 239–246, DOI:10.1021/j100440a003.
- R. Atkinson, Gas-Phase Tropospheric Chemistry of Volatile Organic Compounds: 1. Alkanes and Alkenes, J. Phys. Chem. Ref. Data, 1997, 26(2), 215–290, DOI:10.1063/1.556012.
- M. Kumar, J. Zhong, J. S. Francisco and X. C. Zeng, Criegee Intermediate-Hydrogen Sulfide Chemistry at the Air/Water Interface, Chem. Sci., 2017, 8(8), 5385–5391, 10.1039/c7sc01797a.
- H. L. Huang, W. Chao and J. J. M. Lin, Kinetics of a Criegee Intermediate That Would Survive High Humidity and May Oxidize Atmospheric SO2, Proc. Natl. Acad. Sci. U.S.A., 2015, 112(35), 10857–10862, DOI:10.1073/pnas.1513149112.
- K. T. Kuwata, E. J. Guinn, M. R. Hermes, J. A. Fernandez, J. M. Mathison and K. Huang, A Computational Re-Examination of the Criegee Intermediate–Sulfur Dioxide Reaction, J. Phys. Chem. A, 2015, 119(41), 10316–10335, DOI:10.1021/acs.jpca.5b06565.
- D. B. Menzel, D. A. Keller and K.-H. Leung, Covalent Reactions in the Toxicity of SO2 and Sulfite, in Biological Reactive Intermediates III: Mechanisms of Action in Animal Models and Human Disease, Advances in Experimental Medicine and Biology, ed. J. J. Kocsis, D. J. Jollow, C. M. Witmer, J. O. Nelson and R. Snyder, Springer US, Boston, MA, 1986, pp. 477–492, DOI:10.1007/978-1-4684-5134-4_46.
- Z. Zeng, M. Altarawneh, I. Oluwoye, P. Glarborg and B. Z. Dlugogorski, Inhibition and Promotion of Pyrolysis by Hydrogen Sulfide (H2S) and Sulfanyl Radical (SH), J. Phys. Chem. A, 2016, 120(45), 8941–8948, DOI:10.1021/acs.jpca.6b09357.
- M. R. McGillen, B. F. E. Curchod, R. Chhantyal-Pun, J. M. Beames, N. Watson, M. A. H. Khan, L. McMahon, D. E. Shallcross and A. J. Orr-Ewing, Criegee Intermediate–Alcohol Reactions, A Potential Source of Functionalized Hydroperoxides in the Atmosphere, ACS Earth Space Chem., 2017, 1(10), 664–672, DOI:10.1021/acsearthspacechem.7b00108.
- H.-Y. Lin, Y.-H. Huang, X. Wang, J. M. Bowman, Y. Nishimura, H. A. Witek and Y.-P. Lee, Infrared Identification of the Criegee Intermediates - and Anti-CH3CHOO, and Their Distinct Conformation-Dependent Reactivity, Nat. Commun., 2015, 6, 7012, DOI:10.1038/ncomms8012.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, A. M. Scheer, D. E. Shallcross, B. Rotavera, E. P. F. Lee, J. M. Dyke, D. K. W. Mok, D. L. Osborn and C. J. Percival, Direct Measurements of Conformer-Dependent Reactivity of the Criegee Intermediate CH3CHOO, Science, 2013, 340(6129), 177–180, DOI:10.1126/science.1234689.
- Y.-H. Lin, K. Takahashi and J. J.-M. Lin, Reactivity of Criegee Intermediates toward Carbon Dioxide, J. Phys. Chem. Lett., 2018, 9(1), 184–188, DOI:10.1021/acs.jpclett.7b03154.
- W. Chao, Y. H. Lin, C. Yin, W. H. Lin, K. Takahashi and J. J. M. Lin, Temperature and Isotope Effects in the Reaction of CH3CHOO with Methanol, Phys. Chem. Chem. Phys., 2019, 21(25), 13633–13640, 10.1039/C9CP02534K.
- M. J. Newland, A. R. Rickard, L. Vereecken, A. Muñoz, M. Ródenas and W. J. Bloss, Atmospheric Isoprene Ozonolysis: Impacts of Stabilised Criegee Intermediate Reactions with SO2, H2O and Dimethyl Sulfide, Atmos. Chem. Phys., 2015, 15(16), 9521–9536, DOI:10.5194/acp-15-9521-2015.
- O. Welz, A. J. Eskola, L. Sheps, B. Rotavera, J. D. Savee, A. M. Scheer, D. L. Osborn, D. Lowe, A. Murray Booth, P. Xiao, H. Anwar, M. Khan, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Rate Coefficients of C1 and C2 Criegee Intermediate Reactions with Formic and Acetic Acid Near the Collision Limit: Direct Kinetics Measurements and Atmospheric Implications, Angew. Chem., Int. Ed., 2014, 53(18), 4547–4550, DOI:10.1002/anie.201400964.
- M. J. Newland, C. Mouchel-Vallon, R. Valorso, B. Aumont, L. Vereecken, M. E. Jenkin and A. R. Rickard, Estimation of Mechanistic Parameters in the Gas-Phase Reactions of Ozone with Alkenes for Use in Automated Mechanism Construction, Atmos. Chem. Phys., 2022, 22(9), 6167–6195, DOI:10.5194/acp-22-6167-2022.
- Y. Shi, Y. Xu and L. Jia, Arrhenius Parameters for the Gas-Phase Reactions of O3 with Two Butenes and Two Methyl-Substituted Butenes over the Temperature Range of 295-351 K, Int. J. Chem. Kinet., 2011, 43(5), 238–246, DOI:10.1002/kin.20553.
- E. V. Avzianova and P. A. Ariya, Temperature-Dependent Kinetic Study for Ozonolysis of Selected Tropospheric Alkenes, Int. J. Chem. Kinet., 2002, 34(12), 678–684, DOI:10.1002/kin.10093.
