
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrolyte composition dependent Li-ion binding and degradation of organic radical battery material†
Davis Thomas
Daniel
 *ab,
Emmanouil
Veroutis
a,
P. Philipp M.
Schleker
a,
Rüdiger-A.
Eichel
ac and
Josef
Granwehr
ab
*ab,
Emmanouil
Veroutis
a,
P. Philipp M.
Schleker
a,
Rüdiger-A.
Eichel
ac and
Josef
Granwehr
ab
aInstitute of Energy Technologies (IET-1), Forschungszentrum Jülich GmbH, 52425 Jülich, Germany. E-mail: d.daniel@fz-juelich.de
bInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, 52074 Aachen, Germany
cInstitute of Physical Chemistry, RWTH Aachen University, Aachen 52074, Germany
First published on 24th January 2025
Abstract
Electrolyte composition governs battery design due to its influence on ion dynamics, active material stability, and performance. Using electron paramagnetic resonance (EPR) and nuclear magnetic resonance (NMR), complemented by density functional theory calculations, the impact of electrolyte properties on an organic redox unit, TEMPO methacrylate (TMA), is explored. EPR hyperfine spectroscopy revealed that the amount of TMA bound to Li ions can be altered depending on the solvent used, and a higher fraction of TMA are Li-bound in linear carbonates compared to cyclic carbonates. The active material itself can be involved in the solvation shell of electrolyte ions, and insight into active material–electrolyte interactions from pulsed EPR may enable tuning of ion dynamics in organic radical batteries. Furthermore, the impact of moisture-dependent electrolyte degradation on the stability of TMA, investigated using time-resolved NMR and continuous wave EPR spectroscopy, resulted in the identification of degradation products and a degradation pathway mediated by the electrolyte.
Electrolytes, an essential component of all battery technologies, exert significant influence on ion mobility, charge transport and overall battery performance.1–3 Therefore, the electrolyte composition is a decisive factor in the design of battery systems.
Organic radical batteries (ORBs) utilise organic radical polymers (ORPs) as active materials, where pendant radical moieties are responsible for the redox activity.4–6 While the redox mechanisms of conventional Li-ion batteries with inorganic cathodes feature slow intercalation processes and change in valence state of metal ions, ORBs feature comparatively fast redox reactions, where an organic redox unit undergoes reversible oxidation or reduction. Despite these differences, electrolytes employed in ORBs are largely adopted from conventional Li-ion batteries, which mainly utilise liquid electrolytes.1,7 Specifically tailoring electrolytes for ORB systems requires a fundamental understanding of how electrolytes interact with ORB active materials.
Most liquid electrolytes used in ORBs are composed of a conducting salt dissolved in organic solvents such as carbonate esters.8 Cyclic carbonate solvents feature a high dielectric constant, which results in a separation of the conducting salt into its constituent ions and prevents the formation of ion pairs.9 However, higher viscosity of these solvents hinders ionic mobility, which can be mitigated by addition of less viscous linear carbonate solvents.10 The type of carbonate solvent used in the electrolyte is also known to influence the constituents of the Li-ion solvation shell, which in turn affects the mobility of Li-ions and, therefore, the battery performance. Ion solvation shells with weakly bound constituents are easier to modify and can result in faster ion dynamics.11 This is especially applicable for Li-ORBs with p-type organic cathode materials such as poly(2,2,6,6-tetramethyl-1-piperidinyloxy-4-yl methacrylate) (PTMA) and a Li metal anode.12 During both charging and discharging, the performance limiting factor of the ORB is the migration and mobility of ions.7 During charging, the active material adopts the cationic form and the electrolyte anions migrate to the cathode while Li-ions migrate to the anode. During discharge, the ions are released back into the electrolyte. Therefore, in case of Li-ORBs, a close interaction of the active material and the electrolyte ions raises the possibility of the active material itself influencing the solvation shell of electrolyte ions and, consequently, the ion dynamics. Investigation of active material–electrolyte interactions could reveal ion-mobility related performance bottlenecks and provide insights into influencing ion dynamics through the composition of the ion solvation shell.
In addition to the solvent system, the conducting salt, which provides the ionic species, constitutes another key component of the electrolyte. The choice of conducting salt is determined by compatibility with the electrode materials and, analogous to conventional Li-ion batteries, LiPF6 is commonly used for Li-ORBs.7 The decomposition of the conducting salt not only affects the battery performance but also entails numerous safety concerns as the degradation products are usually toxic.13 Moisture sensitivity of LiPF6 based electrolytes is a known cause of electrolyte decomposition and consequent performance degradation in conventional Li-ion batteries.14,15 However, the impact of such electrolyte degradation on organic active materials in different solvent environments is relatively unexplored. Understanding the effects of electrolyte degradation on organic redox units would enable the optimisation of electrolyte composition with respect to specific ORB active materials.
