
A complete ab initio thermodynamic and kinetic catalogue of the defect chemistry of hematite α-Fe2O3, its cation diffusion, and sample donor dopants†
 ab
Mostafa
Youssef
*b
ab
Mostafa
Youssef
*b
Abstract
This paper studies comprehensively the defect chemistry of and cation diffusion in α-Fe2O3. Defect formation energies and migration barriers are calculated using density functional theory with a theoretically calibrated Hubbard U correction. The established model shows a good agreement with experimental off-stoichiometry and cation diffusivities available in the literature. At any temperature, 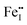 and
and 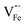 are the predominant ionic defects in hematite at the two extremes of oxygen partial pressure (pO2) range, reducing and oxidizing, respectively. Between these two extremes, an intrinsic electronic regime exists where small polaronic electrons and holes are the dominant charge carriers. The calculated migration barriers show that Fe ions favor the diffusion along the 〈111〉 direction in the primitive cell through an interstitial crowdion-like mechanism. Our model suggests that cation diffusion in hematite is mainly controlled by the migration of
are the predominant ionic defects in hematite at the two extremes of oxygen partial pressure (pO2) range, reducing and oxidizing, respectively. Between these two extremes, an intrinsic electronic regime exists where small polaronic electrons and holes are the dominant charge carriers. The calculated migration barriers show that Fe ions favor the diffusion along the 〈111〉 direction in the primitive cell through an interstitial crowdion-like mechanism. Our model suggests that cation diffusion in hematite is mainly controlled by the migration of 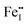 , while
, while 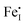 may contribute to cation diffusion at extremely low pO2. Our analysis in the presence of two sample donor dopants Ti and Sn indicates that high temperature annealing at T > 1100 K is needed to prepare n-type hematite at ambient pO2, consistently with prior experimental findings. Alternatively, annealing at lower temperatures requires much lower pO2 to avoid compensating the donors with Fe vacancies. A synergistic comparison of our theoretical model and the experimental results on Ti-doped hematite led us to propose that free electrons and small polarons coexist and both contribute to n-type conductivity. Our validated model of defective hematite is a foundation to study hematite in applications such as corrosion and water splitting.
may contribute to cation diffusion at extremely low pO2. Our analysis in the presence of two sample donor dopants Ti and Sn indicates that high temperature annealing at T > 1100 K is needed to prepare n-type hematite at ambient pO2, consistently with prior experimental findings. Alternatively, annealing at lower temperatures requires much lower pO2 to avoid compensating the donors with Fe vacancies. A synergistic comparison of our theoretical model and the experimental results on Ti-doped hematite led us to propose that free electrons and small polarons coexist and both contribute to n-type conductivity. Our validated model of defective hematite is a foundation to study hematite in applications such as corrosion and water splitting.

- This article is part of the themed collection: 2021 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