- R. Søndergaard, O. J. Nielsen, M. D. Hurley, T. J. Wallington and R. Singh, Atmospheric Chemistry of Trans-CF3CHCHF: Kinetics of the Gas-Phase Reactions with Cl Atoms, OH Radicals, and O3, Chem. Phys. Lett., 2007, 443(4), 199–204, DOI:10.1016/j.cplett.2007.06.084.
- M. P. S. Andersen, E. J. K. Nilsson, O. J. Nielsen, M. S. Johnson, M. D. Hurley and T. J. Wallington, Atmospheric Chemistry of Trans-CF3CHCHCl: Kinetics of the Gas-Phase Reactions with Cl Atoms, OH Radicals, and O3, J. Photochem. Photobiol., A, 2008, 199(1), 92–97, DOI:10.1016/j.jphotochem.2008.05.013.
- A. Soto, B. Ballesteros, E. Jiménez, M. Antiñolo, E. Martínez and J. Albaladejo, Kinetic and Mechanistic Study of the Gas-Phase Reaction of CxF2x+1CHCH2 (X = 1, 2, 3, 4 and 6) with O3 under Atmospheric Conditions, Chemosphere, 2018, 201, 318–327, DOI:10.1016/j.chemosphere.2018.02.183.
- M. P. S. Andersen, O. J. Nielsen, A. Toft, T. Nakayama, Y. Matsumi, R. L. Waterland, R. C. Buck, M. D. Hurley and T. J. Wallington, Atmospheric Chemistry of CxF2x+1CHCH2 (X = 1, 2, 4, 6, and 8): Kinetics of Gas-Phase Reactions with Cl Atoms, OH Radicals, and O3, J. Photochem. Photobiol., A, 2005, 176(1), 124–128, DOI:10.1016/j.jphotochem.2005.06.015.
- O. J. Nielsen, M. S. Javadi, M. P. Sulbaek Andersen, M. D. Hurley, T. J. Wallington and R. Singh, Atmospheric Chemistry of CF3CFCH2: Kinetics and Mechanisms of Gas-Phase Reactions with Cl Atoms, OH Radicals, and O3, Chem. Phys. Lett., 2007, 439(1), 18–22, DOI:10.1016/j.cplett.2007.03.053.
- K. Sato, B. Klotz, T. Taketsugu and T. Takayanagi, Kinetic Measurements for the Reactions of Ozone with Crotonaldehyde and Its Methyl Derivatives and Calculations of Transition-State Theory, Phys. Chem. Chem. Phys., 2004, 6(15), 3969–3976, 10.1039/B402496F.
- K. E. Leather, M. R. McGillen and C. J. Percival, Temperature-Dependent Ozonolysis Kinetics of Selected Alkenes in the Gas Phase: An Experimental and Structure–Activity Relationship (SAR) Study, Phys. Chem. Chem. Phys., 2010, 12(12), 2935–2943, 10.1039/B919731A.
- E. Grosjean and D. Grosjean, Rate Constants for the Gas-phase Reaction of C5-C10 Alkenes with Ozone, Int. J. Chem. Kinet., 1995, 27(11), 1045–1054, DOI:10.1002/kin.550271102.
- L. Jia, Y. Xu, M. Ge, L. Du, G. Wang and G. Zhuang, Kinetic Study of the Gas-Phase Ozonolysis of Propylene, Acta Phys.-Chim. Sin., 2006, 22(10), 1260–1266, DOI:10.1016/S1872-1508(06)60060-0.
- Y. Ren, B. Grosselin, V. Daële and A. Mellouki, Investigation of the Reaction of Ozone with Isoprene, Methacrolein and Methyl Vinyl Ketone Using the HELIOS Chamber, Faraday Discuss., 2017, 200, 289–311, 10.1039/c7fd00014f.
- E. Jiménez, S. González, M. Antiñolo and J. Albaladejo, Atmospheric Implications of the Emission of CF3CF2CHCH2 (HFC-1345fz) as a Consequence of Its Use as Foam Blowing Agents, Curr. Environ. Eng., 2014, 1, 118–125 CrossRef.
- P. K. Rao and S. P. Gejji, Molecular Insights for the HFO-1345fz +X (X = Cl, O3 or NO3) Reaction and Fate of Alkoxy Radicals Initiated by Cl: DFT Investigations, J. Fluorine Chem., 2017, 204, 65–75, DOI:10.1016/j.jfluchem.2017.08.015.
- L. Baptista, R. Pfeifer, E. C. da Silva and G. Arbilla, Kinetics and Thermodynamics of Limonene Ozonolysis, J. Phys. Chem. A, 2011, 115(40), 10911–10919, DOI:10.1021/jp205734h.
- M. H. Almatarneh, I. A. Elayan, M. Altarawneh and J. W. Hollett, A Computational Study of the Ozonolysis of Sabinene, Theor. Chem. Acc., 2019, 138(2), 30, DOI:10.1007/s00214-019-2420-7.
- M. H. Almatarneh, I. A. Elayan, R. A. Poirier and M. Altarawneh, The Ozonolysis of Cyclic Monoterpenes: A Computational Review, Can. J. Chem., 2017, 96(3), 281–292, DOI:10.1139/cjc-2017-0587.
- A. V. Mayorov, B. E. Krisyuk and N. Sokolova, Kinetics and Mechanisms of the First Step of Ozonolysis of Trans-Isoprene, Comput. Theor. Chem., 2020, 1186, 112904, DOI:10.1016/j.comptc.2020.112904.
- C. J. Percival, O. Welz, A. J. Eskola, J. D. Savee, D. L. Osborn, D. O. Topping, D. Lowe, S. R. Utembe, A. Bacak, G. McFiggans, M. C. Cooke, P. Xiao, A. T. Archibald, M. E. Jenkin, R. G. Derwent, I. Riipinen, D. W. K. Mok, E. P. F. Lee, J. M. Dyke, C. A. Taatjes and D. E. Shallcross, Regional and Global Impacts of Criegee Intermediates on Atmospheric Sulphuric Acid Concentrations and First Steps of Aerosol Formation, Faraday Discuss., 2013, 165, 45–73, 10.1039/c3fd00048f.
