
Density-functional calculations of the conversion of methane to methanol on platinum-decorated sheets of graphene oxide
Abstract
By means of calculations based on density-functional theory (DFT), we have investigated the conversion of methane on two platinum atoms supported with a graphene-oxide sheet (Pt2/GO). In our calculations, a CH4 molecule can be adsorbed around the Pt atoms of the Pt2/GO sheet with adsorption energies within −0.11 to −0.53 eV; an elongated C–H bond indicates that Pt atoms on that sheet can activate the C–H bond of a CH4 molecule. The role of the GO sheet in the activation of CH4 was identified according to an analysis of the electronic density: the GO sheet induces the d-band of Pt atoms to generate several specific dz2 state features above the Fermi level, which enabled the activation of the C–H bond of CH4 in generating an evident area of overlap with the hydrogen s orbital of the C–H bond. Upon a dioxygen molecule being added onto the Pt2/GO sheet, this molecule can react with activated CH4 according to mechanisms of form 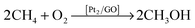 , and restore the original Pt2/GO sheet.
, and restore the original Pt2/GO sheet.

 Please wait while we load your content...
Please wait while we load your content...