Magnetic resonance methods can provide insight into the interactions between species at a microscopic level with high specificity. Nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR) methods have been demonstrated as suitable analytical tools in battery research, as both structural and dynamic information can be obtained.16–18 As active materials and redox processes in ORBs feature paramagnetic states or unpaired electrons, EPR spectroscopy is applicable. In case of ORBs, continuous wave (CW) EPR techniques are routinely applied for quantification of radical content and identification of the paramagnetic species.12,19 Using in operando CW-EPR, investigation of state-of-charge dependent changes in an ORB with a nitroxide based active material was previously reported.20 Pulsed EPR methods have also been applied to nitroxide-containing cathode materials and can be applied to probe specific interactions.21 For instance, pulsed EPR relaxation measurements were demonstrated as a means to study electronic contact in organic battery materials.22 Pulsed EPR methods such as electron–nuclear double resonance (ENDOR) or hyperfine sublevel correlation (HYSCORE) spectroscopy can be used to detect nuclear spins in the vicinity of the unpaired electron, allowing for insight into solvation structures.23–25 Diamagnetic states are not accessible by EPR, and NMR spectroscopy is better suited. While NMR investigations aimed specifically at ORBs are rare,26 NMR techniques have been previously demonstrated to study electrolyte characteristics, solvation structures and intercalation processes27 in conventional batteries. Long-term in operando NMR methods have also been utilised for studying battery degradation and morphological changes of lithium anodes.28,29
In this work, the organic redox unit TEMPO methacrylate (TMA) and its interactions with LiPF6 based electrolytes is studied in two different solvent environments consisting of a linear carbonate ester, dimethyl carbonate (DMC), and a cyclic carbonate ester, propylene carbonate (PC). Moreover, the stability of TMA in the two different electrolyte solvents and its relation to moisture-dependent electrolyte degradation is investigated. The two experimental systems, hereafter referred to as 1 and 2 (see inset of Fig. 1e and f), are composed of 20 mM TMA and 1 M LiPF6 dissolved in either DMC or PC, respectively.
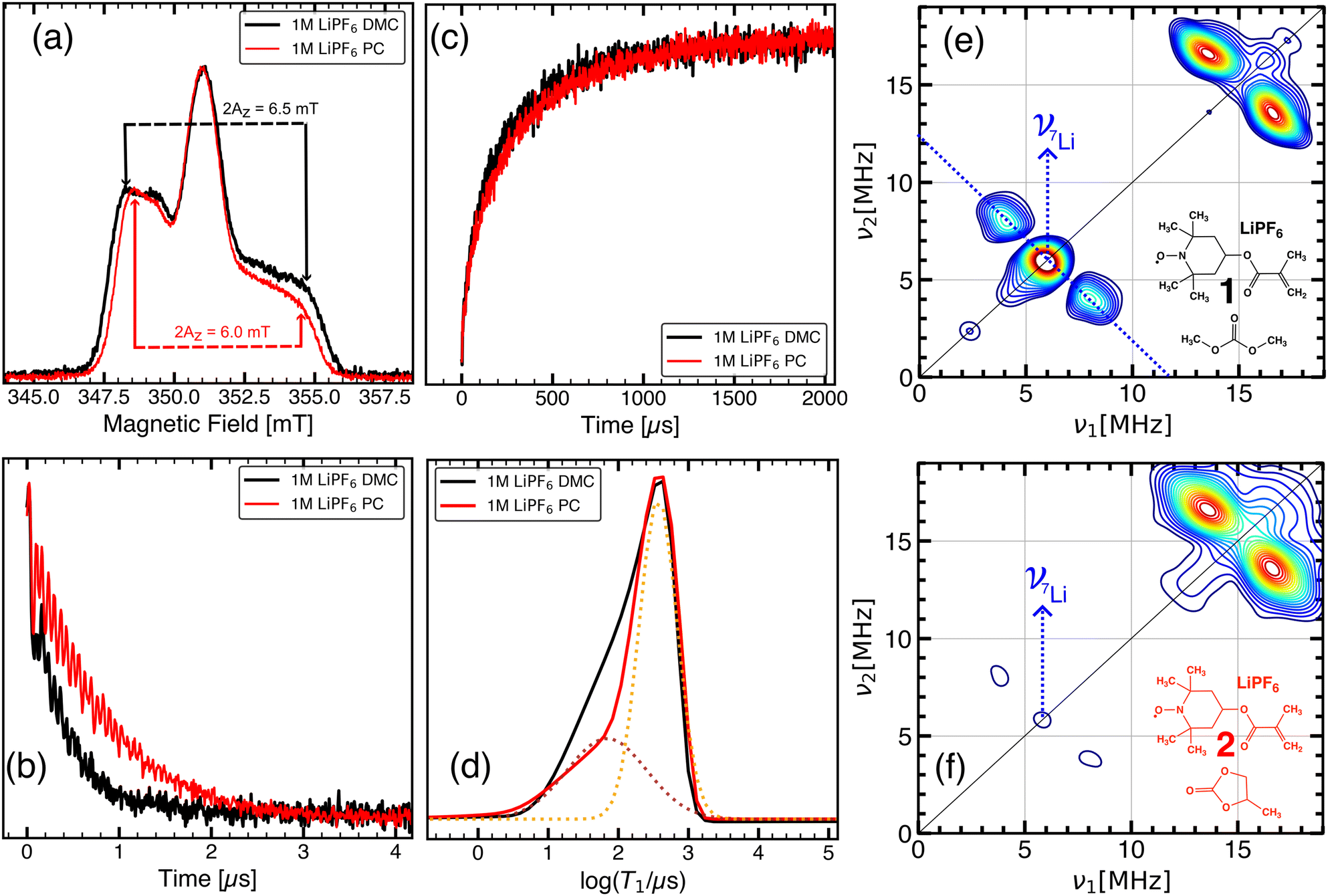 | ||
| Fig. 1 X-band pulsed EPR measurements at 60 K to investigate TMA–electrolyte interactions. (a)–(d) Comparison of 20 mM TEMPO methacrylate in 1 M LiPF6-DMC (black; 1 in inset of (e)) and 1 M LiPF6-PC (red; 2 in inset of (f)). (a) Field-swept echo detected EPR spectra. (b) Spin echo decay time traces (Tm). (c) Inversion recovery (T1) time traces. (d) T1 relaxation time constant distributions. Two relaxation components (brown and orange dotted lines) which were fit using Gaussian functions in log space, is shown for 2 (For 1, see Fig. S16, ESI†). Echo decay and inversion recovery traces are scaled to the same minimum and maximum for comparison. HYSCORE spectrum of (e) 1 and (f) 2 at a magnetic field of 350.9 mT. 7Li cross peaks are indicated by blue dotted lines. Cross peaks at 14.9 MHz correspond to contributions from 1H nuclei. | ||
The mobility of TMA is expected to be higher in 1 than in 2 at room temperature due to the lower viscosity of DMC (0.5 mPa s) than PC (2.2 mPa s).30 This is also evident upon comparison of continuous wave (CW) EPR spectra of 1 and 2 at 298 K (see Fig. S2a in the ESI†). The intensity ratio of the three lines from the nitroxide radical can be used to estimate the rotational correlation time of TMA,31 which was found to be 420 ± 12 ps for 2 and 100 ± 5 ps for 1, consistent with the higher viscosity of PC. In case of 1, LiPF6 considerably affects the spin exchange characteristics and, therefore, the CW EPR spectrum of TMA. In the absence of LiPF6, 20 mM TMA in DMC exhibits an EPR spectrum with spin exchange broadened lines,19 indicative of frequent collisional encounters between the radicals (see Fig. S2b in the ESI†). In contrast, the CW EPR spectrum of 1 exhibits narrower lines, suggesting that the mobility of TMA in 1 is influenced by the addition of LiPF6.