- V. P. Barber, S. Pandit, A. M. Green, N. Trongsiriwat, P. J. Walsh, S. J. Klippenstein and M. I. Lester, Four-Carbon Criegee Intermediate from Isoprene Ozonolysis: Methyl Vinyl Ketone Oxide Synthesis, Infrared Spectrum, and OH Production, J. Am. Chem. Soc., 2018, 140(34), 10866–10880, DOI:10.1021/jacs.8b06010.
- US EPA, Refrigerant Transition & Environmental Impacts, US EPA, https://www.epa.gov/mvac/refrigerant-transition-environmental-impacts, accessed 2019-01-09 Search PubMed.
- European Commission, Press Release – Refrigerants Used in Mobile Air Condition Systems (MAC) – State of Play, https://europa.eu/rapid/press-release_MEMO-14-50_en.htm, accessed 2019-01-09 Search PubMed.
- Z. Wang, Y. Wang, J. Li, S. Henne, B. Zhang, J. Hu and J. Zhang, Impacts of the Degradation of 2,3,3,3-Tetrafluoropropene into Trifluoroacetic Acid from Its Application in Automobile Air Conditioners in China, the United States, and Europe, Environ. Sci. Technol., 2018, 52(5), 2819–2826, DOI:10.1021/acs.est.7b05960.
- M. K. Vollmer, S. Reimann, M. Hill and D. Brunner, First Observations of the Fourth Generation Synthetic Halocarbons HFC-1234yf, HFC-1234ze(E), and HCFC-1233zd(E) in the Atmosphere, Environ. Sci. Technol., 2015, 49(5), 2703–2708, DOI:10.1021/es505123x.
- D. J. Luecken, R. L. Waterland, S. Papasavva, K. N. Taddonio, W. T. Hutzell, J. P. Rugh and S. O. Andersen, Ozone and TFA Impacts in North America from Degradation of 2,3,3,3-Tetrafluoropropene (HFO-1234yf), A Potential Greenhouse Gas Replacement, Environ. Sci. Technol., 2010, 44(1), 343–348, DOI:10.1021/es902481f.
- M. R. McGillen, Z. T. P. Fried, M. A. H. Khan, K. T. Kuwata, C. M. Martin, S. O'Doherty, F. Pecere, D. E. Shallcross, K. M. Stanley and K. Zhang, Ozonolysis Can Produce Long-Lived Greenhouse Gases from Commercial Refrigerants, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(51), e2312714120, DOI:10.1073/pnas.2312714120.
- M. P. Pérez-Peña, J. A. Fisher, C. Hansen and S. H. Kable, Assessing the Atmospheric Fate of Trifluoroacetaldehyde (CF3CHO) and Its Potential as a New Source of Fluoroform (HFC-23) Using the AtChem2 Box Model, Environ. Sci.: Atmos., 2023, 3(12), 1767–1777, 10.1039/D3EA00120B.
- J. S. Campbell, K. Nauta, S. H. Kable and C. S. Hansen, Photodissociation Dynamics of CF3CHO: C–C Bond Cleavage, J. Chem. Phys., 2021, 155(20), 204303, DOI:10.1063/5.0073974.
- J. L. Wang, C. Chew, C. Y. Chang, W. C. Liao, S. C. C. Lung, W. N. Chen, P. J. Lee, P. H. Lin and C. C. Chang, Biogenic Isoprene in Subtropical Urban Settings and Implications for Air Quality, Atmos. Environ., 2013, 79, 369–379, DOI:10.1016/j.atmosenv.2013.06.055.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37(2), 785–789, DOI:10.1103/PhysRevB.37.785.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, Electron Affinities of the First-row Atoms Revisited. Systematic Basis Sets and Wave Functions, J. Chem. Phys., 1992, 96(9), 6796–6806, DOI:10.1063/1.462569.
- A. D. Becke, Density-functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98(7), 5648–5652, DOI:10.1063/1.464913.
- T. H. Dunning, Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen, J. Chem. Phys., 1989, 90(2), 1007–1023, DOI:10.1063/1.456153.
- C. Gonzalez and H. B. Schlegel, Reaction Path Following in Mass-Weighted Internal Coordinates, J. Phys. Chem., 1990, 94(14), 5523–5527, DOI:10.1021/j100377a021.
- K. Ishida, K. Morokuma and A. Komornicki, The Intrinsic Reaction Coordinate. An ab initio Calculation for HNC → HCN and H− + CH4 → CH4 + H−, J. Chem. Phys., 1977, 66(5), 2153–2156, DOI:10.1063/1.434152.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc, Wallingford CT, 2016 Search PubMed.
- T. B. Adler and H. J. Werner, An Explicitly Correlated Local Coupled Cluster Method for Calculations of Large Molecules Close to the Basis Set Limit, J. Chem. Phys., 2011, 135(14), 144117, DOI:10.1063/1.3647565.
- R. Chhantyal-Pun, M. R. McGillen, J. M. Beames, M. A. H. Khan, C. J. Percival, D. E. Shallcross and A. J. Orr-Ewing, Temperature-Dependence of the Rates of Reaction of Trifluoroacetic Acid with Criegee Intermediates, Angew. Chem., Int. Ed., 2017, 56(31), 9044–9047, DOI:10.1002/anie.201703700.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Molpro: A General-Purpose Quantum Chemistry Program Package: Molpro, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2(2), 242–253, DOI:10.1002/wcms.82.
- D. R. Glowacki, C. H. Liang, C. Morley, M. J. Pilling and S. H. Robertson, MESMER: An Open-Source Master Equation Solver for Multi-Energy Well Reactions, J. Phys. Chem. A, 2012, 116(38), 9545–9560, DOI:10.1021/jp3051033.
- S. Canneaux, F. Bohr and E. Henon, KiSThelP: A Program to Predict Thermodynamic Properties and Rate Constants from Quantum Chemistry Results, J. Comput. Chem., 2014, 35(1), 82–93, DOI:10.1002/jcc.23470.
