
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Double layer capacitance as a sensitive metric to monitor the formation of solid electrolyte interphases in Li–ion batteries†
Maximilian
Schalenbach
 *a,
Baolin
Wu
a,
Chih-Long
Tsai
a,
Anna
Windmüller
a,
Luc
Raijmakers
a,
Shicheng
Yu
a,
Hermann
Tempel
a and
Rüdiger-A.
Eichel
ab
*a,
Baolin
Wu
a,
Chih-Long
Tsai
a,
Anna
Windmüller
a,
Luc
Raijmakers
a,
Shicheng
Yu
a,
Hermann
Tempel
a and
Rüdiger-A.
Eichel
ab
aInstitute of Energy Technologies (IET-1), Fundamental Electrochemistry, Forschungszentrum Jülich, Wilhelm-Johnen-Straße, 52425 Jülich, Germany. E-mail: m.schalenbach@fz-juelich.de
bInstitute of Physical Chemistry, RWTH Aachen University, 52062 Aachen, Germany
First published on 31st October 2024
Abstract
In Li–ion batteries with conventional liquid electrolytes, the formation of solid electrolyte interphases (SEIs) at carbonaceous anodes prevents continuous electrochemical decomposition of the electrolyte. Typically, SEI formation and electrolyte decomposition are examined with linear potential scans, where time and potential dependencies are intertwined. Herein, a stepwise potential variation in combination with amperometry and electrochemical impedance spectroscopy (EIS) is used to characterize the impacts of time and potential as individual degrees of freedom on the SEI formation. Based on EIS data, the double layer capacitance (DLC) is introduced as a sensitive in situ metric to monitor the SEI formation. This technique is used to show the similarities and differences in the SEI formation processes with typical Li–ion battery electrolytes consisting of hexafluorophosphate and carbonate solvents. A polished glassy carbon electrode is employed to provide model-like EIS data with reliable interpretation. Changes in the electrochemical interface within only few atomic layers are tracked with DLC, indicating that SEIs are formed below 1.9 V vs. Li/Li+ with the employed electrolytes. Amperometry measurements show that the decomposition of the employed electrolytes starts at approximately 2.7 V vs. Li/Li+, displaying smaller electrochemical windows than those previously reported.
Introduction
The electrochemical window describes the potential range within which an electrolyte is electrochemically stable. Towards lower potentials, the reduction of organic molecules and ions can form solid electrolyte interphases (SEIs), which reduce the rate of further electrochemical decomposition of the electrolyte.1–3 By widening the effective potential window of the electrolyte, SEIs are a decisive factor that enabled commercially successful Li–ion4,5 and Na–ion6–10 battery chemistries. Methods from computational chemistry can be used to estimate electrochemical windows.11–15 However, these methods come with several uncertainties16–18 as predicting the catalytic properties of the electrode towards electrolyte decomposition remains challenging.19–21 Experimentally, electrochemical windows and SEI formation are typically examined with linear potential scans,22–25 leading to ambiguous results and unclear distinctions between time and potential-dependent processes of SEI formations and ongoing electrolyte decompositions.26,27SEIs suppress electrolyte decomposition28–30 by blocking the access of solvent molecules and anions to electrodes’ surfaces.31–33 By being cation-conductive, SEIs allow cations to intercalate into carbonaceous electrodes.34–39 These unique selective transport properties are a key component in the success of Li–ion batteries,4,5 in which electrodes operate at potentials outside the electrochemical windows of common organic solvents.40 Modern cryo-electron tomography enabled to image SEIs,41–46 showing that amorphous parts complicate their chemical and structural characterization. Although Li–ion batteries were commercialized over 30 years ago,4,5 the properties, formation mechanisms, and structures of SEIs remain an area of active and prosperous research.47–49 At the time this article was written, more than ten thousand publications were associated in Clarivate's “Web of Science” with the search term “solid electrolyte interphase”. Similar to carbonaceous anodes, the SEI is a crucial component of batteries with metal anodes.50
Picturing the initial formation of the first few atomic layers of the SEI is challenging as most microscopic techniques cannot resolve such length scales. However, in this spatial regime, ions are adsorbed in the inner Helmholtz-layer of the double layer at the electrode.51–53 The effect of the SEI on the ion distribution in the double layer is expected as being measurable by the capacitance obtained with electrochemical impedance spectroscopy (EIS).54 EIS analyses of standard composite Li–ion battery anodes are limited due to their structural complexities that arise from polymer bound graphite particles,55–57 and the influence of the active materials distribution on the electrochemical intercalation and SEI formation.58 In detail, resistive-capacitive relaxations in porous structures and spatially inhomogeneous effects on the ion transport typically lead to ambiguous interpretation of the EIS data with equivalent circuits.59 Hence, the battery community mostly uses EIS to probe simpler accessible processes, such as the charge transfer processes at the electrodes.60–64
The aim of this study is to characterize the potential and time-dependent dynamics of the SEI formation via the double layer capacitance. Hereto, a newly developed in situ and online monitoring technique is presented, which consists of a stepwise potential variation in combination with amperometry and EIS. To obtain easily interpretable and reproducible EIS data, a polished glassy carbon plate is employed as a plane model electrode. The SEI formation on the glassy carbon electrode is compared for lithium hexafluorophosphate (LiPF6)-based electrolytes with various carbonate solvents, providing a deeper understanding of the SEI formation under the influence of potential and time. The measurements start in the potential region of the open circuit potential (see ESI†), in which the electrolyte is stable and the interface between the electrode and electrolyte is pristine with a distinct interface between the electrode and the liquid phase. By decreasing the potential stepwise, the SEI forms at the interface. Regarding application, the presented approach can be used as a fast-screening method to test the ability of electrolyte compositions to form stable SEIs.
Methods
Experimental setup
A type 2 glassy carbon electrode (Alfa Aesar) was used for all measurements in combination with various standard carbonate-based battery electrolytes (Merck, battery grade) containing 1 M LiPF6. In detail, the following electrolyte compositions were employed: (i) a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ethylene carbonate and dimethyl carbonate (EC–DMC), (ii) a 1:1 mixture of ethylene carbonate and diethyl carbonate (EC–DEC), (iii) dimethyl carbonate (DMC), (iv) a 1:1 mixture of propylene carbonate and diethyl carbonate (PC–DEC), and (v) propylene carbonate (PC). A blueprint of the three-electrode electrochemical cell used for the electrochemical characterization and more detailed experimental information are given in the ESI.† The cell was filled with 4 ml of the examined electrolyte, while a surface area of approximately 1 cm2 of the glassy carbon plate was exposed to the electrolyte by sealing with a perfluoroelastomeric compound (FFKM) O-ring with an inner diameter of 1.1 cm and a thickness of 0.15 cm (Westring GmbH, Germany).
:1 mixture of ethylene carbonate and dimethyl carbonate (EC–DMC), (ii) a 1:1 mixture of ethylene carbonate and diethyl carbonate (EC–DEC), (iii) dimethyl carbonate (DMC), (iv) a 1:1 mixture of propylene carbonate and diethyl carbonate (PC–DEC), and (v) propylene carbonate (PC). A blueprint of the three-electrode electrochemical cell used for the electrochemical characterization and more detailed experimental information are given in the ESI.† The cell was filled with 4 ml of the examined electrolyte, while a surface area of approximately 1 cm2 of the glassy carbon plate was exposed to the electrolyte by sealing with a perfluoroelastomeric compound (FFKM) O-ring with an inner diameter of 1.1 cm and a thickness of 0.15 cm (Westring GmbH, Germany).
Before each measurement, the surface of the glassy carbon plate was prepared on an automated polishing machine (Struers, Tegramin) with 2000 and 4000 grid sandpaper. Hereto, ultrapure water was used as lubricant. After preparing the surface of the glassy carbon plate, it was thoroughly rinsed with pure ethanol and dried on a heating plate. After importing the glassy carbon specimen and the cell into an argon-filled glove box, the glassy carbon plate was again treated on a heating plate at 220 °C for 10 minutes to remove residual adsorbed water or ethanol. The electrochemical cell was then assembled in an argon-filled glove box and filled with the electrolytes. The counter and reference electrodes for the cell were made from titanium wires with lithium metal melted onto their tips.