In frozen solution at 60 K, viscosity differences between DMC and PC are not expected to affect the EPR spectrum significantly, and field-swept echo detected (FSED) EPR spectra of 1 and 2 show the typical EPR spectrum of isolated nitroxide radicals in the static limit (see Fig. 1a). The width of the FSED spectrum allows for an estimation of the Az component of the 14N hyperfine tensor, which characterises the interaction strength between the 14N nuclear spin and the unpaired electron. An increase in Az can be caused by bonding interactions towards the N–O moiety and can be used as an indicator of the polarity of the micro-environment.32,33 However, a significantly higher dielectric constant of PC than DMC does not lead to a higher Az value in case of 2. Instead, Az of 1 was found to be larger than that of 2 by 0.25 mT, which is comparable to Az differences found for nitroxide radicals in protic vs. aprotic environments, respectively, due to different extents of H-bonding interactions.34 As both DMC and PC are aprotic solvents, such an Az difference likely arises from the propensity of the N–O moiety to interact with the constituent ions of LiPF6. As Li-bonding interactions share similarities with H-bonding interactions,35 an Az increase can be caused by the presence of Li nuclei in the micro-environment of TMA. Differing Az values of 1 and 2, therefore, indicate a solvent dependent fraction of TMA with Li ions in their vicinity.
Relaxation characteristics of a paramagnetic centre such as TMA can be used to gain insight into its interaction with its environment.22 Phase-memory times, Tm, which are influenced by spin–spin interactions, showed considerable contrast between 1 and 2, with faster spin echo decay observed for 1 than for 2 (see Fig. 1b). The presence of Li nuclei near TMA may result in faster relaxation through electron–nuclear dipolar interactions.36 However, as methyl rotations of the solvent matrix may contribute to echo dephasing even at cryogenic temperatures,37–39 faster Tm for 1 cannot be solely attributed to the proximity of TMA to Li nuclei. Spin–lattice relaxation times, T1, showed comparatively less pronounced difference between 1 and 2, but followed the same trend as Tm (see Fig. 1c). Moreover, T1 relaxation distributions obtained using Laplace inversion (see Section S5 in the ESI†) of the longitudinal relaxation time traces suggest the presence of two relaxation components with a T1 difference of ≈300 μs in both 1 and 2 (see Fig. 1d). By fitting the relaxation distributions with two Gaussian functions in log space (shown for 1 and 2 in Fig. S16, ESI† and Fig. 1d respectively), relative proportions of the two relaxation components in 1 and 2 were estimated. The relative amount of the fast relaxing component was found to be ≈60% of the total integral intensity in case of 1, which decreased to ≈30% in case of 2. The two distinct relaxation components may originate from two different populations of TMA species such as Li-bound TMA and free TMA. In such a system, as a consequence of dipolar interactions between Li nuclear spins and TMA radicals, Li-bound TMA is expected to relax faster than free TMA.
To explore the micro-environment of TMA and detect Li-bound TMA species, HYSCORE was employed. Fig. 1e and f compare the HYSCORE spectrum of 1 and 2, showing cross peaks from weakly coupled nuclear spins in the vicinity of TMA electron spin. For both 1 and 2, the HYSCORE spectrum consists of 1H cross peaks centred at 14.9 MHz, which corresponds to the nuclear Larmor frequency of 1H at a magnetic field strength of 350.9 mT. In case of 1, intensive cross peaks from 7Li are observed at 5.8 MHz, corresponding to the Larmor frequency of 7Li. Furthermore, an intensive matrix peak from 7Li nuclei that are more distant from TMA is also observed on the diagonal. The intensity of the 7Li peaks decreases substantially in case of 2, indicating a lower amount of Li-bound TMA. For both 1 and 2, the splitting of 7Li cross peaks is similar, suggesting that in both solvent environments, the hyperfine coupling and, therefore, the distance, between the TMA radical and Li ions in Li-bound TMA species is similar. The 7Li cross peaks confirm the presence of Li-bound TMA species in both 1 and 2, yet the amount of Li-bound TMA and free TMA in 1 and 2 differ considerably. Simulation of the 7Li hyperfine tensor of TMA bound to a DMC-solvated Li ion (see Fig. S3a in the ESI†) was in qualitative agreement with features in the experimental HYSCORE spectrum of 1 (see Fig. S3b in the ESI†). It indicates that in 1, the majority of TMA molecules contains Li-ions in proximity to the N–O moiety. For 2, HYSCORE spectrum suggests a considerably lower fraction of Li in the TMA solvation shell, which could be a result of preferential binding of Li ions by PC molecules. Such a result is consistent with the observation that in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of linear and cyclic carbonates, Li ions preferentially coordinate with cyclic carbonate molecules.40 Overall, these results suggest a competition between TMA and the electrolyte solvent for Li coordination, with PC showing a significantly higher affinity for Li than DMC.