- J. M. Gomez and J. M. C. Plane, Reaction Kinetics of CaOH with H and O2 and O2CaOH with O: Implications for the Atmospheric Chemistry of Meteoric Calcium, ACS Earth Space Chem., 2017, 1(7), 431–441, DOI:10.1021/acsearthspacechem.7b00072.
- R. Chhantyal-Pun, R. J. Shannon, D. P. Tew, R. L. Caravan, M. Duchi, C. Wong, A. Ingham, C. Feldman, M. R. McGillen, M. A. H. Khan, I. O. Antonov, B. Rotavera, K. Ramasesha, D. L. Osborn, C. A. Taatjes, C. J. Percival, D. E. Shallcross and A. J. Orr-Ewing, Experimental and Computational Studies of Criegee Intermediate Reactions with NH3 and CH3NH2, Phys. Chem. Chem. Phys., 2019, 21, 14042–14052, 10.1039/C8CP06810K.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, Assessment of Gaussian-3 and Density-Functional Theories on the G3/05 Test Set of Experimental Energies, J. Chem. Phys., 2005, 123(12), 124107, DOI:10.1063/1.2039080.
- Y. Zhao, O. Tishchenko, J. R. Gour, W. Li, J. J. Lutz, P. Piecuch and D. G. Truhlar, Thermochemical Kinetics for Multireference Systems: Addition Reactions of Ozone, J. Phys. Chem. A, 2009, 113(19), 5786–5799, DOI:10.1021/jp811054n.
- S. E. Wheeler, D. H. Ess and K. N. Houk, Thinking Out of the Black Box: Accurate Barrier Heights of 1,3-Dipolar Cycloadditions of Ozone with Acetylene and Ethylene, J. Phys. Chem. A, 2008, 112(8), 1798–1807, DOI:10.1021/jp710104d.
- O. B. Gadzhiev, S. K. Ignatov, B. E. Krisyuk, A. V. Maiorov, S. Gangopadhyay and A. E. Masunov, Quantum Chemical Study of the Initial Step of Ozone Addition to the Double Bond of Ethylene, J. Phys. Chem. A, 2012, 116(42), 10420–10434, DOI:10.1021/jp307738p.
- N. A. I. Watson and J. M. Beames, Bimolecular Sinks of Criegee Intermediates Derived from Hydrofluoroolefins – a Computational Analysis, Environ. Sci.: Atmos., 2023, 3(10), 1460–1484, 10.1039/D3EA00102D.
- S. A. Epstein and N. M. Donahue, The Kinetics of Tetramethylethene Ozonolysis: Decomposition of the Primary Ozonide and Subsequent Product Formation in the Condensed Phase, J. Phys. Chem. A, 2008, 112(51), 13535–13541, DOI:10.1021/jp807682y.
- T. L. Nguyen, H. Lee, D. A. Matthews, M. C. McCarthy and J. F. Stanton, Stabilization of the Simplest Criegee Intermediate from the Reaction between Ozone and Ethylene: A High-Level Quantum Chemical and Kinetic Analysis of Ozonolysis, J. Phys. Chem. A, 2015, 119(22), 5524–5533, DOI:10.1021/acs.jpca.5b02088.
- M. Pfeifle, Y. T. Ma, A. W. Jasper, L. B. Harding, W. L. Hase and S. J. Klippenstein, Nascent Energy Distribution of the Criegee Intermediate CH2OO from Direct Dynamics Calculations of Primary Ozonide Dissociation, J. Chem. Phys., 2018, 148(17), 174306, DOI:10.1063/1.5028117.
- M. Olzmann, E. Kraka, D. Cremer, R. Gutbrod and S. Andersson, Energetics, Kinetics, and Product Distributions of the Reactions of Ozone with Ethene and 2,3-Dimethyl-2-Butene, J. Phys. Chem. A, 1997, 101(49), 9421–9429, DOI:10.1021/jp971663e.
- R. A. Cox and S. A. Penkett, Aerosol Formation from Sulphur Dioxide in the Presence of Ozone and Olefinic Hydrocarbons, J. Chem. Soc., Faraday Trans. 1, 1972, 68, 1735–1753, 10.1039/F19726801735.
- D. H. Stedman, C. H. Wu and H. Niki, Kinetics of Gas-Phase Reactions of Ozone with Some Olefins, ACS Publications, 1973, DOI:10.1021/j100907a004.
- J. T. Herron and R. E. Huie, Rate Constants for the Reactions of Ozone with Ethene and Propene, from 235.0 to 362.0 K, J. Phys. Chem., 1974, 78(21), 2085–2088, DOI:10.1021/j100614a004.
- S. M. Japar, C. H. Wu and H. Nikl, Rate Constants for the Reaction of Ozone with Olefins In the Gas Phase, J. Phys. Chem., 1974, 78(23), 2318–2320, DOI:10.1021/j150671a003.
- S. M. Japar, C. H. Wu and H. Niki, Effect of Molecular Oxygen on the Gas Phase Kinetics of the Ozonolysis of Olefins, J. Phys. Chem., 1976, 80(19), 2057–2062, DOI:10.1021/j100560a002.
- S. A. Adeniji, J. A. Kerr and M. R. Williams, Rate Constants for Ozone–Alkene Reactions under Atmospheric Conditions, Int. J. Chem. Kinet., 1981, 13(2), 209–217, DOI:10.1002/kin.550130210.
- R. Atkinson, S. M. Aschmann, D. R. Fitz, A. M. Winer and J. N. Pitts Jr, Rate Constants for the Gas-Phase Reactions of O3 with Selected Organics at 296 K, Int. J. Chem. Kinet., 1982, 14(1), 13–18, DOI:10.1002/kin.550140103.
- J. Treacy, M. E. Hag, D. O'Farrell and H. Sidebottom, Reactions of Ozone with Unsaturated Organic Compounds, Ber. Bunsenges. Phys. Chem., 1992, 96(3), 422–427, DOI:10.1002/bbpc.19920960337.