Electrochemical measurement protocol
Fig. 1A shows a flowchart that resembles the step protocol used in this study, in which amperometry and EIS are used. First, the electrode open circuit potential (OCP) was measured and six impedance measurements at this potential were conducted in series. The values of the OCPs of the different electrolytes ranged between 3.0 and 3.3 V vs. Li/Li+, as shown in the ESI.† A potential of 3.4 V vs. Li/Li+ was chosen as the start potential for the stepwise potential iteration, as it is close to the measured OCP data. In each potential step, six consecutive repetitions of 10 s of amperometry in combination with impedance spectra (a compromise between the time to reach quasi-steady states and the entire measurement duration) were conducted. Such a potential step takes a total time of approximately 1560 s. After finishing these measurements, the next potential step was initialized by reducing the electrode potential by 0.1 V. The measurement procedure was finished when a potential of −0.4 V vs. Li/Li+ was reached. A Zennium XC potentiostat (Zahner, Germany) controlled with Python script was used for all presented measurements. All impedance measurements presented in this study were conducted with an amplitude of 10 mV and six points per decade in a logarithmically distributed frequency range between 100 kHz and 0.1 Hz. All measurements were performed in a temperature-controlled laboratory at 20 °C.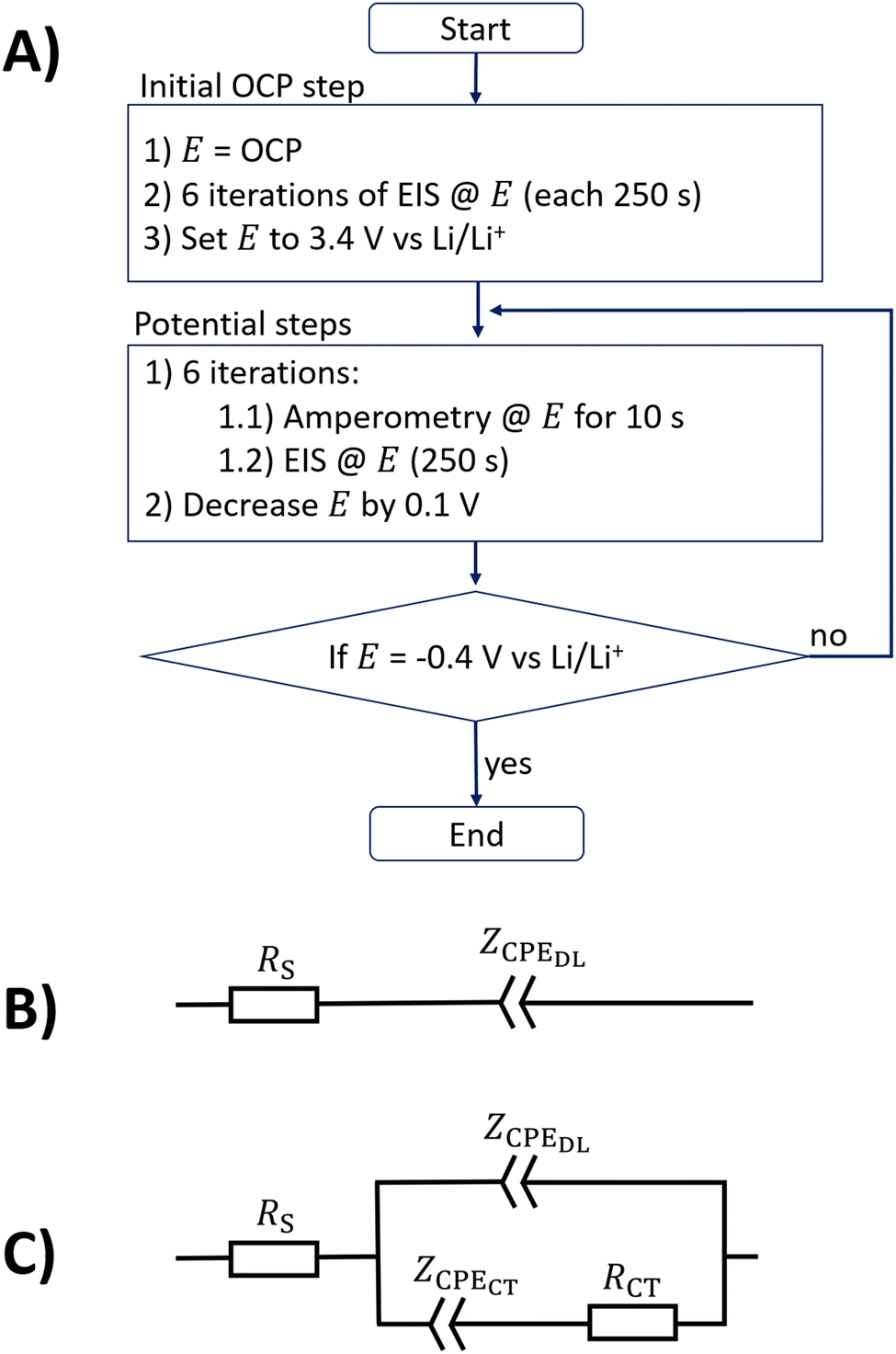 | ||
| Fig. 1 (A) Flow chart of the electrochemical step protocol used in this study. (B) Equivalent circuit model of a blocking electrode, consisting of the electrolyte resistance and a constant phase element (CPE) for double layer (DL) capacitance. (C) Equivalent circuit model including the double layer (DL) and a charge transfer (CT) process with transport limitations that are parameterized by a CPE. | ||
Impedance evaluation
The potential perturbation during EIS periodically rearranges the charge distributions in the double layer. The impedance of the double layer is typically parameterized by a constant phase element (CPE).65–68 The impedance ZCPE of a CPE equals:69,70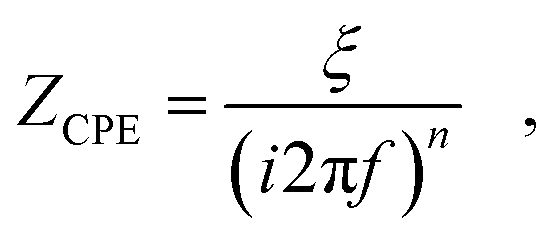 | (1) |
Fig. 1C includes the charge transfer processes of the electrolyte decomposition by the resistance RCT. Transport limitations of the charge transfer are parameterized by the CPE-based impedance ZDLCPE. The CPE with an exponent of n = 0.5 equals the Warburg element, which is typically used to describe diffusion limited charge transfer processes.74,75
The capacitance dispersion CEIS(f) has been used in previous works to evaluate the capacitance of impedance measurements,54,76 and reflects the capacitive contributions to the impedance as a function of the frequency. It is calculated based on the imaginary part of the measured impedance Z′′:
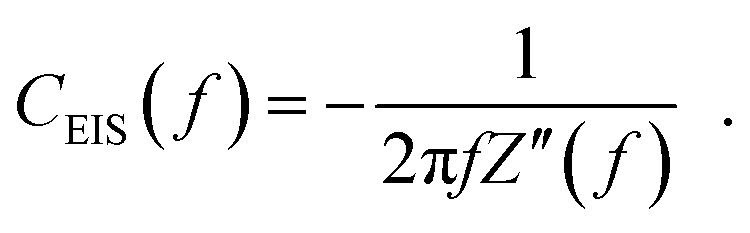 | (2) |
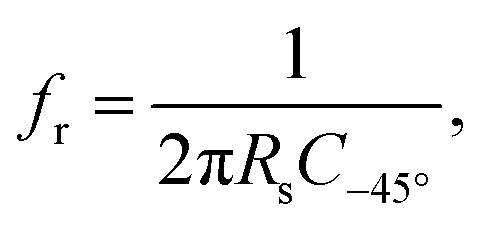 | (3) |
Cyclic voltammetry measurements
The same electrochemical cell as that used for the step protocol is examined with cyclic voltammetry (CV). Every measurement started with pristine electrolyte and freshly polished glassy carbon surfaces (same preparation procedure as discussed above) for the sake of reproducibility. The measurements were conducted with a scan rate of 10 mV s−1, while two full cycles starting from 3.4 V vs. Li/Li+ to a vertex potential of 0 V vs. Li/Li+ were applied. Before starting the CV, the working electrode was equilibrated at 3.4 V vs. Li/Li+ for 30 s. Again, a Zennium XC potentiostat was used for the measurements.Results and discussion
The ESI,† includes repetition measurements that show the reproducibility of the data presented in this study. Moreover, impedance data of all examined electrolytes and codes for their evaluation are shown in the ESI,† for more interested readers. The step protocol shown in Fig. 1A is applied to the five electrolyte solutions (with EC–DMC, EC–DEC, DMC, PC–DMC, and PC solvent) with the glassy carbon electrode. The protocol starts at 3.4 V vs. Li/Li+, which ranges in the potential regimes of the open-circuit potentials measured for these solutions (see ESI†).Example EIS data on the EC–DEC electrolyte
Fig. 2A and B show the impedance spectra (magnitude and phase angle) of the glassy carbon electrode with the EC–DMC electrolyte at four potentials (0, 1, 2, and 3.4 V vs. Li/Li+). These selected data are part of the overall dataset obtained with the electrochemical protocol from Fig. 1A, representing the first of a total of six successive electrochemical impedance spectroscopy (EIS) measurements per voltage step, respectively. By using eqn (2), the capacitance dispersion is extracted from the impedance data, showing the capacitive contributions to the impedance in the dimension of the unit Faraday in Fig. 2C. In the following, the data of the EC–DMC electrolyte are exemplarily discussed, aiming to understand the impedance observed during SEI formation.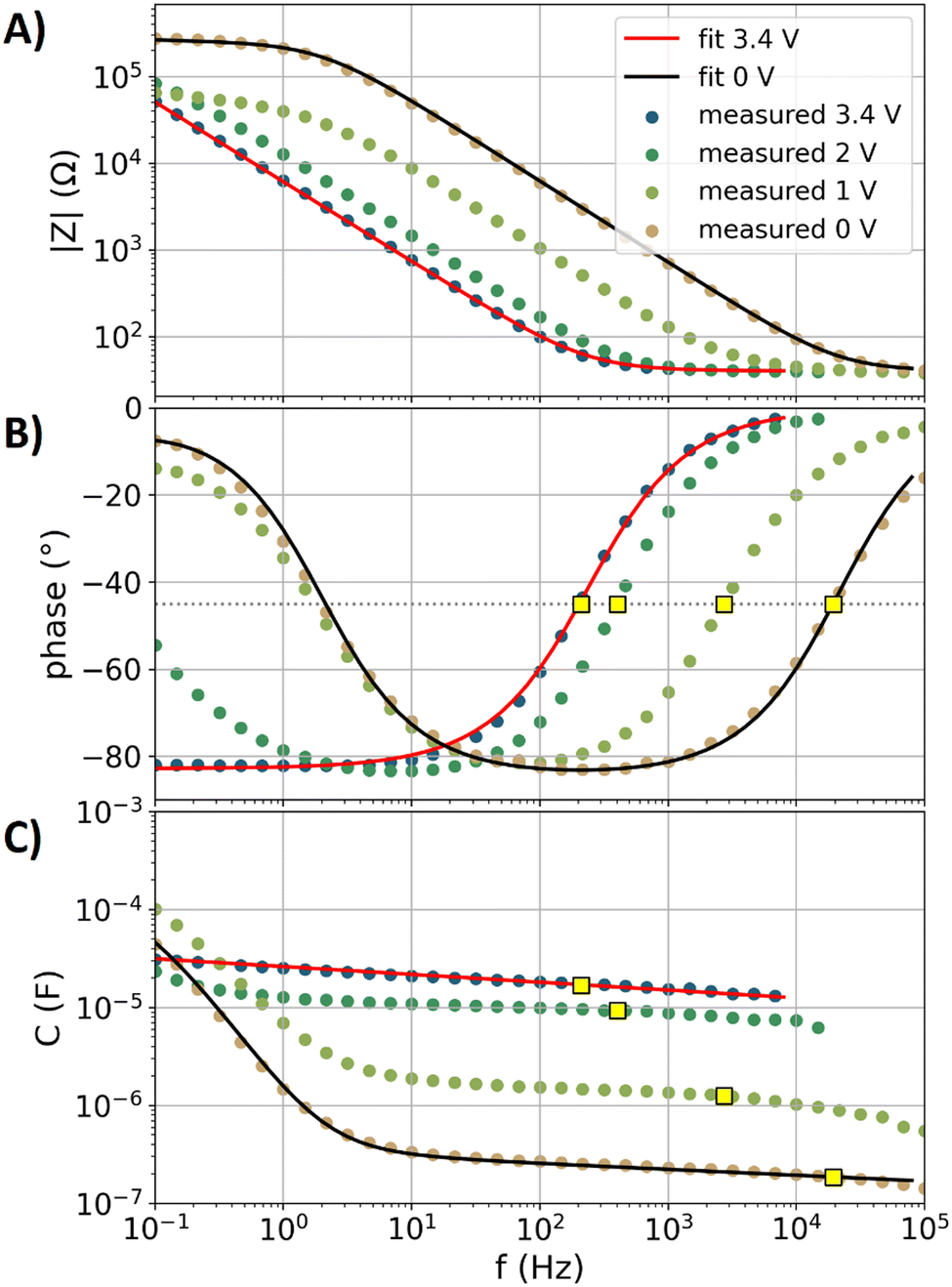 | ||
| Fig. 2 Impedance data of the glassy carbon electrode with the EC–DMC electrolyte at four selected potentials (0, 1, 2, and 3.4 V vs. Li/Li+). The graphed data represent the first iterations of the electrochemical step protocol shown in Fig. 1A. Solid lines: fits. Dots: measured data. Yellow squares: values at the relaxation frequency. (A) Impedance magnitude. (B) Impedance phase angle. The horizontal dotted gray line represents a visual guide with a value of −45° to estimate the relaxation frequency. (C) Capacitance dispersion extracted from the impedance using eqn (2). | ||
