
Interactive network of the dehydrogenation of alkanes, alkenes and alkynes – surface carbon hydrogenative coupling on Ru(111)†

Abstract
To understand the reaction mechanisms of the dehydrogenation and retrosynthesis of alkanes, the consecutive dissociation of methane, ethane, ethene and ethyne, as well as propane, propene and propyne, on the fcc Ru(111) surface has been investigated using periodic density functional theory computations (rPBE). Methane dissociation has the energy minimum path of 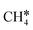 →
→ 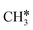 →
→ 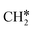 → CH* → C*. Although ethane dissociation does not have ethene and ethyne as intermediates, they have the same final surface species with the minimum energy paths for ethane [
→ CH* → C*. Although ethane dissociation does not have ethene and ethyne as intermediates, they have the same final surface species with the minimum energy paths for ethane [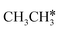 →
→ 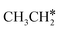 → CH3CH* → CH3C* →
→ CH3CH* → CH3C* → 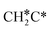 → HC*C* → HC* + C*], ethene [
→ HC*C* → HC* + C*], ethene [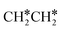 →
→ 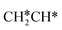 →
→ 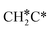 → HC*C* → HC* + C*] and ethyne [CH*CH* → HC*C* → HC* + C*]. Propane dissociation has the competitive routes of n-propyl [
→ HC*C* → HC* + C*] and ethyne [CH*CH* → HC*C* → HC* + C*]. Propane dissociation has the competitive routes of n-propyl [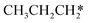 → CH3CH2CH* → CH3CH2C* → CH3CH*C* → CH3C*C* → CH3C* + C* →→ HC* + C*] and isopropyl with propyne as an intermediate [CH3CH*CH3 → CH3C*CH3 →
→ CH3CH2CH* → CH3CH2C* → CH3CH*C* → CH3C*C* → CH3C* + C* →→ HC* + C*] and isopropyl with propyne as an intermediate [CH3CH*CH3 → CH3C*CH3 → 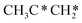 → CH3C*CH* → CH3C*C* →→ HC* + C*], and the n-propyl route has propene as an intermediate for dissociation [
→ CH3C*CH* → CH3C*C* →→ HC* + C*], and the n-propyl route has propene as an intermediate for dissociation [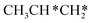 → CH3C*CH2*/CH3CH*CH* → CH3CH*C*/CH3C*CH* → CH3C*C* →→ HC* + C*]. In these reactions, the most stable surface intermediates are HC*, CH3C* and CH3CH2C* as homologs, as found experimentally on other metal surfaces. Our results rationalized the experimentally observed interconversion between
→ CH3C*CH2*/CH3CH*CH* → CH3CH*C*/CH3C*CH* → CH3C*C* →→ HC* + C*]. In these reactions, the most stable surface intermediates are HC*, CH3C* and CH3CH2C* as homologs, as found experimentally on other metal surfaces. Our results rationalized the experimentally observed interconversion between 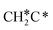 + H* and CH3C* as well as surface HC*C* and CH3C*C* as key intermediates for the first C–C bond dissociation [HC*C* → HC* + C*; CH3C*C* → CH3C* + C*]. On the basis of surface C* and H2 gas, the retrosynthesis of methane, ethane and propane has increasing apparent barriers of 1.08, 1.51 and 1.66 eV, respectively, at 490 K and 1 atm H2 and 0.83, 1.14 and 1.15 eV, respectively, at 19.7 atm H2. Surface carbon coverage changes the formation of alkanes from endergonic to exergonic. This pressure- and coverage-dependency is very important for understanding the reaction mechanism and selectivity. Surface alkynyl groups should be the intermediates for C–C coupling. The computed vibrational frequencies of CH*, CH3C*, CH2C*, HC*C* and
+ H* and CH3C* as well as surface HC*C* and CH3C*C* as key intermediates for the first C–C bond dissociation [HC*C* → HC* + C*; CH3C*C* → CH3C* + C*]. On the basis of surface C* and H2 gas, the retrosynthesis of methane, ethane and propane has increasing apparent barriers of 1.08, 1.51 and 1.66 eV, respectively, at 490 K and 1 atm H2 and 0.83, 1.14 and 1.15 eV, respectively, at 19.7 atm H2. Surface carbon coverage changes the formation of alkanes from endergonic to exergonic. This pressure- and coverage-dependency is very important for understanding the reaction mechanism and selectivity. Surface alkynyl groups should be the intermediates for C–C coupling. The computed vibrational frequencies of CH*, CH3C*, CH2C*, HC*C* and 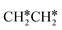 agree with the experiments. The comparison in adsorption energies and reaction barriers and energies shows that the fcc Ru(111) surface is more active than the hcp Ru(0001) surface despite their very similar surface structures.
agree with the experiments. The comparison in adsorption energies and reaction barriers and energies shows that the fcc Ru(111) surface is more active than the hcp Ru(0001) surface despite their very similar surface structures.

 Please wait while we load your content...
Please wait while we load your content...