:1 mixtures of linear and cyclic carbonates, Li ions preferentially coordinate with cyclic carbonate molecules.40 Overall, these results suggest a competition between TMA and the electrolyte solvent for Li coordination, with PC showing a significantly higher affinity for Li than DMC.
The stability of TMA in the two electrolyte systems was studied using time-resolved EPR and NMR spectroscopy. Along with differences in the solvation shell, also the stability of TMA differs in 1 and in 2. In 1, radical degradation occurs at a faster rate than in 2 (Fig. 2). A complete loss of EPR signal intensity was observed after ≈17 hours for TMA in 1, while only 5% decrease of the signal intensity was observed for 2 during the same time period. In the absence of LiPF6, no loss of EPR signal intensity was observed in DMC or in PC. Prolonged drying of TMA (≈1 month) before sample preparation results in a slower degradation, but the stability differences of TMA between DMC and PC based electrolyte are still reproduced (see 1′ and 2′ in Fig. 2). Therefore, the rate of radical degradation is influenced by the amount of water initially present in the active material as well as the type of carbonate solvent.
 | ||
| Fig. 2 Change in integrated EPR signal with time for 20 mM TEMPO methacrylate in 1 M LiPF6–DMC (black, 1) and in 1 M LiPF6–PC (red, 2). Samples 1′ and 2′ were prepared with TMA which was dried for a longer time but are otherwise identical to 1 and 2 in composition respectively. | ||
Residual water found in electrolyte solvents constitutes another source of moisture in the present system. The 1H NMR spectrum of 1 M LiPF6 in DMC without TMA (neat electrolyte, see Fig. S6, ESI†) shows a weak signal at 4.47 ppm with an integral of 0.007% relative to the DMC resonance, indicating trace amounts of water.41,42 In addition, the presence of methanol in the neat electrolyte is indicated by the 1H NMR peak at δ = 3.34 ppm with an integral of 0.004% relative to the DMC resonance (see Fig. S6 in the ESI†).42,43 A quantification of water content for 1 M LiPF6 in PC without TMA was not attempted due to the overlap of resonances. Upon addition of TMA to the electrolyte to form 1, the water content in the overall system increases and as the radical degrades in 1, the amount of water decreases exponentially with time (see Fig. S7 and discussion in Section S4.2 in the ESI†). LiPF6 is known to react even with trace amounts of water, resulting in electrolyte decomposition.9 Additionally, 19F and 31P NMR spectroscopy after complete TMA radical degradation revealed the presence of typical fluorophosphate species (see Fig. S4 in the ESI†), formed due to decomposition of LiPF6 based electrolytes upon reaction with protic impurities (see Fig. 4 and, for the assignment of NMR resonances, Section S4.2 in the ESI†).15,18 The resonance corresponding to hydrofluoric acid (HF), a by-product of LiPF6 decomposition in contact with water or methanol, was also observed as a broad singlet at −187.5 ppm in the 19F NMR spectrum (see Fig. S4b, ESI†), which may result in the disproportionation of nitroxide radicals into diamagnetic species (see Fig. 5).44–46
To test for such a degradation pathway of TMA in 1 and to identify the disproportionation products shown in Fig. 5, time-resolved NMR was employed. Fig. 3 shows the evolution of the 1H NMR spectrum of 1 from time t = 0 to 48 hours in steps of 55 minutes. At t = 0, 1 exhibits signals broadened by paramagnetic relaxation due to the radical species. Chemical shifts of paramagnetic species depend on the hyperfine coupling between the nuclear spins and unpaired electron spin, which in turn depends on the unpaired electron spin density on the nucleus.47,48 Therefore, protons belonging to the methacrylate branch of TMA radical species (protons H-17, H-22, H-23, see Fig. 3) experience less of a paramagnetic effect than protons near the N–O moiety, and the corresponding peaks from the methyl group (H-17, δ = 1.93, 2.00 ppm) and the ethylene group (H-22,23, δ = 5.68, 5.76, 6.12, 6.22 ppm) were resolved even at t =0. By DFT, the respective paramagnetic contributions for the branch protons were estimated to be between 0.01 and 0.03 ppm.