- P. Neeb and G. K. Moortgat, Formation of OH Radicals in the Gas-Phase Reaction of Propene, Isobutene, and Isoprene with O3: Yields and Mechanistic Implications, J. Phys. Chem. A, 1999, 103, 9003–9012, DOI:10.1021/jp9903458.
- R. Wegener, T. Brauers, R. Koppmann, S. R. Bares, F. Rohrer, R. Tillmann, A. Wahner, A. Hansel and A. Wisthaler, Simulation Chamber Investigation of the Reactions of Ozone with Short-Chained Alkenes, J. Geophys. Res., 2007, 112(D13) DOI:10.1029/2006JD007531.
- C. Robinson, L. Onel, J. Newman, R. Lade, K. Au, L. Sheps, D. E. Heard, P. W. Seakins, M. A. Blitz and D. Stone, Unimolecular Kinetics of Stabilized CH3CHOO Criegee Intermediates: Syn-CH3CHOO Decomposition and Anti-CH3CHOO Isomerization, J. Phys. Chem. A, 2022, 126(39), 6984–6994, DOI:10.1021/acs.jpca.2c05461.
- J. Hakala and N. M. Donahue, Carbonyl Oxide Stabilization from Trans Alkene and Terpene Ozonolysis, J. Phys. Chem. A, 2023, 127(41), 8530–8543, DOI:10.1021/acs.jpca.3c03650.
- A. R. Rickard, D. Johnson, C. D. McGill and G. Marston, OH Yields in the Gas-Phase Reactions of Ozone with Alkenes, J. Phys. Chem. A, 1999, 103(38), 7656–7664, DOI:10.1021/jp9916992.
- E. C. Tuazon, S. M. Aschmann, J. Arey and R. Atkinson, Products of the Gas-Phase Reactions of O3 with a Series of Methyl-Substituted Ethenes, Environ. Sci. Technol., 1997, 31(10), 3004–3009, DOI:10.1021/es970258y.
- B. Qi, B. Yang, Z. Wang, H. Yang and L. Liu, Production of Radicals in the Ozonolysis of Propene in Air, Sci. China, Ser. B:Chem., 2009, 52(3), 356–361, DOI:10.1007/s11426-008-0132-2.
- E. Grosjean and D. Grosjean, Gas Phase Reaction of Alkenes with Ozone: Formation Yields of Primary Carbonyls and Biradicals, Environ. Sci. Technol., 1997, 31(8), 2421–2427, DOI:10.1021/es970075b.
- O. B. Gadzhiev, S. K. Ignatov, B. E. Krisyuk, A. V. Maiorov, S. Gangopadhyay and A. E. Masunov, Quantum Chemical Study of the Initial Step of Ozone Addition to the Double Bond of Ethylene, J. Phys. Chem. A, 2012, 116(42), 10420–10434, DOI:10.1021/jp307738p.
- W. T. Chan and I. P. Hamilton, Mechanisms for the Ozonolysis of Ethene and Propene: Reliability of Quantum Chemical Predictions, J. Chem. Phys., 2003, 118(4), 1688–1701, DOI:10.1063/1.1531104.
- A. V. Maiorov, B. E. Krisyuk and A. A. Popov, The Reaction of Ozone with Hexafluoropropylene: Competition of Concerted and Nonconcerted Addition, Russ. J. Phys. Chem. B, 2008, 2(5), 707–710, DOI:10.1134/S1990793108050084.
- M. Kumar, J. Shee, B. Rudshteyn, D. R. Reichman, R. A. Friesner, C. E. Miller and J. S. Francisco, Multiple Stable Isoprene–Ozone Complexes Reveal Complex Entrance Channel Dynamics in the Isoprene + Ozone Reaction, J. Am. Chem. Soc., 2020, 142(24), 10806–10813, DOI:10.1021/jacs.0c02360.
- M. Pfeifle, Y. T. Ma, A. W. Jasper, L. B. Harding, W. L. Hase and S. J. Klippenstein, Nascent Energy Distribution of the Criegee Intermediate CH2OO from Direct Dynamics Calculations of Primary Ozonide Dissociation, J. Chem. Phys., 2018, 148(17), 174306, DOI:10.1063/1.5028117.
- J. M. Anglada, R. Crehuet and B. J. Maria, The Ozonolysis of Ethylene: A Theoretical Study of the Gas-Phase Reaction Mechanism, Chem.–Eur. J., 1999, 5(6), 1809–1822, DOI:10.1002/(SICI)1521-3765(19990604)5:6<1809::AID-CHEM1809>3.0.CO;2-N.
- Y. Li, H. Liu, X. Huang, Z. Li, Y. Sun and C. Sun, Theoretical Study for Ozonolysis of 1,3-Butadiene, J. Mol. Struct.:THEOCHEM, 2010, 945(1), 120–128, DOI:10.1016/j.theochem.2010.01.021.
- R. A. Cox, M. Ammann, J. N. Crowley, H. Herrmann, M. E. Jenkin, V. F. McNeill, A. Mellouki, J. Troe and T. J. Wallington, Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Volume VII – Criegee Intermediates, Atmos. Chem. Phys., 2020, 20(21), 13497–13519, DOI:10.5194/acp-20-13497-2020.
- R. M. Kamens, M. W. Gery, H. E. Jeffries, M. Jackson and E. I. Cole, Ozone–Isoprene Reactions: Product Formation and Aerosol Potential, Int. J. Chem. Kinet., 1982, 14(9), 955–975, DOI:10.1002/kin.550140902.
- P. Neeb, A. Kolloff, S. Koch and G. K. Moortgat, Rate Constants for the Reactions of Methylvinyl Ketone, Methacrolein, Methacrylic Acid, and Acrylic Acid with Ozone, Int. J. Chem. Kinet., 1998, 30(10), 769–776, DOI:10.1002/(SICI)1097-4601(1998)30:10<769::AID-KIN9>3.0.CO;2-T.