Fig. 2 also shows a fit of the equivalent circuit model of Fig. 1B (displaying a blocking electrode59,76) to the measured impedance data at 3.4 V vs. Li/Li+. The glassy carbon electrode shows an open circuit potential of approximately 3.24 V vs. Li/Li+ in EC–DMC, representing a chemically stable state with a negligible amount of charge transfer reactions. The step protocol depicted in Fig. 1A applies a potential 3.4 V vs. Li/Li+ first, which leads to a current of less than 10 nA in the amperometry measurements. The fit of the blocking electrode equivalent circuit (Fig. 1B) precisely represents the measured response at 3.4 V vs. Li/Li+. The parameters of the fitted equivalent circuit are RS = 40 Ω, while the constant phase element (CPE) describing the double layer with the impedance ZCPEDL is characterized by n = 0.92 and ξ = 33210 Ω Hzn. A detailed discussion of the impedance spectra of such a blocking electrode can be found in the literature.54 In brief, a resistively dominated regime with phase angles of 0° at high frequencies blends towards low frequencies into a capacitive dominance with phase angles; here, with a phase angle of approximately −82°. This transition of the phase angle as a function of the frequency describes a resistive–capacitive relaxation.54,76
The EIS data at the potentials of 0, 1, and 2 V vs. Li/Li+ show a significant increase of the phase angles towards frequencies lower than 10 Hz, which are a result of the charge transfer reactions76 that are related to the electrolyte decomposition. This electrolyte decomposition partly contributes to the formation of the SEI. Moreover, liquid decomposition products can diffuse back into the electrolyte, while gaseous products are also not incorporated into the SEI. The data at 0 V vs. Li/Li+ were fitted with the equivalent circuit model of Fig. 1C, which includes the charge transfer processes. In this equivalent circuit model, the effects of transport effects such as diffusion limitations on the charge transfer are described by a CPE with the impedance ZCPECT. The parameters of the fit of the equivalent circuit model are as follows: RS = 40 Ω, RCT = 250 Ω, n = 0.94 and ξ = 2670540 Ω Hzn for ZCPEDL, and n = 0.5 and ξ = 2000 Ω Hzn for ZCPECT. Uncertainties and ambiguities of such parameterization are described in the literature in detail.59 In brief, the three-parameter based description of the charge transfer process is unreliable, as several combinations of the three parameters can represent the measured data, while the effects of spatial gradients in the formed SEI also affect the current distribution in the formed porous structure.59 Owing to this uncertainty, further characterization of the SEI formation will be conducted with impedance-derived metrics rather than equivalent circuit model-based analyses.
In the recent literature,76 capacitance estimations of electrodes have been discussed in detail, showing that the specific capacitance (normalized to the surface area) of electrodes with different morphologies in different electrolytes can be most reliably compared at the relaxation frequency. The relaxation frequency can be estimated54,76 by the resistive–capacitive relaxation of the double layer response at a phase angle of −45°. In Fig. 2B, yellow squares show the frequencies at which the measured phase angle during the relaxation is interpolated to −45°. These frequencies are denoted as f−45°. In Fig. 2C, the yellow squares show the capacitance values at f−45°, which are denoted as C−45°. The thus-determined C−45° will be used as a metric to describe the capacitance of the measured impedance spectra. Below 100 Hz, charge transfer processes and the microstructure of the glassy carbon plate affect the capacitance dispersion.76 The phase angles in Fig. 2B show resistive–capacitive relaxations in the regime between 100 Hz and 10 kHz. Fig. 2 shows that higher values of fr lead to inversely proportional lower values of C−45°, which is in agreement with eqn (3).
Qualitative model for decreasing capacitances by SEIs
Fig. 2C shows that the capacitance decreases towards lower electrode potentials, which is ascribed to the formation of a SEI. The SEI displays a passivation film on the electrode that reduces the rate of the electrochemical electrolyte decomposition. Passivating films on metals in aqueous electrolytes are known to decrease the capacitance,59,76,77 as a result of additional capacitances of semi-conducting or insulating interfaces between the metallic electrodes and the electrolytes. Such a serial combination of the two capacitances C1 and C1 results in a total capacitance Ctotal of 59, 76, 77: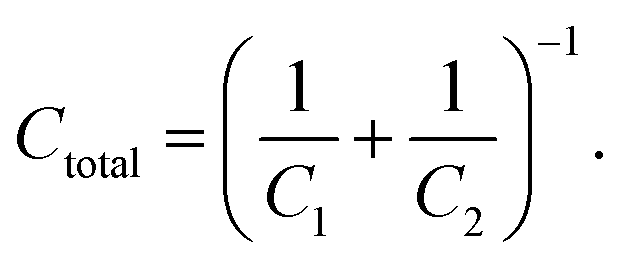 | (4) |
Fig. 3 displays a schematic sketch that shows how the SEI blocks the ion adsorption in the double layer, eventually causing a capacitance drop. At the SEI-free electrochemical interface (Fig. 3A), ions can be absorbed directly at the electrode. In contrast, the SEI in Fig. 3B hampers the direct adsorption of the ions at the electrode. The SEI is conductive to lithium ions.34–38 However, its conductivity is lower than that of the bulk electrolyte for which the rate of the charge accumulation on the electrode is reduced. Moreover, the solubilities of Li–ions in the polymerous and inorganic phases of the SEI are expected to be smaller than that inside the bulk electrolyte. Hence, less ions can accumulate directly at the electrochemical interface with a SEI-covered electrode compared to a non-SEI covered surface. Hence, with a SEI, the distance between the electrode and the ions in the double layer increases, causing a deeper penetration of the electric field into the electrolyte that consequently decreases the capacitance.