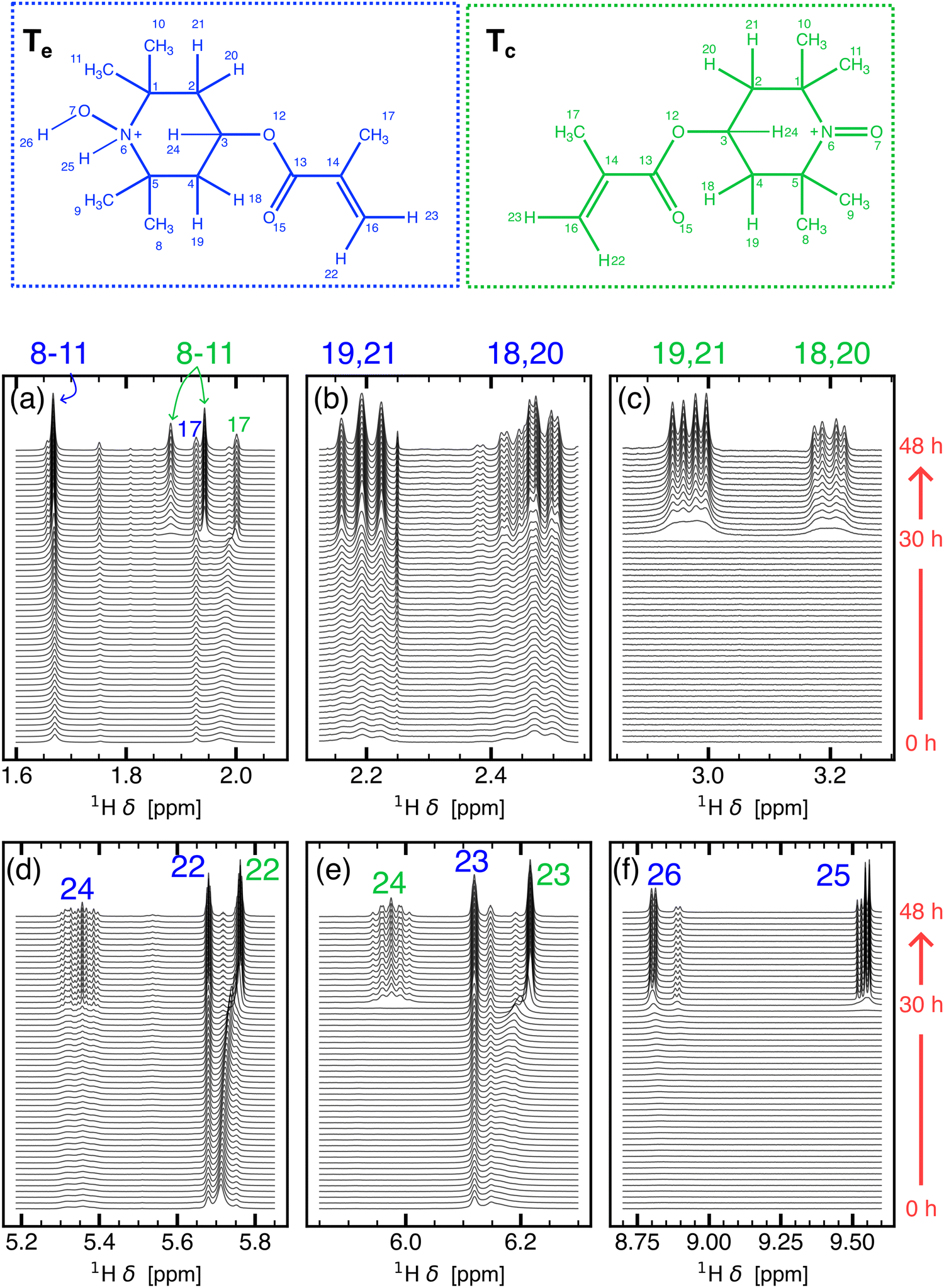 | ||
| Fig. 3 Time resolved 1H NMR spectrum of 1 from t = 0 to 48 hours in steps of 55 minutes. The full spectrum is divided into smaller spectral regions corresponding to (a) methyl protons of Tc and Te, (b) methylene protons of Te, (c) methylene protons of Tc, (d) and (e) methylidyne and ethylene protons of Te and Tc, (f) N–H and O–H protons of Te. Assignment criteria are discussed in Section S4.3 of the ESI.† The structures of Te (blue) and Tc (green) are shown at the top with numbered protons. | ||
At t = 48 hours, the 1H NMR spectrum indicates the presence of at least two diamagnetic species, evident from the observation of qualitatively similar pairs of signals (see Fig. 3b–e) and approximately double the number of methyl protons expected from a single TMA species. (see Fig. S14a, ESI†). The identity of these species can be discerned by following the changes occurring to the N–O moiety and the corresponding NMR signals in the region of 8.7 ppm to 9.6 ppm (see Fig. 3f). At t = 48 hours, two sets of doublets at 8.76 ppm and 9.55 ppm with a J-coupling of ≈ 5.6 Hz are observed. The coupling of the corresponding protons is further verified by 1H–1H correlation spectroscopy (see Fig. S9, ESI†). The chemical shift region, the spatial proximity of the protons, and the splitting pattern of these two peaks is consistent with the 3-bond coupling between H-25 and H-26 (see TeFig. 3) and is in agreement with the nitroxide moeity of Te shown in Fig. 5. Along with Te, the second set of qualitatively similar NMR peaks, which appear more upfield than the resonances from Te, correspond to Tc (see Fig. S8 and, for NMR resonance assignments and the structure elucidation of TMA disproportionation products, Section S4.3 in the ESI†). DFT calculated chemical shifts, δDFT, for Tc and Te agree with experimentally observed chemical shifts, δExp., and the relative chemical shift difference of the two species (see Table S5 in Section S4.4 in the ESI†). J-couplings of the two species are given in Table S3 (Section S4.4 in the ESI†).
