
Predicting nearest neighbor free energies of modified RNA with LIE: results for pseudouridine and N1-methylpseudouridine within RNA duplexes†

Abstract
Pseudouridine (Ψ) and N1-methylpseudouridine (m1Ψ) are among the key modifications in the field of mRNA therapeutics and vaccine research. The accuracy of the design and development of therapeutic RNAs containing such modifications depends on the accuracy of the secondary structure prediction, which in turn depends on the nearest neighbor (NN) thermodynamic parameters for the standard and modified residues. Here, we propose a simple approach based on molecular dynamics simulations and linear interaction energy (LIE) approximation that is able to predict the NN free energy parameters 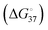 for U–A, Ψ–A and m1Ψ–A pairs in reasonable agreement with the recent experimental reports. We report the NN thermodynamic parameters for different U, Ψ and m1Ψ base pairs, which might be helpful for a deeper understanding of the effect of these modifications in RNA. The predicted NN free energy parameters in this study are able to closely reproduce the folding free energies of duplexes containing internal Ψ for which the thermodynamic data were available. Additionally, we report the predicted folding free energies for the duplexes containing internal m1Ψ.
for U–A, Ψ–A and m1Ψ–A pairs in reasonable agreement with the recent experimental reports. We report the NN thermodynamic parameters for different U, Ψ and m1Ψ base pairs, which might be helpful for a deeper understanding of the effect of these modifications in RNA. The predicted NN free energy parameters in this study are able to closely reproduce the folding free energies of duplexes containing internal Ψ for which the thermodynamic data were available. Additionally, we report the predicted folding free energies for the duplexes containing internal m1Ψ.

 Please wait while we load your content...
Please wait while we load your content...