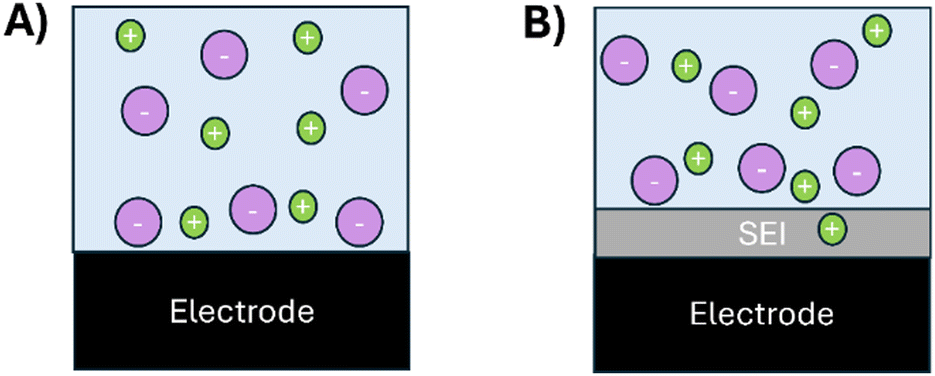 | ||
| Fig. 3 Schematic sketch showing the electrochemical interface on the glassy carbon electrode. Green dots: lithium cations. Violet dots: anions. Light blue shading: electrolyte phase. Gray shading: solid electrolyte interphase (SEI). Black: glassy carbon electrode. (A) At the electrode without SEI coverage, the ions are directly absorbed at the electrode. (B) With SEI coverage, the ion movement is constrained so that ion accumulation and adsorption directly at the electrode are hampered. | ||
Metrics for the dynamics of the SEI formation
The electrochemical decomposition of electrolyte molecules can lead to two cases: (i) the products are at least partly incorporated into the SEI. (ii) The products leave the interface into the electrolyte. In the following, the first of these processes will be characterized with the double layer capacitance. Hereto, the above introduced C−45° will serve as a capacitive measure to monitor the potential and time-dependent dynamics of the SEI formation. The electrochemical electrolyte decomposition is often characterized by the charge transfer resistance extracted from the impedance data.60–64 However, the transport effects involved in the electrolyte decomposition cannot be unambiguously characterized with equivalent circuit models (see discussion for Fig. 2). Hence, the charge transfer resistance cannot be unambiguously determined from the measured impedance data. As an alternative and more solid measure for the characterization of the electrochemical electrolyte decomposition, the currents measured by amperometry are used as metrics to characterize the electrocatalytic electrolyte decomposition. In the following, the bypass current Ibypass is used as a metric to characterize the electrolyte decomposition, defined as the average value of a 10 s long amperometry measurement in the electrochemical protocol (see Fig. 1A). Another metric for the electrolyte decomposition will be the exchange charge Qbypass as the integrated bypass currents over time.Fig. 4 shows the three metrics C−45°, Ibypass and Qbypass of the measurements with the five electrolytes (EC–DMC, EC–DEC, DMC, PC, and PC–DMC) on the glassy carbon electrode. The electrochemical protocol in Fig. 1A uses 37 voltage steps with each of the six repetitions of amperometry and impedance measurements. For each of these 222 measurements, the three metrics are extracted and plotted in Fig. 3. By comparing the different iterations of the amperometry and EIS measurements, the time-dependent dynamics of the SEI formation can be elucidated. In the following, the data of the EC–DMC electrolyte will be discussed in detail to interpret the shown data. Afterwards, the SEI formation-dynamics of the five different electrolytes will be compared.
 | ||
| Fig. 4 Metrics derived from the impedance and amperometry measurements of the step protocol (see Fig. 1A), which show the potential and time-dynamics of the SEI formation. The total time for each measurement was approximately 16 hours. Colors: different electrolytes. Points: values extracted from 222 individual impedance spectra and amperometry measurements (37 different potential stages with each 6 iterations in time) for each electrolyte, respectively. Solid lines: values of the first iteration. Dashed lines: values of the sixth iteration. Shaded colored areas: data-span between the first and sixth iteration of the measurements. (A) Capacitances extracted from the impedance spectra. (B) Bypass currents extracted from the amperometry measurements. (C) Charges derived from integrated bypass currents. | ||
In Fig. 4A, the variation of the potential from 3.4 to 2.4 V vs. Li/Li+ with the EC–DMC electrolyte leads to a capacitance decrease from 17 to 9.7 μF for the first iterations, respectively. The capacitance of the sixth iteration is at 2.4 V vs. Li/Li+ approximately 92% of that of the first iteration. This slight decrease of the capacitance may indicate a change of the surface; for instance, by adsorption of molecules on the surface. From 2.4 to 2 V vs. Li/Li+, a capacitance plateau is reached, which indicates no significant change of the electrochemical interface. A more distinct and continuous decrease of the capacitance can be observed from 1.9 to 0 V vs. Li/Li+, ending with a capacitance of 0.19 μF that is approximately one hundred times smaller than that measured at 3.4 V vs. Li/Li+. This drastic reduction of the capacitance is associated with the SEI formation (see Fig. 3), which hinders the ion adsorption at the surface of the electrode.
In Fig. 4B, −Ibypass with the EC–DMC electrolyte remains below 0.1 μA during the potential reduction from 3.4 and 3 V vs. Li/Li+. By approaching 2.7 V vs. Li/Li+, −Ibypass exceeds 1 μA, showing significant contributions of a charge transfer process such as the electrolyte decomposition. The values of the bypass currents increase until 2 V vs. Li/Li+ is reached, whereas the capacitance remains on a plateau in this potential range. Hence, the electrolyte is decomposed to substances that do not form a stable SEI. Towards lower potentials, −Ibypass remains above 1 μA, while the capacitance alteration indicates that some of the decomposed electrolyte molecules lead to the SEI formation. At −0.2 V vs. Li/Li+, the bypass current significantly increases, which may be caused by significant Li electroplating on the glassy carbon electrode.
The observed bypass current between 2.7 and 0.8 V vs. Li/Li+ results from the electrolyte decomposition. Towards lower potentials, Li–ion intercalation was reported79 based on measured electrochemical measurements. However, the current from the electrochemical electrolyte decomposition and Li-intercalation cannot be easily distinguished, which also applies to the presented data here. Based on density-functional-theory calculations,80 the Li–ion intercalation potential in graphite was reported to start at 0.3 V vs. Li/Li+. In the case of glassy carbon, no 81 or little.82 Li-intercalation was reported. However, owing to the uncertainty of the actual extent of Li-intercalation, the currents below 0.8 V vs. Li/Li+ cannot be unambiguously assigned to the electrolyte decomposition and Li-intercalation. In Fig. 3C, a continuous growth of −Qbypass towards lower potentials is observed, which doubles when descending from 0 to −0.3 V vs. Li/Li+ due to Li electroplating.
Effect of solvents on the SEI formation
In Fig. 4, the metrics C−45°, −Ibypass, and −Qbypass of all examined electrolytes show similar potential dependencies. The potential change from 3.4 to −0.3 V vs. Li/Li+ causes a reduction of C−45° by about two orders of magnitude for all electrolytes. As discussed for the EC–DMC electrolyte in detail, the approximately 50% decrease of C−45° between 3.4 and 2 V vs. Li/Li+ is also observed for all other electrolytes. Below 1.9 V vs. Li/Li+, a drastic decline of the capacitance is observed for all electrolytes, which is attributed to the SEI formation. The profiles of −Ibypass and −Qbypass are comparable for all electrolytes except DMC, which stands out by significantly larger values. These higher currents indicate a larger permeability of the formed SEI to the electrolyte than that for the other electrolytes. The more profound passivation with the PC and EC-containing electrolytes may be attributable to polymerization via ring opening.83,84With PC-based electrolytes, a fully reversible intercalation of Li–ions into graphite is not possible.4,5 Historically, the commercial breakthrough of the Li–ion battery was delayed by decades due to the initial preference of PC-based electrolytes over EC.4 However, the lack of intercalation reversibility with PC-based electrolytes is still debated in the literature, either naming an unstable SEI formed by PC85–87 or exfoliation of graphite by PC intrusion or co-intercalation88–90 as the underlying reason. The unsolved case of this phenomenon is typically referred to as the ‘EC–PC mystery’. In Fig. 3, the similar capacitance drops and exchanged charges −Qbypass for EC–DMC and PC–DMC indicate that both electrolytes form similarly stable SEIs at the glassy carbon electrode. Remarkably, the PC electrolyte without DMC addition even showed the lowest −Qbypass of all measured electrolytes. Hence, the presented results support the exfoliation assumption as the physicochemical reason for the ‘EC–PC mystery’ rather than the SEI stability assumption.