- E. Grosjean and D. Grosjean, Rate Constants for the Gas-Phase Reaction of Ozone with Unsaturated Oxygenates, Int. J. Chem. Kinet., 1998, 30(1), 21–29, DOI:10.1002/(SICI)1097-4601(1998)30:1<21::AID-KIN3>3.0.CO;2-W.
- D. Grosjean, E. L. I. Williams and E. Grosjean, Atmospheric Chemistry of Isoprene and of Its Carbonyl Products, Environ. Sci. Technol., 1993, 27(5), 830–840, DOI:10.1021/es00042a004.
- S. E. Paulson, M. Y. Chung and A. S. Hasson, OH Radical Formation from the Gas-Phase Reaction of Ozone with Terminal Alkenes and the Relationship between Structure and Mechanism, J. Phys. Chem. A, 1999, 103(41), 8125–8138, DOI:10.1021/jp991995e.
- R. Atkinson and S. M. Aschmann, Hydroxyl Radical Production from the Gas-Phase Reactions of Ozone with a Series of Alkenes under Atmospheric Conditions, Environ. Sci. Technol., 1993, 27(7), 1357–1363, DOI:10.1021/es00044a010.
- S. E. Paulson, M. Chung, A. D. Sen and G. Orzechowska, Measurement of OH Radical Formation from the Reaction of Ozone with Several Biogenic Alkenes, J. Geophys. Res.:Atmos., 1998, 103(D19), 25533–25539, DOI:10.1029/98JD01951.
- G. E. Orzechowska and S. E. Paulson, Production of OH Radicals from the Reactions of C4–C6 Internal Alkenes and Styrenes with Ozone in the Gas Phase, Atmos. Environ., 2002, 36(3), 571–581, DOI:10.1016/S1352-2310(01)00445-9.
- C. D. McGill, A. R. Rickard, D. Johnson and G. Marston, Product Yields in the Reactions of Ozone with Z-but-2-ene, E-but-2-ene and 2-methylbut-2-ene, Chemosphere, 1999, 38(6), 1205–1212, DOI:10.1016/S0045-6535(98)00512-8.
- E. Grosjean, J. B. de Andrade and D. Grosjean, Carbonyl Products of the Gas-Phase Reaction of Ozone with Simple Alkenes, Environ. Sci. Technol., 1996, 30(3), 975–983, DOI:10.1021/es950442o.
- B. S. Nelson, Understanding the Chemical Processes Leading to in Situ Ozone Production in Delhi, India. phd, University of York, 2021, https://etheses.whiterose.ac.uk/30545/, accessed 2024-04-12 Search PubMed.
- N. Peter, H. Osamu and G. K. Moortgat, Gas-phase Ozonolysis of Ethene in the Presence of Hydroxylic Compounds, Int. J. Chem. Kinet., 1999, 28(10), 721–730, DOI:10.1002/(SICI)1097-4601(1996)28:10<721::AID-KIN2>3.0.CO;2-P.
- M. P. S. Andersen, T. I. Sølling, L. L. Andersen, A. Volkova, D. Hovanessian, C. Britzman, O. J. Nielsen and T. J. Wallington, Atmospheric Chemistry of (Z)-CF3CHCHCl: Products and Mechanisms of the Cl Atom, OH Radical and O3 Reactions, and Role of (E)–(Z) Isomerization, Phys. Chem. Chem. Phys., 2018, 20(44), 27949–27958, 10.1039/C8CP04903C.
- N. M. Donahue, J. H. Kroll, J. G. Anderson and K. L. Demerjian, Direct Observation of OH Production from the Ozonolysis of Olefins, Geophys. Res. Lett., 1998, 25(1), 59–62, DOI:10.1029/97GL53560.
- O. Horie, P. Neeb and G. K. Moortgat, The Reactions of the Criegee Intermediate CH3CHOO in the Gas-Phase Ozonolysis of 2-butene Isomers, Int. J. Chem. Kinet., 1997, 29(6), 461–468, DOI:10.1002/(SICI)1097-4601(1997)29:6<461::AID-KIN8>3.0.CO;2-S.
- L. Vereecken, B. Aumont, I. Barnes, J. W. Bozzelli, M. J. Goldman, W. H. Green, S. Madronich, M. R. Mcgillen, A. Mellouki, J. J. Orlando, B. Picquet-Varrault, A. R. Rickard, W. R. Stockwell, T. J. Wallington and W. P. L. Carter, Perspective on Mechanism Development and Structure-Activity Relationships for Gas-Phase Atmospheric Chemistry, Int. J. Chem. Kinet., 2018, 50(6), 435–469, DOI:10.1002/kin.21172.
- S. E. Paulson and J. H. Seinfeld, Development and Evaluation of a Photooxidation Mechanism for Isoprene, J. Geophys. Res.:Atmos., 1992, 97(D18), 20703–20715, DOI:10.1029/92JD01914.
- A. A. Chew and R. Atkinson, OH Radical Formation Yields from the Gas-Phase Reactions of O3 with Alkenes and Monoterpenes, J. Geophys. Res.:Atmos., 1996, 101(D22), 28649–28653, DOI:10.1029/96JD02722.
- W. C. D. Rathman, T. A. Claxton, A. R. Rickard and G. Marston, A Theoretical Investigation of OH Formation in the Gas-Phase Ozonolysis of E-but-2-ene and Z-but-2-ene, Phys. Chem. Chem. Phys., 1999, 1(17), 3981–3985, 10.1039/A903186C.
- IUPAC: Subcommittee for Gas Kinetic Data Evaluation, https://www.iupac-kinetic.ch.cam.ac.uk Search PubMed.
- M. S. Alam, A. R. Rickard, M. Camredon, K. P. Wyche, T. Carr, K. E. Hornsby, P. S. Monks and W. J. Bloss, Radical Product Yields from the Ozonolysis of Short Chain Alkenes under Atmospheric Boundary Layer Conditions, J. Phys. Chem. A, 2013, 117(47), 12468–12483, DOI:10.1021/jp408745h.