TMA degradation can be linked to electrolyte degradation by inspecting the kinetic profiles of the species related to electrolyte degradation and TMA disproportionation (see Fig. S7 and S8f–i in the ESI†). Initially, water is consumed exponentially (Fig. S7, ESI†) for the hydrolysis of LiPF6 to form HF through reaction pathways A or A′ (Fig. 4). Simultaneously, methanol (δ = 3.34 ppm), likely resulting from DMC hydrolysis,41,43 shows a slight increase until t = 30 hours followed by a short period of rapid decrease (Fig. S7, ESI†). Electrolyte degradation pathway A/A′–B–C results in the formation of HF which protonates Ta′ to form Tb (R1 in Fig. 5). From t = 0 to 30 hours, 1H peaks corresponding to Tb, for instance the O–H group at 8.83 ppm, exhibit an increase due to this conversion (Fig. S8i, ESI†). At t < 30 hours, peaks originating from Tc are not apparent (see Fig. S8f–h in the ESI†). Furthermore, a degradation pathway involving Ta to Td conversion via a Tb intermediate is not indicated, as protons H-22, H-23 of the two species assigned at t = 48 hours maintain different chemical shifts throughout the experiment (see Fig. 3d and e), suggesting that two distinct species are simultaneously present. Energy profiles obtained using nudged elastic band calculations for conversion of Ta to Tb due to the presence of HF also suggests such a conversion to be energetically unfavourable (see Fig. S1 in the ESI†). Therefore, the paramagnetic species, Ta′ and Ta along with diamagnetic Tb undergo disproportionation to form diamagnetic Tc and Td. (R2 in Fig. 5). At t ≈ 30 hours, a rapid consumption of CH3OH is observed along with Tc and Te formation. This rapid increase in diamagnetic disproportionation products and resolution enhancement is likely a result of the cyclic electrolyte degradation pathways E and F initiated by CH3OH (Fig. 4), resulting in a pronounced acidic environment around TMA. The conversion of Td to Te (R3 in Fig. 5) by a second protonation at the N atom of Td is indicated by the appearance of two sets of doublets at 8.76 ppm (O–H) and 9.55 ppm (N–H). 1H–1H nuclear Overhauser effect spectroscopy (NOESY) of 1 indicates the proximity of HF to the N–H group which suggests that the protonation is caused by HF (see Fig. S10 in the ESI†).
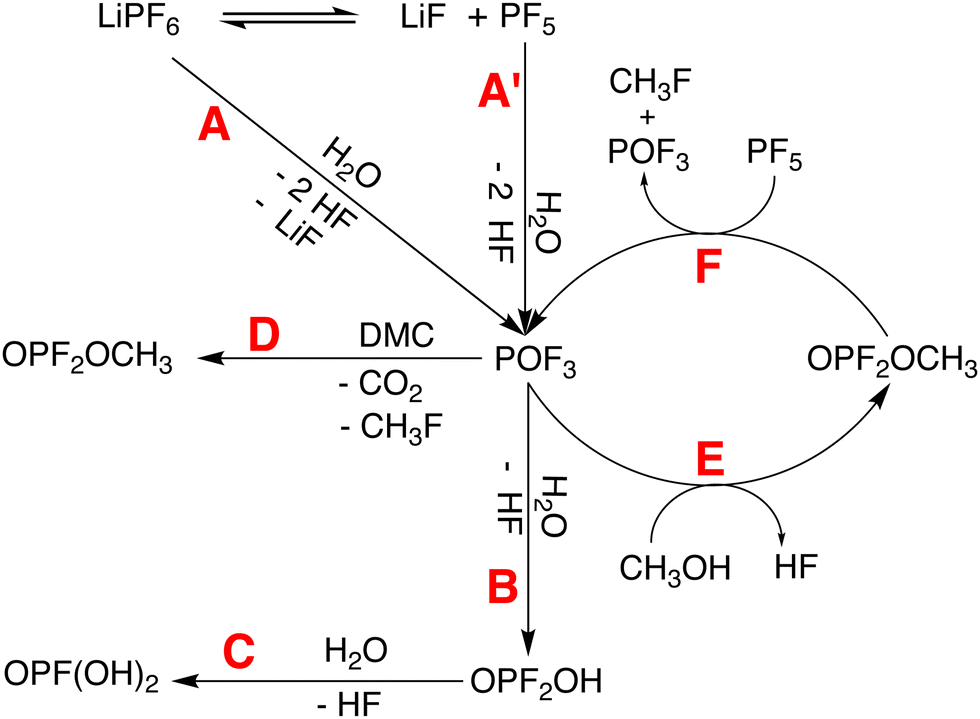 | ||
| Fig. 4 Decomposition of LiPF6 and formation of HF due to the presence of water and methanol in 1. | ||
 | ||
| Fig. 5 Acid disproportionation steps of nitroxide radical into diamagnetic products. Only the nitroxide moiety bonded to two carbon atoms of the six-membered ring of TMA is shown for clarity. | ||
Overall, the kinetic profiles leading from paramagnetic to diamagnetic species resemble a sigmoid curve (see Fig. S8f–i, ESI†), often observed for auto-catalytic reactions characterised by a lag, followed by a rapid and eventually a saturation phase.49 The initially slow increase in diamagnetic species is caused by the linear electrolyte degradation pathways (A/A′–B–C in Fig. 4), which is known to be more favoured than pathway D in the case where water concentration is initially high.18 The rapid phase is likely facilitated by the cyclic electrolyte degradation pathways E and F, which are considered auto-catalytic.50,5119F and 31P NMR did not indicate the presence of POF3 in the final electrolyte degradation products, possibly due to its conversion to OPF2OH and OPF2CH3via pathways B and D, respectively. During the rapid phase, a significant deposition of OPF2CH3 is not apparent, however, formation of OPF2CH3 through pathway D during the initial stages (t < 30 hours) of TMA degradation and subsequent consumption of this species during the rapid phase was found (see Fig. S7, ESI†). The kinetic profile obtained from time-resolved EPR differs from the NMR profile, and an early onset of the rapid phase and an overall faster degradation is indicated by CW EPR (Fig. 2). While HF is known to react with borosilicate glass of NMR tubes18,41,43 and corresponding reaction products are also indicated in the present system (see Fig. S4c, S5 and Section S4.2 in the ESI†), EPR tubes made of quartz glass are more chemically resistant to HF.52 Therefore, the difference likely arises from the different consumption rates of the main acidic species, HF.