Cyclic voltammetry (CV) data
CV and linear sweep voltammetry (LSV) are state-of-the-art measurement techniques to probe electrochemical windows and to characterize the electrochemical SEI formation,91,92 both using a linear potential scan as a function of time. Fig. 5 shows the descending first branches of CV measurements with the different electrolytes, for which a scan rate of 10 mV s−1 was employed. The full CV data of two cycles and reproduction measurements are shown in the ESI.† For all examined electrolytes, the current becomes more negative as the potential decreases.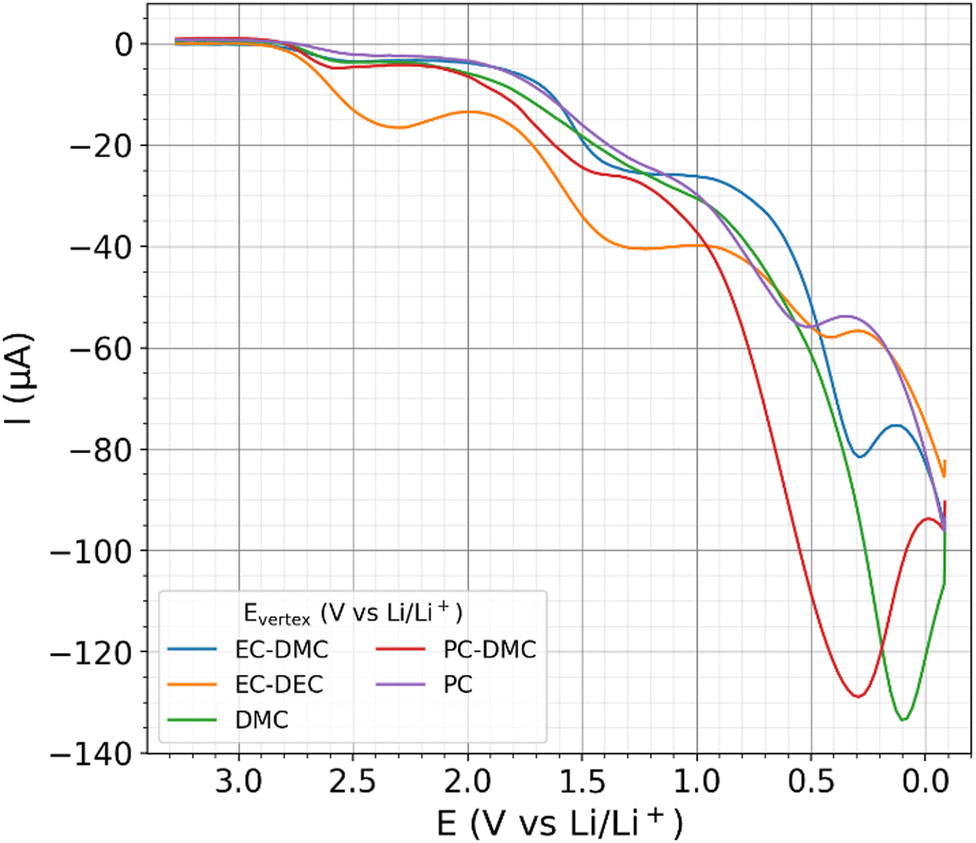 | ||
| Fig. 5 The first descending branch of cyclic voltammetry (CV) from 3.4 to 0 V vs. Li/Li+ on different electrolytes. | ||
Potential and time dependencies of the SEI formation
When probing a blocking electrode with small amplitudes, the current-response of CV and EIS is partly transferable.93 However, the irreversible formation of the SEI hinders such a transfer between both measurement techniques. During the linear potential scan of CV, capacitance, pseudocapacitances, ad/desorption processes, and the electrolyte decomposition contribute to the measured currents. Detangling these individual contributions is not possible. With the stepwise potential reduction, currents of capacitances and ad/desorption processes drop exponentially with time at each potential step. Hence, with the 10 s of amperometry measurements used in this work, electrochemical reactions that decompose the electrolyte dominate the measured currents. By using the step protocol, the driving force for the electrolyte decomposition is constant, while the evolution of a semi-steady state is measured. Thus, the influence of potential and time on the electrolyte decomposition can be considered separately as individual degrees of freedom, while the perturbations probed by EIS observe the evolution of the electrochemical interface. Table 1 shows the total charges involved in the electrolyte decompositions during the potential reduction from 3.4 to 0 V vs. Li/Li+ with the step protocol (data from Fig. 4) and CV (data from Fig. 5), denoted as Q0VSQ and Q0VCV, respectively. These metrics are roughly tenfold larger for the amperometry measurement than that of the CV measurements, whereas the current maxima of both measurement methods show an opposing trend (direct comparison of the currents in Fig. 4 and 5). These differences show the dynamics of the electrolyte decomposition and SEI formation with its dependence on the employed potential-time protocol.| Q 0VSQ (mC) | Q 0VCV (mC) | E 1μASP (V vs. Li/Li+) | E 1μACV (V vs. Li/Li+) | |
|---|---|---|---|---|
| EC–DMC | 92.5 | 8.31 | 2.7 | 2.75 |
| EC–DEC | 62.4 | 10.2 | 2.6 | 2.81 |
| DMC | 386 | 10.6 | 2.8 | 2.71 |
| PC–DMC | 90.4 | 12.7 | 2.6 | 2.73 |
| PC | 56.7 | 7.95 | 2.6 | 2.64 |
The step protocol introduced in Fig. 1A is one example out of an infinite number of possible combinations of potential and time sequences to probe the SEI formation. For instance, by alternating potential change hysteresis effects and irreversibility of the SEI formation can be examined. By varying the step sizes and length, the SEI dynamics can be probed with potential and time as individual degrees of freedoms, overcoming the drawbacks of CV and LSV procedures where both parameters are tangled.
Electrochemical windows and onset potentials
During CV with the employed scan rate of 10 mV s−1, capacitive currents on polished electrodes are typically93 smaller than 1 μA cm−2. With respect to the geometric area of the glassy carbon plate of approximately 1 cm2, a current of 1 μA serves in the following as a threshold that indicates more than just capacitive contributions to the response. For instance, these contributions can be displayed by electrochemical reactions or pseudocapacitive bond cleavages by adsorption. Table 1 show a comparison of the highest potentials at which the current threshold of 1 μA was exceeded, denoted as E1μASP for the step protocol data and E1μACV for the CV data. The values of E1μASP and E1μACV are similar, showing that both methods indicate similar potential regions of significant electrolyte decompositions.In the literature,91,92 a linear fitting procedure of CV/LSV data is reported to determine the onset potential of the electrolyte decomposition and associated electrochemical window. The onset-potential of a catalytic process is not defined with reference to the Butler–Volmer equation that describes catalytic electrochemical processes.21,94 However, the reported linear fitting procedure in non-logarithmic plots91,92 indicates that the Ohmic drop of the electrocatalytic electrolyte decomposition leads to a linear voltage–current relation that overshadows the kinetics of the electrocatalytic reaction. Moreover, a proper Tafel analysis of an electrocatalytic process21 requires a quasi-stationary and stable reaction, which is not the case of the time-dependent reaction of the SEI formation. In the many stages of the SEI formation observed in Fig. 5, it remains unclear how to apply an electrocatalytic analysis and how this shall relate to the electrochemical windows and SEI formation. Hence, the literature CV data95–97 on EC–DEC electrolytes report decomposition between 1 and 2 V vs. Li/Li+, although some of these CV data show significant currents at higher potentials, similar to Fig. 5.95 The proposed measure of a current threshold seems more consistent and precise to determine the lower margin of the electrochemical window. With the step protocol, the current threshold is less volatile towards capacitive effects or adsorption currents as that of the CV data. Hence, the current threshold in combination with the step protocol is here suggested as a solid measure for the lower margin of the electrochemical window.
Relevance of the result for batteries
The results presented in this study can be interpreted as a fundamental scientific examination of the in situ SEI formation on an experimental model electrode in terms of a polished glassy carbon plate. This electrode displays reduced complexity compared to the composite electrodes of real-world Li–ion battery anodes, which typically consist of graphite particles that are attached with a polymer binder to a copper current collector.55–57 The current collector and the polymer binder interact with the SEI formation on the graphite particles.98–101 The mechanical stress of intercalating and de-intercalating at the graphite particles also impacts the SEI formation,102,103 while porous separators influence the transport processes at the electrodes. Due to the porosity of such electrodes and associated resistive–capacitive relaxation processes, the impedance of such electrodes becomes more challenging to interpret and analyze.59By using the glassy carbon electrode, the impedance spectra could be directly evaluated by simple metrics. Hence, the influence of the SEI formation on the double layer capacitance could be shown. Despite the influence of Li–ion intercalation cannot be ruled out for the glassy carbon electrode, for graphite electrode this effect increases, overshadowing the formation process of the SEI. These fundamental scientific analysis on the model glassy carbon surface may be helpful to understand the SEI formation and to evaluate new electrolyte chemistries.104,105 Directly applying this measurement method to real-world battery electrodes may be possible if a more detailed understanding of the double layer capacitance in porous structures is available.59
Conclusion
In summary, the initial formation of a solid electrolyte interphase (SEI) on a glassy carbon electrode in typical Li–ion battery electrolytes (LiPF6 dissolved in carbonate solvents) was characterized with a newly developed electrochemical step protocol. With such step protocols, the influence of the potential and time on the SEI formation can be considered separately as individual degrees of freedom, which is not possible with the state-of-the-art potential-scan-based techniques. In these protocols, electrochemical impedance spectroscopy is used to characterize the double layer capacitance (DLC), while amperometry monitors the current of the electrochemical electrolyte decomposition. The formation of the SEI is discussed to hinder the accumulation of ions at the electrode–electrolyte interface, thus reducing the DLC. Using the sensitive measurement of the DLC, a significant SEI formation below 1.9 V vs. Li/Li+ is measured. The amperometry measurement indicates that electrolyte decomposition already starts at 2.7 V vs. Li/Li+, which means the presence of a significantly narrower electrochemical windows of battery electrolytes than those than previously reported. The presented examinations on the glassy carbon model electrode are highly reproducible and enable fast in situ and online characterization of the SEI formation, which hopefully will find further application for characterizing new electrolyte compositions that enable more stable SEIs.Data availability
The raw measurement data shown in this study are publicly available under https://gitlab.fz-juelich.de/m.schalenbach/double-layer-sei-data.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this study.Acknowledgements
This work was supported by the German Federal Ministry of Education and Research (BMBF) within the Project iNEW2.0 (Inkubator Nachhaltige Elektrochemie, 03SF0627A).References
- C. Fang, T. N. Tran, Y. Zhao and G. Liu, Electrolyte decomposition and solid electrolyte interphase revealed by mass spectrometry, Electrochim. Acta, 2021, 399, 139362 CrossRef CAS.