- T. Braure, V. Riffault, A. Tomas, R. I. Olariu, C. Arsene, Y. Bedjanian and P. Coddeville, Ozonolysis of a Series of Methylated Alkenes: Reaction Rate Coefficients and Gas-Phase Products, Int. J. Chem. Kinet., 2015, 47(9), 596–605, DOI:10.1002/kin.20934.
- M. J. Molina and F. S. Rowland, Stratospheric Sink for Chlorofluoromethanes: Chlorine Atom-Catalysed Destruction of Ozone, Nature, 1974, 249(5460), 810–812, DOI:10.1038/249810a0.
- P. K. Rao and S. P. Gejji, Atmospheric Degradation of HCFO-1233zd(E) Initiated by OH Radical, Cl Atom and O3 Molecule: Kinetics, Reaction Mechanisms and Implications, J. Fluorine Chem., 2018, 211, 180–193, DOI:10.1016/j.jfluchem.2018.05.001.
- B. Zhang, Z. Zhai and J. Zhang, Distribution of Trifluoroacetic Acid in Gas and Particulate Phases in Beijing from 2013 to 2016, Sci. Total Environ., 2018, 634, 471–477, DOI:10.1016/j.scitotenv.2018.03.384.
- M. Kumar and J. S. Francisco, Reactions of Criegee Intermediates with Non-Water Greenhouse Gases: Implications for Metal Free Chemical Fixation of Carbon Dioxide, J. Phys. Chem. Lett., 2017, 8(17), 4206–4213, DOI:10.1021/acs.jpclett.7b01762.
- M. Kumar and J. S. Francisco, H–X (X = H, CH3, CH2F, CHF2, CF3, and SiH3) Bond Activation by Criegee Intermediates: A Theoretical Perspective, J. Phys. Chem. A, 2017, 121(49), 9421–9428, DOI:10.1021/acs.jpca.7b10535.
- K. Xu, W. Wang, W. Wei, W. Feng, Q. Sun and P. Li, Insights into the Reaction Mechanism of Criegee Intermediate CH2OO with Methane and Implications for the Formation of Methanol, J. Phys. Chem. A, 2017, 121(38), 7236–7245, DOI:10.1021/acs.jpca.7b05858.
- L. Vereecken, H. Harder and A. Novelli, The Reactions of Criegee Intermediates with Alkenes, Ozone, and Carbonyl Oxides, Phys. Chem. Chem. Phys., 2014, 16(9), 4039, 10.1039/c3cp54514h.
- M. S. Long, R. Yantosca, J. E. Nielsen, C. A. Keller, A. da Silva, M. P. Sulprizio, S. Pawson and D. J. Jacob, Development of a Grid-Independent GEOS-Chem Chemical Transport Model (v9-02) as an Atmospheric Chemistry Module for Earth System Models, Geosci. Model Dev., 2015, 8(3), 595–602, DOI:10.5194/gmd-8-595-2015.
- M. A. H. Khan, M. C. Cooke, S. R. Utembe, A. T. Archibald, P. Maxwell, W. C. Morris, P. Xiao, R. G. Derwent, M. E. Jenkin, C. J. Percival, R. C. Walsh, T. D. S. Young, P. G. Simmonds, G. Nickless, S. O'Doherty and D. E. Shallcross, A Study of Global Atmospheric Budget and Distribution of Acetone Using Global Atmospheric Model STOCHEM-CRI, Atmos. Environ., 2015, 112, 269–277, DOI:10.1016/j.atmosenv.2015.04.056.
- R. Sommariva, S. Cox, C. Martin, K. Borońska, J. Young, P. K. Jimack, M. J. Pilling, V. N. Matthaios, B. S. Nelson, M. J. Newland, M. Panagi, W. J. Bloss, P. S. Monks and A. R. Rickard, AtChem (Version 1), an Open-Source Box Model for the Master Chemical Mechanism, Geosci. Model Dev., 2020, 13(1), 169–183, DOI:10.5194/gmd-13-169-2020.
- A. Totterdill, T. Kovács, W. Feng, S. Dhomse, C. J. Smith, J. C. Gómez-Martín, M. P. Chipperfield, P. M. Forster and J. M. C. Plane, Atmospheric Lifetimes, Infrared Absorption Spectra, Radiative Forcings and Global Warming Potentials of NF3, CF3CF2Cl (CFC-115), Atmos. Chem. Phys., 2016, 16(17), 11451–11463, DOI:10.5194/acp-16-11451-2016.
- M. P. Sulbaek Andersen, J. A. Schmidt, A. Volkova and D. J. Wuebbles, A Three-Dimensional Model of the Atmospheric Chemistry of E and Z-CF3CHCHCl (HCFO-1233(Zd) (E/Z)), Atmos. Environ., 2018, 179, 250–259, DOI:10.1016/j.atmosenv.2018.02.018.
- V. C. Papadimitriou, K. G. Kambanis, Y. G. Lazarou and P. Papagiannakopoulos, Kinetic Study for the Reactions of Several Hydrofluoroethers with Chlorine Atoms, J. Phys. Chem. A, 2004, 108(14), 2666–2674, DOI:10.1021/jp031081z.
- Q. Liang, M. P. Chipperfield, E. L. Fleming, N. L. Abraham, P. Braesicke, J. B. Burkholder, J. S. Daniel, S. Dhomse, P. J. Fraser, S. C. Hardiman, C. H. Jackman, D. E. Kinnison, P. B. Krummel, S. A. Montzka, O. Morgenstern, A. McCulloch, J. Mühle, P. A. Newman, V. L. Orkin, G. Pitari, R. G. Prinn, M. Rigby, E. Rozanov, A. Stenke, F. Tummon, G. J. M. Velders, D. Visioni and R. F. Weiss, Deriving Global OH Abundance and Atmospheric Lifetimes for Long-Lived Gases: A Search for CH3CCl3 Alternatives, J. Geophys. Res.:Atmos., 2017, 122(21), 11914–11933, DOI:10.1002/2017JD026926.