As the degradation mechanism involves the disproportionation of TMA in an acidic environment caused by the electrolyte degradation, the rate of TMA radical degradation is closely linked to the formation of HF which, in turn, depends on the the stability of LiPF6. Decomposition rates of LiPF6 differ depending on the solvent system.9 In linear carbonates such as DMC, a lower degree of ionisation of LiPF6 is found while in case of cyclic carbonates such as PC with a higher dielectric constant, electrolyte ions are solvent separated.9 A higher degree of LiPF6 ionisation in PC decreases the probability of PF5 formation and the subsequent formation of HF is delayed. In contrast, in case of 1, the presence of neutral LiPF6 increases the probability of electrolyte degradation and the formation of HF through reaction pathways shown in Fig. 4. As the degradation pathway of TMA is LiPF6 mediated, use of a less moisture sensitive conducting salt such as lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in DMC was found to inhibit the degradation of TMA (see Section S4.5 in the ESI†).
In conclusion, interactions between TMA, a common organic redox unit of ORBs, and electrolyte ions were investigated in two different solvent environments of DMC and PC. Pulsed EPR experiments revealed that the active material, TMA, could itself participate in Li solvation, and that the chemical structure of the solvent influences the fraction of Li-bound TMA radicals, i.e. the structure of Li-solvation shells. This implies that through appropriate chemical functionalisation of the organic redox unit, Li solvation shells can be influenced, hence competitive binding of the active material and solvent molecules towards Li ions can be adjusted. Time-resolved CW EPR further demonstrated that the stability of TMA differs in PC and DMC. NMR spectroscopic techniques confirmed that TMA degradation in DMC is closely linked to the decomposition of the conducting salt, LiPF6. The degradation pathway involves the disproportionation of TMA in the presence of HF, a by-product of LiPF6 decomposition. Using DFT-based chemical shift calculations, the degradation products observed in the NMR spectra were identified as disproportionation products of TMA, thereby validating the degradation pathway. The results indicate that the stability of active materials in ORB cathodes can be influenced by the electrolyte composition, without use of customised additives. This work highlights the utility of combined NMR and EPR investigations complemented with theoretical models for the characterisation of organic battery materials.
Author contributions
Davis Thomas Daniel: conceptualisation, methodology, investigation, formal analysis, writing – original draft, Visualisation Emmanouil Veroutis: investigation, writing – review & editing P. Philipp M. Schleker: writing – review & editing Rüdiger-A. Eichel: writing – review & editing, funding acquisition, supervision Josef Granwehr: software, writing – review & editing, funding acquisition, supervision. all authors have read and agreed to the published version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Souvik Mitra (Institute of Physical Chemistry, University of Münster) and Dr. Diddo Diddens (IMD-4, Helmholtz Institute Münster) for helpful discussions regarding the solvation of electrolyte ions. Computational resources from the RWTH Aachen University under project rwth1253 and IET-1 (Forschungszentrum Jülich GmbH) are acknowledged. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within the priority program “Polymer-based batteries” (SPP 2248)-project no. 441255373.References
- M. Li, R. P. Hicks, Z. Chen, C. Luo, J. Guo, C. Wang and Y. Xu, Chem. Rev., 2023, 123, 1712–1773 CrossRef CAS PubMed.
- P. Lennartz, B. A. Paren, A. Herzog-Arbeitman, X. C. Chen, J. A. Johnson, M. Winter, Y. Shao-Horn and G. Brunklaus, Joule, 2023, 7, 1471–1495 CrossRef CAS.
- P. P. M. Schleker, C. Grosu, M. Paulus, P. Jakes, R. Schlögl, R.-A. Eichel, C. Scheurer and J. Granwehr, Commun. Chem., 2023, 6, 113 CrossRef CAS PubMed.
- L. Zhao, A. E. Lakraychi, Z. Chen, Y. Liang and Y. Yao, ACS Energy Lett., 2021, 6, 3287–3306 CrossRef CAS.
- J. Kim, Y. Kim, J. Yoo, G. Kwon, Y. Ko and K. Kang, Nat. Rev. Mater., 2023, 8, 54–70 CrossRef.
- C. Friebe and U. S. Schubert, Electrochem. Energy Storage, 2019, 65–99 Search PubMed.
- S. Muench, A. Wild, C. Friebe, B. Haupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- T. Kawamura, S. Okada and J.-I. Yamaki, J. Power Sources, 2006, 156, 547–554 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- S. Han, Sci. Rep., 2019, 9, 5555 CrossRef PubMed.
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS.
- N. P. Lebedeva and L. Boon-Brett, J. Electrochem. Soc., 2016, 163, A821 CrossRef CAS.
- M. Stich, M. Göttlinger, M. Kurniawan, U. Schmidt and A. Bund, J. Phys. Chem. C, 2018, 122, 8836–8842 CrossRef CAS.
- U. Heider, R. Oesten and M. Jungnitz, J. Power Sources, 1999, 81, 119–122 CrossRef.
- H. Nguyen and R. J. Clement, ACS Energy Lett., 2020, 5, 3848–3859 CrossRef CAS.
- J. P. Allen and C. P. Grey, J. Phys. Chem. C, 2023, 127, 4425–4438 CrossRef CAS PubMed.
- S. Wiemers-Meyer, M. Winter and S. Nowak, Phys. Chem. Chem. Phys., 2016, 18, 26595–26601 RSC.
- D. T. Daniel, S. Oevermann, S. Mitra, K. Rudolf, A. Heuer, R.-A. Eichel, M. Winter, D. Diddens, G. Brunklaus and J. Granwehr, Sci. Rep., 2023, 13, 10934 CrossRef CAS PubMed.