- M. Nie and B. L. Lucht, Role of Lithium Salt on Solid Electrolyte Interface (SEI) Formation and Structure in Lithium Ion Batteries, J. Electrochem. Soc., 2014, 161, A1001–A1006 CrossRef CAS.
- Y. Ein-Eli, New perspective on the foundation and structure of the solid electrolyte interface at the graphite anode of Li–ion cells, Electrochem. Solid-State Lett., 1999, 2, 212–214 CrossRef CAS.
- M. Winter, B. Barnett and K. Xu, Before Li Ion Batteries, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS.
- M. Li, J. Lu, Z. Chen and K. Amine, 30 Years of Lithium–Ion Batteries, Adv. Mater., 2018, 30, 1–24 Search PubMed.
- L. Gao, J. Chen, Q. Chen and X. Kong, The chemical evolution of solid electrolyte interface in sodium metal batteries, Sci. Adv., 2022, 8, 1–8 Search PubMed.
- H. Kumar, E. Detsi, D. P. Abraham and V. B. Shenoy, Fundamental Mechanisms of Solvent Decomposition Involved in Solid–Electrolyte Interphase Formation in Sodium Ion Batteries, Chem. Mater., 2016, 28, 8930–8941 CrossRef CAS.
- M. Ma, H. Cai, C. Xu, R. Huang, S. Wang, H. Pan and Y. S. Hu, Engineering Solid Electrolyte Interface at Nano-Scale for High-Performance Hard Carbon in Sodium–Ion Batteries, Adv. Funct. Mater., 2021, 31, 2100278 CrossRef CAS.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Electrochemical Na insertion and solid electrolyte interphase for hard-carbon electrodes and application to Na–ion batteries, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- M. Á. Muñoz-Márquez, M. Zarrabeitia, S. Passerini and T. Rojo, Structure, Composition, Transport Properties, and Electrochemical Performance of the Electrode–Electrolyte Interphase in Non-Aqueous Na–Ion Batteries, Adv. Mater. Interfaces, 2022, 9, 2101773 CrossRef.
- T. K. Schwietert, A. Vasileiadis and M. Wagemaker, First-Principles Prediction of the Electrochemical Stability and Reaction Mechanisms of Solid-State Electrolytes, JACS Au, 2021, 1, 1488–1496 CrossRef CAS PubMed.
- O. Borodin, X. Ren, J. Vatamanu, A. Von Wald Cresce, J. Knap and K. Xu, Modeling Insight into Battery Electrolyte Electrochemical Stability and Interfacial Structure, Acc. Chem. Res., 2017, 50, 2886–2894 CrossRef CAS.
- S. Lin, Y. Lin, B. He, B. Pu, Y. Ren, G. Wang, Y. Luo and S. Shi, Reclaiming Neglected Compounds as Promising Solid State Electrolytes by Predicting Electrochemical Stability Window with Dynamically Determined Decomposition Pathway, Adv. Energy Mater., 2022, 12, 1–13 Search PubMed.
- K. Kim and D. J. Siegel, Predicting Wettability and the Electrochemical Window of Lithium-Metal/Solid Electrolyte Interfaces, ACS Appl. Mater. Interfaces, 2019, 11, 39940–39950 CrossRef CAS PubMed.
- X. Zhang, J. K. Pugh and P. N. Ross, Computation of Thermodynamic Oxidation Potentials of Organic Solvents Using Density Functional Theory, J. Electrochem. Soc., 2001, 148, E183–E188 CrossRef CAS.
- T. Binninger, A. Marcolongo, M. Mottet, V. Weber and T. Laino, Comparison of computational methods for the electrochemical stability window of solid-state electrolyte materials, J. Mater. Chem. A, 2020, 8, 1347–1359 RSC.
- O. Borodin, Challenges with prediction of battery electrolyte electrochemical stability window and guiding the electrode – electrolyte stabilization, Curr. Opin. Electrochem., 2019, 13, 86–93 CrossRef CAS.
- S. P. Ong, O. Andreussi, Y. Wu, N. Marzari and G. Ceder, Electrochemical windows of room-temperature ionic liquids from molecular dynamics and density functional theory calculations, Chem. Mater., 2011, 23, 2979–2986 CrossRef CAS.
- M. Martins, D. Haering, J. G. Connell, H. Wan, K. L. Svane, B. Genorio, P. Farinazzo Bergamo Dias Martins, P. P. Lopes, B. Gould, F. Maglia, R. Jung, V. Stamenkovic, I. E. Castelli, N. M. Markovic, J. Rossmeisl and D. Strmcnik, Role of Catalytic Conversions of Ethylene Carbonate, Water, and HF in Forming the Solid–Electrolyte Interphase of Li–Ion Batteries, ACS Catal., 2023, 13, 9289–9301 CrossRef CAS.
- Y. Li, M. Liu, X. Feng, Y. Li, F. Wu, Y. Bai and C. Wu, How Can the Electrode Influence the Formation of the Solid Electrolyte Interface, ACS Energy Lett., 2021, 6, 3307–3320 CrossRef CAS.
- M. Schalenbach, R. Tesch, P. M. Kowalski and R. A. Eichel, The electrocatalytic activity for the hydrogen evolution reaction on alloys is determined by element-specific adsorption sites rather than d-band properties, Phys. Chem. Chem. Phys., 2024, 26, 14171–14185 RSC.
- P. Georén and G. Lindbergh, On the use of voltammetric methods to determine electrochemical stability limits for lithium battery electrolytes, J. Power Sources, 2003, 124, 213–220 CrossRef.
- D. Moosbauer, S. Jordan, F. Wudy, S. S. Zhang, M. Schmidt and H. J. Gores, Determination of electrochemical windows of novel electrolytes for double layer capacitors by stepwise cyclic voltammetry experiments, Acta Chim. Slov., 2009, 56, 218–224 CAS.
- G. Yan, D. Alves-Dalla-Corte, W. Yin, N. Madern, G. Gachot and J.-M. Tarascon, Assessment of the Electrochemical Stability of Carbonate-Based Electrolytes in Na–Ion Batteries, J. Electrochem. Soc., 2018, 165, A1222–A1230 CrossRef CAS.
- P. Ruschhaupt, S. Pohlmann, A. Varzi and S. Passerini, Determining Realistic Electrochemical Stability Windows of Electrolytes for Electrical Double-Layer Capacitors, Batteries Supercaps, 2020, 3, 698–707 CrossRef CAS.
- A. Méry, S. Rousselot, D. Lepage and M. Dollé, A critical review for an accurate electrochemical stability window measurement of solid polymer and composite electrolytes, Materials, 2021, 14, 3840 CrossRef PubMed.
- J. Kasnatscheew, B. Streipert, S. Röser, R. Wagner, I. Cekic Laskovic and M. Winter, Determining oxidative stability of battery electrolytes: validity of common electrochemical stability window (ESW) data and alternative strategies, Phys. Chem. Chem. Phys., 2017, 19, 16078–16086 RSC.
- B. S. Parimalam, A. D. MacIntosh, R. Kadam and B. L. Lucht, Decomposition Reactions of Anode Solid Electrolyte Interphase (SEI) Components with LiPF6, J. Phys. Chem. C, 2017, 121, 22733–22738 CrossRef CAS.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Generation and Evolution of the Solid Electrolyte Interphase of Lithium–Ion Batteries, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- H. Adenusi, G. A. Chass, S. Passerini, K. V. Tian and G. Chen, Lithium Batteries and the Solid Electrolyte Interphase (SEI)—Progress and Outlook, Adv. Energy Mater., 2023, 13, 2203307 CrossRef CAS.
- L. Köbbing, A. Latz and B. Horstmann, Growth of the solid–electrolyte interphase: electron diffusion versus solvent diffusion, J. Power Sources, 2023, 561, 232651 CrossRef.
- Z. Huang, S. Choudhury, N. Paul, J. H. Thienenkamp, P. Lennartz, H. Gong, P. Müller-Buschbaum, G. Brunklaus, R. Gilles and Z. Bao, Effects of Polymer Coating Mechanics at Solid–Electrolyte Interphase for Stabilizing Lithium Metal Anodes, Adv. Energy Mater., 2022, 12, 1–11 Search PubMed.
- F. Single, A. Latz and B. Horstmann, Identifying the Mechanism of Continued Growth of the Solid–Electrolyte Interphase, ChemSusChem, 2018, 11, 1950–1955 CrossRef CAS PubMed.
- A. S. Kulathuvayal and Y. Su, Ionic Transport through the Solid Electrolyte Interphase in Lithium–Ion Batteries: A Review from First-Principles Perspectives, ACS Appl. Energy Mater., 2023, 6, 5628–5645 CrossRef CAS.
- S. Shi, P. Lu, Z. Liu, Y. Qi, L. G. Hector, H. Li and S. J. Harris, Direct calculation of Li–ion transport in the solid electrolyte interphase, J. Am. Chem. Soc., 2012, 134, 15476–15487 CrossRef CAS PubMed.
- D. Bedrov, O. Borodin and J. B. Hooper, Li+ Transport and Mechanical Properties of Model Solid Electrolyte Interphases (SEI): Insight from Atomistic Molecular Dynamics Simulations, J. Phys. Chem. C, 2017, 121, 16098–16109 CrossRef CAS.
- R. Jorn, L. Raguette and S. Peart, Investigating the Mechanism of Lithium Transport at Solid Electrolyte Interphases, J. Phys. Chem. C, 2020, 124, 16261–16270 CrossRef CAS.