- M. J. Prather and X. Zhu, Lifetimes and Timescales of Tropospheric Ozone, Elem. Sci. Anth., 2024, 12(1), 00112, DOI:10.1525/elementa.2023.00112.
- D. S. Stevenson, A. Zhao, V. Naik, F. M. O'Connor, S. Tilmes, G. Zeng, L. T. Murray, W. J. Collins, P. T. Griffiths, S. Shim, L. W. Horowitz, L. T. Sentman and L. Emmons, Trends in Global Tropospheric Hydroxyl Radical and Methane Lifetime since 1850 from AerChemMIP, Atmos. Chem. Phys., 2020, 20(21), 12905–12920, DOI:10.5194/acp-20-12905-2020.
- T. L. Malkin, A. Goddard, D. E. Heard and P. W. Seakins, Measurements of OH and HO2 Yields from the Gas Phase Ozonolysis of Isoprene, Atmos. Chem. Phys., 2010, 10(3), 1441–1459, DOI:10.5194/acp-10-1441-2010.
- N. Nishino, J. Arey and R. Atkinson, Rate Constants for the Gas-Phase Reactions of OH Radicals with a Series of C6-C14 Alkenes at 299 +/-2 K, J. Phys. Chem. A, 2009, 113(5), 852–857, DOI:10.1021/jp809305w.
- H. Fuchs, S. Albrecht, I.-H. Acir, B. Bohn, M. Breitenlechner, H.-P. Dorn, G. I. Gkatzelis, A. Hofzumahaus, F. Holland, M. Kaminski, F. N. Keutsch, A. Novelli, D. Reimer, F. Rohrer, R. Tillmann, L. Vereecken, R. Wegener, A. Zaytsev, A. Kiendler-Scharr and A. Wahner, Investigation of the Oxidation of Methyl Vinyl Ketone (MVK) by OH Radicals in the Atmospheric Simulation Chamber SAPHIR, Atmos. Chem. Phys., 2018, 18(11), 8001–8016, DOI:10.5194/acp-18-8001-2018.
- X. Zhu, J. T. Chen and C. W. Zhou, Exploring the Kinetics and Thermochemistry Effects on C2-C6 Alkene Combustion Chemistry by OH Radical; Implications for Combustion Modeling and Simulation, Combust. Flame, 2022, 245, 112302, DOI:10.1016/j.combustflame.2022.112302.
- Q. D. Wang, M. M. Sun and J. H. Liang, Reaction Mechanisms and Kinetics of the Hydrogen Abstraction Reactions of C4–C6 Alkenes with Hydroxyl Radical: A Theoretical Exploration, Int. J. Mol. Sci., 2019, 20(6), 1275, DOI:10.3390/ijms20061275.
- UPAC Subcommittee, IUPAC – Task Group on Atmospheric Chemical Kinetic Data Evaluation - Data Sheet Ox_VOC10 - O3 + methyl vinyl ketone, https://iupac-aeris.ipsl.fr/datasheets/pdf/Ox_VOC10.pdf, accessed 2024-06-22 Search PubMed.
- M. Duncianu, R. I. Olariu, V. Riffault, N. Visez, A. Tomas and P. Coddeville, Development of a New Flow Reactor for Kinetic Studies. Application to the Ozonolysis of a Series of Alkenes, J. Phys. Chem. A, 2012, 116(24), 6169–6179, DOI:10.1021/jp211480x.
- L. Vereecken, H. Harder and A. Novelli, The Reaction of Criegee Intermediates with NO, RO2, and SO2, and Their Fate in the Atmosphere, Phys. Chem. Chem. Phys., 2012, 14(42), 14682–14695, 10.1039/C2CP42300F.
- A. S. Hasson, M. Y. Chung, K. T. Kuwata, A. D. Converse, D. Krohn and S. E. Paulson, Reaction of Criegee Intermediates with Water Vapor: An Additional Source of OH Radicals in Alkene Ozonolysis?, J. Phys. Chem. A, 2003, 107(32), 6176–6182, DOI:10.1021/jp0346007.
- J. M. Anglada and A. Solé, Impact of the Water Dimer on the Atmospheric Reactivity of Carbonyl Oxides, Phys. Chem. Chem. Phys., 2016, 18(26), 17698–17712, 10.1039/C6CP02531E.
- A. Vela, The EU confirms itself the global climate leader on refrigerants, NGOs say, https://eeb.org/the-eu-confirms-itself-the-global-climate-leader-on-refrigerants/, accessed 2024-04-21 Search PubMed.
- Study Links Three HFOs to Super-Pollutant R23 in Ozone Reaction, https://naturalrefrigerants.com/study-links-three-hfos-to-super-pollutant-r23-via-ozone-reaction-in-atmosphere/, accessed 2024-04-21 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A full breakdown of the master equation rate constants (kTHEO) and canonical rate constants (kCAN) through temperature range of 200 K < T < 500 K at p = 760 torr for alkenes 1–19 reactions with O3; calculated product yields for alkenes 1–19 reactions with O3; calculated values for O3 + alkene rate constants at selected temperatures and pressures for direct comparison with experiment; literature tropospheric abundances for ozone and alkenes 1–19; relative energies [kJ mol−1] of stationary point for O3 + alkene reactions; assessment of the impact of alkene interconversion on ozonolysis chemistry; assessment of the impact of POZ interconversion on ozonolysis chemistry; assessment of the impact of CI interconversion on ozonolysis chemistry; assessment on the role of the epoxidation in alkene ozonolysis; calculated values for the effective rate constants for O3 + alkene reactions; Cartesian coordinates and vibrational frequencies of all stationary points, and IRCs of all transition states in this study; an example MEMSER input file for the O3 + alkene 1 reaction (PDF). See DOI: https://doi.org/10.1039/d4va00298a |
| This journal is © The Royal Society of Chemistry 2025 |