
Theoretical studies on the structure and thermochemistry of cyclicparaphenylenediazenes†
 *a
and
Mohammad A.
Alam
*b
*a
and
Mohammad A.
Alam
*b
Abstract
Cyclicparaphenylenediazenes (CPPDs) have drawn great attention due to their potential applications in molecular electronics and solar energy. However, to date, there have been no detailed structure and thermochemistry investigations on CPPDs. Herein, we used an integrated computational approach aimed at providing reliable structural and thermochemical information on novel CPPDs based photoswitchable molecular rings. The approach involves hybrid density functional theory calculation at the B3LYP/6-31+G(d,p) level coupled with homodesmotic reaction approach to calculate strain energies (SE) and standard enthalpies of formation 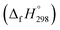 of all cis and all trans isomers of [n]CPPDs (n = 2 to 8). The results show that strain energies and
of all cis and all trans isomers of [n]CPPDs (n = 2 to 8). The results show that strain energies and 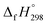 per-unit monomer of all cis-[n]CPPDs (n = 4 to 8) increase with increasing the numbers of Ph–N
per-unit monomer of all cis-[n]CPPDs (n = 4 to 8) increase with increasing the numbers of Ph–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N linkages. However, an opposite trend was observed for all trans-[n]CPPDs (n = 4 to 8). The results were also compared with carbon nanoring structures i.e., cyclicparaphenyleneacetylenes (CPPAs) and cycloparaphenylenes (CPPs). The calculated highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) energy gaps are in the range of 1.98 eV to 2.36 eV, indicating potential material for the construction of solar cells. The reported structures, SE,
N linkages. However, an opposite trend was observed for all trans-[n]CPPDs (n = 4 to 8). The results were also compared with carbon nanoring structures i.e., cyclicparaphenyleneacetylenes (CPPAs) and cycloparaphenylenes (CPPs). The calculated highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) energy gaps are in the range of 1.98 eV to 2.36 eV, indicating potential material for the construction of solar cells. The reported structures, SE, 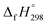 , and electronic properties of CPPDs can be also helpful to synthesize these novel materials.
, and electronic properties of CPPDs can be also helpful to synthesize these novel materials.

 Please wait while we load your content...
Please wait while we load your content...