- T. Hu, J. Tian, F. Dai, X. Wang, R. Wen and S. Xu, Impact of the Local Environment on Li Ion Transport in Inorganic Components of Solid Electrolyte Interphases, J. Am. Chem. Soc., 2023, 145, 1327–1333 CrossRef CAS PubMed.
- J. Kullgren, J. H. Chang, S. Loftager, S. Dhillon, T. Vegge and D. Brandell, Modelling interfacial ionic transport in Li2VO2F cathodes during battery operation, Energy Adv., 2024, 3, 2271–2279 RSC.
- D. Wang, T. He, A. Wang, K. Guo, M. Avdeev, C. Ouyang, L. Chen and S. Shi, A Thermodynamic Cycle-Based Electrochemical Windows Database of 308 Electrolyte Solvents for Rechargeable Batteries, Adv. Funct. Mater., 2023, 33, 2212342 CrossRef CAS.
- S. P. Kühn, K. Edström, M. Winter and I. Cekic-Laskovic, Face to Face at the Cathode Electrolyte Interphase: From Interface Features to Interphase Formation and Dynamics, Adv. Mater. Interfaces, 2022, 9, 2102078 CrossRef.
- W. Huang, P. M. Attia, H. Wang, S. E. Renfrew, N. Jin, S. Das, Z. Zhang, D. T. Boyle, Y. Li, M. Z. Bazant, B. D. McCloskey, W. C. Chueh and Y. Cui, Evolution of the Solid–Electrolyte Interphase on Carbonaceous Anodes Visualized by Atomic-Resolution Cryogenic Electron Microscopy, Nano Lett., 2019, 19, 5140–5148 CrossRef CAS PubMed.
- E. Zhang, M. Mecklenburg, X. Yuan, C. Wang, B. Liu and Y. Li, Expanding the cryogenic electron microscopy toolbox to reveal diverse classes of battery solid electrolyte interphase, iScience, 2022, 25, 105689 CrossRef CAS PubMed.
- V. A. Agubra and J. W. Fergus, The formation and stability of the solid electrolyte interface on the graphite anode, J. Power Sources, 2014, 268, 153–162 CrossRef CAS.
- B. Han, Y. Zou, G. Xu, S. Hu, Y. Kang, Y. Qian, J. Wu, X. Ma, J. Yao, T. Li, Z. Zhang, H. Meng, H. Wang, Y. Deng, J. Li and M. Gu, Additive stabilization of SEI on graphite observed using cryo-electron microscopy, Energy Environ. Sci., 2021, 14, 4882–4889 RSC.
- B. Han, Z. Zhang, Y. Zou, K. Xu, G. Xu, H. Wang, H. Meng, Y. Deng, J. Li and M. Gu, Poor Stability of Li2CO3 in the Solid Electrolyte Interphase of a Lithium-Metal Anode Revealed by Cryo-Electron Microscopy, Adv. Mater., 2021, 33, 1–10 Search PubMed.
- J. D. McBrayer, C. A. Apblett, K. L. Harrison, K. R. Fenton and S. D. Minteer, Mechanical studies of the solid electrolyte interphase on anodes in lithium and lithium ion batteries, Nanotechnology, 2021, 32, 502005 CrossRef CAS.
- H. Wu, H. Jia, C. Wang, J. G. Zhang and W. Xu, Recent Progress in Understanding Solid Electrolyte Interphase on Lithium Metal Anodes, Adv. Energy Mater., 2021, 11, 1–35 Search PubMed.
- N. Takenaka, A. Bouibes, Y. Yamada, M. Nagaoka and A. Yamada, Frontiers in Theoretical Analysis of Solid Electrolyte Interphase Formation Mechanism, Adv. Mater., 2021, 33, 1–15 CrossRef.
- S. T. Oyakhire and S. F. Bent, Interfacial engineering of lithium metal anodes: what is left to uncover?, Energy Adv., 2023, 3, 108–122 RSC.
- J. Wu, Understanding the Electric Double-Layer Structure, Capacitance, and Charging Dynamics, Chem. Rev., 2022, 122, 10821–10859 CrossRef CAS PubMed.
- W. Schmickler and D. Henderson, New models for the structure of the electrochemical interface, Prog. Surf. Sci., 1986, 22, 323–419 CrossRef CAS.
- D. Henderson and D. Boda, Insights from theory and simulation on the electrical double layer, Phys. Chem. Chem. Phys., 2009, 11, 3822–3830 RSC.
- M. Schalenbach, Y. E. Durmus, S. Robinson, H. Tempel, H. Kungl and R. Eichel, The Physicochemical Mechanisms of the Double Layer Capacitance Dispersion and Dynamics: An Impedance Analysis, J. Phys. Chem. C, 2021, 125, 5870–5879 CrossRef CAS.
- S. L. Chou, Y. Pan, J. Z. Wang, H. K. Liu and S. X. Dou, Small things make a big difference: binder effects on the performance of Li and Na batteries, Phys. Chem. Chem. Phys., 2014, 16, 20347–20359 RSC.
- X. Zhong, J. Han, L. Chen, W. Liu, F. Jiao, H. Zhu and W. Qin, Binding mechanisms of PVDF in lithium ion batteries, Appl. Surf. Sci., 2021, 553, 149564 CrossRef CAS.
- F. Zou and A. Manthiram, A Review of the Design of Advanced Binders for High-Performance Batteries, Adv. Energy Mater., 2020, 10, 1–28 Search PubMed.
- S. Prykhodska, K. Schutjajew, E. Troschke, L. Kaberov, J. Eichhorn, F. H. Schacher, F. Walenszus, D. Werner and M. Oschatz, The impact of templating and macropores in hard carbons on their properties as negative electrode materials in sodium–ion batteries, Energy Adv., 2024, 3, 1342–1353 RSC.
- M. Schalenbach, L. Raijmakers, H. Tempel and R. Eichel, How Microstructures, Oxide Layers, and Charge Transfer Reactions influence Double Layer Capacitances. Part 2: Equivalent Circuit Models, Electrochem. Sci. Adv., 2024, e202400010, DOI:10.1002/elsa.202400010 , In Print.
- P. Wang, D. Yan, C. Wang, H. Ding, H. Dong, J. Wang, S. Wu, X. Cui, C. Li, D. Zhao and S. Li, Study of the formation and evolution of solid electrolyte interface via in situ electrochemical impedance spectroscopy, Appl. Surf. Sci., 2022, 596, 1–11 Search PubMed.
- K. Van Havenbergh, S. Turner, K. Driesen, J. S. Bridel and G. Van Tendeloo, Solid–Electrolyte Interphase Evolution of Carbon-Coated Silicon Nanoparticles for Lithium–Ion Batteries Monitored by Transmission Electron Microscopy and Impedance Spectroscopy, Energy Technol., 2015, 3, 699–708 CrossRef CAS.
- T. P. Heins, N. Harms, L. S. Schramm and U. Schröder, Development of a new Electrochemical Impedance Spectroscopy Approach for Monitoring the Solid Electrolyte Interphase Formation, Energy Technol., 2016, 4, 1509–1513 CrossRef CAS.
- K. Xu, S. Zhang and R. Jow, Electrochemical impedance study of graphite/electrolyte interface formed in LiBOB/PC electrolyte, J. Power Sources, 2005, 143, 197–202 CrossRef CAS.
- X. Wang, Z.-G. Shao, G. Li, L. Zhang, Y. Zhao, W. Lu and B. Yi, Preparation and characterization of partial-cocrystallized catalyst-coated membrane for solid polymer electrolyte water electrolysis, Int. J. Hydrogen Energy, 2013, 38, 9057–9064 CrossRef CAS.
- T. Pajkossy, Capacitance dispersion on solid electrodes: anion adsorption studies on gold single crystal electrodes, Solid State Ionics, 1997, 94, 123–129 CrossRef CAS.
- P. Iurilli, C. Brivio and V. Wood, On the use of electrochemical impedance spectroscopy to characterize and model the aging phenomena of lithium–ion batteries: a critical review, J. Power Sources, 2021, 505, 229860 CrossRef CAS.
- N. Meddings, M. Heinrich, F. Overney, J. S. Lee, V. Ruiz, E. Napolitano, S. Seitz, G. Hinds, R. Raccichini, M. Gaberšček and J. Park, Application of electrochemical impedance spectroscopy to commercial Li–ion cells: a review, J. Power Sources, 2020, 480, 228742 CrossRef CAS.
- J. Wehbe and N. Karami, Battery equivalent circuits and brief summary of components value determination of lithium ion: a review, 2015 3rd Int. Conf. Technol. Adv. Electr. Electron. Comput. Eng. TAEECE 2015, 2015, 45–49.
- G. J. Brug, A. L. G. van den Eeden, M. Sluyters-Rehbach and J. H. Sluyters, The analysis of electrode impedances complicated by the presence of a constant phase element, J. Electroanal. Chem., 1984, 176, 275–295 CrossRef CAS.
- S. M. Gateman, O. Gharbi, H. Gomes de Melo, K. Ngo, M. Turmine and V. Vivier, On the use of a constant phase element (CPE) in electrochemistry, Curr. Opin. Electrochem., 2022, 36, 101133 CrossRef CAS.
- J. C. Wang, Realizations of Generalized Warburg Impedance with RC Ladder Networks and Transmission Lines, J. Electrochem. Soc., 1987, 134, 1915–1920 CrossRef CAS.
- M. Schalenbach, Y. E. Durmus, H. Tempel, H. Kungl and R. A. Eichel, A Dynamic Transmission Line Model to Describe the Potential Dependence of Double-Layer Capacitances in Cyclic Voltammetry, J. Phys. Chem. C, 2021, 125, 27465–27471 CrossRef CAS.