- I. Kulikov, N. A. Panjwani, A. A. Vereshchagin, D. Spallek, D. A. Lukianov, E. V. Alekseeva, O. V. Levin and J. Behrends, Energy Environ. Sci., 2022, 15, 3275–3290 RSC.
- I. Kulikov, A. A. Vereshchagin, D. A. Lukianov, O. V. Levin and J. Behrends, J. Magn. Reson. Open, 2023, 16, 100134 CrossRef.
- D. T. Daniel, C. Szczuka, P. Jakes, R.-A. Eichel and J. Granwehr, Phys. Chem. Chem. Phys., 2023, 25, 12767–12776 RSC.
- P. Höfer, A. Grupp, H. Nebenführ and M. Mehring, Chem. Phys. Lett., 1986, 132, 279–282 CrossRef.
- J. P. Allen, C. Szczuka, H. E. Smith, E. Jónsson, R.-A. Eichel, J. Granwehr and C. P. Grey, Phys. Chem. Chem. Phys., 2024, 26, 19505–19520 RSC.
- C. Szczuka, P. Jakes, R.-A. Eichel and J. Granwehr, Adv. Energy Sustainability Res., 2021, 2, 2100121 CrossRef CAS.
- H. A. López-Peña, L. S. Hernández-Muñoz, B. A. Frontana-Uribe, F. J. González, I. González, C. Frontana and J. Cardoso, J. Phys. Chem. B, 2012, 116, 5542–5550 CrossRef PubMed.
- O. Pecher, J. Carretero-González, K. J. Griffith and C. P. Grey, Chem. Mater., 2017, 29, 213–242 CrossRef CAS.
- S. A. Kayser, A. Mester, A. Mertens, P. Jakes, R.-A. Eichel and J. Granwehr, Phys. Chem. Chem. Phys., 2018, 20, 13765–13776 RSC.
- K. Märker, C. Xu and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 17447–17456 CrossRef PubMed.
- A. Ponrouch, E. Marchante, M. Courty, J.-M. Tarascon and M. R. Palacin, Energy Environ. Sci., 2012, 5, 8572–8583 RSC.
- P. Knowles, D. Marsh and H. Rattle, An Introduction to the Theory and Practice of NMR and ESR in Biological Systems, 1976, pp. 1–207 Search PubMed.
- D. Marsh and C. Toniolo, J. Magn. Reson., 2008, 190, 211–221 CrossRef CAS PubMed.
- D. Marsh, J. Magn. Reson., 2002, 157, 114–118 CrossRef CAS PubMed.
- R. Owenius, M. Engström, M. Lindgren and M. Huber, J. Phys. Chem. A, 2001, 105, 10967–10977 CrossRef CAS.
- A. Sannigrahi, T. Kar, B. G. Niyogi, P. Hobza and P. V. R. Schleyer, Chem. Rev., 1990, 90, 1061–1076 CrossRef CAS.
- S. S. Eaton and G. R. Eaton, Relaxation Mechanisms, John Wiley & Sons, Ltd, 2016, pp. 1543–1556 Search PubMed.
- G. Jeschke, Appl. Magn. Reson., 2022, 53, 635–651 CrossRef CAS PubMed.
- A. Zecevic, G. R. Eaton, S. S. Eaton and M. Lindgren, Mol. Phys., 1998, 95, 1255–1263 CrossRef CAS.
- M. Lindgren, G. R. Eaton, S. S. Eaton, B.-H. Jonsson, P. Hammarström, M. Svensson and U. Carlsson, J. Chem. Soc., Perkin Trans. 2, 1997, 2549–2554 RSC.
- M. G. Giorgini, K. Futamatagawa, H. Torii, M. Musso and S. Cerini, J. Phys. Chem. Lett., 2015, 6, 3296–3302 CrossRef CAS.
- B. L. Rinkel, J. P. Vivek, N. Garcia-Araez and C. P. Grey, Energy Environ. Sci., 2022, 15, 3416–3438 RSC.
- G. R. Fulmer, A. J. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- B. L. D. Rinkel, D. S. Hall, I. Temprano and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 15058–15074 CrossRef CAS PubMed.
- V. D. Sen and V. A. Golubev, J. Phys. Org. Chem., 2009, 22, 138–143 CrossRef CAS.
- H. Park, B. Uluca-Yazgi, S. Heumann, R. Schlögl, J. Granwehr, H. Heise and P. P. M. Schleker, J. Magn. Reson., 2020, 312, 106688 CrossRef CAS PubMed.
- S.-y Kishioka, T. Ohsaka and K. Tokuda, Electrochim. Acta, 2003, 48, 1589–1594 CrossRef CAS.
- A. J. Pell, G. Pintacuda and C. P. Grey, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 111, 1–271 CrossRef CAS PubMed.
- W. Van den Heuvel and A. Soncini, J Chem. Phys., 2013, 138, 5 CrossRef PubMed.
- A. I. Hanopolskyi, V. A. Smaliak, A. I. Novichkov and S. N. Semenov, ChemSystemsChem, 2021, 3, e2000026 CrossRef CAS.
- C. L. Campion, W. Li, W. B. Euler, B. L. Lucht, B. Ravdel, J. F. DiCarlo, R. Gitzendanner and K. Abraham, Electrochem. Solid-State Lett., 2004, 7, A194 CrossRef CAS.
- C. L. Campion, W. Li and B. L. Lucht, J. Electrochem. Soc., 2005, 152, A2327 CrossRef CAS.
- V. Leko and L. Komarova, Glass Ceram., 1973, 30, 754–755 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00612g |
| This journal is © The Royal Society of Chemistry 2025 |