- M. Schalenbach, Y. E. Durmus, H. Tempel, H. Kungl and R. A. Eichel, The role of the double layer for the pseudocapacitance of the hydrogen adsorption on platinum, Sci. Rep., 2022, 12, 1–10 CrossRef.
- D. D. MacDonald, Reflections on the history of electrochemical impedance spectroscopy, Electrochim. Acta, 2006, 51, 1376–1388 CrossRef CAS.
- J. Huang, Diffusion impedance of electroactive materials, electrolytic solutions and porous electrodes: Warburg impedance and beyond, Electrochim. Acta, 2018, 281, 170–188 CrossRef CAS.
- M. Schalenbach, L. Raijmakers, V. Selmert, A. Kretzschmar, Y. E. Durmus, H. Tempel and R.-A. Eichel, How Microstructures, Oxide Layers, and Charge Transfer Reactions influence Double Layer Capacitances. Part 1: Impedance Spectroscopy and Cyclic Voltammetry to estimate Electrochemically Active Surface Areas (ECSAs), Phys. Chem. Chem. Phys., 2024, 20, 1 Search PubMed.
- S. L. Medway, C. A. Lucas, A. Kowal, R. J. Nichols and D. Johnson, In situ studies of the oxidation of nickel electrodes in alkaline solution, J. Electroanal. Chem., 2006, 587, 172–181 CrossRef CAS.
- W. Schmickler, Electrochemical Theory: Double Layer, Elsevier Inc., Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, 2014 Search PubMed.
- Y. Wu, T. Okajima and T. Ohsaka, Lithium intercalation into graphene ribbons of glassy carbon, Int. J. Electrochem. Sci., 2017, 12, 1004–1013 CrossRef CAS.
- Y. Okamoto, Density functional theory calculations of alkali metal (Li, Na, and K) graphite intercalation compounds, J. Phys. Chem. C, 2014, 118, 16–19 CrossRef CAS.
- G. Zampardi, F. La Mantia and W. Schuhmann, Determination of the formation and range of stability of the SEI on glassy carbon by local electrochemistry, RSC Adv., 2015, 5, 31166–31171 RSC.
- S. Pérez-Villar, P. Lanz, H. Schneider and P. Novák, Characterization of a model solid electrolyte interphase/carbon interface by combined in situ Raman/Fourier transform infrared microscopy, Electrochim. Acta, 2013, 106, 506–515 CrossRef.
- R. Abdul-Karim, A. Hameed and M. I. Malik, Ring-opening polymerization of propylene carbonate: microstructural analysis of the polymer and selectivity of polymerization by 2D-NMR techniques, Eur. Polym. J., 2018, 105, 95–106 CrossRef CAS.
- J. C. Lee and M. H. Litt, Ring-opening polymerization of ethylene carbonate and depolymerization of poly(ethylene oxide-co-ethylene carbonate), Macromolecules, 2000, 33, 1618–1627 CrossRef CAS.
- H.-L. Zhang, C.-H. Sun, F. Li, C. Liu, J. Tan and H.-M. Cheng, New Insight into the Interaction between Propylene Carbonate-Based Electrolytes and Graphite Anode Material for Lithium Ion Batteries, J. Phys. Chem. C, 2007, 111, 4740–4748 CrossRef CAS.
- J. Zhang, J. Yang, L. Yang, H. Lu, H. Liu and B. Zheng, Exploring the redox decomposition of ethylene carbonate-propylene carbonate in Li–ion batteries, Mater. Adv., 2021, 2, 1747–1751 RSC.
- T. Melin, R. Lundström and E. J. Berg, Revisiting the Ethylene Carbonate–Propylene Carbonate Mystery with Operando Characterization, Adv. Mater. Interfaces, 2022, 9, 2101258 CrossRef CAS.
- L. J. Hardwick, H. Buqa, M. Holzapfel, W. Scheifele, F. Krumeich and P. Novák, Behaviour of highly crystalline graphitic materials in lithium–ion cells with propylene carbonate containing electrolytes: an in situ Raman and SEM study, Electrochim. Acta, 2007, 52, 4884–4891 CrossRef CAS.
- J. O. Besenhard, M. Winter, J. Yang and W. Biberacher, Filming mechanism of lithium–carbon anodes in organic and inorganic electrolytes, J. Power Sources, 1995, 54, 228–231 CrossRef CAS.
- L. Xing, X. Zheng, M. Schroeder, J. Alvarado, A. Von Wald Cresce, K. Xu, Q. Li and W. Li, Deciphering the Ethylene Carbonate–Propylene Carbonate Mystery in Li–Ion Batteries, Acc. Chem. Res., 2018, 51, 282–289 CrossRef CAS PubMed.
- E. J. Olson and P. Bühlmann, Unbiased Assessment of Electrochemical Windows: Minimizing Mass Transfer Effects on the Evaluation of Anodic and Cathodic Limits, J. Electrochem. Soc., 2013, 160, A320–A323 CrossRef CAS.
- M. P. S. Mousavi, A. J. Dittmer, B. E. Wilson, J. Hu, A. Stein and P. Bühlmann, Unbiased Quantification of the Electrochemical Stability Limits of Electrolytes and Ionic Liquids, J. Electrochem. Soc., 2015, 162, A2250–A2258 CrossRef CAS.
- M. Schalenbach, Y. E. Durmus, H. Tempel, H. Kungl and R.-A. Eichel, Double Layer Capacitances Analysed with Impedance Spectroscopy and Cyclic Voltammetry: Validity and Limits of the Constant Phase Element Parameterization, Phys. Chem. Chem. Phys., 2021, 23, 21097–21105 RSC.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Insight on Tafel slopes from a microkinetic analysis of aqueous electrocatalysis for energy conversion, Sci. Rep., 2015, 5, 1–21 Search PubMed.
- X. Zhang, J. E. Soc, X. Zhang, R. Kostecki, T. J. Richardson, J. K. Pugh and P. N. Ross, Electrochemical and Infrared Studies of the Reduction of Organic Carbonates Electrochemical and Infrared Studies of the Reduction of Organic Carbonates, J. Electrochem. Soc., 2001, 148, A1341 CrossRef CAS.
- K. Abe, H. Yoshitake, T. Kitakura, T. Hattori, H. Wang and M. Yoshio, Additives-containing functional electrolytes for suppressing electrolyte decomposition in lithium–ion batteries, Electrochem. Acta, 2004, 49, 4613–4622 CrossRef CAS.
- M. Tang, S. Lu and J. Newman, Experimental and Theoretical Investigation of Solid–Electrolyte-Interphase Formation Mechanisms on Glassy Carbon, J. Electrochem. Soc., 2012, 159, A1775–A1785 CrossRef CAS.
- K. Ui, D. Fujii, Y. Niwata, T. Karouji, Y. Shibata, Y. Kadoma, K. Shimada and N. Kumagai, Analysis of solid electrolyte interface formation reaction and surface deposit of natural graphite negative electrode employing polyacrylic acid as a binder, J. Power Sources, 2014, 247, 981–990 CrossRef CAS.
- T. Zhang, I. de Meatza, X. Qi and E. Paillard, Enabling steady graphite anode cycling with high voltage, additive-free, sulfolane-based electrolyte: role of the binder, J. Power Sources, 2017, 356, 97–102 CrossRef CAS.
- C. Forestier, S. Grugeon, C. Davoisne, A. Lecocq, G. Marlair, M. Armand, L. Sannier and S. Laruelle, Graphite electrode thermal behavior and solid electrolyte interphase investigations: role of state-of-the-art binders, carbonate additives and lithium bis(fluorosulfonyl)imide salt, J. Power Sources, 2016, 330, 186–194 CrossRef CAS.
- E. M. C. Jones, Ö. Ö. Çapraz, S. R. White and N. R. Sottos, Reversible and Irreversible Deformation Mechanisms of Composite Graphite Electrodes in Lithium–Ion Batteries, J. Electrochem. Soc., 2016, 163, A1965–A1974 CrossRef CAS.
- N. Iqbal, Y. Ali and S. Lee, Analysis of mechanical failure at the interface between graphite particles and polyvinylidene fluoride binder in lithium–ion batteries, J. Power Sources, 2020, 457, 228019 CrossRef CAS Contents.
- M. Yoo, C. W. Frank, S. Mori and S. Yamaguchi, Effect of poly(vinylidene fluoride) binder crystallinity and graphite structure on the mechanical strength of the composite anode in a lithium ion battery, Polymer, 2003, 44, 4197–4204 CrossRef CAS.
- W. Chen, D. Zhang, H. Fu, J. Li, X. Yu, J. Zhou and B. Lu, Restructuring Electrolyte Solvation by a Partially and Weakly Solvating Cosolvent toward High-Performance Potassium–Ion Batteries, ACS Nano, 2024, 18, 12512–12523 CrossRef CAS PubMed.
- D. Zhang, H. Fu, X. Ma, X. Yu, F. Li, J. Zhou and B. Lu, Nonflammable Phosphate-Based Electrolyte for Safe and Stable Potassium Batteries Enabled by Optimized Solvation Effect, Angew. Chemie, 2024, 136, e202405153 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The supporting information to this article contains: Blueprint of the cell used for the measurements. OCP data evaluation. All raw impedance-data on the electrolytes. Reproduction measurements. Full CV data. Python codes used for the data evaluation. See DOI: https://doi.org/10.1039/d4ya00524d |
| This journal is © The Royal Society of Chemistry 2025 |